Synergistic effect between ZnCo2O4 and Co3O4 induces superior electrochemical performance as anodes for lithium-ion batteries
Received
19th December 2023
, Accepted 23rd March 2024
First published on 5th April 2024
Abstract
The current work describes a facile synthesis of spinel-type ZnCo2O4 along with an additional phase, Co3O4, by simply maintaining a non-stoichiometric ratio of Zn and Co precursors. Pure ZnCo2O4 and Co3O4 were also synthesized using the same method to compare results. The obtained morphologies of samples show that small-sized nanoparticles are interconnected and form a porous nanosheet-like structure. When used as anode materials for Li-ion batteries, the ZnCo2O4/Co3O4 nanocomposite electrode exhibits a highly stable charge capacity of 1146.2 mA h g−1 at 0.5C after 350 cycles, which is superior to those of other two pure electrodes, which can be attributed to its optimum porosity, synergistic effect of ZnCo2O4 and Co3O4, increased active sites for Li+ ion diffusion, and higher electrical conductivity. Although the pure Co3O4 electrode displayed a much higher rate capability than the ZnCo2O4/Co3O4 nanocomposite electrode at all investigated current rates, the Co3O4 morphology apparently could not withstand long-term cycling, and the electrode became pulverized due to the repeated volume expansion/contraction, resulting in a rapid decrease in the capacity.
1 Introduction
Transition metal oxides have been widely studied as promising anode materials to replace the conventional anode graphite, whose theoretical capacity is much inferior (only ∼372 mA h g−1), due to the growing demand for large-scale energy storage devices. Since transition metal oxides exhibit great promise, such as earth abundance, large specific capacity, suitable discharge voltages and synthetic versatility, it is believed that they might fulfill the immediate demands of advance LIB applications. In particular, the binary metal oxide Co3O4 possesses various intriguing features, such as high theoretical capacity (∼890 mA h g−1), good thermal/chemical stability, excellent catalytic activity, easy production, low toxicity and high redox activity.1–10 However, Co3O4 unfortunately has numerous issues including poor ionic and electronic conductivity and drastic volume expansion/contraction during charge/discharge, resulting in unsatisfactory rate performance and unstable cycling performance, which ultimately restricts its commercial applications in LIBs.11,12 Among various strategies that have been adopted to overcome the above issues of Co3O4 anodes, the fabrication of nanoscale porous architectures with tailored morphology was suggested as an effective method to reduce the diffusion path length of ions and buffer volume variations that occur during the Li+ insertion/de-insertion process.2,13–20 However, this proposed strategy was also unable to provide Co3O4 anodes with excellent electrochemical performance.15 Thereafter, a novel research effort was made to fabricate ternary metal oxide anodes by partially replacing the cobalt (Co) of Co3O4 using various elements to alleviate above-mentioned issues via the complementary behavior and synergistic effect between the two different metal elements.21,22 Among all ternary metal oxides, the synthesis and/or use of ZnCo2O4 spinel has been abundantly reported for energy conversion and storage applications due to its low cost, low toxicity, numerous novel morphologies, excellent electrical conductivity, high theoretical capacity (∼903 mA h g−1) and use of inexpensive and nontoxic Zn.23–26 In addition, the conversion and alloying/de-alloying reactions occurred in the ZnCo2O4 anode because of the further reaction of Zn as a conversion product with Li and formation of the LiZn alloy, which eventually offers extra capacity.23 However, like other anodes, ZnCo2O4 suffers from intrinsically low conductivity and large volume variations (∼200%) during the lithium insertion/de-insertion process, resulting in electrode cracking, poor cycling stability and low rate capability.27 Recently, it was reported that the framing of two different active materials in one frame was found to be promising to improve physical/chemical properties and provide high electrochemical reactivity and excellent mechanical stability due to the synergistic effect of each component.7–10 Since ZnCo2O4 and Co3O4 individually exhibit severe limitations as an anode for LIBs, it would be novel to manufacture a single advanced electrode that combined the characteristics of the electrodes by offering high capacity due to the contribution of each electrode and that nullified their individual limitations during repeated charge/discharge cycles. Although Zn and Co have electrochemical activity toward lithium, their mutual favorable buffering matrices and complementary behavior may not only alleviate the large mechanical stress derived from the severe volume change during cycling but also contribute to enhancing the lithium-storage performance.
Thus, in the current work, a nanocomposite of ZnCo2O4 and Co3O4 was synthesized by simply controlling the molar ratio of the Zn and Co precursors. It is believed that the ZnCo2O4/Co3O4 nanocomposite electrode may exhibit superior electrochemical performance due to the intimate interconnection between ZnCo2O4 and Co3O4, which can relieve stresses caused by volume variations due to the different working voltages of the two electrochemically active metal oxides. Additionally, the coexisting Co3O4 may participate in electrochemical reactions and contribute to the total capacity of the host material by storing 8Li+ ions per formula unit (Co3O4 + 8Li+ + 8e− ↔ 3Co + 4Li2O).
2 Experimental
2.1 Materials and methods
Scheme 1 presents a schematic illustration of the synthesis procedure that was used to prepare the samples. In a typical synthesis, 0.3 g of urea and nonstoichiometric amounts of the precursors, namely, 2 mM zinc nitrate (Zn(NO3)2·6H2O, Sigma Aldrich, 98%, Germany) and 6 mM cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, Sigma Aldrich, ≥98%, United Kingdom) were first combined. Then, all reagents were dissolved in a mixed solution of 40 mL of deionized water and 30 mL of ethylene glycol (EG) under constant stirring at room temperature. Thereafter, 0.5 g of polyvinyl pyrrolidone (PVP, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Alfa Aesar, China) was added to the above solution under continuous magnetic stirring to achieve a homogeneous solution. Then, the obtained homogeneous solution was transferred into a 100-mL Teflon-lined stainless-steel autoclave and heated to 150 °C for 14 h. After natural cooling to room temperature, the as-obtained product was washed with de-ionized water and absolute ethanol several times and later dried in an oven at 60 °C overnight. The dried product was further calcined at 400 °C for 3 h in air, with a ramping rate of 2 °C min−1 to ensure the purity of the sample. For comparison, pure ZnCo2O4 was synthesized with same experimental conditions except using a Zn:Co stoichiometric ratio of 2:4. In addition, pure Co3O4 was synthesized using same parameters but without the precursor Zn(NO3)2·6H2O.
000 g mol−1, Alfa Aesar, China) was added to the above solution under continuous magnetic stirring to achieve a homogeneous solution. Then, the obtained homogeneous solution was transferred into a 100-mL Teflon-lined stainless-steel autoclave and heated to 150 °C for 14 h. After natural cooling to room temperature, the as-obtained product was washed with de-ionized water and absolute ethanol several times and later dried in an oven at 60 °C overnight. The dried product was further calcined at 400 °C for 3 h in air, with a ramping rate of 2 °C min−1 to ensure the purity of the sample. For comparison, pure ZnCo2O4 was synthesized with same experimental conditions except using a Zn:Co stoichiometric ratio of 2:4. In addition, pure Co3O4 was synthesized using same parameters but without the precursor Zn(NO3)2·6H2O.
