
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1H and 13C chemical shift–structure effects in anhydrous β-caffeine and four caffeine–diacid cocrystals probed by solid-state NMR experiments and DFT calculations†
Debashis
Majhi‡
,
Baltzar
Stevensson
 ,
Tra Mi
Nguyen
and
Mattias
Edén
*
,
Tra Mi
Nguyen
and
Mattias
Edén
*
Department of Materials and Environmental Chemistry, Stockholm University, SE-106 91, Stockholm, Sweden. E-mail: mattias.eden@mmk.su.se
First published on 8th April 2024
Abstract
By using density functional theory (DFT) calculations, we refined the H atom positions in the structures of β-caffeine (C), α-oxalic acid (OA; (COOH)2), α-(COOH)2·2H2O, β-malonic acid (MA), β-glutaric acid (GA), and I-maleic acid (ME), along with their corresponding cocrystals of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (2C–OA, 2C–MA) or 1:1 (C–GA, C–ME) stoichiometry. The corresponding 13C/1H chemical shifts obtained by gauge including projector augmented wave (GIPAW) calculations agreed overall very well with results from magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy experiments. Chemical-shift/structure trends of the precursors and cocrystals were examined, where good linear correlations resulted for all COO1H sites against the H⋯O and/or H⋯N H-bond distance, whereas a general correlation was neither found for the aliphatic/caffeine-stemming 1H sites nor any 13C chemical shift against either the intermolecular hydrogen- or tetrel-bond distance, except for the 13COOH sites of the 2C–OA, 2C–MA, and C–GA cocrystals, which are involved in a strong COOH⋯N bond with caffeine that is responsible for the main supramolecular stabilization of the cocrystal. We provide the first complete 13C NMR spectral assignment of the structurally disordered anhydrous β-caffeine polymorph. The results are discussed in relation to previous literature on the disordered α-caffeine polymorph and the ordered hydrated counterpart, along with recommendations for NMR experimentation that will secure sufficient 13C signal-resolution for reliable resonance/site assignments.
:1 (2C–OA, 2C–MA) or 1:1 (C–GA, C–ME) stoichiometry. The corresponding 13C/1H chemical shifts obtained by gauge including projector augmented wave (GIPAW) calculations agreed overall very well with results from magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy experiments. Chemical-shift/structure trends of the precursors and cocrystals were examined, where good linear correlations resulted for all COO1H sites against the H⋯O and/or H⋯N H-bond distance, whereas a general correlation was neither found for the aliphatic/caffeine-stemming 1H sites nor any 13C chemical shift against either the intermolecular hydrogen- or tetrel-bond distance, except for the 13COOH sites of the 2C–OA, 2C–MA, and C–GA cocrystals, which are involved in a strong COOH⋯N bond with caffeine that is responsible for the main supramolecular stabilization of the cocrystal. We provide the first complete 13C NMR spectral assignment of the structurally disordered anhydrous β-caffeine polymorph. The results are discussed in relation to previous literature on the disordered α-caffeine polymorph and the ordered hydrated counterpart, along with recommendations for NMR experimentation that will secure sufficient 13C signal-resolution for reliable resonance/site assignments.
1 Introduction
Efficient administration of active pharmaceutical ingredients (APIs) is often challenging because the powders are either unstable or feature a low solubility in water/body fluids. One option for improving the solubility and/or other physical or chemical properties without recourse to ionic salts is the usage of a cocrystal, i.e., a supramolecular compound of an API and another molecular species (or even another API, a so-called drug–drug cocrystal).1–3 Consequently, intense efforts have been spent during the past two decades to explore synthetic routes to cocrystal preparation as well as their structures and formation mechanisms.2–5One widely used API is the stimulant caffeine (1,3,7-trimethylpurine-2,6-dione), whose storage and precise quantitative (physiological) administration is complicated by its highly hygroscopic nature.6–8 At room temperature (RT) in the absence of humidity, anhydrous caffeine exists as the low-temperature “β” modification, which at temperatures >150 °C converts into the metastable high-temperature “α” form, which transforms very slowly into β-caffeine at RT and dry air.9 Hence, commercial anhydrous caffeine powders often comprise a mixture of both modifications. Caffeine also exists in a monohydrate form, which unfortunately is very unstable at RT and converts either into phases with <1 water molecule/caffeine moiety or anhydrous β-caffeine (depending on the relative humidity).7,9 Hence, large-scale storage, processing and other handling of caffeine powder with a well-defined stoichiometry is not trivial.
Yet, as demonstrated by Trask et al.,8 cocrystals of caffeine with diacids, such as oxalic acid (OA), malonic acid (MA), glutaric acid (GA), and maleic acid (ME), are stable across wide humidity ranges. Several studies are reported on their formation, stability, and physical properties.8,10–14 The present investigation targeted the cocrystal structures obtained from β-caffeine (β-C) with each of OA, MA, GA, and ME, abbreviated as 2C–OA, 2C–MA, C–GA, and C–ME, respectively, according to the corresponding 2:1 and 1:1 caffeine:diacid stoichiometry. Although a fixed stoichiometry would be desirable, the C–ME cocrystal is metastable,10 while 2C–OA, 2C–MA, and C–GA cocrystals were hitherto not reported. Each X-ray diffraction (XRD) derived cocrystal structure is reported,8 encompassing the main caffeine–diacid motifs governing their supramolecular organization via hydrogen bonds (Section 3). The well-known limitation of XRD to locate precise H-atom positions, however, restricts its capacity of unveiling quantitative details on weak non-covalent interactions, such as H⋯O/N hydrogen and C⋯O/N tetrel bonds that account for the intermolecular interactions and structural organization of cocrystals, as well as numerous other supramolecular systems. Then, high-resolution magic-angle-spinning (MAS) 1H and 13C nuclear magnetic resonance (NMR) spectroscopy offer valuable complementary structural insight,15–20 in particular for structurally disordered (in)organic phases,18,20,21 encompassing β-caffeine.14,22–24
Herein, we present a comprehensive combined MAS NMR and density functional theory (DFT) study, reporting on DFT refinements of previous XRD-derived structures of each 2C–OA, 2C–MA, C–GA, and C–ME cocrystal8 together with each β-caffeine,9 α-OA,25 α-OA·2H2O,26 β-MA,27 β-GA,28 and I-ME29 precursor phase. 1H and 13C isotropic chemical shifts calculated by the gauge including projector augmented wave (GIPAW) method30–33 were contrasted with those from MAS NMR experiments, aiming at improved insight into the shift–structure trends, notably the hydrogen bond (HB)34–46—and for the methyl moieties also tetrel bond (TB)47–49—effects on the chemical shifts. While the complete sets of 13C chemical shifts/NMR-peak assignments of the 2C–MA and C–GA cocrystals were reported earlier by Vigilante and Mehta,50 out of the four cocrystals considered herein, a majority of the 1H chemical shifts are reported for the first time, along with nearly half of their 13C counterparts, such as those for 2C–OA and C–ME. We also present the first complete 13C NMR spectral assignment of the ten resonances from the eight unique 13C sites of the structurally disordered anhydrous β-caffeine polymorph. The results are discussed in relation to previous 13C MAS NMR reports on the ordered hydrated and anhydrous disordered α/β-caffeine and polymorphs, highlighting the role of the magnetic field for unambiguously discriminating between the α- and β-caffeine forms.
2 Materials and Methods
2.1 Cocrystal preparation
Anhydrous β-caffeine (98% purity), oxalic acid (98%), malonic acid (99%), glutaric acid (99%) and chloroform were obtained from Sigma-Aldrich along with maleic acid (99%) from Thermo Fisher. All cocrystals were prepared by solvent drop grinding,8,13 using ball milling (Mixer Mill 500 Vario; Retsch) of the two precursor powders that were placed in a 25 mL stainless steel jar and ground with 7 mm stainless steel balls at a rate of 30 Hz for 90 min. All cocrystals were prepared from stoichiometric amounts of the precursors. The 2C–OA (space group P21/c) and 2C–MA (Fdd2) cocrystals8 with a 2:1 caffeine:diacid stoichiometry resulted by mixing 250 mg of β-caffeine with either 60 mg of α-OA or 70 mg of β-MA, respectively, along with 5 drops of chloroform. Likewise, the 1:1 stoichiometric C–GA (form II;8 space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) cocrystal was prepared by grinding 510 mg of β-caffeine with 350 mg of β-GA and a few drops of chloroform, and likewise for the C–ME (P21/n) counterpart (250 mg of β-MA and 150 mg of I-ME).8
) cocrystal was prepared by grinding 510 mg of β-caffeine with 350 mg of β-GA and a few drops of chloroform, and likewise for the C–ME (P21/n) counterpart (250 mg of β-MA and 150 mg of I-ME).8
2.2 Powder X-ray diffraction
Powder XRD (PXRD) patterns were collected from all cocrystals and precursors with a Bruker D8 Discover diffractometer equipped with a LYNXEYE position-sensitive detector, using Cu Kα1,2 radiation (λ1 = 154.06 pm; λ2 = 154.44 pm) and a focusing Göbel mirror. Each powder was filled in a 0.7 mm borosilicate glass capillary. All diffractograms were collected at RT over a 2θ range of 5°–50°, employing a step size of 0.02° with either 0.25 s per step (for β-caffeine, OA, β-MA, β-GA, I-ME, 2C–MA, and C–ME) or 0.75 s per step (for 2C–OA and C–GA), giving total measurement times of around 10 and 30 min per sample, respectively. The XRD patterns of the precursors and cocrystals are shown in Fig. S1 and S2 (ESI†), respectively. Rietveld refinements and Pawley fits, performed with the TOPAS software,51 verified a near-100% purity of all specimens, except for the oxalic acid powder, which comprised a mixture of α-OA (96.1 wt%), α-OA·2H2O (3.5%), and β-OA (0.39 wt%). The anhydrous β-caffeine purity was ≥99.6%, along with a (statistically unascertained) minute amount of α-caffeine (0.37 wt%). All refined unit cell parameters are listed in Table S1 (ESI†).2.3 Solid-state NMR
All solid-state NMR experiments were performed at ambient temperature with a Bruker Avance-III spectrometer and a magnetic field (B0) of 14.1 T, which provided the respective 1H and 13C Larmor frequencies of −600.1 MHz and −150.9 MHz. 1H and 13C chemical shifts are quoted relative to neat tetramethylsilane (TMS).1H→13C cross polarization (CP) MAS NMR (CPMAS) experimentation was performed with filled 4.0 mm zirconia rotors spinning at the MAS rate (νr) of 12.00 kHz, using contact periods (τCP) of 1.25 ms (except for β-MA; 833 μs and νr = 9.00 kHz), and the modified Hartmann–Hahn condition νH = νC + νr, where the 13C nutation frequency (νC) was ramped linearly52 by ±4.7 kHz around νC = 47 kHz. Throughout, the 90° 1H pulse prior to CP operated at νH ≈ 100 kHz and SPINAL-6453 proton decoupling at νH ≈ 78 kHz (6.4 μs pulses) was applied during signal detection. The relaxation delays (τrelax) varied between 15 s and 30 s. The signal averaging involved 2048–3328 co-added signal transients, except for MA and GA (1024). Single-pulse (“Bloch decay”) 1H MAS NMR spectra were recorded with 1.3 mm zirconia rotors undergoing fast MAS at 60.00 kHz with 90° rf pulses operating at the 1H nutation frequency νH ≈ 80 kHz. 256–512 accumulated NMR-signal transients were recorded with τrelax = 3 s. These relaxation delays were not sufficiently long to ensure NMR intensities that quantitatively reflect the relative H/C site populations in the structures but do not affect any conclusions from our analysis, which only concerned the chemical shifts. NMR spectral deconvolutions were performed with in-house developed software.54
2.4 DFT/GIPAW calculations
DFT energy optimizations were performed on the following published PXRD-derived structures: β-caffeine;9 α-OA;25 α-OA·2H2O;26 β-MA;27 β-GA;28 I-ME,29 whereas all cocrystal structures were from ref. 8; see Table S2 (ESI†). The first-principle DFT energy minimizations were performed with the CASTEP software55 (version 22.11) along current standard and well-developed protocols,19,32,33 encompassing usage of the local density approximation (LDA) functional56 with on-the-fly-generated ultrasoft pseudopotentials57 and a plane-wave basis set.58 The Tkatchenko and Scheffler method was employed for dispersion corrections.59 All structures were initially optimized by solely adjusting the proton positions with fixed positions of all heavier atoms and unit-cell parameters. However, the agreement between the experimental and GIPAW-derived chemical shifts improved slightly for the β-caffeine structure by optimizing all cell parameters and atom positions (Table S2 and Section 4.3, ESI†). The 1H and 13C magnetic shielding values were calculated with the GIPAW method19,30–33 for all DFT-optimized structures. For both the DFT energy optimizations and the GIPAW shielding-parameter calculations, a Monkhorst–Pack k-point grid60 was used with a maximum spacing of 0.05 Å−1 in the reciprocal space, and a 1000 eV plane-wave energy cutoff to ensure convergence.The DFT/GIPAW-derived principal values, {σHjxx, σHjyy, σHjzz} and {σCjxx, σCjyy, σCjzz}, of the respective second-rank magnetic shielding tensor of each unique 1Hj and 13Cj site in the structure were converted into the corresponding chemical shift values, {δSjxx, δSjyy, δSjzz}, by the expression19,32,33
| δSjαα = σSref − σSjαα, with αα = {xx, yy, zz} and S = {1H, 13C}. | (1) |
 | (2) |
3 Overview of cocrystals and their precursor structures
Fig. 1 shows the structures of caffeine and the four diacid molecules, where each number represents the 13C site index, giving the label “Cj” with 1 ≤ j ≤ 22, while all its directly bonded H, O, and N atoms carry the same index j. Note that although the C14/C18 sites are crystallographically equivalent in the β-GA structure, they are inequivalent in the C–GA cocrystal. For simplicity, we employ a strict C1–C22 labelling because all C/H sites are crystallographically distinct in either/both the diacid/cocrystal structure, except for C9/C10 (H9/H10), which remain equivalent in each α-OA, α-OA·2H2O, and 2C–OA structure. | ||
| Fig. 1 Molecular structure with atom numbering of (a) caffeine, and the (b) oxalic, (c) malonic, (d) glutaric, and (e) maleic diacids. Each label/index j refers to carbon atom “Cj”, whose directly bonded O, N, and H atoms feature the same label (Oj, Nj, and Hj, respectively), except for the nitrogen atom of the imidazolium ring (cyan color) in (a), which is referred to simply as “N”. | ||
Fragments from the by DFT energy-minimized crystal structures (Section 2.4) are shown in Fig. 2 for the diacids (a, b, d, f, h) and their respective cocrystals with caffeine (c, e, g, i). All pristine diacid structures involve strong intermolecular H bonds between the carboxy groups of neighboring molecules, except for I-ME, which besides one expected C19OOH⋯O22 contact also features a short intramolecular C22OOH⋯O19 HB (Fig. 2h), which remains intact also in the C–ME cocrystal shown in Fig. 2i, within a minute (2 pm) lengthening.
