
Elucidation of the catalytic mechanism of a single-metal dependent homing endonuclease using QM and QM/MM approaches: the case study of I-PpoI†
Rajwinder
Kaur
 ,
Angela
Frederickson
and
Stacey D.
Wetmore
*
,
Angela
Frederickson
and
Stacey D.
Wetmore
*
Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta T1K 3M4, Canada. E-mail: stacey.wetmore@uleth.ca
First published on 1st March 2024
Abstract
Homing endonucleases (HEs) are highly specific DNA cleaving enzymes, with I-PpoI having been suggested to use a single metal to accelerate phosphodiester bond cleavage. Although an I-PpoI mechanism has been proposed based on experimental structural data, no consensus has been reached regarding the roles of the metal or key active site amino acids. This study uses QM cluster and QM/MM calculations to provide atomic-level details of the I-PpoI catalytic mechanism. Minimal QM cluster and large-scale QM/MM models demonstrate that the experimentally-proposed pathway involving direct Mg2+ coordination to the substrate coupled with leaving group protonation through a metal-activated water is not feasible due to an inconducive I-PpoI active site alignment. Despite QM cluster models of varying size uncovering a pathway involving leaving group protonation by a metal-activated water, indirect (water-mediated) metal coordination to the substrate is required to afford this pathway, which renders this mechanism energetically infeasible. Instead, QM cluster models reveal that the preferred pathway involves direct Mg2+–O3′ coordination to stabilize the charged substrate and assist leaving group departure, while H98 activates the water nucleophile. These calculations also underscore that both catalytic residues that directly interact with the substrate and secondary amino acids that position or stabilize these residues are required for efficient catalysis. QM/MM calculations on the solvated enzyme–DNA complex verify the preferred mechanism, which is fully consistent with experimental kinetic, structural, and mutational data. The fundamental understanding of the I-PpoI mechanism of action, gained from the present work can be used to further explore potential uses of this enzyme in biotechnology and medicine, and direct future computational investigations of other members of the understudied HE family.
Introduction
Homing endonucleases (HEs), which are also known as meganucleases, are highly specific DNA cleaving enzymes that are prevalent among bacteria, archaea, and eukarya.1,2 HEs are known for their ability to self-propagate genetic elements, specifically moving or transferring the genes that encode them into the host genome.3 In particular, a homing endonuclease gene (HEG) is transcribed and spliced, and then translated into a HE, which generates double-strand DNA breaks in the uninterrupted host genome that does not contain the intervening sequence.1 Subsequently, the HEG is incorporated into the host genome through homologous recombination repair in combination with DNA replication, completing the homing process.1,3 The human genome is known to be populated with a large number of such mobile genetic elements (∼40%).4 Although these insertion elements may help shape the human genome, their insertion has also been linked to human diseases such as hemophilia,5 breast cancer,6 and muscular dystrophy.7Due to their ability to create double-strand breaks in complex genomes, HEs have been used for genome engineering,8 including applications such as gene targeting,9 cloning,10 and therapy.11,12 HE-directed gene therapy has been applied in various applications such as correcting the human XPC gene to treat xeroderma pigmentosum,13–15 and the mutated Duchenne muscular dystrophy (DMD) gene associated with DMD.16 Engineered HEs have also been used to target human RAG1 genes, presenting a viable option for treating severe combined immunodeficiency.17,18 Gene disruption by HEs has been exploited to downregulate the expression of chemokine receptor 5, which carries the HIV infection into cells.19 HE-based strategies for genetic control of vector-borne diseases have also proven useful for creating transgenic mosquitoes with a high level of sterility, thus confirming the suitability of this approach for controlling diseases like malaria.20–22 The vast applications of HEs warrant a deeper understanding of their catalytic function.
Like most endonucleases, HEs use metals to facilitate catalysis.3,23 Although the majority of nucleases have been proposed to use two metals to cleave phosphodiester bonds24,25 and therefore the two-metal mediated reaction has been widely studied,26–34 evidence has grown that some endonucleases use only one metal for the chemical step.35–45 Among members of the HE family, I-PpoI (intron-encoded endonuclease from Physarum polycephalum) is an example of an enzyme that may use only a single metal for catalysis.23,39 Indeed, kinetic studies have demonstrated that Mg2+ is essential for I-PpoI catalytic activity46 and X-ray crystallographic data reveal a single Mg2+ bound in the I-PpoI active site (Fig. 1a–b).38,39 Nevertheless, despite being one of the most well-characterized members of HE family through experimental biochemical and structural techniques,20,21,39,46–60 no consensus has been reached regarding the I-PpoI catalytic mechanism.
 | ||
| Fig. 1 I-PpoI active site from an X-ray crystal structure of the (a) Mg2+-containing RC for the H98A mutant and (b) Mg2+-containing PC for the wild-type enzyme. (c) The experimentally proposed phosphodiester bond cleavage pathway for I-PpoI.39 | ||
I-PpoI recognizes and binds to the DNA major groove and cleaves across the minor groove to generate 3′-overhangs that are 4-nucleotides long.3,61 In the experimentally-proposed I-PpoI mechanism of action, the single metal directly coordinates to the O3′-leaving group and non-bridging oxygen of the scissile phosphate, and thereby promotes leaving group departure through simultaneous substrate stabilization and leaving group protonation by a metal-activated water (Fig. 1c).39,57 Because of the position of H98 with respect to the substrate in the product complex (PC) for wild type I-PpoI (Fig. 1b)38 and the complete loss of enzyme activity upon H98A mutation,57 H98 was proposed to be the general base that activates the water nucleophile to initiate the reaction (Fig. 1c).39 The N119A mutation also kills catalytic activity as N119 is the only amino acid coordinated to Mg2+ (Fig. 1a and b). Furthermore, N119 is within hydrogen-bonding distance of the O5′-bridging oxygen of the scissile phosphate in the reactant complex (RC) analogue of the H98A I-PpoI mutant (Fig. 1a and Fig. S1a, ESI†), and therefore has been proposed to stabilize the scissile phosphate through hydrogen bonding.57 Although the R61A mutation leads to only a slight reduction in I-PpoI activity, R61 has been suggested to stabilize the substrate39,57 and may inhibit the reverse reaction by hydrogen bonding with the cleaved phosphate backbone (Fig. 1b and Fig. S1b, ESI†).38,39 Indeed, I-PpoI binds tightly to cleaved DNA, with product release being the proposed rate-determining step.57 Finally, although the H78A mutation does not significantly impact I-PpoI catalytic activity,57 H78 is within hydrogen-bonding distance of the water nucleophile in the X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant39 and close to H98 in the PC for wild type I-PpoI,39 which suggests that H78 may facilitate the overall P–O bond cleavage process (Fig. 1 and Fig. S1, ESI†). Collectively, this data serves as an excellent starting point for modeling, which can provide further insights into the roles of various active site residues, the metal, and metal-ligated water, in the phosphodiester bond cleavage mediated by a representative HE.
Since computational techniques are powerful tools to investigate enzyme-catalyzed reaction mechanisms, characterize high energy intermediates, shed light on the roles of amino acids in the reaction, and compare multiple mechanisms to predict preferred pathways,25,62,63 this study uses a combined quantum mechanics (QM) and quantum mechanics-molecular mechanics (QM/MM) approach within the ONIOM formulism to provide atomic-level details of the catalytic pathway used by I-PpoI. Specifically, starting from a high-resolution X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant,39 several cluster models of varying size are used to map the phosphodiester bond cleavage mechanism using density functional theory (DFT). Subsequently, QM/MM (ONIOM) is used to characterize the phosphodiester bond hydrolysis pathway within the context of the solvated enzyme–DNA complex, which permits comparison of the obtained Gibbs energy barriers to the experimental values (∼86–95 kJ mol−1)56,57 and thereby identification of the preferred catalytic pathway. The fundamental understanding about I-PpoI function obtained from the present work can be used to further explore its potential use in the areas of genome engineering for biotechnological and therapeutic solutions.8,11 Furthermore, our improved mechanistic understanding can direct future computational investigations on other HEs, such as I-HmuI41 and Hpy188I,44 which share the active site metal-binding arrangement of I-PpoI, but differ in the composition and arrangement of active site amino acids. In addition, this work contributes to the growing body of literature supporting that one metal is enough to facilitate phosphodiester bond cleavage in nucleic acids,35–37 with comparison to the literature on other single metal-dependent nucleases40–42,64–67 highlighting that this chemistry can be achieved in diverse ways.