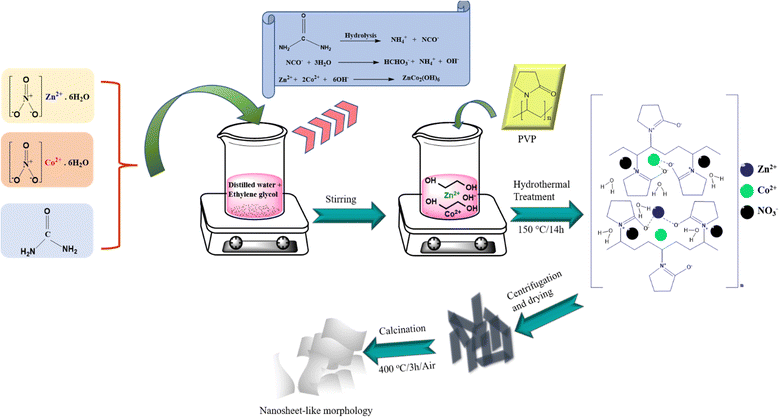 |
| | Scheme 1 Schematic illustration of the synthesis of the ZnCo2O4 sample. | |
2.2 Material characterization
To confirm the crystal structure and phase purity of calcined products, X-ray diffraction (XRD) analysis was performed using a Bruker D8 discover high-resolution X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å, Karlsruhe, Germany). Detailed microstructural analysis was carried out using field-emission scanning electron microscopy (FE-SEM, Zeiss Gemini SEM 500 Thermal field emission type, Zeiss) equipped with energy dispersive spectrometry (EDS) and field emission transmission electron microscopy (FE-TEM, FEI Tecnai G2-20 at 200 kV). The specific surface area of calcined samples, including the pore size distribution, was determined using the Brunauer–Emmett–Teller (BET, Micromeritics Instrument Corp. Gemini-V) using the N2 adsorption/desorption isotherm. X-ray photoelectron spectroscopy (XPS) was also used to investigate the chemical composition and oxidation states of the elements present in the samples using M/s Thermo Fischer Scientific Instrument UK with a monochromatic Al Kα X-ray source.
2.3 Electrode fabrication
CR2032 coin-type half-cells were fabricated using lithium metal foil as the counter and reference electrode to measure the electrochemical properties of the calcined samples. The slurries were first prepared by mixing of 60 wt% of the active material, 20 wt% of carbon black as a conductive agent and 20 wt% of polyvinylidene difluoride (PVdF) as a binder in N-methyl-2-pyrrolidone (NMP). The obtained paste was then cast on a copper foil (current collector) using a doctor blade, which was then dried in a vacuum oven at 120 °C overnight and pressed through a twin roller hot press for better contact between the copper foil and slurry. The cell assembly was then performed in an argon (Ar)-filled glovebox (H2O and O2 content less than 1 ppm). Glass fibre was used as a separator. A 1 M solution of the salt LiPF6 in an ethylene carbonate (EC):dimethyl carbonate (DMC) mixture with a volume ratio of 1:1 was used as the electrolyte. The electrochemical testing of assembled half-cells was performed using a BTS-2004H, Nagano, Japan electrochemical workstation within the potential range of 0.01–3.0 V vs. Li+/Li at various current rates.
3 Results and discussion
3.1 Crystal structure and morphology
First, powder XRD analysis was performed to confirm the purity and crystal structure of pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples. All thethree samples displayed identical diffraction patterns that could be well indexed to the standard JCPDS data of ZnCo2O4 (JCPDS no. 01-081-2299) and Co3O4 (JCPDS no. 00-043-1003).28 Since the diffraction peaks of Co3O4 are perfectly overlapped with those of ZnCo2O4 due to their similar structural parameters, it is quite difficult to differentiate them in the nanocomposite sample. More importantly, there were no other impurity peaks observed in XRD patterns, indicating the high purity of the prepared samples. The actual lattice parameters were calculated using Rietveld refinement with the program Fullprof.29 The Rietveld refinement fit for ZnCo2O4/Co3O4 ZnCo2O4 and Co3O4 in the 2θ range of 20° to 80° using a cubic structure in the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group is shown in Fig. 1(a)–(c). During the refinement, a pseudo-Voigt function and linear interpolation between a set background point with refinable heights was considered for the profile shape and background, respectively. The lattice parameters obtained from the Rietveld refinement were 8.1309(8), 8.1359(5) and 8.0842(3) Å for ZnCo2O4/Co3O4, ZnCo2O4 and Co3O4, respectively. The lattice parameter of ZnCo2O4 is slightly larger than that of Co3O4 due to presence of Zn ions, which have a slightly larger ionic radius than Co, in the Co site. Further, in the case of ZnCo2O4/Co3O4, there is a minute decrease in the lattice parameter because of the increase in the concentration of Co ions, which have a smaller ionic radius than Zn ions. There is a significant possibility that in the formation of ZnCo2O4/Co3O4 composite, some of Zn sites are occupied by the Co ions and behave like a solid solution. However, very-high-resolution synchrotron data would be required to clearly identify this phenomenon.
m space group is shown in Fig. 1(a)–(c). During the refinement, a pseudo-Voigt function and linear interpolation between a set background point with refinable heights was considered for the profile shape and background, respectively. The lattice parameters obtained from the Rietveld refinement were 8.1309(8), 8.1359(5) and 8.0842(3) Å for ZnCo2O4/Co3O4, ZnCo2O4 and Co3O4, respectively. The lattice parameter of ZnCo2O4 is slightly larger than that of Co3O4 due to presence of Zn ions, which have a slightly larger ionic radius than Co, in the Co site. Further, in the case of ZnCo2O4/Co3O4, there is a minute decrease in the lattice parameter because of the increase in the concentration of Co ions, which have a smaller ionic radius than Zn ions. There is a significant possibility that in the formation of ZnCo2O4/Co3O4 composite, some of Zn sites are occupied by the Co ions and behave like a solid solution. However, very-high-resolution synchrotron data would be required to clearly identify this phenomenon.