 | ||
| Fig. 2 Fragments from the DFT-refined crystal structures of (a) α-OA, (b) α-OA·2H2O, (c) 2C–OA, (d) β-MA, (e) 2C–MA, (f) β-GA, (g) C–GA, (h) I-ME, and (i) C–ME. The dotted lines mark intermolecular H and tetrel bonds, with the accompanying number specifying the distance (in pm). The red and green lines/numbers distinguish tetrel bonds between caffeine–diacid and caffeine–caffeine molecules, respectively. Note that each methyl group undergoes a rapid rotation around the C–N bond and that only one H bond is indicated for each of the three equivalent protons. | ||
As discussed by Trask et al.,8 both 2C–OA and 2C–MA cocrystals feature a heteromeric synthon where one diacid molecule is sandwiched between two caffeine units by a strong COOH⋯N hydrogen bond and a weaker C4H⋯OOC counterpart (Fig. 2c and e). The alternating caffeine–diacid–caffeine moieties of the supramolecular structure are stabilized primarily by those H bonds.8 The 1:1 caffeine:diacid stoichiometries of C–GA and C–ME, however, yield different intermolecular interactions. Owing to the intact intramolecular C22H⋯O19 bond in the C–ME cocrystal (Fig. 2i), its structure involves strictly alternating caffeine–ME interconnections, with a similar C4H⋯O19OC and C19OOH⋯N bond constellation as in the 2C–OA/MA cocrystals, yet with only one diacid-COOH group H-bonded to caffeine. Moreover, while the C–GA structure is also stabilized by two N⋯H14OOC and C4H⋯O14OC bonds between caffeine and one GA molecule (Fig. 2g), the C4H atom also involves another HB with O18 of a second diacid molecule, where moreover C18H is bonded to O14 of the first diacid unit. Hence, each caffeine molecule in C–GA features three different H bonds to two distinct GA molecules, both of which are additionally interlinked by a HB between their carboxy moieties, as in the parent GA structure.
Although the N⋯H bond between caffeine and a COOH group of a diacid unit constitutes the strongest intermolecular contact of the cocrystal,8 a further minor structure stabilization is arranged by H bonds between the CH3 groups of caffeine with either diacid carboxy moieties or the O1/O2 atoms of neighboring caffeine molecules (Fig. 2c, e, g and i). The CH3 moieties may furthermore form weak CH3⋯O tetrel bonds47–49 with the O1/O2 atoms of caffeine, or with the CO counterparts of the diacids, as marked in green and red color, respectively, in Fig. 2c, e, g and i (see Section 4.7). The methyl groups also form intermolecular tetrel and H bonds in the structurally disordered β-caffeine structure (Table S3, ESI†). We refer to ref. 9 and 22 for details about the anhydrous β-caffeine structure, whose unit cell comprises five inequivalent molecules, and thereby five NMR signal-contributions to each detected 13Cj and 1Hj resonance.
4 Results and discussion
4.1. 13C MAS NMR spectra from cocrystal precursors
Because the chemical shift of a given 13C or 1H nuclear site reflects its electronic environment in the structure, it is often a sensitive probe of the precise location of neighboring atoms in close vicinity. Fig. 3 displays the 13C CPMAS NMR spectra recorded from the diacids and β-caffeine cocrystal precursors. They involve resonances ranging from the most deshielded 13C nuclei (high chemical shifts) of carboxy groups with δC ≳ 160 ppm from the diacids (Fig. 3a–d) to the more shielded (lower shifts) aliphatic 13C sites with chemical shifts δC ≲ 40 ppm from β-MA, β-GA, and β-caffeine (Fig. 3b, c and e). The NMR spectral region intermediate of these extreme 13C shifts encompasses the comparatively deshielded 13C20 and 13C21 sites of I-ME (Fig. 3d), together with the 13C1–13C5 sites of β-caffeine resonating between 105–155 ppm (Fig. 3e).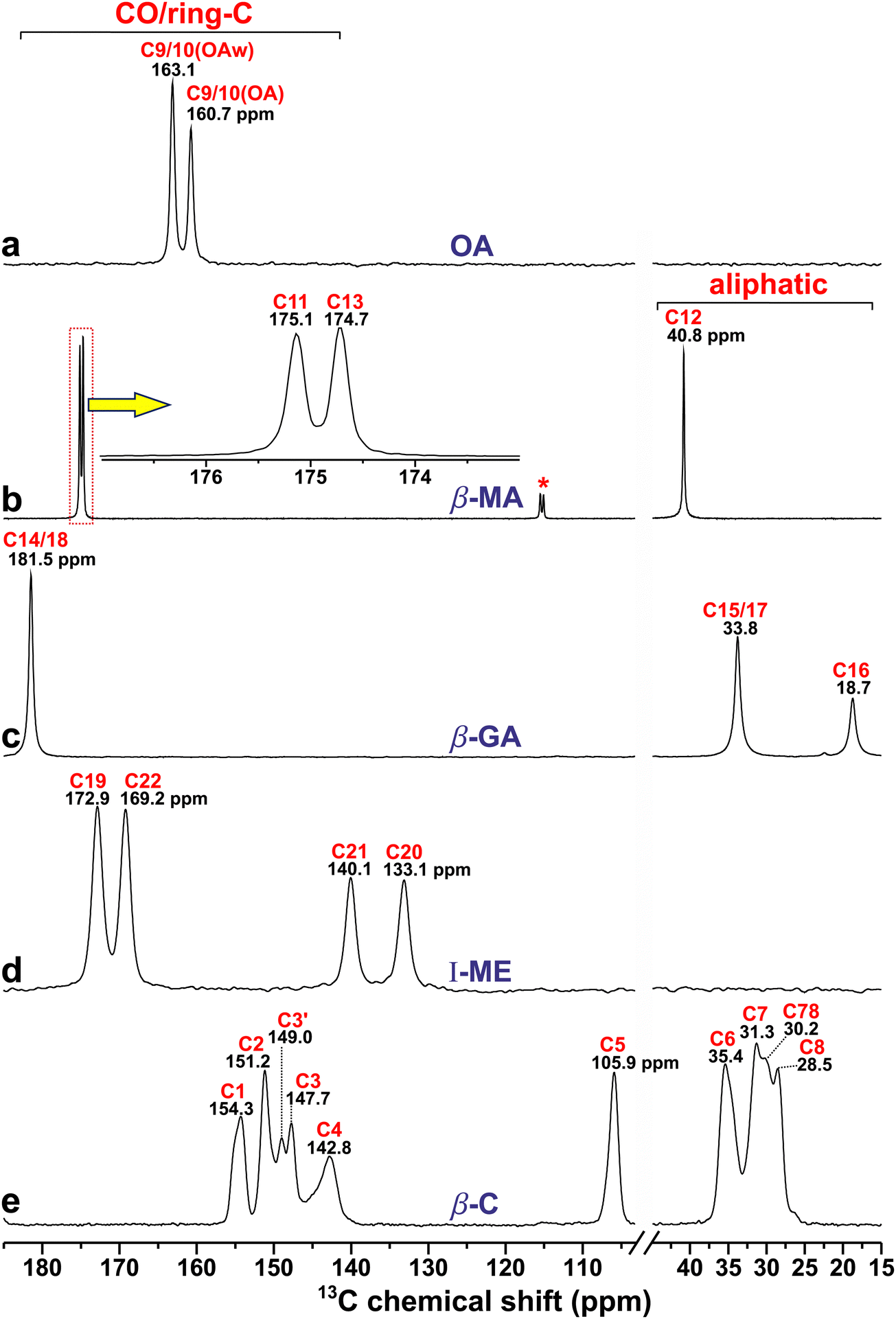 | ||
| Fig. 3 13C CPMAS NMR spectra recorded at 14.1 T and a MAS rate of 12.00 kHz from (a) α-OA/α-OA·2H2O mixture (“OA”), (b) malonic acid (β-MA), (c) glutaric acid (β-GA), (d) maleic acid (I-ME), and (e) anhydrous β-caffeine (β-C). Here and in other figures: NMR signals from the α-OA and α-OA·2H2O phases are abbreviated by “OA” and “OAw”, respectively, whereas the red and black number above each NMR peak marks the C label of Fig. 1 and the chemical shift (in ppm) at the peak maximum, respectively. The inset of (b) is a zoom of the NMR spectral region marked by the dotted rectangle, and the asterisk marks a spinning sideband from the use of a lower MAS rate (9.00 kHz) for this experiment. | ||
All cocrystal precursor powders (but OA) were phase pure, where the two crystallographically inequivalent 13COOH sites of MA and ME reveal chemical-shift differences of 0.4 ppm and 3.7 ppm, respectively, while both 13COOH sites of GA resonate at 181.5 ppm. In contrast, the two 13C NMR signals at 160.7 ppm and 163.1 ppm from OA derive from anhydrous α-OA and α-OA·2H2O, respectively, each featuring one crystallographically unique 13COOH site.25,26 From the integrated 13C NMR intensities of Fig. 3a, and confirmed by integrating the corresponding 1H MAS NMR spectrum (Section 4.5.1), we estimated relative OA:OA·2H2O amounts of approximately 0.7:1.0 in the oxalic acid mixture. (Although that estimate is only approximate, its precise value is immaterial for our subsequent analyses. The reason for the markedly higher OA·2H2O content derived by NMR relative to PXRD is unknown but likely stems from a water uptake of the OA powder prior to the NMR experiments). The 13C chemical shifts obtained from Fig. 3 agree well with previous reports from α-OA,36,64 α-OA·2H2O,36 MA,14,65 GA,36,65,66 and ME.65–67
4.2. 13C NMR-peak assignments of anhydrous β-caffeine
Notwithstanding several 13C NMR publications on caffeine-based cocrystals,23,24,41,46,49,50 encompassing complete {δC} reports thereof,23,50 ambiguities prevail about the precise set of 13C shifts and their assignments of the anhydrous β-caffeine structure itself, which produces two hitherto unassigned 13C resonances, none of which are present in MAS NMR spectra from either the structurally ordered caffeine monohydrate (henceforth referred to as C·H2O) or disordered anhydrous α-caffeine modifications,22 each of which manifests one 13C resonance per C1–C8 site. Enright et al.22 presented 13C MAS NMR spectra from all α-/β-caffeine and C·H2O forms, but precise chemical shifts were only reported for the latter.22 Likewise, δC values were not provided along with the 13C MAS NMR spectra from the anhydrous caffeine modifications studied in ref. 12, 24, 50 and 68. To the best of our knowledge, Table 1 presents the precise 13C chemical shifts and complete peak assignments for the anhydrous β-caffeine polymorph for the first time, where all further references herein to “α/β-caffeine” imply the anhydrous polymorphs. The 13C MAS NMR spectrum from β-caffeine of Fig. 3e appears to match very well that reported in ref. 22 while moreover our PXRD analysis confirmed a phase-pure specimen (Section 2.2). Notwithstanding that all eight {δnC} values of both α/β-caffeine modifications are (very) similar,22 the presence of two additional 13C NMR peaks at 149 ppm and 30 ppm in β-caffeine (Fig. 3e) distinguishes it from its “α” and C·H2O counterparts, which Enright et al.22 attributed to 13C sites of crystallographically distinct molecules in the β-caffeine unit cell. That very plausible suggestion is confirmed below.| Site | δ j C[NMR] (ppm) |
![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) j
C[NMR] (ppm)
j
C[NMR] (ppm) |
j
C[DFT] (ppm) |
δ j C[DFT] (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
a
13C chemical shift at the NMR peak maximum, δjC[NMR], along with the center-of-gravity (average) chemical shift obtained by deconvolution of the MAS NMR spectrum (Fig. S3, ESI), jC[NMR], or by DFT calculations, δjC[DFT]; the latter values are averages over the 13C chemical shift values of five distinct caffeine molecules in the unit cell, each of which is given in the five rightmost columns.