Computational methodology
QM calculations
QM cluster models are a computationally efficient approach that have provided valuable insights into the chemistry of P–O bond cleavage reactions, including the roles of amino acids and preferred metal-binding architectures, and thereby can direct large-scale QM/MM modeling.65,67–71 Each QM cluster model was built from a 1.93 Å resolution X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant (PDB ID: 1CYQ; Fig. 1a), with the alanine mutation at position 98 manually reverted to histidine using PyMOL 2.5.72 The smallest QM cluster model (Model 1, 92 atoms, Fig. 2a) includes residues that have been proposed to be essential for catalysis.46,57 Specifically, the nucleotides containing the scissile P–O bond (dA189 and dG190), H98 (the proposed general base), and a nucleophilic water were included. Additionally, the model incorporated Mg2+ that is directly coordinated to N119 and three water molecules, S97, and a water molecule that may facilitate maintenance of the octahedral metal coordination geometry by simultaneously hydrogen bonding to a Mg2+-coordinated water molecule and a non-bridging phosphate oxygen. An additional water molecule was either hydrogen bonding to another Mg2+-coordinated water molecule (Fig. 2a) or directly coordinated to Mg2+ (Fig. S2a, ESI†). In the smallest model, the substrate was truncated at O3′ of dG190 and O5′ of dA189, with the truncation points frozen to the crystallographic coordinates and capped with hydrogen atoms. The associated nucleobases (A on the 5′-side and G on the 3′-side of the scissile bond) were also replaced with hydrogen atoms. Each amino acid was truncated at the α carbon, meaning the Cα–C/N(backbone) bonds were cut, the unsaturated C(backbone) replaced with a hydrogen atom, and the location of Cα fixed to the crystallographic coordinates, as previously recommended based on a QM cluster study of a one-metal dependent nuclease.67 Subsequent cluster models were built from the smallest model. All larger models include an extended substrate that contains the nucleobases in the nucleosides on either side of the the scissile bond (dA189 and dG190). The 134-atom model also contains H78 (Model 2), the 141-atom model includes R61 (Model 3), and the 163-atom model contains H78, R61, and the 5′-phosphate of dA191 on the 3′-side of dG190 (Model 4, in which the 5′-phosphate is capped and frozen at C5′; Fig. 2a and Fig. S2a, ESI†). | ||
| Fig. 2 (a) Schematic of QM cluster models considered in this work: Model 1 (black), Model 2 (Model 1 + H78 (blue) + expanded substrate (red)), Model 3 (Model 1 + R61 (green) + expanded substrate), Model 4 (Model 1 + H78 + R61 + 5′ phosphate moiety of dA191 on the 3′-side of dG190 (orange)). (b) The enzyme–DNA QM/MM (ONIOM) model (left) and the corresponding QM region (black box, right). | ||
Since the inclusion of implicit solvent during optimizations has been shown to minimally impact the energetics of enzyme-catalyzed reactions for cluster models larger than 100 atoms,67,73–75 each QM cluster RC was optimized in the gas phase using B3LYP-D3(BJ)/6-31G(d,p) calculations. Subsequently, a guess for a concerted TS was generated by constraining the key reaction parameters involved in the attack of nucleophilic water on the phosphorus reaction center (r(P⋯Ow) = 1.9–2.2 Å) and cleavage of phosphodiester bond (r(P–O3′) = 1.9–2.2 Å). The distance constraints for the key reaction parameters were chosen based on values reported for a similar phosphodiester bond hydrolysis reaction catalyzed by one-metal dependent APE1.67 For a stepwise mechanism, the P⋯Ow distance was constrained (r(P⋯Ow) = 1.9–2.2 Å) in the first calculation phase to obtain a guess for TS1. After optimizing the corresponding intermediate complex (IC), the P–O3′ distance was then constrained (r(P–O3′) = 1.9–2.2 Å) to obtain a guess for TS2. After fully optimizing each TS, intrinsic reaction coordinate (IRC) calculations76 were performed to obtain the associated RC, IC, and/or PC, which were then fully optimized. For the one stepwise pathway characterized in the present work, the IRCs from TS1 and TS2 gave rise to structurally and energetically similar ICs (Fig. S3, ESI†), verifying the pathway is connected. The nature of the optimized stationary points were confirmed using frequency calculations at same level of theory (i.e., minima have all positive and TSs have one negative frequency), which also afforded the thermal corrections to the Gibbs energy. Furthermore, to account for the surrounding enzymatic environment, IEF-PCM-M06-2X/6-311+G(2df,p) single-point energy calculations were performed on the QM cluster models using a dielectric constant of ε = 4 that provides an accurate representation of the enzyme surroundings.77 Since ωB97M-V was recently suggested to be among the most reliable functionals for describing enzymatic reactions,78 single-point calculations were carried out using CPCM-ωB97M-V/6-311+G(2df,2p) and a dielectric constant of ε = 4.9 as available in ORCA 5.0.4. We find negligible differences upon changing the functional, dielectric, and basis set (Fig. S4 and Table S1, ESI†).
QM/MM calculations
QM/MM calculations were performed within the ONIOM formulism due to previous successes of this methodology in providing accurate structural and energetic descriptions for other enzymatic reactions.63,65,66,79–81 The QM/MM model was built using the X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant (PDB ID: 1CYQ, Fig. 1a) as calculations initiated from an X-ray crystal structure have been successfully used to elucidate the reaction mechanisms for several enzymes.82–86 The alanine mutation at position 98 in the crystal structure was manually reverted to H98 using PyMOL based on the residue orientation in the crystal structure of the wild type I-PpoI PC (1.80 Å resolution; PDB ID: 1A73, Fig. 1b). The enzyme–DNA complex was solvated in a 10 Å TIP3P octahedral water box and PROPKA was used to assign the standard protonation states of titratable amino acids.87 All water molecules with any atom further than 6 Å from the enzyme–substrate complex were removed. The solvated enzyme–DNA complex was minimized using AMBER18.88,89The QM region for the QM/MM model includes Mg2+ along with the directly coordinated three water molecules and N119. Furthermore, the QM layer contains S97, H98, H78, R61, a nucleophilic water, an additional water molecule simultaneously hydrogen bonded to a Mg2+-coordinated water and a non-bridging phosphate oxygen, the nucleotides containing the scissile P–O bond (dA189 and dG190), and the phosphate from dA191 on the 3′-side of dG190. Finally, depending on the metal–substrate binding configuration, an additional water molecule was incorporated into the QM region that either hydrogen bonded to a Mg2+-coordinated water (Fig. 2b) or directly coordinates to Mg2+ (Fig. S2b, ESI†). The QM/MM model contains 137 atoms in the QM region, which has a charge of +1. The MM region includes the remaining I-PpoI–DNA complex and the surrounding water molecules. The overall charge of the entire model is –23. The QM/MM boundary was placed between Cα and Cβ of each amino acid, and C4′ and C5′ of dA189 and dA191 of the substrate. Mechanical embedding (ME) was used to describe the boundary in all QM/MM models, while electrostatic embedding (EE) was subsequently employed for the preferred mechanism.
Each RC was fully optimized using B3LYP-D3(BJ)/6-31G(d,p) for the QM region67 and the AMBER force field (OL15 for DNA89 and ff14SB for the enzyme88) for the MM region. Next, scans of key reaction distances were performed to obtain initial guesses for TSs, where each parameter was increased/decreased in 0.10–0.15 Å increments. Specifically, a TS guess was obtained by successively decreasing the P⋯Ow distance. The bond was then frozen to the distance corresponding to a maximum on the potential energy surface, and the P–O3′ bond distance was subsequently increased. Full TS optimizations were then performed and IRC calculations run to obtain the associated RC and PC. Frequency calculations were conducted at the same level of theory to confirm the nature of the stationary points (i.e., minima have all positive and TSs have one imaginary frequency) and obtain thermal corrections to the Gibbs energies. Finally, single-point calculations at the ONIOM(M06-2X/6-311+G(2df,p):AMBER) level of theory were used to calculate the relative Gibbs energies.