 |
| | Fig. 1 Rietveld refinement fit on the XRD patterns of (a) ZnCo2O4/Co3O4, (b) ZnCo2O4 and (c) Co3O4 in the 2θ range 20° to 80° using a cubic structure in the Fdm space group. In the figure, black dots, red line and cyan line represent experimental, calculated and difference profiles, respectively, and vertical tick marks above the difference profile show Bragg peak positions. | |
To further explore the surface morphology of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples, FE-SEM, FE-TEM, HR-TEM and EDS mapping were carried out, and the obtained results are displayed in Fig. 2. Fig. 2(a) shows the nanosheet-like morphology of Co3O4, which is actually made up of small-sized interconnected nanoparticles. Numerous voids can easily be seen on the surface of nanosheets, which confirmed the existence of porosity within the sample. Conversely, the FE-SEM images of the pure ZnCo2O4 (Fig. 2d) and ZnCo2O4/Co3O4 nanocomposite (Fig. 2g) samples clearly showed a 2D ultrathin nanosheet-like morphology. As can be observed, the nanosheets of the pure ZnCo2O4 sample exhibit rough surfaces with high porosity, which can be attributed to the loss of organic moieties, PVP and gases during the calcination process. However, the ZnCo2O4/Co3O4 nanocomposite sample shows a relatively smooth and intact surface morphology, which may be due to the presence of the additional phase Co3O4, promoting better sustainability of the morphology and suppression of the high porosity. Detailed microstructural analysis of all the three samples was further conducted using FE-TEM and HR-TEM analysis. It is evident from Fig. 2(b) that the interconnected nanoparticles have sizes in the range of ∼20–35 nm for the Co3O4 sample. The white pores between the nanoparticles can be clearly visualized in the FE-TEM image. In contrast, the FE-TEM images of the pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples revealed that both the samples were mainly composed of a large number of small interconnected nodal-shaped nanoparticles with sizes of ∼5–40 nm and ∼6–15 nm, respectively, as shown in Fig. 2(e) and (h). It can be noticed that the nanocomposite sample has a high density of nanoparticles that are closely interconnected to form a solid network-like structure in comparison to the pure ZnCo2O4 sample. In contrast, the nanoparticles of the pure ZnCo2O4 sample are comparatively less intact with poor interconnection and larger pores, resulting in a fragile and broken morphology, as presented in Fig. 2(e). The corresponding HR-TEM images of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples are displayed in Fig. 2(c), (f) and (i), respectively. The HR-TEM image of the Co3O4 nanosheet exhibits two lattice fringes of ∼0.28 nm and ∼0.46 nm corresponding to the (220) and (111) planes of Co3O4, respectively, which is in accordance with the XRD result, as shown in Fig. 1(c). Moreover, the interplanar distances were measured to be ∼0.15 nm and ∼0.28 nm for pure ZnCo2O4 and ∼0.14 nm and ∼0.22 nm for the nanocomposite sample, which also matched well with the (511), (220), (440) and (222) planes of the cubic spinel of ZnCo2O4, respectively. Furthermore, representative energy-dispersive spectroscopy (EDS) of the ZnCo2O4/Co3O4 nanocomposite sample was carried out as shown in Fig. 2(j)–(l), which confirmed the existence of only the elements Zn, Co, and O.
 |
| | Fig. 2 FE-SEM micrographs (a), (d) and (g), FE-TEM images (b), (e) and (h), and HR-TEM images (c), (f) and (i) of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples, respectively. (j)–(l) Representative energy dispersive X-ray elemental mapping images of the ZnCo2O4/Co3O4 sample. | |
To investigate the specific surface area and pore sizes of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples, BET measurement was performed using N2 adsorption/desorption isotherms. Fig. 3(a)–(c) clearly show that all the three samples exhibit a typical type-IV isotherm with an H3 hysteresis loop, which demonstrates the existence of mesoporous characteristics in the samples.15 The BET specific surface areas of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite samples were found to be ∼24.8 m2 g−1, ∼61.4 m2 g−1 and ∼56.7 m2 g−1, respectively. It is obvious that the pure ZnCo2O4 sample has a high surface area, which may be due to the existence of numerous pores, as was evident from the FE-SEM and FE-TEM images. The insets of Fig. 3(a)–(c) depict pore size distribution plots generated using Barret–Joyner–Halenda (BJH) analysis. The obtained plots demonstrate a wide distribution of pores in the samples with an average pore diameter of ∼15 nm for the pure Co3O4 sample, ∼9.4 nm for the pure ZnCo2O4 sample and ∼1.4 nm for the ZnCo2O4/Co3O4 nanocomposite sample, which is in agreement with the microstructural analysis. It is believed that the obtained porosity within the samples may provide several surface-active sites, which can allow the passage of electrolytes for better electrode/electrolyte interface and eventually promote fast Li+ ion insertion/de-insertion reactions in the sample.
 |
| | Fig. 3 N2 adsorption–desorption isotherm with corresponding BJH pore size distribution curve (inset) of the pure Co3O4, (a) pure ZnCo2O4 (b) and ZnCo2O4/Co3O4 nanocomposite (c) samples. | |
A representative XPS analysis was further used to investigate the surface composition and oxidation state of metal ions in the ZnCo2O4/Co3O4 nanocomposite sample, and the obtained results are presented in Fig. 4. The survey spectrum of the sample (Fig. 4a) confirmed the presence of only four elements, i.e., Zn, Co, O and C. The obtained spectra of Zn-2p, Co-2p, and O-1s were fully fitted against a linear-type background using the software XPSPEAK 41. Fig. 4(b) depicts the high resolution spectrum of Zn-2p, which exhibits two prominent peaks located at binding energies of ∼1021.4 eV and ∼1044.5 eV, corresponding to the spin doublet of Zn 2p3/2 and Zn 2p1/2, respectively, indicating the +2 oxidation state of Zn in the nanocomposite sample.30,31 The high-resolution spectrum of Co-2p shows two spin orbit doublets along with satellite peaks (denoted as Sat.), which mainly belong to Co 2p3/2 and Co 2p1/2 components, as shown in Fig. 4(c). The individual components deconvoluted at binding energies of ∼779.9 eV and ∼794.9 eV confirmed the existence of Co3+, while the obtained binding energies of ∼781.3 eV and ∼796.5 eV verified the presence of Co2+ species.30–32 Moreover, the high-resolution spectra of O-1s (Fig. 4(d)) displays three peaks at binding energies of ∼529.6 eV (O1), ∼531.5 eV (O2) and ∼532.3 eV (O3), which can be assigned to the metal–oxygen bond and physically/chemically adsorbed water in the surface region. In conclusion, the obtained results confirmed the existence of Zn2+, Co3+ and Co2+ ions within the ZnCo2O4/Co3O4 nanocomposite sample, which is in agreement with previous literature.30–32