b The C3 sites produces two resonances at 149.0 ppm and 147.7 ppm, which are labelled by “C3′” and “C3” in Fig. 3e.
c The “C78” 13C resonance intensity at 30.2 ppm (Fig. 3e) involves equal contributions from the 13C7 and 13C8 sites.
|
||||||||||
| C1 | 154.3 | 154.5 ± 0.2 | 153.0 ± 0.3 | 152.5 | 152.7 | 152.8 | 152.9 | 154.1 | ||
| C2 | 151.2 | 150.8 ± 0.2 | 149.4 ± 0.3 | 148.3 | 148.9 | 149.5 | 149.7 | 150.5 | ||
| C3b | 149.0/147.7 | 148.0 ± 0.2 | 147.5 ± 0.6 | 146.5 | 146.7 | 147.4 | 147.7 | 149.4 | ||
| C4 | 142.8 | 143.5 ± 0.4 | 146.0 ± 1.3 | 142.9 | 143.9 | 145.0 | 148.9 | 149.3 | ||
| C5 | 105.9 | 106.1 ± 0.2 | 107.8 ± 0.6 | 106.2 | 106.9 | 107.9 | 108.1 | 109.7 | ||
| C6 | 35.4 | 35.0 ± 0.2 | 35.3 ± 0.6 | 33.7 | 34.8 | 35.4 | 35.7 | 36.7 | ||
| C7c | 31.3/30.2 | 30.7 ± 0.2 | 29.6 ± 0.9 | 28.0 | 28.3 | 29.3 | 30.5 | 32.2 | ||
| C8c | 30.2/28.5 | 29.6 ± 0.2 | 29.5 ± 0.6 | 27.7 | 28.6 | 29.2 | 29.9 | 32.0 | ||
Previous solid-state 13C NMR-peak assignments of all anhydrous and monohydrate caffeine polymorphs derive either from (i) employing DFT/GIPAW calculations of a caffeine-based cocrystal to assign its 13C sites of the caffeine moiety23,24,50 or from (ii) the solution-NMR work of Sitkowski et al.,69 which was exploited by Enright et al.22 for the 13C-site/NMR-peak identifications of the ordered C·H2O modification in the solid state (which is greatly facilitated by its narrow 13C resonances). Those peak assignments were subsequently assumed in ref. 14 and 70. Although such straightforward 13C site/shift-mappings are also sufficiently reliable for application to α-caffeine, which involves one resonance per 13C site, the peak-assignment strategies (i) and (ii) are precluded for the two additional resonances at around 149/30 ppm from β-caffeine and labelled as C3′/C78 in Fig. 3e. To the best of our knowledge, both remain unidentified in current literature. Nonetheless, for each 13C1–13C8 resonance assignment of Fig. 3e, the DFT/GIPAW-derived 13C chemical shifts of Table 1 corroborate previously employed NMR-peak assignments made for both C·H2O and α-caffeine modifications.14,22–24,70 The modeled chemical shifts, which were averaged over the 5 distinct caffeine molecules in the unit cell, reproduce the 13C NMR results from β-caffeine (very) well (Table 1), which for every asymmetric peakshape represents the center-of-gravity (CG) shift (jC) obtained from NMR-peak deconvolutions into two components (Fig. S3, ESI†) rather than the shift at the peak-maximum amplitude (δjC). Note that (I) jC and δjC represent the average and the most probable value over the chemical-shift distribution of 13Cj, respectively; (II) invoking CG shifts is only required for the structurally disordered β-caffeine structure, as opposed to those of any cocrystal or diacid precursor considered herein, for which jC = δjC throughout.
Despite an overall good agreement between the experimental and modeled 13C chemical shifts of β-caffeine, the DFT/GIPAW calculations alone cannot uniquely identify the hitherto unassigned resonances at 149.0 ppm and 30.2 ppm (Fig. 3e). By spectral deconvolution, however, the integrated 13C NMR-peak intensities at around {154.3, 151.2, 149.0, 147.7} ppm were found to relate as 1.00:1.44:0.27:0.74 (Fig. S3, ESI†). That observation naturally attributes the resonance at 149.0 ppm to C3 (it is therefore referred to as C3′ in Fig. 3e), leading to jC values of {154.5(C1), 150.8(C2), 148.0(C3)} ppm with relative intensities 1.00:1.44:1.01. The deviations from the expected exact 1:1:1 ratios (where the discrepancy is only significant for C2) mainly reflect that integrated 13C NMR-peak intensities obtained by 1H → 13C CPMAS NMR experiments do generally not exactly reproduce the corresponding 13C site populations in the structure. Likewise, the 13CH3-associated resonance at δjC = 30.2 ppm, which is labelled “C78” in Fig. 3e, was deduced to involve equal contributions from 13C7 and 13C8. Upon distributing half of the NMR-signal intensity at δjC = 30.2 ppm to each of 13C7 (δjC = 31.3 ppm) and 13C8 (δjC = 28.5 ppm), the as-obtained relative integrated NMR-signal intensities of the C6:C7:C8 sites become 1.00:1.03:0.96, which is in excellent agreement with the expected 1:1:1 ratios (Fig. S3, ESI†).
Further support for the spectral-deconvolution-derived attribution of the “C3′” and “C78” NMR peaks to the 13C3 site and equal contributions to 13C7/13C8, respectively, is provided by the resulting very close {3C, 7C, 8C} = {148.0, 30.7, 29.6} ppm values observed relative to the {δ3C, δ7C, δ8C} = {147.8, 30.6, 29.0} ppm counterparts of the structurally ordered C·H2O polymorph.22 The excellent agreement between the average 13C chemical shifts (i.e., jC) of the disordered β-caffeine structure obtained by “down-projecting” its 10 13C NMR peaks to the unique 8 13C resonance-mapping of the C·H2O polymorph22 is very gratifying. Moreover, almost all jC/δjC discrepancies between β-caffeine and C·H2O are well within the experimental uncertainties. For instance, the {1C, 5C, 6C} values of Table 1 agree within 0.2 ppm with the {δ1C, δ5C, δ6C} counterparts of C·H2O.22 Significant chemical-shift differences between the two polymorphs are only observed for the 13C2 and 13C4 carbonyl sites, both of which are ≈1.0 ppm higher for β-caffeine than those of C·H2O, which is attributed to distinct HB scenarios between the two structures. DFT calculations (not shown) reveal that the crystallographically unique C4H site of C·H2O involves one C4H⋯OH2 HB (201 pm), whereas the five distinct H4 sites of β-caffeine feature a range of HB distances to the O1 (198; 235; 250 pm), O2 (212 pm), and N (236 pm) sites of neighboring molecules. They contribute strongly to the “local” disorder of the 13C4 environment, and thereby to its sizable fwhm value relative to any other 13Cj site of β-caffeine, while also explaining the accompanying larger 4C–δ4C difference (Table 1).
4.3 NMR/GIPAW-derived 13C chemical shifts of cocrystals and precursors
Fig. 4 contrasts the 13C CPMAS NMR spectra recorded from β-caffeine and its four cocrystals. The successful completion of each cocrystal reaction is evidenced both from our PXRD analyses (Section 2.2) and by the unique 13Cj site/chemical-shift mapping resulting for all cocrystals, for which δjC of each 13C1–13C22 site differs only slightly relative to that of its precursor phases (Table 2). In particular, each of the 8 caffeine-moiety-related 13C resonances is readily identified for each cocrystal, meaning that no cocrystal manifests NMR peaks traceable to either “C3′” or “C78” of Fig. 4e, thereby eliminating all NMR-peak assignment ambiguities of β-caffeine. Hence, all 13C resonance identifications for each cocrystal (Fig. 4) readily follow from those established for its parent phases (Fig. 3), as corroborated further by contrasting the experimental and DFT/GIPAW-generated {δjC} values listed in Table 2. | ||
| Fig. 4 13C CPMAS NMR spectra acquired at 14.1 T and 12.00 kHz MAS from cocrystals of β-caffeine and (a) oxalic acid (2C–OA), (b) malonic acid (2C–MA), (c) glutaric acid (C–GA), and (d) maleic acid (C–ME), along with the spectrum from (e) β-caffeine (β-C) for reference. | ||
| Coformer | Site | Coformer δC (ppm) | 2C–OA δC (ppm) | 2C–MA δC (ppm) | C–GA δC (ppm) | C–ME δC (ppm) | ||
|---|---|---|---|---|---|---|---|---|
|
a
13C chemical shifts (δjC) for the 13Cj site labels of Fig. 1, obtained either experimentally from the shift at the NMR-peak maximum, or by DFT/GIPAW calculations (values within parentheses). The δC uncertainties are around ±0.30 ppm (DFT/GIPAW) and ±0.15 ppm (NMR), except for β-caffeine (Table 1).
b For β-caffeine (only), center-of-gravity 13C chemical shifts (jC) are reported rather than δjC. The experimental {jC} data were obtained by deconvolution of the MAS NMR spectrum, while each DFT/GIPAW-derived jC value is the average chemical shift over five crystallographically distinct sites/molecules (see Table 1).
