
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Can Duvelisib and Eganelisib work for both cancer and COVID-19? Molecular-level insights from MD simulations and enhanced samplings†
Saroj Kumar
Panda
a,
Shaswata
Karmakar
a,
Parth Sarthi
Sen Gupta
b and
Malay Kumar
Rana
 *a
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Berhampur, Odisha 760010, India. E-mail: mrana@iiserbpr.ac.in
bSchool of Biosciences and Bioengineering, D Y Patil International University, Akurdi, Pune, India
First published on 12th March 2024
Abstract
SARS-CoV-2 has caused severe illness and anxiety worldwide, evolving into more dreadful variants capable of evading the host's immunity. Cytokine storms, led by PI3Kγ, are common in cancer and SARS-CoV-2. Naturally, there is a yearning to see whether any drugs could alleviate cytokine storms for both. Upon investigation, we identified two anticancer drugs, Duvelisib and Eganelisib, that could also work against SARS-CoV-2. This report is the first to decipher their synergic therapeutic effectiveness against COVID-19 and cancer with molecular insights from atomistic simulations. In addition to PI3Kγ, these drugs exhibit specificity for the main protease among all SARS-CoV-2 targets, with significant negative binding free energies and small time-dependent conformational changes of the complexes. Complexation makes active sites and secondary structures highly mechanically stiff, with barely any deformation. Replica simulations estimated large pulling forces in enhanced sampling to dissociate the drugs from Mpro's active site. Furthermore, the radial distribution function (RDF) demonstrated that the therapeutic molecules were closest to the His41 and Cys145 catalytic dyad residues. Finally, analyses implied Duvelisib and Eganelisib as promising dual-purposed anti-COVID and anticancer drugs, potentially targeting Mpro and PI3Kγ to stop virus replication and cytokine storms concomitantly. We also distinguished hotspot residues imparting significant interactions.
1. Introduction
In late December 2019, Wuhan Municipal Health Commission reported a cluster of 27 cases of pneumonia, later diagnosed as caused by SARS-CoV-2, a novel coronavirus (COVID-19).1 The world health organization (WHO) declared COVID-19 a pandemic in March 2020.2 SARS-CoV-2 is an enveloped, positive-sensed, and single-stranded RNA beta-coronavirus causing respiratory diseases and is one of the seven pathogenic members of the Coronaviridae family.3 Other members of this family, such as hCoV-OC43, HKU, 229E, etc., are responsible for causing the mild common cold.4,5 The severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV-1) discovered in 2002 and the Middle East respiratory syndrome (MERS) coronavirus (MERS-CoV) discovered in 2012, with fatality rates of 10% and 36%, respectively, are also members of this family.5–7 Compared to MERS-CoV and SARS-CoV, SARS-CoV-2 is more fatal and contagious, raising a serious global concern.5,8The viral spike protein (S) allows virus entry by binding to lung cells’ angiotensin-converting enzyme 2 (ACE2) receptors. The host body's immune system recognizes this spike trimer as a foreign substance, triggering antibody production in response. However, the genetic coding sequence for the spike protein has repeatedly been evolving to create new distinct versions as a defense against the immune system. The most mutated SARS-CoV-2 variant, at the moment, is Omicron, with high transmissibility and immune evasion capability of growing concern. Consequently, Omicron has quickly displaced Delta as the predominant variant. With evasiveness from vaccine-induced neutralizing antibodies, the receptor-binding domain (RBD) of the mutated S protein binds to ACE2 with improved infectivity.9 Because of this, virus tracking and effective vaccine or therapeutic developments have become more challenging. However, scientists have revealed that some nonstructural proteins (NSPs) are conserved compared to spikes in SARS genomes, vindicating that the former could be a more suitable target to defeat this microscopic demon.