 |
| | Fig. 4 High resolution XPS spectra of the ZnCo2O4/Co3O4 nanocomposite sample: (a) survey spectrum, (b) Zn-2p, (c) Co-2p and (d) O-1s. | |
3.2 Electrochemical performance
To further analyze the Li+ ion diffusion mechanism in the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes, cyclic voltammetry (CV) tests were conducted at various scan rates, as depicted in Fig. 5. The reduction peak observed at ∼0.96 V during the cathodic sweep of the Co3O4 electrode can be ascribed to the multistep electrochemical reduction (lithiation) of Co3O4 to metallic cobalt (Co3+ → Co2+ → Co) embedded in the Li2O matrix (conversion mechanism), followed by the formation of a solid electrolyte interphase (SEI) film on the electrode surface.33,34 However, it can be seen that the pure and nanocomposite electrodes exhibit a sharp irreversible cathodic peak at ∼0.75 V and ∼0.81 V, respectively, at a lower scan rate of 0.1 mV s−1, which can be attributed to the reduction of ZnCo2O4 to Zn0 and Co0, followed by SEI layer formation on electrode surfaces. Additionally, a weak peak appeared at ∼0.41 V for the pure ZnCo2O4 and ∼0.38 V for the ZnCo2O4/Co3O4 nanocomposite electrode, which is in fact due to the further lithiation of Zn0 to form a Li–Zn alloy, as described in eqn (iii).30,35–37 However, the cathodic peaks became wide during the subsequent scanning and shifted to ∼0.86 V and ∼0.95 V for the pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes, respectively, which corresponds to the reduction of ZnO to metallic Zn and Co3O4/CoO to metallic Co.30,36 The notable disparity between the first and subsequent scans indicates a change in lithium-ion insertion reactions. On the contrary, two oxidation peaks were observed at ∼1.6 V and ∼2.1 V during the anodic sweep for all three electrodes, which are due to the reversible oxidation (de-lithiation) of the metallic Co to Co2+/Co3+ of the Co3O4 and Zn of ZnCo2O4 to Zn2+ and Co3+, respectively, along with the decomposition of the SEI film (eqn (iv)–(vi)).38,39 The binary metal oxide Co3O4 generally shows a normal spinel structure in which the Co3+ and Co2+ ions are octahedrally and tetrahedrally coordinated in a cubic close-packed oxide anion lattice, respectively, which demonstrates multistep redox reactions of Co3+/Co2+/Co during the lithiation/de-lithiation process.11 As can be seen, the oxidation peaks of the pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes shifted to ∼1.81 V and ∼1.74 V during subsequent anodic scans, respectively. At various subsequent scan rates ranging from 0.2 to 2 mV s−1, all CV curves exhibit a similar shape with nominal shift of reduction and oxidation peaks, indicating high reversibility with small polarization.40,41
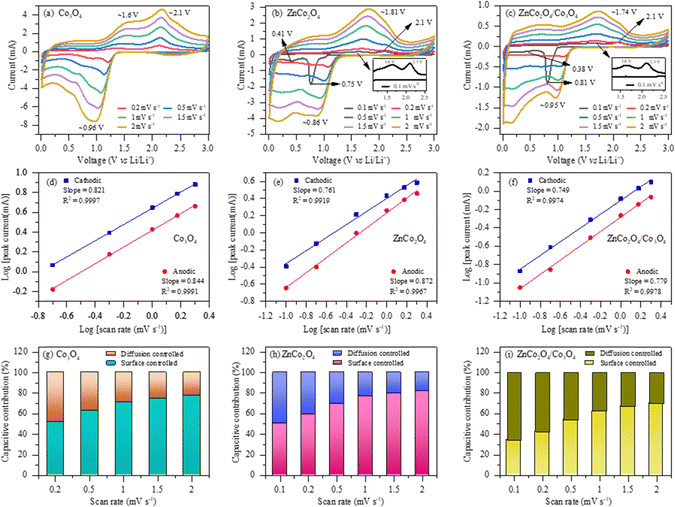 |
| | Fig. 5 (a)–(c) Cyclic voltammetry curves at various scan rates; (d)–(f) linear relationship between the logarithm of peak current and scan rates; and (g)–(i) corresponding capacitive and diffusion contribution ratios at different scan rates for the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes, respectively. | |
According to previous reports,30,36,39 the electrochemical reaction mechanism involved in the active materials of Co3O4 and ZnCo2O4 during the Li+ ion insertion and de-insertion processes can be summarized as follows:
| | | Co3O4 + 8Li+ + 8e− → 4Li2O + 3Co0 | (i) |
| | | ZnCo2O4 + 8Li+ + 8e− → Zn + 2Co + 4Li2O | (ii) |
| | | Zn + Li+ + e− → LiZn | (iii) |
| | | Zn + Li2O ↔ ZnO + 2Li+ + 2e− | (iv) |
| | | Co + Li2O ↔ CoO + 2Li+ + 2e− | (v) |
| | | 3CoO + Li2O ↔ Co3O4 + 2Li+ + 2e− | (vi) |
Moreover, the kinetic behavior of lithium storage in all three electrodes can be examined by applying the power law, which is a widely applied approach to determine the storage mechanism to be either a surface-induced capacitance process or diffusion-controlled insertion process.
42,43 Typically, the relationship between the measured peak current (
i) and scan rate (
v) can be expressed using
eqn (vii) and (viii).
| | | log(i) = log(a) + blog(v) | (viii) |
Here, ‘
a’ and ‘
b’ refer to adjustable empirical constants.
44,45 The ‘
b’ values can be obtained from the slope of the linear relationship between log(
i) and log(
v), as shown in
Fig. 5(d)–(f). The ‘
b’ values range between 0.5 and 1; it has been reported that the diffusion-controlled process is significant if the ‘
b’ value is close to or equal to 0.5. However, if the ‘
b’ values are close to or equal to 1, the electrode will exhibit ideal surface-induced capacitive behavior.
45,46 The ‘
b’ values were calculated after plotting the graph between log(
i)
versus log(
v) for all the three electrodes during anodic and cathodic processes and were calculated to be ∼0.844 and ∼0.821 for the pure Co
3O
4 electrodes, ∼0.761 and ∼0.872 for the pure ZnCo
2O
4 electrodes and ∼0.749 and ∼0.779 for the ZnCo
2O
4/Co
3O
4 nanocomposite electrodes, suggesting that the surface-induced capacitive behavior is dominant in the Li
+ ion diffusion mechanism. Thereafter, the contribution ratios of diffusion-controlled and surface-controlled capacitive mechanisms were quantified using following equations:
45,47| | | i(V)/v1/2 = k1v1/2 + k2, | (x) |
where
v and
i(
V) represent the scan rate and total current at constant potential, respectively, while
k1v and
k2v1/2 stand for surface-controlled capacitive behavior and diffusion-controlled contributions, respectively. Furthermore,
eqn (x), which is a simple rearrangement of
eqn (ix), was used to calculate the values of constants
k1 and
k2. These constants were utilized to determine the slope and intercept of the linear fitting of the plot of
i(
V)/
v1/2vs. v1/2 at a constant potential. The percentage contribution ratio of the surface-controlled capacitive and diffusion-controlled mechanisms at various scan rates of all the three electrodes are summarized in
Fig. 5(g)–(i). It can be noticed that the surface-controlled capacitive contribution increases with increasing scan rate, which indicates that the electrode surfaces are highly active for Li
+ ion storage at high scan rates. More importantly, the surface-controlled capacitive mechanism is higher for the pure ZnCo
2O
4 electrode than the nanocomposite electrode.