|
||||||||
| β-Caffeineb | C1 | 154.5(153.0) | 154.3(153.1) | 154.6(154.8) | 155.5(154.5) | 154.1(153.1) | ||
| C2 | 150.8(149.4) | 152.0(150.4) | 152.1(150.1) | 150.6(148.9) | 151.8(150.5) | |||
| C3 | 148.0(147.5) | 146.0(147.0) | 146.4(146.6) | 146.7(148.5) | 145.4(145.1) | |||
| C4 | 143.5(146.0) | 143.3(145.2) | 144.1(146.6) | 143.0(145.0) | 142.6(143.2) | |||
| C5 | 106.1(107.8) | 107.7(110.9) | 108.0(110.8) | 107.4(110.6) | 107.6(110.2) | |||
| C6 | 35.0(35.3) | 36.5(36.2) | 34.0(33.0) | 34.3(33.5) | 36.5(36.6) | |||
| C7 | 30.7(29.6) | 31.1(28.5) | 31.6(30.0) | 31.5(29.1) | 31.4(29.7) | |||
| C8 | 29.6(29.5) | 27.9(25.2) | 28.3(25.8) | 27.8(24.2) | 29.0(27.4) | |||
| α-OA | C9/10 | 160.7(162.9) | 159.7(162.2) | |||||
| α-OA·2H2O | C9/10 | 163.1(164.5) | 159.7(162.2) | |||||
| β-MA | C11 | 175.1(179.0) | 168.2(170.1) | |||||
| C12 | 40.8(39.7) | 43.7(44.3) | ||||||
| C13 | 174.7(175.4) | 168.2(170.1) | ||||||
| β-GA | C14 | 181.5(186.6) | 179.8(183.4) | |||||
| C15 | 33.8(33.8) | 36.2(36.0) | ||||||
| C16 | 18.7(16.5) | 22.2(22.0) | ||||||
| C17 | 33.8(33.8) | 32.7(32.2) | ||||||
| C18 | 181.5(186.6) | 177.0(182.8) | ||||||
| I-ME | C19 | 172.9(173.4) | 172.4(174.2) | |||||
| C20 | 133.1(141.4) | 129.8(137.9) | ||||||
| C21 | 140.1(148.1) | 138.9(146.4) | ||||||
| C22 | 169.2(168.5) | 164.2(167.2) | ||||||
While hitherto no report on any even partially complete set of {δjC} data or 13C NMR-peak assignments appears to exist for the 2C–OA and C–ME cocrystals, very notable is the excellent agreement between the 13C chemical shifts obtained herein for 2C–MA and C–GA with those reported earlier by Vigilante and Mehta:50 the truly marginal discrepancies of ≤0.2 ppm are well within the experimental uncertainties throughout, where systematic 13C MAS NMR shift-referencing errors often yield δjC discrepancies exceeding 0.5 ppm between studies. For instance, the two 13C chemical shifts reported by Bryce and coworkers for the 13C4 (142.9 ppm)46 and 13C8 (28.3 ppm)49 sites of C–ME accord with those of Table 2 within 0.3 ppm and 0.7 ppm, respectively.
Table 2 reveals a typical agreement of 2–3 ppm between the experimental and modeled {δjC} values across all diacid precursors and their corresponding cocrystals with caffeine, where deviations ≤1 ppm and ≤3 ppm result for 24 and 56 sites out of the entire ensemble of 68 sites/shifts, respectively. Out of the 12 13Cj sites manifesting a >3.0 ppm δjC discrepancy between the model and experiment, however, 8 sites concern the GA and ME diacid precursors and their cocrystals, where the GIPAW calculations of the GA-associated phases consistently overestimate the 13COOH shifts, whereas the 13CH2 counterparts are in excellent agreement with experiments. That scenario is reversed for the ME/C–ME structures, for which the NMR/GIPAW-derived 13COOH chemical shifts accord well, whereas substantial deviations are observed for the 13CH sites (Table 2). Similar systematic errors—but of overall much smaller magnitudes—are observed for the 2C–OA and 2C–MA cocrystals and their OA, OA·2H2O, and MA precursors, where the modeled {δjC} values are consistently higher that their experimental counterparts. While Table 2 also reveals minor systematic discrepancies for the C1–C8 sites of the caffeine moiety in the cocrystals relative to β-caffeine, the GIPAW-derived {δjC} values are typically lower than those from NMR, except for C4/C5 that conform to the more typical trend of overestimated δjC values by the calculations.
Because most deviations between the NMR/GIPAW-derived 13C chemical shifts are systematic, they have little/no bearings on our analyses below that target the shift-difference between the cocrystals and precursors (Section 4.6). Nonetheless, the very significant discrepancies between a few experimental/modeled δjC values for the GA and ME moieties in both precursor and cocrystal structures are reasons for concern. We remind that our δjC values of Table 2 originated from DFT energy minimizations where only the H positions were adjusted, except for β-caffeine, for which all unit-cell parameters and atom coordinates were optimized (Table S2, ESI†). That significantly improved the chemical-shift predictions, as reflected in a root-mean-square deviation (rmsd) of 1.4 ppm relative to the experimental shifts, which may be contrasted with the rmsd(DFT/NMR) = 2.0 ppm outcome that resulted by only adjusting the H atom positions of β-caffeine, or with the corresponding rmsd(DFT/NMR) = 3.0 ppm obtained across the entire {δjC} ensemble from all other phases (60 data points). Yet, there were no improvements by employing full-atom/cell optimizations of the {GA, C–GA} and {ME, C–ME} structures (which manifest the globally largest δjC discrepancies), whose {δjC} sets revealed an even larger rmsd(DFT/NMR) = 6.5 ppm relative to its experimental counterpart, which exceeded that of rmsd(DFT/NMR) = 4.5 ppm resulting by solely optimizing the H atom positions (Table 2). We have no satisfactory explanation for the large discrepancies (≳5 ppm) between the models and experiments for a few 13C sites of the GA/ME-related structures, which must originate from unaccounted structural effects.
4.4. Factors governing the 13C resonance widths
Here we discuss three factors expected to primarily govern the full width at half maximum (fwhm) height of the 13C signals observed from the cocrystals and their precursors (Table S4, ESI†), leading to some recommendations about the choice of external magnetic field (B0) for arranging (sub)optimal 13C MAS NMR spectral resolution. The two primary spectral-resolution-limiting factors involve so-called “inhomogeneous broadening”,61i.e., 13C chemical-shift dispersions from either the anisotropic bulk magnetic susceptibility (ABMS)24,71–74 or static structural disorder.18,20,21 Both scale linearly with B0 on a frequency scale (in Hz), which complicates their discrimination if only having a 13C (CP)MAS NMR spectrum available.24,73,74 Yet, the ABMS-stemming broadening in ppm is shared among all nuclear sites in the structure, regardless of their nuclide type or structural origin, meaning that the ABMS remains constant for all {13Cj} and {1Hj} sites in the sample,24,71–74 in contrast to the local structural disorder that varies among crystallographically distinct 13Cj sites, and thereby giving variable 13C fwhm values (in ppm). Hence, contrasting the (lack of) variations within the {13Cj} fwhm set listed in Table S4 (ESI†) within each precursor/cocrystal helps gauging the potential presence of structural disorder.Fig. 3 reveals that the 13C resonance widths vary markedly among the various well-ordered cocrystal precursors, which most likely reflects variable ABMS effects. The polycrystalline MA powder produces fwhm values of <30 Hz (≤0.2 ppm at 14.1 T; Table S4, ESI†), which are typical for organic molecules in well-ordered crystals with negligible ABMS broadening. In contrast, the OA, OA·2H2O, and GA powders reveal 3–4 times broader 13C resonances than MA, whereas the 13C sites of the ME and β-caffeine specimens manifest substantially larger fwhm values of ≈240 Hz (≈1.6 ppm) and 200–435 Hz (1.3–2.9 ppm), respectively. Here, the near-constant peakwidths observed for ME suggests that ABMS broadening mainly limits its 13C NMR spectral resolution, whereas structural disorder is mainly degrading that for β-caffeine, notably so in the CH3 region (Fig. 3e). Indeed, the markedly larger 13Cj fwhm values of β-caffeine relative to all other specimens (Fig. 3 and 4) primarily stems from a significant (static) structural disorder,22,24 where the lowest fwhm value of 1.3 ppm observed for the 13C5 resonance sets an upper limit of the ABMS contribution to all peak widths.
Despite the larger supramolecular aggregate of the cocrystal units, their 13C peak widths remain consistently narrower than those of β-caffeine (Table S4, ESI†), which reflects a higher degree of local structural order of each cocrystal.12,14,24 The relative 13C fwhm values increase along the series 2C–MA ≪ 2C–OA < C–GA ≲ C–ME, roughly following those of each respective pristine diacids (Table S4, ESI†) but with 1.2–6 times wider 13C resonances. Only the peakwidths from C–ME break that trend, where narrower peaks are observed relative to both the ME and β-caffeine crystallites for which ABMS and structural disorder primarily govern the respective 13C fwhm values, both apparently being reduced in the C–ME crystallites.
A third factor known to broaden 13C resonances from molecules featuring direct C–N bonds is the spin-1 14N nuclide (99.6% natural abundance) that may induce additional NMR-peak splittings/broadenings of nearby 13C sites by 14N–13C dipolar interactions, whose effects are incompletely suppressed by MAS and scale as B0−1 (in Hz) and B0−2 (in ppm).75,76 Indeed, while demonstrated to vastly dominate the 13C MAS NMR-peak widths and the spectral resolution from C·H2O at a low magnetic field of 4.7 T but being negligible at B0 = 21 T,22 we expect marginal signal broadenings for our experimentation at B0 = 14.1 T, as was also concluded in ref. 50. The data of Table S4 (ESI†) confirm those expectations, suggesting a <30 Hz 13C resonance broadening from the 14N–13C dipolar interactions, as is also supported by the 13C NMR peak-width analysis of ref. 24 performed at B0 = 11.7 T, at which one expects slightly larger effects from 14N–13C interactions. From the marginal dipolar broadening to the net 13C fwhm values, we conclude that inhomogeneous ABMS and structural-disorder stemming 13C chemical-shift distributions dominate the NMR peak-widths throughout all specimens considered herein, as also suggested by our 1H NMR results (Section 4.5).
At least for anhydrous β-caffeine, however, 13C NMR experimentation at B0 ≥ 14.1 T is advantageous for achieving sufficient resolution. Recalling the two additional C3′ and C78 NMR peaks (Section 4.2) of anhydrous β-caffeine relative to its α counterpart,22 a puzzling feature is their apparent absence in several 13C MAS NMR spectra reported from β-caffeine in the literature,14,23,24 incidentally rendering those spectra closer to that observed from α-caffeine,22 encompassing some results from “anhydrous caffeine” of unspecified phase identity.68,70 Yet all those NMR spectra were recorded at lower magnetic fields of either 9.4 T (ref. 14 and 23) or 11.7 T (ref. 24) relative to those acquired at 14.1 T herein (Fig. 4e) and in ref. 12 along with the high-field result by Enright et al.22 at B0 = 21 T. Evidently, only MAS NMR spectra recorded at B0 ≥ 14.1 T give discernible 13C resonances at ≈30 ppm (C78) and ≈149 ppm (C3′), whereas those signals apparently coalesce with those from 13C7/13C8 and 13C3, respectively, in NMR spectra obtained at B0 < 14.1 T.14,23,24 This effect must stem from the resolution-degradation associated with lower-B0 experimentation alone and/or its accompanying emphasized 13C resonance broadening from 14N–13C interactions. Likewise, the 13C MAS NMR spectrum obtained from “anhydrous caffeine” at 9.4 T by Nonappa and Kolehmainen68 appears very similar to those reported from β-caffeine in ref. 14, 23 and 24.
We conclude that “high-field” NMR experimentation is beneficial for enabling complete 13C site/NMR-peak assignments of anhydrous β-caffeine, where B0 = 14.1 T appears to be the minimum magnetic field admitting resonance-discrimination between all crystallographically inequivalent 13C sites, potentially enabling 13C NMR quantifications of the α/β-caffeine contributions in a powder mixture without resorting to PXRD.