Each characterized catalytic mechanism was carefully compared to experimental structural,38,39 mutational,46,57 and kinetic data.56,57 Specifically, the geometries of each optimized RC and PC were assessed in relation to the crystal structures of the RC analogue of the H98A I-PpoI mutant (PDB ID: 1CYQ)39 and PC for wild type I-PpoI (PDB ID: 1A73), respectively.38 The observed roles of active site amino acids were correlated with experimental mutational data, including the complete loss of I-PpoI catalytic activity for the H98A and N119A mutants57 and slight reduction in catalytic activity for the H78A46,57 and R61A mutants.57 The predicted QM/MM activation barriers were compared to the barrier for the overall process (∼86–95 kJ mol−1) calculated using the experimental kcat (0.046–1 min−1) measured at 37 °C and pH 7.5, with 10–50 nM of a DNA substrate (42-base pair oligonucleotide) and 5–100 pM of I-PpoI.56,57
The Gaussian 16 program (revision B.01) was used to perform all QM cluster and QM/MM calculations,90 with the exception of ωB97M-V QM cluster calculations which were performed using ORCA (version 5.0.4).91
Results and discussion
The smallest QM cluster and large-scale QM/MM models suggest that the experimentally-proposed pathway in which the metal aids leaving group departure through direct coordination and simultaneous protonation by a metal-activated water is not feasible
As discussed in the computational details, the smallest QM cluster model of I-PpoI includes all essential residues for catalysis (denoted Model 1). The X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant (Fig. 1a) was the basis for the proposal that Mg2+ assists leaving group departure through direct coordination to O3′ coupled with protonation of the leaving group by a metal-activated water during phosphodiester bond cleavage (Fig. 1c).39 Indeed, a water molecule falls within hydrogen-bonding distance of O3′ (3.4 Å, Fig. S1a, ESI†). However, the corresponding TS could not be characterized using the smallest QM cluster model (Model 1; 92 atoms, +1 charge) as the hydrogen-bond distance and angle between the Mg2+-ligated water and the substrate is unfavorable in the optimized RC (Fig. S5a, ESI†). To ensure that the inability to characterize the experimentally-proposed mechanism was not due to insufficient model size, attempts were made to map the same reaction mechanism using our large-scale QM/MM model. As found for Model 1, there remains a poor relative alignment of the active site water and substrate in the QM/MM optimized RC (Fig. S5b, ESI†), which prevents successful characterization of the corresponding TS. Together, QM cluster Model 1 and the QM/MM model highlight that simultaneous direct metal coordination to O3′ and leaving group protonation by a metal activated water is not possible during the P–O bond cleavage step within the confines of the I-PpoI active site. Nevertheless, we acknowledge that protonation of the leaving group by metal-activated or bulk water likely occurs after the chemical step as metal migration and active site rearrangement commonly occur in conjunction with product release for both one- and two-metal dependent nucleases.24,29,35,37,92–95The smallest QM cluster model indicates that leaving group protonation by a metal-ligated water during P–O bond cleavage is infeasible
When the experimentally-proposed pathway was investigated using QM cluster Model 1 and QM/MM models, a water molecule in the second metal coordination sphere migrated between the metal and O3′ during TS optimizations, while direct metal coordination to the non-bridging oxygen was maintained. This suggests that a water may be aligned to facilitate leaving group protonation during P–O bond cleavage as put forward in the experimentally-proposed pathway.39 Indeed, a similar indirect (water-mediated) metal–substrate binding architecture has been reported for one-metal dependent EcoRI (Fig. S6a, ESI†)96 and two-metal dependent BamHI (Fig. S6b, ESI†).26,97 Therefore, the reaction was explored using QM Model 1 from a I-PpoI RC that positions a water molecule between Mg2+ and O3′ of the scissile phosphate (Table 1, Fig. S2a, ESI†). In the optimized RC, H98 hydrogen bonds to a potential water nucleophile, while Mg2+ aids substrate stabilization through direct coordination to a non-bridging oxygen of the scissile phosphate in the optimized RC (Fig. 3). Further substrate stabilization is supplied by a Mg2+-coordinated water through a water chain to the other non-bridging phosphate oxygen (Fig. S7a, ESI†). The water nucleophile attacks the phosphorus reaction center (r(Ow⋯P) = 3.549 Å, Fig. 3b) through a concerted TS. Proton transfer from the nucleophilic water to H98 occurs simultaneously, as well as P–O3′ bond lengthening (2.126 Å) and partial proton transfer from the water bound to Mg2+ and O3′. Together, these structural features lead to a barrier of 182.3 kJ mol−1 (Fig. 4 and Table S2, ESI†). Although direct comparison of QM cluster barriers to experimental data is not recommended due to the missing enzymatic environment,67 our predicted phosphodiester bond cleavage barrier involving indirect metal coordination to the leaving group is much larger than other P–O bond cleavage barriers evaluated using QM cluster models (∼50–95 kJ mol−1).65,68,69,71 Therefore, an alternate mechanism must be considered.| Model (no. of atoms; charge) | H98 | H78 | R61 | 3′-PO42− | Indirect Mg2+–O3′ coordinationc | Direct Mg2+–O3′ coordinationd | Model strengths | Model limitations |
|---|---|---|---|---|---|---|---|---|
| a All models contain a nucleophilic water, Mg2+ directly coordinated to N119 and three water molecules, two additional water molecules, S97, dA189, and dG190. b The number of atoms and the charge correspond to the QM region of the QM/MM model. c Indirect (water-mediated) coordination permits a Mg2+–activated water to protonate the leaving group (Fig. 3). d The pathway in which Mg2+ promotes leaving group departure through direct coordination (Fig. 5). | ||||||||
| Model 1 (92; +1) | X | Nonpreferred | Preferred | H98 activates nucleophile | H98 interacts with S97 (not new P–O bond) | |||
| Model 2 (134; +1) | X | X | Nonpreferred | Preferred | H78 positions nucleophile + H98 | H78 interacts with neighboring dG190 | ||
| Model 3 (141; +2) | X | X | Nonpreferred | Preferred | H98 interacts with new P–O bond | R61 interacts with neighboring dG190 | ||
| Model 4 (163; +1) | X | X | X | X | Nonpreferred | Preferred | H78 or R61 interactions with dG190 eliminated | H78 interacts with R61 and 3′-phosphate |
| QM/MM Model (137; +1)b | X | X | X | X | Nonpreferred | Preferred | crystal structure geometry maintained | None |
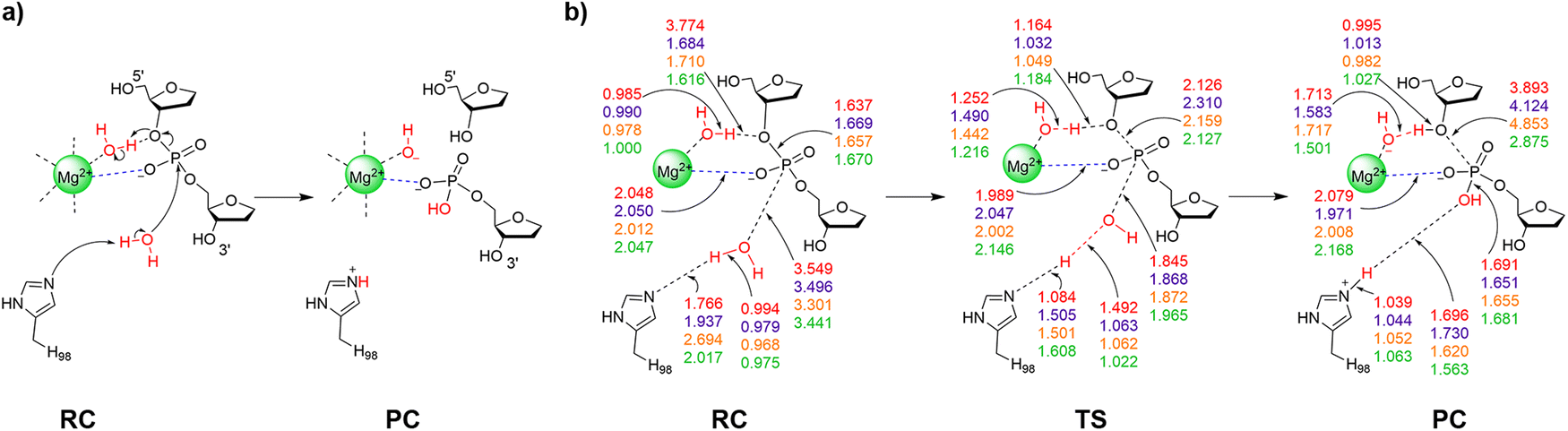 | ||
| Fig. 3 (a) The proposed reaction pathway and (b) key reaction parameters (Å) for each stationary point of the I-PpoI catalyzed phosphodiester bond cleavage involving indirect Mg2+ coordination to the leaving group characterized in the present work using QM cluster Model 1 (red), Model 3 (purple), Model 4 (orange), or QM/MM (green). See Fig. S7–S10 (ESI†) for additional structural parameters for QM cluster Models 1–4. | ||
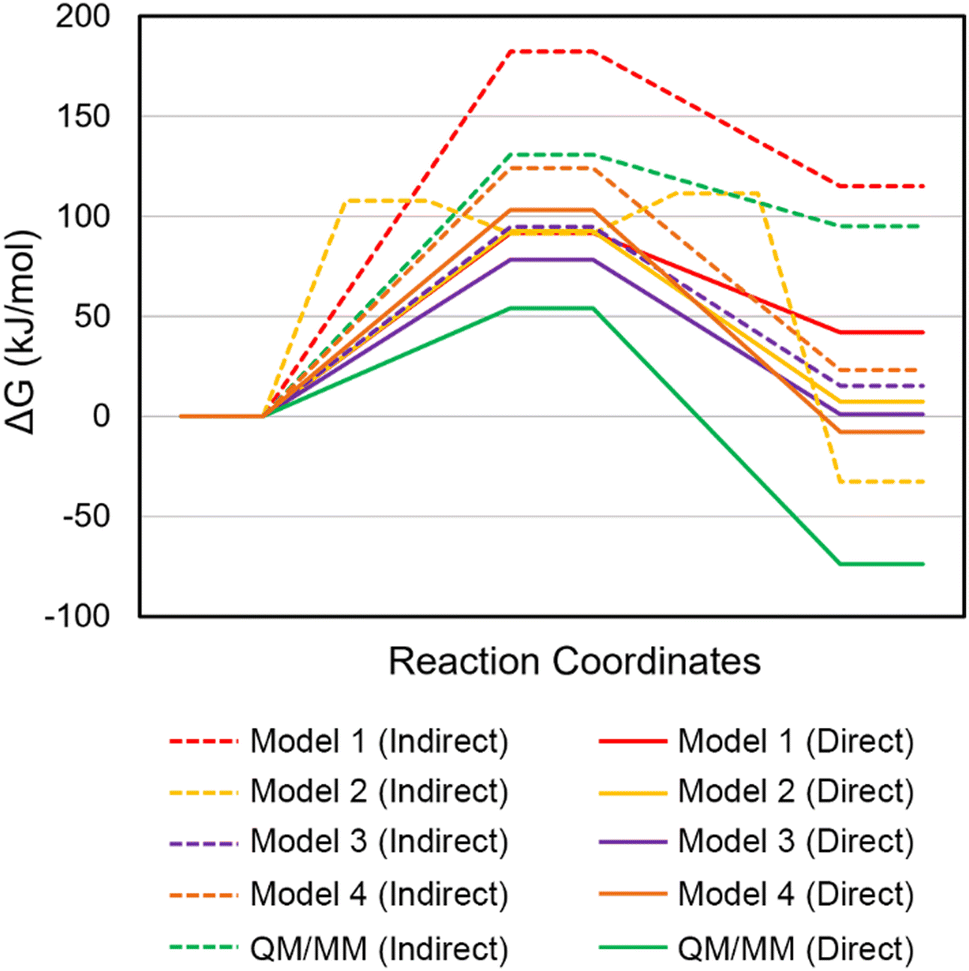 | ||