Whole NSPs constitute a single chain with two genomic regions: polyproteins 1a (pp1a) and 1b (pp1b). NSP 1–11 are represented by pp1a, while NSP 12–16 are included in pp1ab. In the SARS-CoV-2 genome, the 1945-residues long NSP 3 and 306-residues long NSP 5 regions, respectively called papain-like proteinase (PLpro) and main proteinase (Mpro), cleave the other nonstructural proteins. PLpro cuts the N-terminal region of pp1a to release NSP 1, NSP 2, and NSP 3, whereas Mpro cleaves the rest to produce mature and intermediate nonstructural proteins.10
One of the most significant intracellular pathways, which may be viewed as a master regulator for cancer, is phosphoinositide 3-kinase (PI3K)/Protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling. PI3K/AKT/mTOR signaling controls cell growth, motility, survival, metabolism, and angiogenesis. Almost all forms of human cancer, including breast cancer, colorectal cancer, and hematologic malignancies, have been found to have a dysregulated PI3K/AKT/mTOR pathway. This pathway is likely to be a crucial therapeutic target for cancer treatment. Unlike other PI3K isoforms, the overexpression of the δ and γ isoforms is predominantly associated with hematological malignancies, inflammatory illnesses, and autoimmune disorders.11 Interestingly, the PI3K/AKT signaling pathway is also one of the factors that regulates SARS-CoV-2 entry into the cell. The angiotensin II level rises in the lungs as SARS-CoV-2 enters the cell via ACE2 receptors. Additionally, during SARS-CoV-2 infection, the thrombin level in the blood and inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1 (IL-1), and transforming growth factor-β (TGF-β) are elevated. These elements can stimulate platelet cells’ PI3K/AKT signaling pathway,12 causing hyperactivation of PI3K and also clotting. In the case of a cancer patient, this signaling is already hyper-activated, producing cytokine storms. COVID-19 infection in a cancer patient can lead to organ failure and death.
It is evident, therefore, that PI3Kγ/AKT/mTOR is a common pathway in both cancer and COVID-19. Suppression of PI3Kγ could stop the cytokine storm. A well-known cancer drug, Duvelisib, is currently in a clinical trial for COVID-19 patients. Clinical evidence suggests that inhibiting PI3Kγ by Duvelisib could improve patient conditions by potentially suppressing innate immune system hyperactivation, polarizing macrophages, reducing lung inflammation, and limiting viral persistence.13 AI-based drug repurposing revealed that the inhibitory activity of Duvelisib against a coronavirus, feline infectious peritonitis virus FIPV, is approximately 50 μM.14 Duvelisib was also subjected to a clinical trial against COVID-19 to restore immune homeostasis and inhibit viral replication. Overall survival against COVID-19 is the primary end point.15 Recently, Jia et al. performed a comparative binding study of Eganelisib, Duvelisib, and Idelalisib against the PI3Kγ receptor, which has an important role in cancer. They reported that among all the drugs, Eganelisib binds most strongly to PI3Kγ.16 However, the mechanistic and molecular details of the action of these cancer drugs against the different COVID-19 targets are still unknown. Our hunch is that any of these drugs inhibiting SARS-CoV-2 could simultaneously treat cancer and COVID-19, i.e., act as multitarget drugs.
In this study, we have considered two different PI3Kγ inhibitors, the clinical drug Duvelisib and the potent inhibitor Eganelisib, for screening against multiple SARS-CoV-2 targets (total of 13). Because of black box warnings, Idelalisib is not considered here.17 Interestingly, both drugs show a strong binding affinity to Mpro, an essential nonstructural protein performing a pivotal role in virus replication. We subjected the Mpro bound to these drugs to long-term molecular dynamics (MD) simulations to examine the stability and conformational changes. For comparison, we also performed MD simulations of the natural host receptor PI3Kγ following docking with both drugs. The binding free energies determined by post-MD analyses, including molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) and Molecular Mechanics Generalized Born Surface Area (MM-GBSA) methods, reveal that both drugs have significant binding and stability with Mpro as well as PI3Kγ. Additionally, Umbrella sampling was performed to compute the forces for dissociating these two drugs from their Mpro complexes. The mechanical stiffness of both complexes was also estimated to explore the resistance of the residues to external tension.