4.5. 1H NMR and DFT/GIPAW results
 | ||
| Fig. 5 1H MAS NMR spectra recorded at 14.1 T and 60.0 kHz MAS from the (a) α-OA (“OA”) and α-OA·2H2O (“OAw”) mixture together with powders from the other (b) β-MA, (c) β-GA, and (d) I-ME diacids. The red traces in (b) are deconvolution results of the two overlapping OH11/OH13 resonances. The low-ppm resonance region marked by an asterisk in (a) and (d) reveal background signals from the NMR probehead, and the peak marked by “?” in (b) and (c) stems from an unknown impurity phase of the MA and GA precursors. | ||
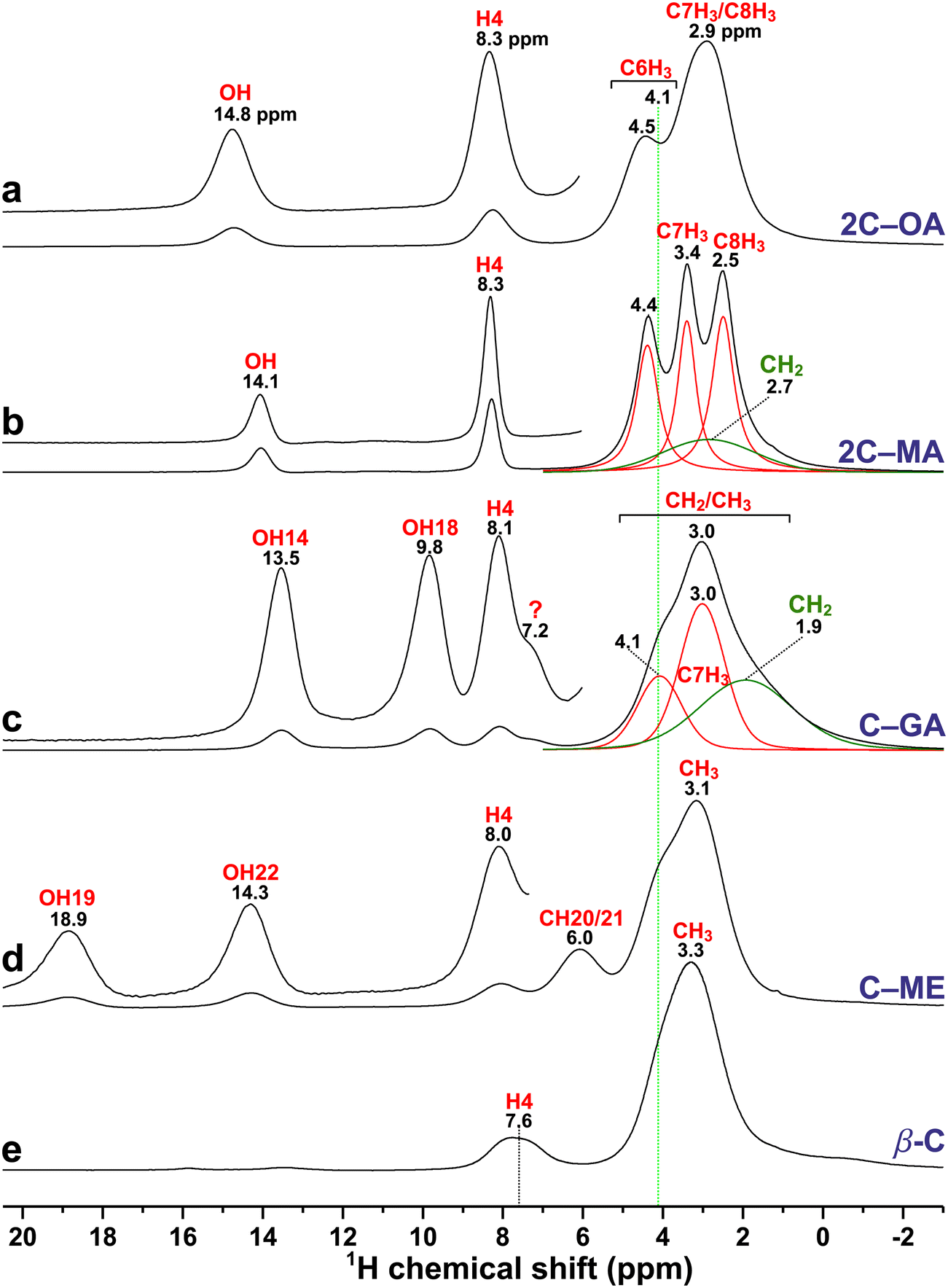 | ||
| Fig. 6 1H MAS NMR spectra recorded at 14.1 T and 60.0 kHz MAS from the (a) 2C–OA, (b) 2C–MA, (c) C–GA, (d) C–ME cocrystals, and (e) anhydrous β-caffeine. The insets show vertically expanded zooms across the high-ppm spectral region. The red/green traces in (b) and (c) are deconvolution results of the overlapping C1H3 (red) and C1H2 (green) NMR signals. The vertical dotted line at δH = 4.1 ppm marks the approximate chemical shift of the C61H3 resonance of the caffeine molecule for the C–GA, C–ME, and β-caffeine specimens in (c)–(e), which differ slightly from those of 2C–OA and 2C–MA in (a) and (b). The NMR peak marked by “?” in (c) stems from an unknown impurity of the GA precursor (see Fig. 5c). | ||
| Coformer | Site | Coformer δH/ppm | 2C–OA δH/ppm | 2C–MA δH/ppm | C–GA δH/ppm | C–ME δH/ppm | ||
|---|---|---|---|---|---|---|---|---|
|
a
1H chemical shifts (δjH) for the Cj1H site labels of Fig. 1, obtained either experimentally from the shift at the NMR-peak maximum, or by DFT/GIPAW calculations (values within parentheses). The δH uncertainties are around ±0.30 ppm (GIPAW) and around ±0.15 ppm for the experimental values for all well-resolved NMR peaks (±0.5 ppm for those obtained by spectral deconvolution; Fig. 6).
b All GIPAW-derived shifts for β-caffeine are averages of the five {δjH} values of the inequivalent molecules in the unit cell.
c Owing to the complete overlap among the C6H3, C7H3, and C8H3 resonances in β-caffeine, only the average chemical-shift values are reported. However, the NMR spectra from the cocrystals admitted further site/signal-resolution by spectral deconvolution, yielding the following NMR-derived δjH (or average CG jH chemical shifts), with the corresponding GIPAW-derived data given within parentheses: for 2C–OA, δ6H = 4.5 (3.7) ppm; 7/8H = 2.9 (2.1) ppm; for 2C–MA, δ6H = 4.4 (3.8) ppm; δ7H =3.4 (2.8) ppm; δ8H = 2.5 (1.8) ppm; for C–GA, δ6H = 4.1 (3.5) ppm; 7/8H = 3.0 (2.3) ppm; for C–ME, δ6H = 4.1 (3.2) ppm; 7/8H = 3.1 (2.3) ppm.
|
||||||||
| β-Caffeineb | H4 | 7.6(8.4) | 8.3(7.2) | 8.3(7.6) | 8.1(7.3) | 8.0(6.7) | ||
| H6/7/8c | 3.3(3.8) | 3.3(2.6) | 3.4(2.8) | 3.3(2.7) | 3.4(2.6) | |||
| α-OA | HO9/10 | 11.1(11.2) | 14.8(15.0) | |||||
| α-OA·2H2O | HO9/10 | 16.6(17.8) | 14.8(15.0) | |||||
| H2O | 5.6(5.6) | |||||||
| β-MA | HO11 | 12.7(13.7) | 14.1(14.8) | |||||
| H12a/b | 3.4(2.6) | 2.7(1.7) | ||||||
| HO13 | 12.3(13.4) | 14.1(14.8) | ||||||
| β-GA | HO14 | 12.7(13.7) | 13.5(14.0) | |||||
| H15/16/17 | 1.8(1.2) | 1.9(1.0) | ||||||
| HO18 | 12.7(13.7) | 9.8(10.6) | ||||||
| I-ME | HO19 | 13.1(13.6) | 18.9(20.2) | |||||
| H20/21 | 6.9(6.2) | 6.0(4.9) | ||||||
| HO22 | 15.8(16.2) | 14.3(14.6) | ||||||
Nonetheless, the reduced 1H NMR spectral resolution in the aliphatic region, for which broadening from residual 1H–1H interactions is most pronounced, compromises accurate site-specific assessments of the 1H chemical-shift alterations upon cocrystal formation. Table 3 collects the NMR and DFT/GIPAW derived 1H chemical shifts, which constitute δjH data for all well-resolved signals but CG shifts (jH) for all heavily overlapping resonances. Hence, in what follows, we focus on the 1Hj resonances that are well resolved in the NMR spectra from both the precursors (Fig. 5) and their corresponding cocrystals (Fig. 6), namely COO1H of the diacid molecules, C41H of caffeine, and the equivalent C1H20/21 sites of ME.
The COO1H chemical shifts of the pristine diacids presented in Fig. 5 correlate qualitatively well with the H⋯O distances of their crystal structures (Fig. 2): the two α-OA·2H2O and α-OA structures reveal the highest (16.6 ppm) and lowest (11.1 ppm) chemical shifts, respectively, as expected from their corresponding shortest (146 pm) and longest (175 pm) HB lengths among all diacids (see caveat below). All COO1H sites of MA and GA along with H19 of ME feature essentially identical H-bond distances of 162–165 ppm (Fig. 2), as is mirrored in very similar δjH values between 12.3–13.1 ppm (Fig. 5). Notably, the 1H NMR signals from the two crystallographically inequivalent OH11 and OH13 sites of MA are discernible in Fig. 5b, despite their very small chemical-shift separation of 0.4 ppm stemming from a minute H⋯O bond-length difference of 1 pm (Fig. 2d), as also reproduced near-perfectly by the GIPAW calculations (0.3 ppm shift-difference).
Within a consistent but minor overestimation of the DFT/GIPAW-derived 1H chemical shift within typically ≲1 ppm (Table 3), the calculated {δjH} values match the experimental counterparts very well for all COO1H sites. Note that while all diacid structures feature H⋯O bonds, all cocrystals involve H⋯N contacts with the “N” atom of caffeine, except for the OH18⋯O14 bond in C–GA and OH22⋯O19 in C–ME (Fig. 2). Each NMR/GIPAW-derived {δjH} dataset gave a good linear correlation with the shortest H⋯O or H⋯N distance, denoted r(H⋯O/N), throughout all COO1H sites. Fig. 7 plots the experimental and calculated 1H chemical shifts along with the corresponding best-fit results given by
| δjH[NMR]/ppm = 47.0 − 0.208r(H⋯O/N)/pm (R2 = 0.912), | (3) |
| δjH[DFT]/ppm = 50.0 − 0.222r(H⋯O/N)/pm (R2 = 0.919), | (4) |
 | ||
| Fig. 7 Experimental (black symbols) and DFT/GIPAW-generated (red) 1H chemical shifts of the carboxy moieties of the pristine diacids (solid symbols) and the cocrystals (open) plotted against the H⋯O/H⋯N distance, r(H⋯O/N), given in Fig. 2. The number around each data-point represents the proton label of Fig. 1. The lines are best-fit results, eqn (3) and (4), obtained by omitting one outlier data point (δ22H) and yielding the as-indicated R2 correlations coefficients. All chemical-shift uncertainties are within the symbol sizes. | ||
| ΔjH = δjH[C–X] − δjH[X], with 1 ≤ j ≤ 22, | (5) |
| Coformer | Site | 2C–OA ΔjH/ppm | 2C–MA ΔjH/ppm | C–GA ΔjH/ppm | C–ME ΔjH/ppm | |
|---|---|---|---|---|---|---|
| a Chemical-shift differences (ΔjH) defined by eqn (5) and calculated from δjH data with two decimals. b Average chemical-shift values over all C6H3, C7H3, and C8H3 sites of the caffeine moiety; see Table 3. | ||||||
| β-Caffeine | H4 | 0.6(−1.2) | 0.7(−0.8) | 0.5(−1.1) | 0.4(−1.7) | |
| H6/7/8b | 0.0(−1.2) | 0.1(−1.0) | 0.0(−1.1) | 0.1(−1.2) | ||
| α-OA | HO9/10 | 3.8(3.8) | ||||
| α-OA·2H2O | HO9/10 | −1.8(−2.8) | ||||
| β-MA | HO11 | 1.3(1.1) | ||||
| H12a/b | −0.7(−0.9) | |||||
| HO13 | 1.8(1.4) | |||||
| β-GA | HO14 | 0.8(0.3) | ||||
| H15/16/17 | 0.1(−0.2) | |||||
| HO18 | −2.9(−3.1) | |||||
| I-ME | HO19 | 5.8(6.6) | ||||
| H20/21 | −0.9(−1.3) | |||||
| HO22 | −1.5(−1.6) | |||||
For the COO1H chemical-shift changes (ΔjH), Table 4 reveals an overall trend of higher δjH values of the cocrystals (i.e., ΔjH > 0), as is witnessed by Fig. 7 and rationalized from the typically shorter H⋯O distance encountered in the cocrystal relative to that of the pristine diacid (Fig. 2). Such HB-length effects also account for the higher δ9H value of 2C–OA relative to its α-OA counterpart, as well as for the increased δ11H and δ13H values upon 2C–MA cocrystal formation. Likewise, the lower δ9H value of 2C–OA relative to α-OA·2H2O—along with the lower chemical shift of 1H18 of C–GA compared to the identical values δ14H = δ18H of the crystallographically equivalent COO1H sites of β-GA—are readily understood from the longer H⋯O distances in the cocrystals.