| Fig. 4 Relative Gibbs energies (kJ mol−1) for the I-PpoI catalyzed phosphodiester bond cleavage characterized using QM cluster (Models 1 – 4) or QM/MM models. Dashed lines refer to the pathways involving indirect (water-mediated) Mg2+ coordination to the leaving group, while solid lines refer to the pathways involving direct Mg2+ coordination to the leaving group. | ||
The smallest QM model suggests that direct metal coordination to the leaving group is necessary for P–O bond cleavage
In the crystal structure of the RC analogue of the H98A I-PpoI mutant, Mg2+ adopts bidentate coordination to the substrate through the O3′-leaving group and a non-bridging oxygen of the scissile phosphate (Fig. 1a). A similar coordination has been observed for other one-metal (T4 endonuclease VII (Fig. S6c, ESI†)42 and I-HmuI (Fig. S6d, ESI†)41) and two-metal (Aquifex aeolicus RNase III (AaRNase III, Fig. S6e, ESI†)98) dependent endonucleases. In these cases, direct coordination to the metal has been proposed to aid leaving group departure without the necessity of simultaneous protonation during P–O bond cleavage.41,42,98 An I-PpoI RC based on Model 1 can be optimized which preserves the direct Mg2+ coordination to the O3′-bridging oxygen as seen in the crystal structure of the RC analogue of the H98A I-PpoI mutant (Fig. 1 and 5a), while N119 interacts with the other bridging oxygen of the scissile phosphate (O5′ of dG190, Fig. S7b, ESI†). The water nucleophile sits closer to the phosphorus reaction center (r(Ow⋯P) is 0.6 Å shorter than in the corresponding RC involving indirect metal coordination to leaving group, Fig. 3b and 5b). The RC evolves into a concerted TS in which the P–O bond cleavage happens earlier than in the first mechanism (r(P⋯O3′) is 0.3 Å shorter, Fig. 3 and 5). In fact, these structural changes coupled with a strong Lewis acid stabilizing the leaving group (direct Mg2+ coordination) reduces the barrier by 90.7 kJ mol−1 compared to the pathway involving indirect (water-mediated) Mg2+–O3′ coordination (Fig. 4 and Table S3, ESI†). As a result, the new pathway leads to a barrier (Δ‡G = 91.6 kJ mol−1) that falls within the range of those calculated for other metalloenzyme-facilitated phosphodiester bond cleavage reactions using QM cluster models (∼50–95 kJ mol−1).65,68,69,71Although the experimental proposal that direct coordination to the leaving group and O3′ protonation occur at the same time is not feasible, pathways for the chemical step involving either element can be characterized. The smallest QM-based I-PpoI model reveals that the mechanism involving direct metal–O3′ coordination as seen in the X-ray crystal structure of the RC analogue of the H98A I-PpoI mutant39 is preferred over the water-mediated coordination necessary for leaving group protonation by a metal-activated water (Table 1). Nevertheless, H98 activates the water nucleophile and N119 stabilizes the substrate as proposed based on experimental data.38,39,57 Despite suggestions from structural (Fig. S1b, ESI†) and mutational studies that H98 helps stabilize the cleaved product,57 H98 interacts with S97 rather than the newly formed P–O bond in the PC (Fig. S7b, ESI†). This is likely an artifact caused by the absence of the broader surrounding enzymatic environment in Model 1, suggesting the need to include additional residue(s) that may help align H98. To ensure the conclusion reached regarding the relative importance of direct metal–O3′ coordination versus leaving group protonation is not the result of model size, both mechanisms successfully characterized for Model 1 will be considered in expanded models moving forward.
H78 primarily plays a structural role in I-PpoI catalysis, positioning the water nucleophile and the general base in QM Model 2
X-ray crystallographic data reveals that H78 is within hydrogen-bonding distance of the nucleophilic water in the RC analogue of the H98A I-PpoI mutant39 and H98 in the PC for wild type I-PpoI (Fig. S1, ESI†),38 while mutational data shows a slight reduction in I-PpoI catalytic activity upon H78 mutation to alanine.57 This suggests that H78 may be important for catalysis and this residue was added to Model 1 (Model 2, 134 atoms). Upon consideration of indirect (water-mediated) Mg2+–O3′ coordination that permits leaving group protonation using Model 2, H78 hydrogen bonds to H98 throughout the reaction, which situates the general base further from the nucleophile in the RC compared to Model 1 (r(NH98⋯Hw) is 0.8 Å longer, Fig. 3b and 6 and Fig. S8a, ESI†). The corresponding phosphodiester bond cleavage progresses through a stepwise mechanism, where TS1 involves nucleophilic attack to form a stable phosphorane intermediate (16.6 kJ mol−1 below TS1, Fig. 4 and Table S2, ESI†), while TS2 involves P–O bond dissociation. Although the rate-determining barrier is 70.8 kJ mol−1 lower for Model 2 than Model 1 (Fig. 4 and 6 and Table S2, ESI†), the barrier involving indirect metal coordination to O3′ remains high (Δ‡G = 111.5 kJ mol−1, Fig. 4 and Table S2, ESI†). | ||
| Fig. 5 (a) The proposed reaction pathway and (b) key reaction parameters (Å) for each stationary point of the I-PpoI catalyzed phosphodiester bond cleavage involving direct Mg2+ coordination to the leaving group characterized in the present work using QM cluster Model 1 (red), Model 2 (yellow), Model 3 (purple), Model 4 (orange), or QM/MM (green). See Fig. S7–S10 (ESI†) for additional structural parameters for QM cluster Models 1–4. | ||
 | ||
| Fig. 6 Key reaction parameters (Å) for each stationary point of the I-PpoI catalyzed phosphodiester bond cleavage involving indirect Mg2+ coordination to the leaving group characterized in the present work using QM cluster Model 2. See Fig. S8a (ESI†) for additional structural parameters for QM cluster Model 2. | ||
When direct Mg2+ coordination to the leaving group is considered with Model 2, H78 hydrogen bonds to the water nucleophile in the RC, which is consistent with experimental structural data (Fig. S1a and S8b, ESI†).39 This results in a 0.3 Å longer nucleophilic distance in the RC compared to Model 1 (Fig. 3b and 5b). In the concerted TS, H78 reorients to hydrogen bond to the general base (H98) as water attacks the phosphorus reaction center (Fig. S8b, ESI†). Although the energy barriers for Models 1 and 2 are nearly equal (within 1 kJ mol−1, Fig. 4 and Table S3, ESI†), the PC for Model 2 contains a hydrogen bond between (protonated) H98 and the newly formed P–O bond (Fig. 6), which agrees with the experimental proposal that H98 stabilizes the cleaved product (Fig. S1b, ESI†).38,57
Overall, despite a similar activation barrier as Model 1, Model 2 highlights that direct metal coordination to the leaving group is preferred (Table 1) and H78 can position the water nucleophile and the general base (H98), which is consistent with structural38,39 and mutational data.57 Nevertheless, H78 interacts with a substrate nucleobase in the 3′ direction with respect to the scissile P–O bond (O6 of dG190) throughout the reaction for Model 2 (Fig. S8b, ESI†), which is not consistent with crystallographic data (r(NH78⋯OdG190) ∼11 Å and r(NH78⋯Onon-bridging) ∼5 Å, PDB ID: 1CYQ and 1A73)38,39 due to the presence of an active site residue (R61) between H78 and the substrate (Fig. S1, ESI†). This suggests further model refinement is required.