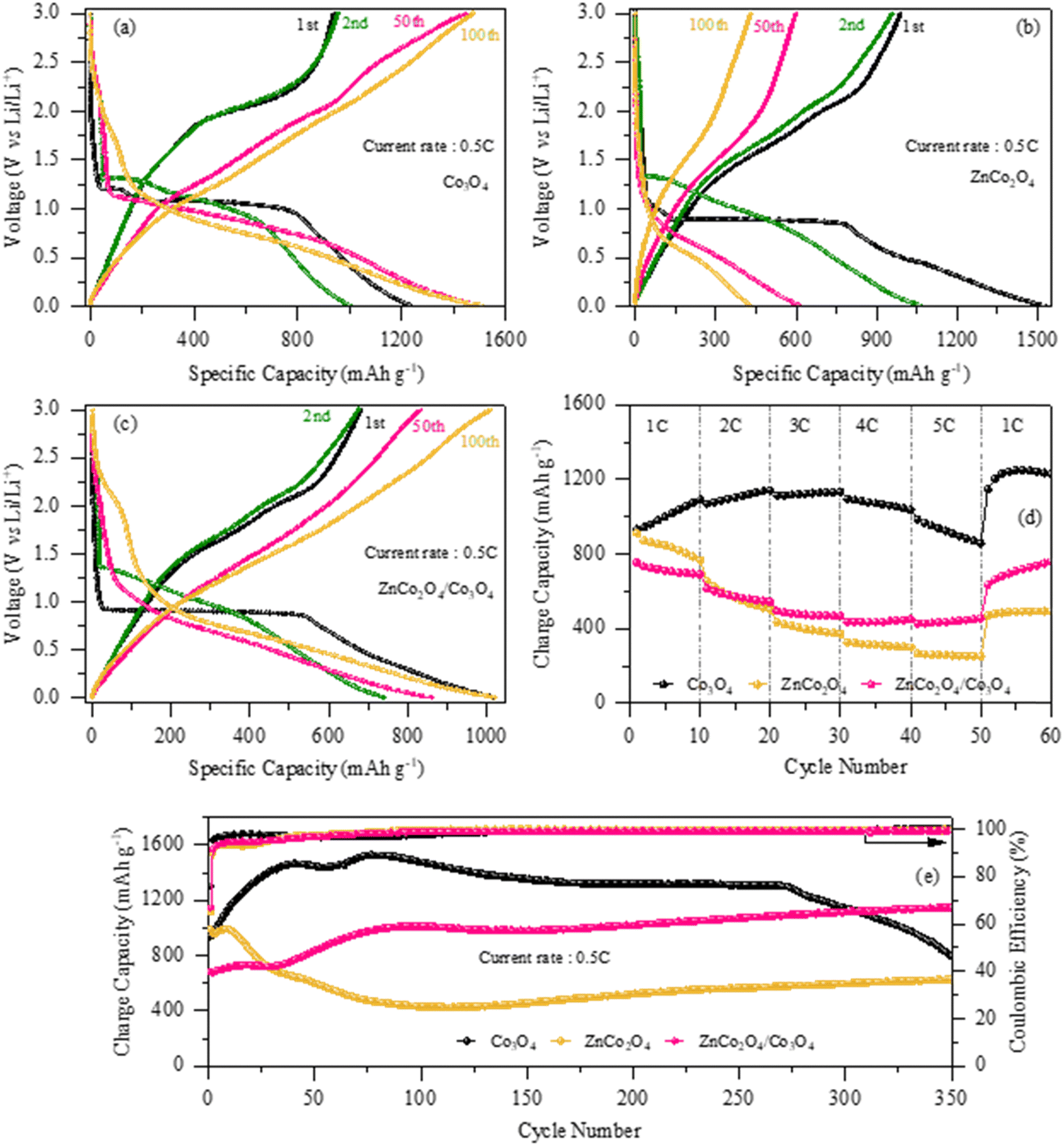
Fig. 6(a)–(c) depicts the galvanostatic discharge/charge profiles of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes for the 1st, 2nd, 50th and 100th cycles at 0.5C within the potential range of 0.01–3.0 V (vs. Li/Li+). During the first discharge of the Co3O4 electrode, two voltage plateaus at ∼1.2 V and ∼1.1 V are clearly seen, followed by a gradual sloping region until the cutoff voltage of ∼0.01 V, which can be ascribed to the reduction of Co3O4 to metallic cobalt and formation of the SEI film, respectively.44 The other two ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes exhibit a short and long voltage plateau at ∼0.6 V and ∼0.9 V during the 1st discharge and later gradually decline until the cut-off potential of ∼0.01 V, which is obviously due to the reduction of ZnCo2O4 to Zn and Co followed by the formation of the Li–Zn alloy.48 The obtained discharge/charge voltage plateaus of the electrodes matched well with redox peaks observed in CV results. The initial discharge/charge capacities of the pure Co3O4, ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes were found to be 1233.4/938.8 mA h g−1, 1529/989 mA h g−1 and 1020/681 mA h g−1 with Coulombic efficiencies of 76%, 64% and 66%, respectively. The significant capacity loss in the first charge cycle can be attributed to the formation of the SEI layer caused by the electrolyte degradation, which prevents the complete de-insertion of Li+ ions from the active materials, resulting in the irreversible capacity loss. From the 2nd cycle onwards, the discharge/charge capacities of the pure Co3O4 electrode started to increase and reached 1001.8/954.6 mA h g−1 for the 2nd cycle and 1509.6/1474.5 mA h g−1 for the 100th cycle with a Coulombic efficiency of 95.2% and 97.6%, respectively, which may be due to the continuous activation of the electrode because of the large surface area and high porosity. However, the discharge/charge capacity of the pure ZnCo2O4 electrode decreased in the 2nd, 50th and 100th cycles. In contrast, the capacity of the nanocomposite electrode significantly increased after the 1st cycle and reached a capacity of 1010/1024 mA h g−1 at the 100th cycle, which is not only greater than the capacity of the pure ZnCo2O4 electrode (431/429 mA h g−1 at the 100th cycle) but also higher than the theoretical capacity of ZnCo2O4 (∼903 mA h g−1). Although the ZnCo2O4/Co3O4 nanocomposite electrode shows high capacity with better electrochemical reversibility (100% Coulombic efficiency) than pure ZnCo2O4, it is believed that the synergistic effect between ZnCo2O4 and Co3O4 offers more active sites for lithium ion storage.
 |
| | Fig. 6 (a)–(c) Charge/discharge profiles at 0.5C; (d) rate performance at various C-rates; and (e) cycling performances at 0.5C over 350 cycles of the pure Co3O4, pure ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes, respectively. | |
Fig. 6(d) presents a comparative analysis of the rate performances of the pure Co3O4, ZnCo2O4 and ZnCo2O4/Co3O4 nanocomposite electrodes at various C-rates. From the overall profile, it can be observed that Co3O4 shows excellent performance at all current rates with specific capacities of 1116.5, 1164.2, 1158.7, 1069.1 and 889.7 mA h g−1 at 1C, 2C, 3C, 4C and 5C, respectively, which are much higher than the capacities of the ZnCo2O4/Co3O4 nanocomposite electrode (691 mA h g−1, 543 mA h g−1, 468 mA h g−1, 449 mA h g−1 and 454 mA h g−1) and pure ZnCo2O4 electrode (767 mA h g−1, 499 mA h g−1, 372 mA h g−1, 299 mA h g−1 and 250 mA h g−1) at same current rates. More importantly, it should be noted that the obtained capacity of the Co3O4 electrode, especially at the high current rate of 5C, is almost same to the theoretical capacity of the Co3O4 electrode (∼890 mA h g−1), which clearly demonstrates the promising potential characteristics of the Co3O4 anode. Additionally, when the current rate was reverted to the initial rate of 1C, the charge capacities of the pure Co3O4 electrode and ZnCo2O4/Co3O4 nanocomposite electrode remained considerably higher after 60 cycles (1229.3 mA h g−1 and 759.8 mA h g−1, respectively) than that of the pure ZnCo2O4 electrode (493.7 mA h g−1), which may be due to the higher structural stability and reduced charge transfer resistance within these two electrodes. It can be seen that the capacity started to decrease with increasing current rates in all the electrodes, which is generally due to the slow diffusion rate of Li+ ions into the electrode material at high current rates.