4.6. 13C chemical-shift changes upon cocrystal formation
In direct analogy with the 1H chemical-shift difference between the cocrystal and its precursors [eqn (5)], we define| ΔjC = δjC[C–X] − δjC[X], with 1 ≤ j ≤ 22, and X = {β-C, OA, OA·2H2O, MA, GA, ME}, | (6) |
| Coformer | Site | 2C–OA ΔjC/ppm | 2C–MA ΔjC/ppm | C–GA ΔjC/ppm | C–ME ΔjC/ppm | |
|---|---|---|---|---|---|---|
| a Chemical-shift differences (ΔjC) defined by eqn (6) and calculated from δjC data with two decimals. | ||||||
| β-Caffeine | C1 | −0.2(0.1) | 0.0(1.8) | 1.0(1.5) | −0.5(0.1) | |
| C2 | 1.2(1.0) | 1.3(0.7) | −0.2(−0.5) | 1.0(1.1) | ||
| C3 | −2.0(−0.6) | −1.6(−0.9) | −1.3(1.0) | −2.7(−2.4) | ||
| C4 | −0.1(−0.8) | 0.6(0.6) | −0.4(−1.0) | −0.9(−2.8) | ||
| C5 | 1.7(3.2) | 1.9(3.1) | 1.4(2.8) | 1.5(2.5) | ||
| C6 | 1.4(0.9) | −1.0(−2.3) | −0.7(−1.7) | 1.5(1.4) | ||
| C7 | 0.4(−1.1) | 0.9(0.3) | 0.8(−0.6) | 0.7(0.0) | ||
| C8 | −1.7(−4.3) | −1.3(−3.7) | −1.8(−5.3) | −0.7(−2.0) | ||
| α-OA | C9/10 | −1.1(−0.7) | ||||
| α-OA·2H2O | C9/10 | −3.5(−2.3) | ||||
| β-MA | C11 | −6.9(−8.8) | ||||
| C12 | 2.9(4.6) | |||||
| C13 | −6.5(−5.3) | |||||
| β-GA | C14 | −1.6(−3.3) | ||||
| C15 | 2.4(2.2) | |||||
| C16 | 3.4(5.5) | |||||
| C17 | −1.1(−1.6) | |||||
| C18 | −4.5(−3.9) | |||||
| I-ME | C19 | −0.4(0.7) | ||||
| C20 | −3.3(−3.5) | |||||
| C21 | −1.1(−1.7) | |||||
| C22 | −5.0(−1.3) | |||||
In contrast with the 13CH2 shifts of MA and GA, which deshield slightly by a few ppm, significantly decreased δC values are observed for the 13COOH diacid sites, whose adjacent O atom form an HB with protons of neighboring caffeine/diacid molecules, which along with COO− → COOH conversions are well known to decrease the 13C shift in various molecular systems.37,38,78–81 However, that trend has remained qualitative without any firm quantitative relationship having thus far been established between the isotropic13C chemical shift and the 13COH⋯O or 13CO⋯HO distance (the principal value δjyy of the chemical-shift tensor, however, correlates linearly with the HB distance/angle parameters36–38). Indeed, the plot of δjC against r(H⋯O/N) in Fig. 8 reveals a significant scatter for the diacid/cocrystal structures. Although a weak trend of 13COOH shift-reduction with a shortened COO⋯H distance is discernible, our data do not reveal any reasonable δjC/r(H⋯O/N) or ΔjC/Δr(H⋯O/N) correlation for either the experimental or modeled data. The only exceptions are the 13COOH sites of the 2C–OA (δ9C), 2C–MA (δ11C), and C–GA (δ14C) cocrystals, all of which share the feature of a strong H⋯N bond to the N atom of the caffeine molecule (which is expected to constitute the primary stabilization of the supramolecular structure8): they establish an excellent linear δC/r(H⋯N) relationship (Fig. 8) for both the NMR and GIPAW-derived shifts but with the caveat that only three data points underlie the correlation.
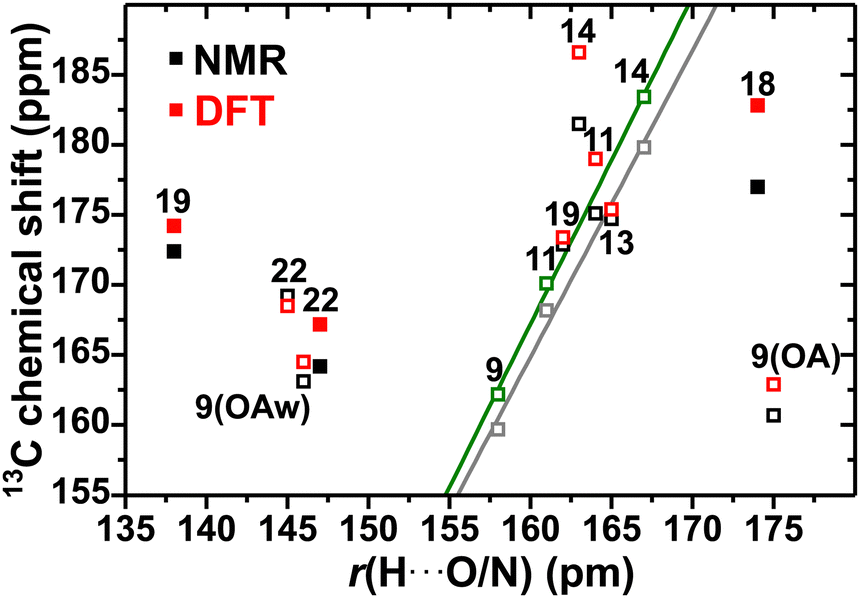 | ||
| Fig. 8 Experimental (black symbols) and DFT/GIPAW-generated (red) 13C chemical shifts of the carboxy moieties of the pristine diacids (solid symbols) and the cocrystals (open) plotted against the H⋯O or H⋯N distance, r(H⋯O/N), within each structure (Fig. 2). Each number represents the corresponding Cj label of Fig. 1. The symbols set in green and gray color for the NMR and DFT/GIPAW data, respectively, correspond to the results obtained from the 2C–OA (δ9C), 2C–MA (δ11C), and C–GA (δ14C) cocrystals, all of which are H-bonded to the “N” atom of caffeine. The corresponding lines are best-fit results to the expressions δjC[NMR]/ppm = −185.6 + 2.1905r(H⋯N)/pm (R2 = 0.990) and δjC[DFT]/ppm = −206.5 + 2.336r(H⋯N)/pm (R2 = 0.998), respectively. All chemical-shift uncertainties are within the symbol sizes. | ||
Turning to the δjC changes of the caffeine moiety in the cocrystals relative to β-caffeine, Table 5 reveals the overall largest observed |ΔjC| values for the {C3, C5, C8} sites upon cocrystal formation (and to a lesser extent, C2 and C6), thereby suggesting that those local 13C electronic environments alter the most by the intermolecular caffeine–coformer interactions in the cocrystals structure (Fig. 2) relative to the caffeine–caffeine contacts in β-caffeine. The C3 and C5 atoms constitute a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C fragment between the pyriminedione and imidazolium rings (Fig. 1). We attribute the (globally largest) chemical-shift alteration of the C3 site of the caffeine molecule to its close proximity to the N atom, whose N⋯HOOC HB constitutes the primary intermolecular interaction in all cocrystal structures;8 see Section 3 and Fig. 2. However, while the 13C3 site shields upon cocrystal formation (Δ3C < 0), 13C5 deshields (Δ5C > 0) to a comparable extent, for unknown reasons when considering its remoteness to any atom of a neighboring molecule in either the β-caffeine or cocrystal structures. The 13CH3 shift alterations are discussed in Section 4.7.
C fragment between the pyriminedione and imidazolium rings (Fig. 1). We attribute the (globally largest) chemical-shift alteration of the C3 site of the caffeine molecule to its close proximity to the N atom, whose N⋯HOOC HB constitutes the primary intermolecular interaction in all cocrystal structures;8 see Section 3 and Fig. 2. However, while the 13C3 site shields upon cocrystal formation (Δ3C < 0), 13C5 deshields (Δ5C > 0) to a comparable extent, for unknown reasons when considering its remoteness to any atom of a neighboring molecule in either the β-caffeine or cocrystal structures. The 13CH3 shift alterations are discussed in Section 4.7.
As commented in Section 4.5, the C41H chemical shift remains near-constant throughout all cocrystals, which is attributed to Δ4C cancellation-effects from variable and complex H-bonding scenarios. Likewise, only minor chemical-shift alterations are observed for 13C4H, which besides being directly linked to the H-bonded “N” site of caffeine, additionally experiences weak H bonds to the CO atoms of the diacid carboxy groups upon cocrystal formation (Fig. 2). We speculate that the chemical-shift effects from those two competing H bonds might partially cancel each other. We remark that even slightly more negative Δ4C values of {−1.3, −0.5, −1.6, −2.0} ppm apply if the respective {2C–OA, 2C–MA, C–GA, C–ME} cocrystals would be prepared from the ordered C·H2O phase that features δ4C = 144.6 ppm22 and one C4H⋯OH2 bond.
4.7 Tetrel-bonding effects on methyl 13C/1H chemical shifts
Besides the possibility of a weak HB, the electrophilic nature of methyl groups offer the option of a H3C⋯O TB to a proximate electronegative and nucleophilic atom.47–49 From our as-observed ΔjH/ΔjC data for the three 13CH3 groups of the caffeine moiety upon cocrystal formation, we discuss the complex HB/TB interplay on their 13C/1H chemical shifts in relation to current literature, which is both sparse and inconclusive. Likewise, our data may only be discussed qualitatively.Both DFT calculations48,49 and NMR experiments49 suggest consistently larger δC than δH alterations when a TB is formed. Scheiner et al.48 investigated the 13CH3 and C1H3 chemical-shift displacements upon CH⋯O HB or HC⋯O TB formation by DFT calculations, inferring that both 1H and 13C chemical shifts increase for either scenario, with the precise deshielding depending on the TB-length and N–C–O bond angle (θTB). The largest chemical-shift alterations occurred for a linear TB constellation, for which ΔjC ≤ 6 ppm was predicted, whereas the 1H chemical shift only increased marginally (≲1 ppm).48 Formation of a weak CH⋯O HB, however, is reported to produce a comparably larger 1H deshielding than for 13C.43,46,48 Currently no firm/general experimental correlation is reported of either the 1H or 13C chemical shifts against the CH3⋯O or H3C⋯O distance. A recent compilation by Southern et al.49 of experimental 13C/1H chemical shifts from the literature along with GIPAW calculations, suggested very scattered 13C/1H chemical-shift-structure correlations of CH3 groups involved in tetrel bonds:49 although experiments confirm that δjC is often increased for decreasing HC⋯O distance, no significant dependence was observed versus θTB, nor of δjH against either the TB distance or θTB. Moreover, when HB and TB bonds coexist for a CH3 group, their effects on δjC may either reinforce or counteract each other.49
The 13C and 1H chemical-shift data from the current cocrystals accord qualitatively with previous findings of a smaller δjH than δjC change when the TB scenario alters between β-caffeine and its cocrystals. The NMR-derived value Δ6/7/8H ≈ 0 apply throughout all cocrystals. Although the δjH/TB distance-accuracy is compromised by the lack of NMR-signal discrimination among the 1H6–1H8 methyl resonances from β-caffeine, the near-constant 1H chemical shift is also confirmed by the DFT calculations (within a constant shift-displacement of Δ6/7/8H ≈ −1 ppm), for which each δjH and ΔjH value is readily determined (Table S5, ESI†). Consistently larger 13C chemical-shift displacements occur on cocrystal formation, where Table 2 reveals the largest changes for 13C8H3, all of which are negative and conforming to the experimental range of −1.8 ≤ Δ8C/ppm ≤ −1.3, whereas more negative values are predicted by the GIPAW calculations: −5.3 ≤ |Δ8C|/ppm ≤ −2.0. Somewhat lower shift-differences (0.7–1.5 ppm) are observed for the 13C6H3 sites, where 2C–OA and C–ME yield positive Δ6C values, while those for 2C–MA and C–GA are negative (Table 5). Besides slightly larger magnitudes predicted by the GIPAW calculations, all positive/negative Δ6C trends are reproduced, as well as the experimental finding that 13C7H3 consistently manifests the smallest shift-change for each cocrystal.