R61 hydrogen bonds with the 3′-nucleobase with respect to the scissile P–O bond in QM Model 3
In the crystal structures of the RC analogue of the H98A I-PpoI mutant (PDB ID: 1CYQ)39 and the PC for wild type I-PpoI (PDB ID: 1A73),38 R61 hydrogen bonds to the substrate and thereby has been suggested to stabilize the TS and the cleaved phosphate group in the PC (Fig. S1b, ESI†), which may inhibit the reverse reaction.38 This proposal is supported by the R61A mutant having slightly reduced I-PpoI catalytic activity.57 Therefore, prior to expanding Model 2, R61 was added to Model 1 (Model 3, 141 atoms, Fig. 1). In Model 3, R61 hydrogen bonds to the nucleobase of dG190 regardless of whether indirect or direct Mg2+ coordination to the leaving group is considered (Fig. S9, ESI†). When indirect (water-mediated) Mg2+ coordination to the leaving group is considered, N119 hydrogen bonds to a metal-ligated water (Fig. S9a, ESI†), which in turn provides significant stabilization to O3′ in the RC compared to Model 1 (r(Ow⋯Hw) decreases by 2.1 Å, Fig. 3b) and substantially decreases the barrier compared to Model 1 (by 87.5 kJ mol−1, Fig. 4 and Table S2, ESI†). Despite the reasonable activation barrier (94.8 kJ mol−1), there is a 13.2 kJ mol−1 decrease in the Gibbs energy barrier when direct bidentate metal–substrate coordination is considered (Tables S2 and S3, ESI†), with most reaction parameters observed for Model 1 retained (Fig. 4). Although the pathway involving leaving group stabilization through direct Mg2+ coordination remains the preferred mechanism for Model 3 (Fig. 4, Table 1 and Tables S2 and S3, ESI†), an interaction between R61 and the 3′-nucleobase (N7 and O6 of dG190) with respect to the scissile P–O bond arises (Fig. S9b, ESI†) that is absent in the I-PpoI crystal structures (Fig. 1 and Fig. S1, ESI†),38,39 suggesting that the model needs to be further expanded.Inclusion of both H78 and R61 as well as the phosphate on the 3′-side of the scissile P–O bond affords correct positioning of the general base and stabilization of the substrate in QM Model 4
Models 2 and 3 indicate that H78 and R61 each play important roles in the phosphodiester bond cleavage by positioning the general base, water nucleophile, and/or substrate, as well as stabilizing charges in the active site. Furthermore, the crystal structure of the RC analogue of the H98A I-PpoI mutant reveals that R61 is within hydrogen-bonding distance of the phosphate on the 3′-side of the scissile P–O bond (dA191, Fig. S1a, ESI†).39 Therefore, H78, R61, and the additional phosphate moiety were simultaneously added to Model 1 (Model 4, 163 atoms) and the reaction pathways were remapped. Regardless of the metal coordination geometry considered using Model 4, R61 hydrogen bonds to the 3′-phosphate and H78 positions the general base (H98) through hydrogen bonding as implied by experimental structural data (Fig. 1 and Fig. S1 and S10a, ESI†).39 These new structural factors reduce the activation barrier by 58.1 kJ mol−1 compared to Model 1 (Fig. 4 and Table S2, ESI†). Nevertheless, the P–O bond cleavage barrier for this pathway is high (Δ‡G = 124.3 kJ mol−1). On the other hand, when Model 4 with direct metal–O3′ coordination is considered, the majority of Model 1 structural parameters are maintained (Fig. S7b and S10b, ESI†), with the exception of a slightly increased nucleophilic attack distance (r(P⋯Ow) is 0.2 Å longer, Fig. 5b), which raises the activation barrier for P–O bond cleavage by 11.6 kJ mol−1 (Fig. 4 and Table S3, ESI†).Overall, inclusion of H78, R61, and the 3′-phosphate overcomes some of the artifacts of the previous QM models including eliminating interactions between H78 or R61 and the substrate nucleobase (dG190, Fig. S10b, ESI†). However, due to the absence of the surrounding environment, interactions between H78 and R61, and between H78 and the 3′-phosphate exist in Model 4, despite these residues being separated by >5 Å in the X-ray crystal structures (r(NH78⋯NR61) and r(NH78⋯OP1dG190), PDB ID: 1CYQ and 1A73).38,39 This underscores the need to characterize the phosphodiester bond cleavage pathway in the presence of the broader enzymatic environment. Indeed, QM cluster models containing multiple shells of amino acids (300–400 atoms) have been shown to be necessary for accurate descriptions of some enzymatic reactions.86,99–102 Nonetheless, our QM cluster calculations provide valuable insights into the residues that must be included in the QM region of QM/MM models of I-PpoI.
QM/MM calculations further highlight that the preferred mechanism in the presence of the surrounding enzymatic environment involves direct metal coordination to the leaving group
As discussed in the computational details, the QM region of the QM/MM model of I-PpoI contains all residues included in QM cluster Model 4. The QM/MM RC involving indirect (water-mediated) Mg2+–O3′ coordination maintains all interactions observed in the I-PpoI active site based on the crystal structures (Fig. 1 and 7a and Fig. S1a, ESI†).38,39 Specifically, unlike any QM cluster model, H78 simultaneously positions the water nucleophile and orients the general base (H98), N119 provides substrate stabilization by hydrogen bonding to the O5′-bridging oxygen of dG190, and R61 hydrogen bonds with the 3′-phosphate with respect to the scissile phosphate (Fig. 7a and Fig. S7a–S10a, ESI†). As a result, most reaction parameters of Model 4 are preserved (Fig. 3) and the activation barriers are similar (within 6.6 kJ mol−1, Fig. 4 and Table S2, ESI†). Therefore, the QM/MM barrier associated with O3′ protonation by a metal-ligated water (130.9 kJ mol−1) is significantly higher than the experimental value for I-PpoI (∼86–95 kJ mol−1)56,57 and typical experimental barriers for metallonucleases catalyzed reactions (∼58–96 kJ mol−1).45,103–106 This supports the conclusions from all QM cluster models that the water-mediated metal–substrate coordination necessary for leaving group protonation cannot effectively catalyze the P–O bond cleavage reaction. | ||
| Fig. 7 Key distances, reaction parameters, and metal coordination distances (Å) in the I-PpoI active site for the phosphodiester bond cleavage pathway characterized using QM/MM with (a) indirect or (b) direct Mg2+ coordination to the leaving group. | ||
When the pathway involving direct coordination of the metal to the leaving group is considered using QM/MM, H78 interacts with both the water nucleophile and the general base (H98), while R61 hydrogen bonds to the phosphate 3′ to the scissile P–O bond in the RC (Fig. 7b). Despite a smaller nucleophilic attack distance (r(Ow⋯P) = 2.757 Å) and the metal being closer to the leaving group (2.096 Å, Fig. 5b) in the QM/MM RC than any QM cluster RC, the proton has not transferred from the water nucleophile to H98 and the P–O bond cleavage is more advanced (2.347 Å) in the QM/MM TS compared to any QM model. Nevertheless, the barrier for the mechanism involving direct metal–leaving group coordination (54.1 kJ mol−1) is lower than that for any other model (by 24.3–38.5 kJ mol−1, Fig. 4 and Table S3, ESI†). Furthermore, the direct bidentate metal–substrate binding architecture observed in the crystal structure of the RC analogue of the H98A I-PpoI mutant (Fig. 1a) results in a 76.8 kJ mol−1 lower barrier than water-mediated metal coordination to the leaving group (Fig. 4 and Tables S2 and S3, ESI†). Direct metal–O3′ coordination is consistent with X-ray crystallographic data for I-PpoI38,39 as well as other one-metal dependent endonucleases (e.g., T4 EndoVII (Fig. S6c, ESI†),42 I-HmuI (Fig. S6d, ESI†),41 and Hpy188I (Fig. S6f, ESI†)44). Furthermore, the alignments of key active site amino acids in the QM/MM stationary points are consistent with the crystal structures of the RC analogue of the H98A I-PpoI mutant and the PC for wild-type I-PpoI (Fig. S11, ESI†). We also note that minimal changes occur in the reaction parameters (Fig. S12a, ESI†) and orientations of active site residues (Fig. S12b, ESI†) upon reoptimization of the preferred pathway using EE. Although the activation barrier slightly increases (by 23.3 kJ mol−1, Table S3, ESI†) for the EE characterized pathway, the computed Gibbs activation barrier for the mechanism involving direct Mg2+ coordination to the leaving group (Δ‡G = 77.4 kJ mol−1) remains below the experimental value (∼86–94 kJ mol−1) for the overall process,56,57 with product release being the rate-determining step.57 Thus, our QM/MM models corroborate the conclusion from QM cluster calculations (Table 1) that direct metal-leaving group coordination is critical for the chemical step facilitated by I-PpoI.