To further examine the long-term cycle life of all the three electrodes, cyclability tests were performed at 0.5C for 350 cycles, and the obtained results are presented in Fig. 6(e). At first sight, it can be seen that the charge capacity of the pure Co3O4 electrode significantly increased to 1462.8 mA h g−1 during the first 40 cycles, which can be attributed to the formation/decomposition of a reversible polymeric gel-like film on the electrode surface due to electrolyte degradation.49,50 Thereafter, the charge capacity was found to decrease slightly from the 40th to 60th cycle, at which the capacity was 1447.7 mA h g−1, which may be due to the restriction of Li+ ion extraction during charging due to the formation of an unstable SEI film, indicating that the conversion reaction may not be fully reversible after a certain cycle number, and therefore, capacity loss is observed in the successive cycles. Furthermore, the Co3O4 electrode exhibits a slow drop in capacity up to 270 cycles, at which it retained a charge capacity of 1304.1 mA h g−1 with a Coulombic efficiency of nearly 100%. Then, the charge capacity rapidly begins to decline, and retained a maximum capacity of only 794.7 mA h g−1 after 350 cycles, which clearly indicates that the obtained morphology of the porous nanosheet-like Co3O4 could not be withstand being used as an anode during long-term cycling. It is believed that the porous nanosheet-like structure of the Co3O4 anode, which is mainly composed of small-sized interconnected nanoparticles with numerous pores, becomes pulverized during long-range cycling due to the repeated volume expansion/contraction during charge and discharge. Unfortunately, the obtained decreasing trend of the Co3O4 anode clearly indicates that the capacity will reach zero in further consecutive cycles. More importantly, the obtained zig-zag trend is common and usually occurs in transition-metal-oxide anode materials for LIBs.51–53 Similarly, the charge capacity of the pure ZnCo2O4 electrode drops quickly over the first 114 cycles with a capacity retention of only ∼424 mA h g−1. Thereafter, the capacity began to increase and a charge capacity of 628.8 mA h g−1 was observed after 350 cycles with a Coulombic efficiency of 100%. Conversely, the ZnCo2O4/Co3O4 nanocomposite electrode exhibits a significant stepwise increase in the capacity as the cycle number increases and demonstrates a charge capacity of 1011 mA h g−1 after 100 cycles with 100% Coulombic efficiency. After 125 cycles, it can be noted that the charge capacity of the nanocomposite electrode is improved, and it retained a capacity of 1146.2 mA h g−1 after 350 cycles, which is nearly double of the capacity of pure electrode. Since the capacity of the nanocomposite electrode is remarkably higher than that of the pure Co3O4 and ZnCo2O4 electrodes, it is believed that the sustainable morphology with optimum porosity as well as the strong synergistic effect between the two electrochemically active materials, i.e., ZnCo2O4 and Co3O4, offered not only more active sites for Li+ ion diffusion but also effectively buffered the dramatic volume change and maintained the mechanical integrity during Li+ ion insertion/extraction. However, in the case of the pure ZnCo2O4 electrode, the high porosity and weak connectivity between the nanoparticles lead to fast capacity fading and poor structural stability during prolonged cycling. In addition, it is reasonable to suggest that the additional phase Co3O4 may provide superior surface stability and high structural stability to prevent the detachment of the ZnCo2O4 active materials during lithiation/de-lithiation processes. At the same time, Co3O4 may also accelerate the electrochemical reactions by effectively storing eight lithium ions per formula unit due to its high electrochemical activity (Co3O4 + 8Li+ + 8e− ↔ 3Co + 4Li2O) and contribute to the total capacity of the host material.
Fig. 7 shows the galvanostatic intermittent titration technique (GITT) analysis, which exhibits the lithium-ion diffusion kinetics of the electrode materials (Co3O4, ZnCo2O4, and ZnCo2O4/Co3O4) during discharge and charge processes. All the three electrodes were discharged and charged at a C-rate of 0.5C for 10 minutes, then left open for 30 minutes in the voltage range of 0.01–3.0 V for GITT analysis. The GITT investigation consists of a sequence of current pulses followed by relaxation periods, in which no current flows through the cell. During the positive current pulse, the cell potential rapidly increases to a level proportional to the iR drop. Thereafter, the potential gradually increases due to the galvanostatic charge pulse, which maintains a constant concentration gradient. When the current pulse is interrupted during the relaxation period, the composition in the electrode tends to become homogeneous due to Li+ diffusion, resulting in the sudden drop in the potential to a level proportional to the iR drop, followed by a gradual decrease until equilibrium is achieved. During a negative current pulse, the opposite sequence occurs. The cell potential quickly decreases to a level proportional to the iR drop, followed by a slow decrease during the galvanostatic discharge pulse. During the relaxation period, the potential experiences a sudden increase proportional to the iR drop, followed by a gradual rise until equilibrium is attained. As can be seen from the GITT profiles of all the three electrodes, the time required for discharge/charge processes is less for the ZnCo2O4/Co3O4 nanocomposite electrode than the other two, which clearly indicates high Li+ diffusion kinetics for this sample as well justifying the obtained high electrochemical performances of the nanocomposite electrode.
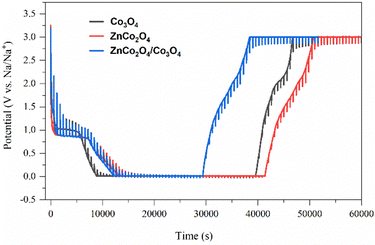 |
| | Fig. 7 GITT analysis of the pure Co3O4, ZnCo2O4, and ZnCo2O4/Co3O4 nanocomposite electrodes. | |
The electrochemical behavior of all the three electrodes was further investigated through electrochemical impedance spectroscopy (EIS) analysis, as depicted in Fig. 8. The Nyquist plot of each electrode consists of a depressed semicircle in the high-frequency region and a sloping line in the low-frequency region. The semicircle observed in the high frequency range signifies the charge transfer resistance (RCT) at the electrode/electrolyte interface, while the inclined line in the low-frequency region represents the Warburg impedance, reflecting electrolyte diffusion within the electrode. A smaller diameter of the semicircle indicates lower RCT and higher Li+ diffusion kinetics. The observed EIS spectrum demonstrates that the ZnCo2O4/Co3O4 nanocomposite electrode exhibits the lowest RCT compared to the pure ZnCo2O4 and pristine Co3O4 electrodes. These results are in good accordance with the cycling performance data of all the electrodes.
 |
| | Fig. 8 EIS analysis of the pure Co3O4, ZnCo2O4, and ZnCo2O4/Co3O4 nanocomposite electrodes. | |
To explain long-term cycling performances, ex situ SEM images of the pure Co3O4, ZnCo2O4, and ZnCo2O4/Co3O4 nanocomposite electrodes were taken after 350 cycles, and the results are displayed in Fig. 9. It is clearly evident that the ZnCo2O4/Co3O4 nanocomposite electrode retains its structural integrity to a certain extent during long-range cycling compared to the other two electrodes, which further verifies the enhanced electrochemical performance of the nanocomposite electrode.
 |
| | Fig. 9
Ex situ SEM images of the (a) pure Co3O4, (b) pure ZnCo2O4 and (c) ZnCo2O4/Co3O4 nanocomposite electrodes after 350 cycles. | |
Since numerous reports have been published on the different nanostructures and composites of ZnCo2O4, a comprehensive table has been compiled to facilitate a clear comparison between previous and current studies. As can be seen from Table 1, the current 2D ultrathin nanosheet-like ZnCo2O4/Co3O4 nanocomposite electrode delivered superior electrochemical performance among all reported ZnCo2O4 data.