Unfortunately, the ΔjC trends identified above for the three 13CH3 sites of the caffeine moiety are for several reasons very difficult to rationalize: (i) δjC is expected to depend on both H⋯O/N and C⋯O/N distance-changes between β-caffeine and each cocrystal, whose effects on the chemical shift may augment or cancel partially. (ii) Even if only each shortest HB/TB distance per 13CH3 site of the cocrystal is considered (which is expected to be most influential on δjC), the presence of five inequivalent molecules in the β-caffeine unit cell implies a complex reference scenario with a range of HB/TB lengths relative to the structurally ordered cocrystals. (iii) The current absence of any firm δjC/r(CHn⋯O) or δjC/r(CHn⋯O) correlation in conjunction with (i) and (ii), complicate the establishment of even semiquantitative ΔjC correlations with the HB/TB-distance alterations accompanying the {2C–OA, 2C–MA, C–GA, C–ME} formation.
The shortest HB and TB distances of the methyl groups in β-caffeine and the cocrystals are compiled in Table S3 (ESI†). We first consider the most pronounced 13C chemical-shift change upon cocrystal formation—i.e., Δ8C, assuming that the HB effects are minor because the range of C8H3⋯O1/O2 distances (236–294 pm) in β-caffeine span those of the cocrystals (and is thereby expected to give, on the average, a very similar 8C contribution as for the cocrystals). Hence, the corresponding TB-length alterations are expected to primarily govern Δ8C. Notably, the TB distances are overall shorter in the cocrystals than in β-caffeine (except for 2C–OA). Then given that a TB contraction is expected to increase δ8C,48,49 positive values of Δ8C are anticipated throughout all cocrystals, in stark contrast to the de facto observed experimental and modeled results. The lack of an even qualitative ΔC/Δr(CH3⋯O/N) correlation also applies to the 13C6H3 site: again, the wide HB-distance range in β-caffeine (207–276 pm; Table S3, ESI†) relative to those of the cocrystals (223–249 pm) suggest minor effects on Δ6C. Notably, however, the C6H3⋯O bond-lengths in the β-caffeine structure is consistently shorter than those of any cocrystal, from which negative Δ6C values are predicted throughout,48,49 in clear contradiction to our NMR and DFT results. Moreover, no correlation was observed for the set of 13CHn chemical shifts against the TB angle (data not shown), despite wide θTB ranges within 71°–90° (4δjC values) and 147°–178° (8δjC values).
Notwithstanding the uncertainties accompanying (i)–(iii) above, we believe that our 13C chemical-shift data suggest that a TB-length contraction may not necessarily produce an increased 13C chemical shift, along previous difficulties to establish a general δjC/r(CH3⋯O/N) correlation. Such a relationship is likely obscured for similar reasons as some inconclusive experimental δjC/r(CHn⋯O) correlations, for which 13C shift-alterations upon CH⋯O bond formation depend both on the precise molecular fragments involved and (longer-range) crystal-structure effects,44,45 whose complex interplay may result in 13C deshielding,44,46 shielding,43 or either/both.42,44
5 Conclusion
The present comprehensive NMR/DFT study encompassed altogether 68 distinct 13C sites and 33 unique 1H resonances from altogether four caffeine-based cocrystals and their six precursor structures, with an overall very good agreement between the experimental and modeled chemical shifts. We believe that the results herein may serve as accurate benchmark values for further MAS NMR investigations, given the very good accordance with previous literature for the 1H/13C chemical shifts—such as the δC data of 2C–MA and C–GA of ref. 50 along with both 1H and 13C chemical shifts of the diacid precursors—and because a significant fraction of all δH/δC data are presented herein for the first time.Despite our large 13C and 1H chemical-shift ensembles and an encouraging NMR/DFT agreement, the herein established linear chemical-shift/distance correlations were confined to the COO1H ⋯O/N diacid–diacid and diacid–caffeine hydrogen bonds, along with the COO1H ⋯N counterparts, which nonetheless are those most instrumental for the supramolecular cocrystal organization. We also examined possible CH3⋯O/N tetrel-bond effects on the 1H/13C chemical shifts, which were overall uncertain and inconclusive but suggesting that previously reported (weak) chemical-shift correlations against tetrel-bond parameters might not hold in general, thereby underscoring the need for more research in this very sparsely investigated field. Further efforts towards better confining the H-atom positions of the caffeine–diacid cocrystals by direct 1H–1H and 1H–13C internuclear-distance measurements are underway by employing a recent NMR crystallography method.81,82
Except for the methylene groups of MA/GA and their cocrystals, the compromised 1H NMR spectral resolution (and for β-caffeine also the 13C NMR counterpart) originates mainly from chemical-shift dispersions stemming from anisotropic bulk magnetic susceptibility and structural disorder rather than the often dominant peak-broadening from 1H–1H interactions. Hence, modest spectral resolution enhancements are expected from faster-MAS (>60 kHz) 1H NMR experiments, and let alone by utilizing higher fields (B0 > 14.1 T) because the shift-dispersion scales linearly with B0. That sharply contrasts with the 13C NMR scenario, however, where B0 ≥ 14.1 T experimentation appears mandatory for resolving all 10 resonances from the 8 distinct 13C sites of anhydrous β-caffeine, where the previously unassigned 13C NMR peaks at 30.2 ppm (C7/C8) and 149.0 ppm (C3) are spectral markers for discriminating the disordered anhydrous β-caffeine polymorph from both its disordered α-caffeine and ordered hydrated C·H2O counterparts. This insight facilitates further α/β-phase quantifications of mixtures of both polymorphs by 13C MAS NMR alone, which has hitherto only proven possible by PXRD analyses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Knut and Alice Wallenberg Foundation (project 2019.0124), and in part by the Swedish Research Council (project VR 2022-03652). The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS), partially funded by the Swedish Research Council through grant agreement no. 2022-06725. We thank an anonymous reviewer for helpful comments on NMR-peak broadening factors.References
- H. G. Brittain, Cocrystal systems of pharmaceutical interest: 2010, Cryst. Growth Des., 2012, 12, 1046–1054 CrossRef CAS.
- R. Shaikh, R. Singh, G. M. Walker and D. M. Croker, Pharmaceutical cocrystal drug products: an outlook on product development, Trends Pharmacol. Sci., 2018, 39, 1033–1048 CrossRef CAS PubMed.
- G. Bolla, B. Sarma and A. K. Nangia, Crystal engineering of pharmaceutical cocrystals in the discovery and development of improved drugs, Chem. Rev., 2022, 122, 11514–11603 CrossRef CAS PubMed.
- T. Friščić and W. Jones, Recent advances in understanding the mechanism of cocrystal formation via grinding, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef.
- M. Rodrigues, B. Baptista, J. A. Lopes and M. C. Sarraguça, Pharmaceutical cocrystallization techniques. Advances and challenges, Int. J. Pharm., 2018, 547, 404–420 CrossRef CAS PubMed.
- U. J. Griesser and A. Burger, The effect of water vapor pressure on desolvation kinetics of caffeine 4/5-hydrate, Int. J. Pharm., 1995, 120, 83–93 CrossRef CAS.
- H. G. M. Edwards, E. Lawson, M. de Matas, L. Shields and P. York, Metamorphosis of caffeine hydrate and anhydrous caffeine, J. Chem. Soc., Perkin Trans., 1997, 2, 1985–1990 RSC.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Pharmaceutical cocrystallization: engineering a remedy for caffeine hydration, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS.
- C. W. Lehmann and F. Stowasser, The crystal structure of anhydrous β-caffeine as determined from X-ray powder-diffraction data, Chem. – Eur. J., 2007, 13, 2908–2911 CrossRef CAS PubMed.
- K. Guo, G. Sadiq, C. Seaton, R. Davey and Q. Yin, Co-crystallization in the caffeine/maleic acid system: lessons from phase equilibria, Cryst. Growth Des., 2010, 10, 268–273 CrossRef CAS.
- T. Leyssens, G. Springuel, R. Montis, N. Candoni and S. Veesler, Importance of solvent selection for stoichiometrically diverse cocrystal systems: Caffeine/maleic acid 1:1 and 2:1 cocrystals, Cryst. Growth Des., 2012, 12, 1520–1530 CrossRef CAS.
- V. S. Mandala, S. J. Loewus and M. A. Mehta, Monitoring cocrystal formation via in situ solid-state NMR, J. Phys. Chem. Lett., 2014, 5, 3340–3344 CrossRef CAS PubMed.
- M. Mukaida, K. Sugano and K. Terada, Stability order of caffeine co-crystals determined by co-crystal former exchange reaction and its application for the validation of in silico models, Chem. Pharm. Bull., 2015, 63, 18–24 CrossRef CAS PubMed.
- K. P. Nartowski, Y. Z. Khimyak and D. J. Berry, Tuning the spontaneous formation kinetics of caffeine: malonic acid co-crystals, CrystEngComm, 2016, 18, 2617–2620 RSC.
- S. P. Brown, Probing proton-proton proximities in the solid state, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 199–251 CrossRef CAS.
- T. Le Marchand, T. Schubeis, M. Bonaccorsi, P. Paluch, D. Lalli, A. J. Pell, L. B. Andreas, K. Jaudzems, J. Stanek and G. Pintacuda, 1H-detected biomolecular NMR under fast magic-angle spinning, Chem. Rev., 2022, 122, 9943–10018 CrossRef CAS PubMed.
- M. K. Dudek, S. Kazmierski, M. Kostrzewa and M. J. Potrzebowski, Solid-state NMR studies of molecular crystals, Annu. Rep. NMR Spectrosc., 2018, 95, 1–81 CrossRef CAS.
- I. Schnell, Dipolar recoupling in fast-MAS solid-state NMR spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 145–207 CrossRef CAS.
- P. Hodgkinson, NMR crystallography of molecular organics, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- M. Li, W. Xu and Y. Su, Solid-state NMR spectroscopy in pharmaceutical sciences, Trac., Trends Anal. Chem., 2021, 135, 116152 CrossRef CAS.
- M. Edén, Probing oxide-based glass structures by solid-state NMR: opportunities and limitations, J. Magn. Reson. Open, 2023, 16–17, 100112 CrossRef.
- G. D. Enright, V. V. Terskikh, D. H. Brouwer and J. A. Ripmeester, The structure of two anhydrous polymorphs of caffeine from single-crystal diffraction and ultrahigh-field solid-state 13C NMR spectroscopy, Cryst. Growth Des., 2007, 7, 1406–1410 CrossRef CAS.
- S. Bordignon, P. C. Vioglio, E. Priola, D. Voinovich, R. Gobetto, Y. Nishiyama and M. R. Chierotti, Engineering codrug solid forms: mechanochemical synthesis of an indomethacin-caffeine system, Cryst. Growth Des., 2017, 17, 5744–5752 CrossRef CAS.
- H. E. Kerr, H. E. Mason, H. A. Sparkes and P. Hodgkinson, Testing the limits of NMR crystallography: the case of caffeine-citric acid hydrate, CrystEngComm, 2016, 18, 6700–6707 RSC.
- S. Bhattacharya, Thermal expansion and dimensionality of a hydrogen bond network: a case study on dimorphic oxalic acid, CrystEngComm, 2020, 22, 7896–7902 RSC.
- N. Casati, P. Macchi and A. Sironi, Hydrogen migration in oxalic acid di-hydrate at high pressure?, Chem. Commun., 2009, 2679–2681 RSC.