The roles of the amino acid residues uncovered from the preferred QM/MM mechanism are also in line with mutational data.46,57 Specifically, H98 initiates the reaction by activating the water nucleophile, which agrees with the experimentally observed complete loss of catalytic activity upon H98A mutation.57 Our in silico H98A mutational results further support this proposal. Specifically, although the reaction parameters and orientation of active site residues are preserved upon H98A mutation (Fig. 5 and 7 and Fig. S13, ESI†), the water nucleophile repositions to be activated by H78 in the QM/MM RC, which substantially increases the activation barrier compared to the wild-type mechanism (by 79.8 kJ mol−1, Fig. S13 and Table S3, ESI†). In our proposed wild-type I-PpoI mechanism, N119 stabilizes the substrate through hydrogen bonding, while holding the metal in place to facilitate the reaction, which correlates with experimental mutational data showing complete enzyme inactivation upon N119 mutation.57 Furthermore, H78 and R61 play structural roles including positioning and stabilizing the nucleophilic water, the general base (H98), and the substrate for efficient catalysis, which correlates with reported slight reductions in enzymatic activity upon mutating these residues.46,57 Overall, our proposed phosphodiester bond hydrolysis pathway involving direct Mg2+ coordination to the leaving group is fully consistent with experimental kinetic,56,57 structural,38,39 and mutational data.46,57
Our proposed pathway for one-metal mediated I-PpoI catalysis draws similarities, yet differences from those facilitated by other one- and two-metal dependent nucleases
For enzyme-catalyzed phosphodiester bond hydrolysis reactions to proceed, a water nucleophile must be activated, the leaving group must depart, and the charge on the substrate must be stabilized.94,107 A metal-ligated water has commonly been proposed to act as the general acid, protonating the leaving group of the substrate for several nucleases (e.g., one-metal dependent EcoRI (Fig. S6a, ESI†),96 APE1,65 and Vvn,40 and two-metal dependent EcoRV28 and BamHI (Fig. S6b, ESI†)26). Despite an equivalent proposal in the literature for I-PpoI based on crystallographic data,39 our QM cluster and QM/MM calculations show that the I-PpoI pathway involving leaving group protonation by a metal-activated water is infeasible. Nevertheless, leaving group departure has also commonly been proposed to be facilitated though direct coordination of the metal to the substrate for various one-metal dependent nucleases (e.g., T4 EndoVII (Fig. S6c, ESI†),42 I-HmuI (Fig. S6d, ESI†),41 and Hpy188I (Fig. S6f, ESI†)44). Furthermore, experimental structural data for two-metal mediated AaRNase III (Fig. S6e, ESI†)98 and computational studies on CRISPR-Cas934 have suggested one metal (MgB2+) directly aids leaving group departure. Our calculations reveal the preferred P–O bond cleavage pathway for I-PpoI also involves direct Mg2+–O3′ coordination, with the single metal in the I-PpoI active site being equivalently positioned with respect to substrate as the second metal in two-metal mediated endonucleases (MgB2+, Fig. S14, ESI†).The charge on a non-bridging oxygen of the scissile phosphate has generally been suggested to be stabilized by both metals (MA2+ and MB2+) for two-metal mediated nucleases (e.g., BamHI26 and AaRNase III,98 Fig. S6b and e, ESI†), while diverse residues have been proposed to adopt this role for single-metal dependent enzymes in addition to the metal. For example, I-HmuI contains Y39,41 while Hpy188I contains K73 and R8444 appropriately positioned with respect to the substrate for charge stabilization (Fig. S6d and e, ESI†). Despite sharing similar metal-binding architectures with I-HmuI and Hpy188I (Fig. 1, and S6d and f, ESI†), our calculations reveal that I-PpoI uniquely uses a water chain between Mg2+ and a non-bridging phosphate oxygen to complement the substrate charge stabilization provided by the metal directly coordinated to the other non-bridging oxygen of the scissile phosphate (Fig. 2a). In terms of nucleophile activation, the first metal (MA2+) is generally proposed to facilitate nucleophilic attack in the well-accepted mechanism for two-metal dependent nucleases.34,98 In contrast, a histidine residue (H98) acts as the general base for I-PpoI, fulfilling the role of the first metal (MA2+) in a two-metal mediated reaction. The proposed role of histidine as the general base in the I-PpoI mechanism of action is in agreement with the experimental structural38,39 and mutational data for I-PpoI,57 as well as experimental crystallographic studies on other single-metal dependent nucleases such as T4 EndoVII (H41, Fig. S6c, ESI†),42 I-HmuI (H75, Fig. S6d, ESI†),41 and Vvn (H80),40 and mutational studies on Serratia nuclease (H89).64 Therefore, our calculations coupled with kinetic46 and structural data38,39 on I-PpoI supports the proposal that one metal is enough for enzymatic cleavage of phosphodiester bonds.
This is only the second time the feasibility of single-metal mediated catalysis of phosphodiester bond cleavage has been demonstrated for an endonuclease using computational techniques, with previous work focused on one-metal dependent APE1.65–67 In contrast to I-PpoI, QM cluster calculations on single-metal dependent APE1 revealed that direct bidentate metal–substrate coordination is not feasible.65 Instead, as confirmed using QM/MM calculations, an indirect metal-binding architecture is preferred in the APE1 active site. Differences between APE1 and I-PpoI arise because of the distinct active site compositions (Fig. 8). Specifically, the presence of a strong negatively charged general base (D240) coupled with a positively charged histidine (H309) positioned to stabilize the charged substrate allows APE1 to use a metal-coordinated water to facilitate leaving group departure. In contrast, the neutral general base (H98) and the absence of a positively charged residue close to the scissile phosphate forces the single metal of I-PpoI to play a more active role in promoting leaving group departure and substrate charge stabilization through direct bidentate coordination. This comparison underscores how a single metal can play diverse roles depending on the active site environment of the enzyme and how the same phosphodiester bond hydrolysis can be achieved in slightly different ways. The fundamental understanding of the unique I-PpoI chemistry afforded by the present work may inspire studies on other single-metal dependent enzymes (e.g., T4 EndoVII (Fig. S6c, ESI†),42 Vvn,40 and Serratia nuclease64), as well as other HEs (e.g., I-HmuI41 and Hpy188I,44 Fig. S6d and f, ESI†). Such future work is necessary to understand how nature cleaves the P–O bond in unique ways, depending on the identity of the proposed general base, mode of charge stabilization, and/or number of metals in the active site.
 | ||
| Fig. 8 Comparison of the proposed phosphodiester bond cleavage pathways for (a) APE165–67 and (b) I-PpoI (present work). | ||
Conclusion
In the present study, a combined QM and QM/MM approach was used to provide atomic-level details of the catalytic pathway employed by I-PpoI to cleave phosphodiester bonds in DNA. In the experimentally-proposed I-PpoI mechanism of action,39 a single metal facilitates P–O bond cleavage through direct coordination to the substrate coupled with simultaneous leaving group protonation via a metal-activated water. However, this previously proposed pathway could not be characterized using our smallest QM cluster or QM/MM model due to geometrical constraints imposed by the I-PpoI active site. Although QM cluster and QM/MM models of varying size permitted the characterization of a pathway involving leaving group protonation by a metal-activated water, an indirect (water-mediated) metal binding configuration to the leaving group is required to achieve this chemistry and this mechanism is not energetically feasible regardless of the model size. Instead, QM cluster calculations uncovered a preferred mechanism for phosphodiester bond hydrolysis in which H98 activates the water nucleophile, while the metal provides substrate charge stabilization and promotes leaving group departure through direct coordination. QM/MM calculations verified the feasibility of this preferred pathway within the context of the solvated enzyme–DNA complex, yielding a mechanism that is fully consistent with experimental structural,38,39 kinetic,56,57 and mutational data.46,57 Despite our calculations highlighting that simultaneous direct metal coordination to O3′ and leaving group protonation by a metal activated water is not possible during the P–O bond cleavage step within the confines of the I-PpoI active site, we acknowledge that protonation of the leaving group by metal-activated or bulk water likely occurs after the chemical step as metal migration and active site rearrangement are common events in conjunction with product release for both one- and two-metal dependent nucleases.24,29,35,37,92–95 The fundamental understanding gained about I-PpoI function from the present work can be used to further explore its potential in the areas of gene therapy and genome engineering for biotechnological and therapeutic solutions. Furthermore, our new mechanistic understanding of I-PpoI can direct computational investigations on other members of the HE family (e.g., I-HmuI41 and Hpy188I44) which share the metal-binding configuration of I-PpoI, but differ in the identities and arrangements of active site amino acids. Additionally, our work will inspire studies on other one-metal dependent nucleases (e.g., T4 EndoVII,42 Vvn,40 and Serratia nuclease64), with the goal to better understand how nature cleaves extremely stable phosphodiester bonds.Author contributions
R. Kaur: conceptualization, project administration, investigation, methodology, data curation, formal analysis, validation, visualization, and writing – original draft. A. Frederickson: investigation and formal analysis. S. D. Wetmore: conceptualization, methodology, project administration, supervision, resources, funding acquisition, and writing – original draft. All authors also contributed to writing – reviewing and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. D. W. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC; 2016-04568) and the Canada Research Chairs program (2021-00484) for financial support. R. K thanks Alberta Innovates – Technology Futures, and the University of Lethbridge Board of Governors and School of Graduate Studies for student scholarships. A. F. thanks NSERC (USRA) and the RNA Innovation program for undergraduate student scholarships. Computer resources were provided by Digital Research Alliance of Canada, and Advanced Research Computing (ARC) at the University of Calgary.References
- B. L. Stoddard, Mobile DNA, 2014, 5, 1–16 CrossRef PubMed.
- M. Belfort and R. J. Roberts, Nucleic Acids Res., 1997, 25, 3379–3388 CrossRef CAS PubMed.
- M. Jurica and B. Stoddard, Cell. Mol. Life Sci., 1999, 55, 1304–1326 CrossRef CAS PubMed.
- N. Guhan and K. Muniyappa, Crit. Rev. Biochem. Mol. Biol., 2003, 38, 199–248 CrossRef CAS PubMed.
- H. H. Kazazian Jr, C. Wong, H. Youssoufian, A. F. Scott, D. G. Phillips and S. E. Antonarakis, Nature, 1988, 332, 164–166 CrossRef CAS PubMed.
- D. C. Hancks and H. H. Kazazian, Mobile DNA, 2016, 7, 1–28 CrossRef PubMed.