Table 1 Comparison of the electrochemical performances of the current 2D ultrathin nanosheet-like ZnCo2O4/Co3O4 electrode with those of previously reported ZnCo2O4 electrodes based on various morphologies/composites
| Material |
Morphology |
Specific capacity/cycle no./current density/rate |
Ref. |
| ZnCo2O4 |
Nanoparticle |
801 mA h g−1/100/100 mA g−1 |
54
|
| ZnCo2O4 |
Nanorods |
767.15 mA h g−1/50/0.2 mA cm−2 |
55
|
| ZnCo2O4 |
Flower-like |
940 mA h g−1/100/0.1C |
56
|
| ZnCo2O4 |
Sliced orange-shaped |
890 mA h g−1/130/200 mA g−1 |
57
|
| ZnCo2O4 |
Nanoparticle |
900 mA h g−1/60/60 mA g−1 |
58
|
| ZnCo2O4 |
Nano-structured Spinel |
1000 mA h g−1/120/0.5C |
59
|
| ZnCo2O4 |
3D Plum like |
746 mA h g−1/110/500 mA g−1 |
60
|
| ZnCo2O4 |
Pineapple shaped |
1132 mA h g−1/120/100 mA g−1 |
61
|
| ZnCo2O4/Graphene |
Nanoparticle |
299.8 mA h g−1/70/0.1C |
62
|
| ZnCo2O4/Graphene |
Sheet-like |
791 mA h g−1/1000/1 A g−1 |
63
|
| Co3O4@ZnCo2O4@NC |
Porous nanowires |
931 mA h g−1/50/100 mA g−1 |
64
|
| ZnCo2O4/Co3O4 |
2D ultrathin nanosheet-like |
1146.2 mA h g−1/350/0.5C |
This work |
4 Conclusions
In summary, we have successfully synthesized a ZnCo2O4/Co3O4 nanocomposite sample using a facile hydrothermal approach, which is comprised of a large number of small interconnected nodal-shaped nanoparticles. To obtain the additional phase Co3O4, a nonstoichiometric ratio between Zn and Co was simply maintained. To accommodate two electrochemically active anode materials in one frame rather than one anode was the motive behind introducing the additional phase Co3O4 along with ZnCo2O4 in this work. With the introduction of Co3O4, the ZnCo2O4/Co3O4 nanocomposite sample exhibits visibly enhanced electrochemical properties compared to the pure ZnCo2O4 and pure Co3O4 samples. It is believed that the additional phase Co3O4 provides a smooth and intact surface morphology, which helps in alleviating the mechanical stress induced by the Li+ insertion/extraction process and preserves the integrity of the nanocomposite morphology. Furthermore, Co3O4 may participate in the electrochemical reactions due to its highly electrochemical active nature and thereby enhance the electrochemical performance of the ZnCo2O4 anode. Based on these structural features and the synergistic effect between ZnCo2O4 and Co3O4, the nanocomposite electrode exhibits superior performance with long-term cycling stability, such as a charge capacity of 1146.2 mA h g−1 at 0.5C after 350 cycles. Hence, it can be concluded that the ZnCo2O4/Co3O4 nanocomposite holds significant potential as an anode material for LIBs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Anubha Tomar would like to express her sincere gratitude to CSIR, New Delhi, India for the award of senior research fellowship (09/045(1687)/2019-EMR-1). Dr Alok Kumar Rai is grateful to University Grants Commission (UGC) for the position under UGC-FRP scheme (FRP ID: 57304). This work was mainly supported by the Faculty Research Programme Grant (Ref. No./IoE/2023-24/12/FRP), sanctioned by Institution of Eminence, University of Delhi and partially supported by the National Research Foundation of Korea (NRF) through a grant funded by the Korea government (MSIT) (NRF-2018R1A5A1025224).
References
- A. Pan, Y. Wang, W. Xu, Z. Nie, S. Liang, Z. Nie, C. Wang, G. Cao and J. G. Zhang, J. Power Sources, 2014, 255, 125–129 CrossRef CAS.
- Y. Tan, C. Yang, W. Qian, X. Sui, C. Teng, Q. Li and Z. Lu, J. Alloys Compd., 2021, 855, 157387 CrossRef CAS.
- L. Zhang, B. R. Lu, F. H. Cao, Z. L. Yu, H. P. Cong and S. H. Yu, J. Mater. Chem. A, 2018, 6, 12962–12968 RSC.
- L. Kong, L. Wang, D. Sun, S. Meng, D. Xu, Z. He, X. Dong, Y. Li and Y. Ji, Molecules, 2019, 24, 3149 CrossRef CAS PubMed.
- Z. Li, B. Li, L. Yin and Y. Qi, ACS Appl. Mater. Interfaces, 2014, 6, 8098–8107 CrossRef CAS PubMed.
- H. Xiao, Y. Li, R. Chen, T. Xie, P. Xu, H. Zhu, J. He, W. Zheng and S. Huang, eScience, 2023, 3, 100134 CrossRef.
- Z. Song, H. Liu, B. Chen, Q. Jiang, F. Sui, K. Wu, Y. Cheng and B. Xiao, Phys. Chem. Chem. Phys., 2024, 26, 1406–1427 RSC.
- Y. Fang, D. Luan and X. W. Lou, Adv. Mater., 2020, 32, 2002976 CrossRef CAS PubMed.
- Y. Fang, D. Luan, Y. Chen, S. Gao and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 2644 CrossRef CAS PubMed.
- Y. Fang, B. Y. Guan, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 7739 CrossRef CAS PubMed.
- H. S. Jadhav, A. K. Rai, J. Y. Lee, J. Kim and C. J. Park, Electrochim. Acta, 2014, 146, 270–277 CrossRef CAS.
- S. Abouali, M. A. Garakani, B. Zhang, H. Luo, Z. L. Xu, J. Q. Huang, J. Huang and J. K. Kim, J. Mater. Chem. A, 2014, 2, 16939–16944 RSC.
- H. Du, K. Huang, M. Li, Y. Xia, Y. Sun, M. Yu and B. Geng, Nano Res., 2018, 11, 1490–1499 CrossRef CAS.
- J. Wang, H. Wang, F. Li, S. Xie, G. Xu, Y. She, M. K. H. Leung and T. Liu, J. Mater. Chem. A, 2019, 7, 3024–3030 RSC.
- S. Baji, S. V. Nair and A. K. Rai, J Solid State Electr., 2017, 21, 2869–2875 CrossRef.
- G. Zhang and X. W. Lou, Angew. Chem., 2014, 126, 9187–9190 CrossRef.
- M. Chen, X. Xia, J. Yin and Q. Chen, Electrochim. Acta, 2015, 160, 15–21 CrossRef CAS.
- J. S. Chen, T. Zhu, Q. H. Hu, J. Gao, F. Su, S. Z. Qiao and X. W. Lou, ACS Appl. Mater. Interfaces, 2010, 2, 3628–3635 CrossRef CAS PubMed.
- X. Yao, X. Xin, Y. Zhang, J. Wang, Z. Liu and X. Xu, J. Alloys Compd., 2012, 521, 95–100 CrossRef CAS.
- Y. Ding, P. Zhang, Z. Long, Y. Jiang, J. Huang, W. Yan and G. Liu, Mater. Lett., 2008, 62, 3410–3412 CrossRef CAS.
- A. Tomar, E. Adhitama, M. Winter, T. Placke and A. K. Rai, Batteries Supercaps, 2023, 6, e202200465 CrossRef CAS.
- J. Singh, S. Lee, P. Yadav, S. Kim, J. Kim and A. K. Rai, ACS Appl. Energy Mater., 2022, 5, 126–136 CrossRef CAS.
- A. K. Rai, T. V. Thi, B. J. Paul and J. Kim, Electrochim. Acta, 2014, 146, 577–584 CrossRef CAS.
- L. Yu, G. Zhang, C. Yuan and X. W. Lou, Chem. Commun., 2013, 49, 137–139 RSC.