- S. Bhattacharya, V. G. Saraswatula and B. K. Saha, Thermal expansion in alkane diacids—another property showing alternation in an odd–even series, Cryst. Growth Des., 2013, 13, 3651–3656 CrossRef CAS.
- V. R. Thalladi, M. Nüsse and R. Boese, The melting point alternation in α,ω-alkanedicarboxylic acids, J. Am. Chem. Soc., 2000, 122, 9227–9236 CrossRef CAS.
- D. Rychkov, S. Arkhipov and E. Boldyreva, Structure-forming units of amino acid maleates. Case study of L-valinium hydrogen maleate, Acta Crystallogr., 2016, B72, 160–163 Search PubMed.
- F. Mauri, B. G. Pfrommer and S. G. Louie, Ab initio theory of NMR chemical shifts in solids and liquids, Phys. Rev. Lett., 1996, 77, 5300–5303 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, All-electron magnetic response with pseudopotentials: NMR chemical shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- T. Charpentier, The PAW/GIPAW approach for computing NMR parameters: a new dimension added to NMR of solids, Solid State Nucl. Magn. Reson., 2011, 40, 1–20 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, First-principles calculation of NMR parameters using the gauge including projector augmented wave method: a chemist's point of view, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- R. K. Harris, P. Jackson, L. H. Merwin, B. J. Say and G. Hägele, Perspectives in high-resolution solid-state nuclear magnetic resonance, with emphasis on combined rotation and multiple-pulse spectroscopy, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 3649–3672 RSC.
- U. Sternberg and E. Brunner, The influence of short-range geometry on the chemical shift of protons in hydrogen bonds, J. Magn. Reson., Ser. A, 1994, 108, 142–150 CrossRef CAS.
- N. R. Jagannathan, Carbon-13 chemical shielding tensors in alkanedicarboxylic acids. Influence of molecular geometry on the carboxyl carbon tensors in alkanedicarboxylic acids and related compounds, Magn. Reson. Chem., 1989, 27, 941–946 CrossRef CAS.
- Z. Gu and A. McDermott, Chemical shielding anisotropy of protonated and deprotonated carboxylates in amino acids, J. Am. Chem. Soc., 1993, 115, 4282–4285 CrossRef CAS.
- Z. Gu, R. Zambrano and A. McDermott, Hydrogen bonding of carboxyl groups in sold-state amino acids and peptides: comparison of chemical shielding, infrared frequencies, and structures, J. Am. Chem. Soc., 1994, 116, 6368–6372 CrossRef CAS.
- R. Gobetto, C. Nervi, M. R. Chierotti, D. Braga, L. Maini, F. Grepioni, R. K. Harris and P. Hodgkinson, Hydrogen bonding and dynamic behaviour in crystals and polymorphs of dicarboxylic-diamine adducts: a comparison between NMR parameters and x-ray diffraction studies, Chem. – Eur. J., 2005, 11, 7461–7471 CrossRef CAS PubMed.
- X. Xue and M. Kanzaki, Proton distributions and hydrogen bonding in crystalline and glassy hydrous silicates and related inorganic materials: insights from high-resolution solid-state nuclear magnetic resonance spectroscopy, J. Am. Ceram. Soc., 2009, 92, 2803–2830 CrossRef CAS.
- F. G. Vogt, J. S. Clawson, M. Strohmeier, A. J. Edwards, T. N. Pham and S. A. Watson, Solid-state NMR analysis of organic cocrystals and complexes, Cryst. Growth Des., 2009, 9, 2620–2626 CrossRef.
- S. Scheiner, Identification of spectroscopic patterns of CH⋯O H-bonds in proteins, J. Phys. Chem. B, 2009, 113, 10421–10427 CrossRef CAS PubMed.
- J. R. Yates, T. N. Pham, C. J. Pickard, F. Mauri, A. M. Armado, A. M. Gil and S. P. Brown, An investigation of weak CH⋯O hydrogen bonds in maltose anomers by a combination of calculation and solid-state NMR spectroscopy, J. Am. Chem. Soc., 2005, 127, 10216–10220 CrossRef CAS PubMed.
- A.-C. Uldry, J. M. Griffin, J. R. Yates, M. Pérez-Torralba, M. D. Santa Maria, A. L. Webber, M. L. L. Beaumont, A. Samoson, R. M. Claramunt, C. J. Pickard and S. P. Brown, Quantifying weak hydrogen bonding in uracil and 4-cyano-4′-ethynylphenyl: a combined computational and experimental investigation of NMR chemical shifts in the solid state, J. Am. Chem. Soc., 2008, 130, 945–954 CrossRef CAS PubMed.
- K. Bouzková, M. Babinský, L. Novosadorá and R. Marek, Intermolecular interactions in crystalline theobromine as reflected in electron deformation density and 13C NMR chemical shift tensors, J. Chem. Theory Comput., 2013, 9, 2629–2638 CrossRef PubMed.
- S. A. Southern and D. L. Bryce, To what extent do bond length and angle govern the 13C and 1H NMR response to weak CH⋯O hydrogen bonds? A case study of caffeine and theophylline cocrystals, Solid State Nucl. Magn. Reson., 2022, 119, 101795 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Tetrel-bonding interaction: rediscovered supramolecular force?, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed.
- S. Scheiner, Ability of IR and NMR spectral data to distinguish between a tetrel bond and a hydrogen bond, J. Phys. Chem. A, 2018, 122, 7852–7862 CrossRef CAS PubMed.
- S. A. Southern, M. S. West, M. J. Z. Bradshaw and D. L. Bryce, Experimental 13C and 1H solid-state NMR response in weakly tetrel-bonded methyl groups, J. Phys. Chem. C, 2021, 125, 2111–2123 CrossRef CAS.
- N. J. Vigilante and M. A. Mehta, A 13C solid-state NMR investigation of four cocrystals of caffeine and theophylline, Acta Crystallogr., 2017, C73, 234–243 Search PubMed.
- A. Coelho, TOPAS-academic v6, Coelho Software, 2016 Search PubMed.
- G. Metz, X. L. Wu and S. O. Smith, Ramped-amplitude cross polarization in magic-angle-spinning NMR, J. Magn. Reson., Ser. A, 1994, 110, 219–227 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, An improved broadband decoupling sequence for liquid crystals and solids, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- E. Leonova, A. S. Hakeem, K. Jansson, B. Stevensson, Z. Shen, J. Grins, S. Esmaeilzadeh and M. Edén, Nitrogen-rich La–Si–Al–O–N oxynitride glass structures probed by solid state NMR, J. Non-Cryst. Solids, 2008, 354, 49–60 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- J. R. Yates, C. J. Pickard and F. Mauri, Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. M. Maricq and J. S. Waugh, NMR in rotating solids, J. Chem. Phys., 1979, 70, 3300–3316 CrossRef CAS.
- U. Haeberlen, High Resolution NMR in Solids. Selective Averaging, Academic Press, New York, 1976 Search PubMed.
- J. Mason, Conventions for the reporting of nuclear magnetic shielding (or shift) tensors suggested by participants in the NATO ARW on NMR shielding constants at the University of Maryland, College Park, July 1992, Solid State Nucl. Magn. Reson., 1993, 2, 285–288 CrossRef CAS PubMed.
- C. López, R. Claramunt and J. Elguero, Oxalic acid/phenols and oxalic acid/cholesterol co-crystals: a solid state 13C CPMAS NMR study, ARKIVOC, 2008, 33–46 Search PubMed.
- E. Pindelska, A. Sokal, L. Szeleszczuk, D. M. Pisklak and W. Kolodziejski, Solid-state NMR studies of theophylline co-crystals with dicarboxylic acids, J. Pharm. Biomed. Anal., 2014, 100, 322–328 CrossRef CAS PubMed.
- J. Kastelic, Ž. Hodnik, P. Šket, J. Plavec, N. Lah, I. Leban, M. Pajk, O. Planinšek and D. Kikelj, Fluconazole cocrystals with dicarboxylic acids, Cryst. Growth Des., 2010, 10, 4943–4953 CrossRef CAS.
- M. Ilczyszyn, D. Godzisz and M. M. Ilczyszyn, Sarcosine-maleic acid (1:1) crystal: structure, 13C NMR and vibrational properties, protonation character, Spectrochim. Acta, Part A, 2003, 59, 1815–1828 CrossRef CAS PubMed.
- Nonappa and E. Kolehmainen, Caffeine as a gelator, Gels, 2016, 2, 9 CrossRef PubMed.
- J. Sitkowski, L. Stefaniak, L. Nicol, M. L. Martin, G. J. Martin and G. A. Webb, Complete assignments of 1H, 13C, and 15N NMR spectra of caffeine, Spectrochim. Acta, Part A, 1995, 51, 839–2841 CrossRef.
- N. Liédana, A. Galve, C. Rubio, C. Téllez and J. Coronas, CAFZIF-8: one-step encapsulation of caffeine in MOF, ACS Appl. Mater. Interfaces, 2012, 4, 5016–5021 CrossRef PubMed.
- D. L. VanderHart, W. L. Earl and A. N. Garroway, Resolution in 13C NMR of organic solids using high-power proton decoupling and magic-angle sample spinning, J. Magn. Reson., 1981, 44, 361–401 CAS.
- M. Alla and E. Lippmaa, Resolution limits in magic-angle rotation NMR spectra of polycrystalline solids, Chem. Phys. Lett., 1982, 87, 30–33 CrossRef CAS.
- A. J. Robbins, W. T. K. Ng, D. Jochym, T. W. Keal, S. J. Clark, D. J. Tozer and P. Hodgkinson, Combining insights from solid-state NMR and first principles calculation: applications to the 19 F NMR of octafluoronaphthalene, Phys. Chem. Chem. Phys., 2007, 9, 2389–2396 RSC.
- M. P. Hanrahan, A. Venkatesh, S. L. Carnahan, J. L. Calahan, J. W. Lubach, E. J. Munson and A. J. Rossini, Enhancing the resolution of 1H and 13C solid-state NMR spectra by reduction of anisotropic bulk magnetic susceptibility broadening, Phys. Chem. Chem. Phys., 2017, 19, 28153–28162 RSC.
- J. G. Hexem, M. H. Frey and S. J. Opella, Molecular and structural information from 14N–13C NMR spectra of solids, J. Chem. Phys., 1982, 77, 3847–3856 CrossRef CAS.
- A. C. Olivieri, L. Frydman and L. E. Diaz, A simple approach for relating molecular structural information to the dipolar coupling in 13C–14N CP MAS NMR, J. Magn. Reson., 1987, 75, 50–62 CAS.
- B. Berglund and R. W. Vaughan, Correlations between proton chemical shift tensors, deuterium quadrupole couplings, and bond distances in solids, J. Chem. Phys., 1980, 73, 2037–2043 CrossRef CAS.
- R. G. Griffin and D. J. Ruben, Carbon-13 chemical shielding in ammonium hydrogen oxalate hemihydrate, J. Chem. Phys., 1975, 63, 1272–1275 CrossRef CAS.
- R. G. Griffin, A. Pines, S. Pausak and J. S. Waugh, 13C chemical shielding in oxalic acid, oxalic acid dihydrate, and diammonium oxalate, J. Chem. Phys., 1975, 63, 1267–1271 CrossRef CAS.
- C. Gardiennet-Doucet, X. Assfeld, B. Henry and P. Tekely, Revealing successive steps of deprotonation of L-phosphoserine through 13C and 31P chemical shielding tensor fingerprints, J. Phys. Chem. A, 2006, 110, 9137–9144 CrossRef CAS PubMed.
- R. Mathew, B. Stevensson and M. Edén, Refined structures of O-phospho-L-serine and its calcium salt by new multinuclear solid-state NMR crystallography methods, J. Phys. Chem. B, 2021, 125, 10985–11004 CrossRef CAS PubMed.
- Y. Yu, B. Stevensson, M. Pujari-Palmer, H. Guo, H. Engqvist and M. Edén, The monetite structure probed by advanced solid-state NMR experimentation at fast magic-angle spinning, Int. J. Mol. Sci., 2019, 20, 6356 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06197c |
| ‡ Present address: Department of Chemistry, National Institute of Technology Tiruchirappalli, Tiruchirappalli-620015, Tamil Nadu, India. |
| This journal is © the Owner Societies 2024 |