- K. Kobayashi, Y. Nakahori, M. Miyake, K. Matsumura, E. Kondo-Iida, Y. Nomura, M. Segawa, M. Yoshioka, K. Saito and M. Osawa, Nature, 1998, 394, 388–392 CrossRef CAS PubMed.
- M. Belfort and R. P. Bonocora, Methods Mol. Biol., 2014, 1123, 1–26 CrossRef PubMed.
- M. Jasin, Trends Genet., 1996, 12, 224–228 CrossRef CAS PubMed.
- J. Z. Dalgaard, M. Banerjee and M. J. Curcio, Genetics, 1996, 143, 673–683 CrossRef CAS PubMed.
- M. J. Marcaida, I. G. Muñoz, F. J. Blanco, J. Prieto and G. Montoya, Cell. Mol. Life Sci., 2010, 67, 727–748 CrossRef CAS PubMed.
- O. Humbert, L. Davis and N. Maizels, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 264–281 CrossRef CAS PubMed.
- P. Redondo, J. Prieto, I. G. Munoz, A. Alibés, F. Stricher, L. Serrano, J.-P. Cabaniols, F. Daboussi, S. Arnould and C. Perez, Nature, 2008, 456, 107–111 CrossRef CAS PubMed.
- S. Arnould, C. Perez, J.-P. Cabaniols, J. Smith, A. Gouble, S. Grizot, J.-C. Epinat, A. Duclert, P. Duchateau and F. Pâques, J. Mol. Biol., 2007, 371, 49–65 CrossRef CAS PubMed.
- A. Dupuy, J. Valton, S. Leduc, J. Armier, R. Galetto, A. Gouble, C. Lebuhotel, A. Stary, F. Pâques and P. Duchateau, PLoS One, 2013, 8, e78678 CrossRef PubMed.
- L. Popplewell, T. Koo, X. Leclerc, A. Duclert, K. Mamchaoui, A. Gouble, V. Mouly, T. Voit, F. Pâques and F. Cédrone, Hum. Gene Ther., 2013, 24, 692–701 CrossRef CAS PubMed.
- S. Grizot, J. Smith, F. Daboussi, J. Prieto, P. Redondo, N. Merino, M. Villate, S. Thomas, L. Lemaire and G. Montoya, Nucleic Acids Res., 2009, 37, 5405–5419 CrossRef CAS PubMed.
- I. G. Munoz, J. Prieto, S. Subramanian, J. Coloma, P. Redondo, M. Villate, N. Merino, M. Marenchino, M. D'Abramo and F. L. Gervasio, Nucleic Acids Res., 2011, 39, 729–743 CrossRef CAS PubMed.
- P. Cannon and C. June, Curr. Opin. HIV AIDS, 2011, 6, 74 CrossRef PubMed.
- T. Klein, N. Windbichler, A. Deredec, A. Burt and M. Benedict, Pathog. Global Health, 2012, 106, 20–31 CrossRef CAS PubMed.
- R. Galizi, L. A. Doyle, M. Menichelli, F. Bernardini, A. Deredec, A. Burt, B. L. Stoddard, N. Windbichler and A. Crisanti, Nat. Commun., 2014, 5, 3977 CrossRef CAS PubMed.
- Y.-S. Chan, R. Takeuchi, J. Jarjour, D. S. Huen, B. L. Stoddard and S. Russell, PLoS One, 2013, 8, e74254 CrossRef CAS PubMed.
- E. A. Galburt and B. L. Stoddard, Biochemistry, 2002, 41, 13851–13860 CrossRef CAS PubMed.
- W. Yang, J. Y. Lee and M. Nowotny, Mol. Cell, 2006, 22, 5–13 CrossRef CAS PubMed.
- G. Palermo, A. Cavalli, M. L. Klein, M. Alfonso-Prieto, M. Dal Peraro and M. De Vivo, Acc. Chem. Res., 2015, 48, 220–228 CrossRef CAS PubMed.
- T. Mordasini, A. Curioni and W. Andreoni, J. Biol. Chem., 2003, 278, 4381–4384 CrossRef CAS PubMed.
- M. De Vivo, M. Dal Peraro and M. L. Klein, J. Am. Chem. Soc., 2008, 130, 10955–10962 CrossRef CAS PubMed.
- P. Imhof, S. Fischer and J. C. Smith, Biochemistry, 2009, 48, 9061–9075 CrossRef CAS PubMed.
- E. Rosta, M. Nowotny, W. Yang and G. Hummer, J. Am. Chem. Soc., 2011, 133, 8934–8941 CrossRef CAS PubMed.
- A. J. M. Ribeiro, M. J. Ramos and P. A. Fernandes, J. Am. Chem. Soc., 2012, 134, 13436–13447 CrossRef CAS PubMed.
- A. R. Araujo, A. J. Ribeiro, P. A. Fernandes and M. J. Ramos, J. Chem. Theory Comput., 2014, 10, 5458–5466 CrossRef CAS PubMed.
- T. Rungrotmongkol, A. J. Mulholland and S. Hannongbua, MedChemComm, 2014, 5, 593–596 RSC.
- S. Xiao, M. L. Klein, D. N. LeBard, B. G. Levine, H. Liang, C. M. MacDermaid and M. Alfonso-Prieto, J. Phys. Chem. B, 2014, 118, 873–889 CrossRef CAS PubMed.
- L. Casalino, L. Nierzwicki, M. Jinek and G. Palermo, ACS Catal., 2020, 10, 13596–13605 CrossRef CAS PubMed.
- C. M. Dupureur, Metallomics, 2010, 2, 609–620 CrossRef CAS PubMed.
- V. Pingoud, W. Wende, P. Friedhoff, M. Reuter, J. Alves, A. Jeltsch, L. Mones, M. Fuxreiter and A. Pingoud, J. Mol. Biol., 2009, 393, 140–160 CrossRef CAS PubMed.
- F. Xie, S. H. Qureshi, G. A. Papadakos and C. M. Dupureur, Biochemistry, 2008, 47, 12540–12550 CrossRef CAS PubMed.
- K. E. Flick, M. S. Jurica, R. J. Monnat and B. L. Stoddard, Nature, 1998, 394, 96–101 CrossRef CAS PubMed.
- E. A. Galburt, B. Chevalier, W. Tang, M. S. Jurica, K. E. Flick, R. J. Monnat and B. L. Stoddard, Nat. Struct. Biol., 1999, 6, 1096–1099 CrossRef CAS PubMed.
- C. L. Li, L. I. Hor, Z. F. Chang, L. C. Tsai, W. Z. Yang and H. S. Yuan, EMBO J., 2003, 22, 4014–4025 CrossRef CAS PubMed.
- B. W. Shen, M. Landthaler, D. A. Shub and B. L. Stoddard, J. Mol. Biol., 2004, 342, 43–56 CrossRef CAS PubMed.
- C. Biertümpfel, W. Yang and D. Suck, Nature, 2007, 449, 616–620 CrossRef PubMed.
- B. Dalhus, A. S. Arvai, I. Rosnes, Ø. E. Olsen, P. H. Backe, I. Alseth, H. Gao, W. Cao, J. A. Tainer and M. Bjørås, Nat. Struct. Mol. Biol., 2009, 16, 138–143 CrossRef CAS PubMed.
- M. Sokolowska, H. Czapinska and M. Bochtler, Nucleic Acids Res., 2011, 39, 1554–1564 CrossRef CAS PubMed.
- B. D. Freudenthal, W. A. Beard, M. J. Cuneo, N. S. Dyrkheeva and S. H. Wilson, Nat. Struct. Mol. Biol., 2015, 22, 924–931 CrossRef CAS PubMed.
- J. H. Eastberg, J. Eklund, R. Monnat and B. L. Stoddard, Biochemistry, 2007, 46, 7215–7225 CrossRef CAS PubMed.
- E. L. Ellison and V. M. Vogt, Mol. Cell. Biol., 1993, 13, 7531–7539 CAS.
- K. E. Flick, D. McHUGH, B. L. Stoddard, J. D. Heath, K. M. Stephens and R. J. Monnat Jr, Protein Sci., 1997, 6, 2677–2680 CrossRef CAS PubMed.
- G. M. Argast, K. M. Stephens, M. J. Emond and R. J. Monnat Jr, J. Mol. Biol., 1998, 280, 345–353 CrossRef CAS PubMed.
- J. Lin and V. M. Vogt, Mol. Cell. Biol., 1998, 18, 5809–5817 CrossRef CAS PubMed.
- P. K. Wittmayer, J. L. McKenzie and R. T. Raines, Gene, 1998, 206, 11–21 CrossRef CAS PubMed.
- R. J. Monnat Jr, A. F. Hackmann and M. A. Cantrell, Biochem. Biophys. Res. Comm., 1999, 255, 88–93 CrossRef CAS PubMed.
- E. A. Galburt, M. S. Chadsey, M. S. Jurica, B. S. Chevalier, D. Erho, W. Tang, R. J. Monnat Jr and B. L. Stoddard, J. Mol. Biol., 2000, 300, 877–887 CrossRef CAS PubMed.
- C. B. McCarthy and V. Romanowski, Biochem. Genet., 2006, 44, 58–65 CrossRef PubMed.
- J. L. Eklund, U. Y. Ulge, J. Eastberg and R. J. Monnat Jr, Nucleic Acids Res., 2007, 35, 5839–5850 CrossRef CAS PubMed.