- G. Zhang, B. Y. Xia, C. Xiao, L. Yu, X. Wang, Y. Xie and X. W. Lou, Angew. Chem., 2013, 125, 8805–8809 CrossRef.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609–4613 CrossRef CAS PubMed.
- J. Liu, Q. Xia, Y. Wang and H. Xia, Mater. Lett., 2017, 193, 220–223 CrossRef CAS.
- H. Li, L. Wang, Y. Guan, Y. Su, J. Mu, H. Che, A. Liu and Z. Guo, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 1–10 CrossRef.
-
J. Rodriguez-Carvajal, FullProf; Laboratoire Leon Brillouin, CEA-CNRS CEA/Saclay:91191, Gif sur Yvette Cedex, France, 2008 Search PubMed.
- H. Xu, Y. Zhang, X. Song, X. Kong, T. Ma and H. Wang, J. Alloys Compd., 2020, 821, 153289 CrossRef CAS.
- M. Carbone, J. Electroanal. Chem., 2018, 815, 151e157 CrossRef.
- S. Cheng, Q. Ru, P. Liu, H. Yan, Z. Shi, X. Hou, S. Su, L. Zhao and F. C. C. Ling, J. Alloys Compd., 2019, 809, 151703 CrossRef CAS.
- J. Qiu, M. Yu, Z. Zhang, X. Cai and G. Guo, J. Alloys Compd., 2019, 775, 366–371 CrossRef CAS.
- Y. Jiang, X. Yan, P. Mei, Y. Zhang, W. Xiao and H. Tang, J. Alloys Compd., 2018, 764, 80–87 CrossRef CAS.
- Z. Zhao, G. Tian, V. Trouillet, L. Zhu, J. Zhu, A. Missiul, E. Welter and S. Dsoke, Inorg. Chem. Front., 2019, 6, 1861–1872 RSC.
- X. Song, Q. Ru, Y. Mo, S. Hu and B. An, J. Alloys Compd., 2014, 606, 219–225 CrossRef CAS.
- M. Du, D. He, Y. Lou and J. Chen, J. Energy Chem., 2017, 26, 673–680 CrossRef.
- Y. Jiang, X. Yan, P. Mei, Y. Zhang, W. Xiao and H. Tang, J. Alloys Compd., 2018, 764, 80–87 CrossRef CAS.
- Z. Wang, Q. Ru, X. Chen, Q. Guo, B. Wang, X. Hou and S. Hu, ChemElectroChem, 2017, 4, 2218–2224 CrossRef CAS.
- P. Gurunathan, P. M. Ette, N. Lakshminarasimhan and K. Ramesha, ACS Omega, 2017, 2, 7647–7657 CrossRef CAS PubMed.
- M. Zhong, W. W. He, W. Shuang, Y. Y. Liu, T. L. Hu and X. H. Bu, Inorg. Chem., 2018, 57, 4620–4628 CrossRef CAS PubMed.
- W. Bai, H. Tang, J. Zhai, C. Cui, W. Wang, C. Cheng, E. Ren, H. Xiao, M. Zhou, R. Guo and S. Lin, Ind. Eng. Chem. Res., 2022, 61, 12555–12566 CrossRef CAS.
- J. Deng, X. Yu, Y. He, B. Li, Q. H. Yang and F. Kang, Energy Storage Mater., 2017, 6, 61–69 CrossRef.
- Y. H. Lu, J. H. Li, Z. F. Xu, J. M. Liu, S. J. Liu and R. X. Wang, J. Mater. Sci., 2021, 56, 2451–2463 CrossRef CAS.
- W. Wang, M. Zhang, R. Mi, Y. Liu and J. Chen, J. Alloys Compd., 2021, 888, 161615 CrossRef CAS.
- B. Sun, S. Lou, W. Zheng, Z. Qian, C. Cui, P. Zuo, C. Du, J. Xie, J. Wang and G. Yin, Nano Energy, 2020, 78, 105366 CrossRef CAS.
- Z. Chen, S. Fei, C. Wu, P. Xin, S. Huang, L. Selegård, K. Uvdal and Z. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 19768–19777 CrossRef CAS PubMed.
- B. Jiang, C. Han, B. Li, Y. He and Z. Lin, ACS Nano, 2016, 10, 2728–2735 CrossRef CAS PubMed.
- J. Mujtaba, H. Sun, G. Huang, K. Mølhave, Y. Liu, Y. Zhao, X. Wang, S. Xu and J. Zhu, Sci. Rep., 2016, 6, 20592 CrossRef CAS PubMed.
- C. Yan, Y. Zhu, Y. Li, Z. Fang, L. Peng, X. Zhou, G. Chen and G. Yu, Adv. Funct. Mater., 2018, 28, 1705951 CrossRef.
- J. Wang, S. Xie, L. Li, Z. Li, A. M. Asiri, H. M. Marwani, X. Han and H. Wang, Part. Part. Syst. Charact., 2019, 36, 1900109 CrossRef.
- B. Wu, Y. Xie, Y. Meng, C. Qian, Y. Chen, A. Yuan, X. Guo, H. Yang, S. Wan and S. Lin, J. Mater. Chem. A, 2019, 7, 6149–6160 RSC.
- L. Zhu, F. Li, T. Yao, T. Liu, J. Wang, Y. Li, H. Lu, R. Qian, Y. Liu and H. Wang, Energy Fuels, 2020, 34, 11574–11580 CrossRef CAS.
- W. Wang, Y. Yang, S. Yang, Z. Guo, C. Feng and X. Tang, Electrochim. Acta, 2015, 155, 297–304 CrossRef CAS.
- H. Liu and J. Wang, Electrochim. Acta, 2013, 92, 371–375 CrossRef CAS.
- S. Hao, B. Zhang, S. Ball, M. Copley, Z. Xu, M. Srinivasan, K. Zhou, S. Mhaisalkar and Y. Huang, J. Power Sources, 2015, 294, 112–119 CrossRef CAS.
- J. Deng, X. Yu, Y. He, B. Lia, Q. H. Yanga and F. Kang, Energy Storage Mater., 2017, 6, 61–69 CrossRef.
- Y. Sharma, N. Sharma, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2855–2861 CrossRef CAS.
- R. A. Adams, V. G. Pol and A. Varma, Ind. Eng. Chem. Res., 2017, 56, 7173–7183 CrossRef CAS.
- B. Fan, X. Chen, A. Hu, Q. Tang, H. Fan, Z. Liu and K. Xiao, RSC Adv., 2016, 6, 79971–79977 RSC.
- L. Guo, Q. Ru, X. Song, S. Hu and Y. Mo, J. Mater. Chem. A, 2015, 3, 8683–8692 RSC.
- A. K. Rai, T. V. Thi, B. J. Paul and J. Kim, Electrochim. Acta, 2014, 146, 577–584 CrossRef CAS.
- H. Cao, X. Zhou, W. Deng, Z. Ma, Y. Liu and Z. Liu, Chem. Eng. J., 2018, 343, 654–661 CrossRef CAS.
- H. Wang, Y. Zheng, Z. Peng, X. Liu, C. Qu, Z. Huang, Z. Cai, H. Fan and Y. Zhang, Dalton Trans., 2021, 50, 7277–7283 RSC.
Footnote |
| † These authors contributed equally to this paper. |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Jay
Singh
a,
Satendra Pal
Singh
b,
Jay
Singh
a,
Satendra Pal
Singh