- P. K. Wittmayer and R. T. Raines, Biochemistry, 1996, 35, 1076–1083 CrossRef CAS PubMed.
- S. J. Mannino, C. L. Jenkins and R. T. Raines, Biochemistry, 1999, 38, 16178–16186 CrossRef CAS PubMed.
- V. Turkki, D. Schenkwein, O. Timonen, T. Husso, H. P. Lesch and S. Ylä-Herttuala, Biomed Res. Int., 2014, 2014, 379340 Search PubMed.
- N. Windbichler, P. A. Papathanos and A. Crisanti, PLoS Genet., 2008, 4, e1000291 CrossRef PubMed.
- P. Friedhoff, I. Franke, K. L. Krause and A. Pingoud, FEBS Lett., 1999, 443, 209–214 CrossRef CAS PubMed.
- B. S. Chevalier and B. L. Stoddard, Nucleic Acids Res., 2001, 29, 3757–3774 CrossRef CAS PubMed.
- R. Kaur, D. J. Nikkel and S. D. Wetmore, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1471 CAS.
- I. Geronimo, P. Vidossich, E. Donati and M. De Vivo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1534 CAS.
- S. Shlyapnikov, V. Lunin, M. Perbandt, K. Polyakov, V. Y. Lunin, V. Levdikov, C. Betzel and A. Mikhailov, Acta Crystallogr., Sect. D, 2000, 56, 567–572 CrossRef CAS PubMed.
- M. M. Aboelnga and S. D. Wetmore, J. Am. Chem. Soc., 2019, 141, 8646–8656 CrossRef CAS PubMed.
- R. Kaur, M. M. Aboelnga, D. J. Nikkel and S. D. Wetmore, Phys. Chem. Chem. Phys., 2022, 24, 29130–29140 RSC.
- R. Kaur, D. J. Nikkel, M. M. Aboelnga and S. D. Wetmore, J. Phys. Chem. B, 2022, 126, 10672–10683 CrossRef CAS PubMed.
- X. Zhang, H. Gao, H. Xu, J. Xu, H. Chao and C. Zhao, J. Mol. Catal. A: Chem., 2013, 368, 53–60 Search PubMed.
- H. Gao, Z. Ke, N. J. DeYonker, J. Wang, H. Xu, Z.-W. Mao, D. L. Phillips and C. Zhao, J. Am. Chem. Soc., 2011, 133, 2904–2915 CrossRef CAS PubMed.
- G. Sharma, V. M. Jayasinghe-Arachchige, Q. Hu, G. Schenk and R. Prabhakar, ACS Catal., 2020, 10, 3684–3696 CrossRef CAS.
- M. E. Alberto, G. Pinto, N. Russo and M. Toscano, Chem. Eur. J., 2015, 21, 3736–3745 CrossRef CAS PubMed.
- W. L. DeLano, CCP4 Newsl. Protein Crystallogr., 2002, 40, 82–92 Search PubMed.
- P. E. M. Siegbahn and F. Himo, J. Biol. Inorg. Chem., 2009, 14, 643–651 CrossRef CAS PubMed.
- M. Kazemi, X. Sheng, W. Kroutil and F. Himo, ACS Catal., 2018, 8, 10698–10706 CrossRef CAS.
- C. Acosta-Silva, J. Bertran, V. Branchadell and A. Oliva, J. Phys. Chem. A, 2017, 121, 8525–8534 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- D. A. Wappett and L. Goerigk, J. Chem. Theory Comput., 2023, 19, 8365–8383 CrossRef CAS PubMed.
- J. Castro-Amorim, A. Oliveira, A. K. Mukherjee, M. J. Ramos and P. A. Fernandes, J. Chem. Inf. Model., 2023, 63, 4056–4069 CrossRef CAS PubMed.
- E. F. Gérard, T. Mokkawes, L. O. Johannissen, J. Warwicker, R. R. Spiess, C. F. Blanford, S. Hay, D. J. Heyes and S. P. de Visser, ACS Catal., 2023, 13, 8247–8261 CrossRef PubMed.
- J. Shirazi, S. Jafari, U. Ryde and M. Irani, J. Phys. Chem. B, 2023, 127, 4480–4495 CrossRef CAS PubMed.
- A. G. Ritacca, A. Rovaletti, G. Moro, U. Cosentino, U. Ryde, E. Sicilia and C. Greco, ACS Catal., 2022, 12, 7336–7343 CrossRef CAS.
- H. S. Fernandes, S. F. Sousa and N. M. Cerqueira, Mol. Diversity, 2022, 26, 1373–1381 CrossRef CAS PubMed.
- M. Prejanò, F. E. Medina, M. J. Ramos, N. Russo, P. A. Fernandes and T. Marino, ACS Catal., 2020, 10, 2872–2881 CrossRef PubMed.
- L. Sánchez, F. E. Medina, F. Mendoza, C. Febres-Molina and G. A. Jaña, J. Chem. Inf. Model., 2022, 63, 270–280 CrossRef PubMed.
- D. Bím, M. Navrátil, O. Gutten, J. Konvalinka, Z. Kutil, M. Culka, V. Navrátil, A. N. Alexandrova, C. Barinka and L. Rulisek, J. Phys. Chem. B, 2022, 126, 132–143 CrossRef PubMed.
- M. Rostkowski, M. H. M. Olsson, C. R. Søndergaard and J. H. Jensen, BMC Struct. Biol., 2011, 11, 1–6 CrossRef PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- R. Galindo-Murillo, J. C. Robertson, M. Zgarbova, J. Sponer, M. Otyepka, P. Jurecka and T. E. Cheatham III, J. Chem. Theory Comput., 2016, 12, 4114–4127 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, 2016.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- N. Oezguen, C. H. Schein, S. R. Peddi, T. D. Power, T. Izumi and W. Braun, Proteins, 2007, 68, 313–323 CrossRef CAS PubMed.
- C. M. Dupureur, Curr. Opin. Chem. Biol., 2008, 12, 250–255 CrossRef CAS PubMed.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS PubMed.
- H. He, Q. Chen and M. M. Georgiadis, Biochemistry, 2014, 53, 6520–6529 CrossRef CAS PubMed.
- A. Jeltsch, J. Alves, H. Wolfes, G. Maass and A. Pingoud, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8499–8503 CrossRef CAS PubMed.
- H. Viadiu and A. K. Aggarwal, Nat. Struct. Biol., 1998, 5, 910–916 CrossRef CAS PubMed.
- J. Gan, G. Shaw, J. E. Tropea, D. S. Waugh, D. L. Court and X. Ji, Mol. Microbiol., 2007, 67, 143–154 CrossRef PubMed.
- R. Balhara, R. Chatterjee and G. Jindal, Phys. Chem. Chem. Phys., 2021, 23, 9500–9511 RSC.
- Q. Cheng and N. J. DeYonker, J. Phys. Chem. B, 2021, 125, 3296–3306 CrossRef CAS PubMed.
- F. Planas, M. J. McLeish and F. Himo, ACS Catal., 2021, 11, 12355–12366 CrossRef CAS.
- T. J. Summers, Q. Cheng, M. A. Palma, D.-T. Pham, D. K. Kelso, C. E. Webster and N. J. DeYonker, Biophys. J., 2021, 120, 3577–3587 CrossRef CAS PubMed.
- H. Feng, A. M. Klutz and W. Cao, Biochemistry, 2005, 44, 675–683 CrossRef CAS PubMed.
- J. Wu, N. L. Samara, I. Kuraoka and W. Yang, Mol. Cell, 2019, 76, 44–56 CrossRef CAS PubMed.
- S. Gong, H. H. Yu, K. A. Johnson and D. W. Taylor, Cell Rep., 2018, 22, 359–371 CrossRef PubMed.
- H. Wu, W. F. Lima and S. T. Crooke, J. Biol. Chem., 2001, 276, 23547–23553 CrossRef CAS PubMed.
- F. Eckstein, Annu. Rev. Biochem., 1985, 54, 367–402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information for crystal structures of I-PpoI (Fig. S1), as well as EcoRI, BamHI, T4 EndoVII, Hpy188I, I-HmuI, and AaRNase III (Fig. S6), QM cluster and QM/MM models involving indirect metal–substrate coordination (Fig. S2), comparison of ICs for Model 2 (Fig. S3), comparison of M06-2X and ωB97M-V Gibbs activation energies (Fig. S4), QM and QM/MM optimized RCs for the experimentally-proposed mechanism (Fig. S5), QM optimized stationary points for Models 1–4 (Fig. S7–S10), overlays of QM/MM stationary points with crystal structures (Fig. S11), comparison of ME and EE-optimized QM/MM stationary points (Fig. S12), data for H98A mutant (Fig. S13), overlay of QM/MM I-PpoI and X-ray AaRNase III PC (Fig. S14), M06-2X and ωB97M-V Gibbs energies (Table S1), Gibbs energies for models involving indirect (Table S2) and direct (Table S3) metal–leaving group coordination. Cartesian coordinates for each stationary point from all wild-type and in silico H98A I-PpoI mutant pathways as a zip file. See DOI: https://doi.org/10.1039/d3cp06201e |
| This journal is © the Owner Societies 2024 |