2. Materials and methods
2.1. DFT optimization of drugs and screening of targets
Duvelisib and Eganelisib were retrieved from the DrugBank and subjected to gas-phase geometry optimization at the B3LYP/6-311G+(d,p) level of theory using Gaussian 16.18–22 All the quantum parameters, such as HOMO, LUMO, band gap, dipole moment, ionization potential, electron affinity, electronegativity, chemical potential, chemical hardness, chemical softness, and electrophilicity index, of both molecules were calculated using our home-built script.22 Having downloaded the structures of the targets from the RCSB protein data bank (PDB), missing residues were fixed using MODELLER 9.19.23 The Modeller 9.19 tool is an automated comparative modelling method that satisfies spatial constraints. MODELLER predicts the 3-D structure of a given target protein sequence based primarily on its alignment to one or more template proteins of known structure. The prediction process comprises fold assignment, target-template alignment, model building, and model evaluation. For the model evaluation, the discrete optimized protein energy (DOPE) score was used to choose five modeled structures; the best model is the one that has the lowest DOPE score. All missing H atoms were added using UCSF Chimera.24 The active site of all targets was ascertained based on the published literature. The protein targets, duly prepared, underwent reverse screening employing optimized Duvelisib. The optimal docking pose was determined from the ensemble of 10 poses, discerned based on the docking score criterion. Duvelisib shows a strong affinity for the main protease (Mpro). Eganelisib, a potent inhibitor of the PI3Kγ receptor and member of the same drug family as Duvelisib,23 was docked with Mpro. Similarly, both drug molecules were docked with the host natural receptor, PI3Kγ. All docking studies were performed using AutoDock Vina.25 Because of their high accuracy, AutoDock tools are widely used; a plethora of literature indicates this.26 Despite several advantages, AutoDock has certain limitations; for example, it often leads to unreliable results when small molecules are docked into flexible binding sites. Also, it does not consider the flexibility of cyclic and macro-cyclic ligands. Another limitation is that the receptor in such methods is rigid.25,26 In the present work, the protein and ligands were, respectively, considered as rigid and flexible during docking. The best docking poses of the drugs against different targets were analyzed using Discovery Studio Visualizer 4.5. Among all, Duvelisib and Eganelisib yield the lowest negative binding energies against the Mpro and Helicase proteins. To validate the docking outcomes, we extensively conducted docking investigations for the two molecules across a spectrum of 13 distinct SARS-CoV-2 targets, employing the SwissDock27 and DockThor28 software platforms. Hereafter, we performed MD simulations of the Mpro, Helicase, and PI3Kγ complexes of Duvelisib and Eganelisib, including Mpro-α-Ketoamide 13b for comparison.2.2. MD simulations
MD simulations are essential for revealing the stability, hidden states, or any short-lived configurations of the protein–ligand complexes at the atomistic level, which are otherwise difficult to access experimentally. The Mpro-Duvelisib, Mpro-Eganelisib, Mpro-α-Ketoamide 13b, Helicase NCB site-Duvelisib, Helicase NCB site-Eganelisib, PI3Kγ-Duvelisib, and PI3Kγ-Eganelisib complexes were subjected to 500 ns MD runs using Gromacs 2021.22,29–31 The protein topology was produced using the AMBER99SB force field, whereas small molecule topologies were produced using ACPYPE's antechamber tool.32,33 According to previous benchmark and literature studies, the amber99sb force field is more accurate and compatible with biomolecular simulations.33–36 Each complex was solvated in a cubical box of minimum distance of 1 nm from the protein surface to the edges of the box with the Simple Point Charge (SPC) water model. Counter-ions were added to make the systems electrically neutral at a salt concentration of 0.15 mol L−1. Each system underwent energy minimization prior to the MD run by using the steepest descent integrator for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps with a force convergence of <1000 kcal mol−1 nm−1.
000 steps with a force convergence of <1000 kcal mol−1 nm−1.
First, the protein–ligand complexes were equilibrated for 5 ns using the canonical (NVT) and isothermal–isobaric (NPT) ensembles. During equilibration, each system was coupled with the Berendsen temperature and Parrinello–Rahman pressure controllers, to maintain a temperature of 300 K and pressure of 1 bar, respectively. A time step of 2 fs was used. The Particle Mesh Ewald (PME) algorithm37 was employed to deal with the long-range Coulomb interactions with a Fourier grid spacing of 0.12 nm. The short-range van der Waals interactions were treated with the Lennard-Jones potential with a cut-off distance of 1 nm. All bond lengths were constrained by the linear constraint solver (LINCS) method.38
Subsequently, a 500 ns production run was carried out for all simulations. During the production run, the coordinates were saved every 10 ps. As the TIP3P water model is more compatible with the AMBER force field,36,39,40 some systems were simulated for 500 ns again in such water. In contrast to SPC, the TIP3P water model possesses a larger dipole moment and has a bond angle and dielectric constant closer to the experimental values.
For structural, dynamical, and energetics analyses, the resultant MD trajectories were analyzed using the built-in modules of GROMACS and visual molecular dynamics (VMD 1.9.1).41 2D plots depicting the intrinsic dynamical stabilities by the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), and principal component analysis (PCA) of the complexes were generated by Origin 2021. RMSD was obtained using eqn (1):
 | (1) |
 and ri(t) is the position of atom i at time t after a least-squares fitting of the structure to the reference structure.
and ri(t) is the position of atom i at time t after a least-squares fitting of the structure to the reference structure.
The RMSF was obtained as in eqn (2):
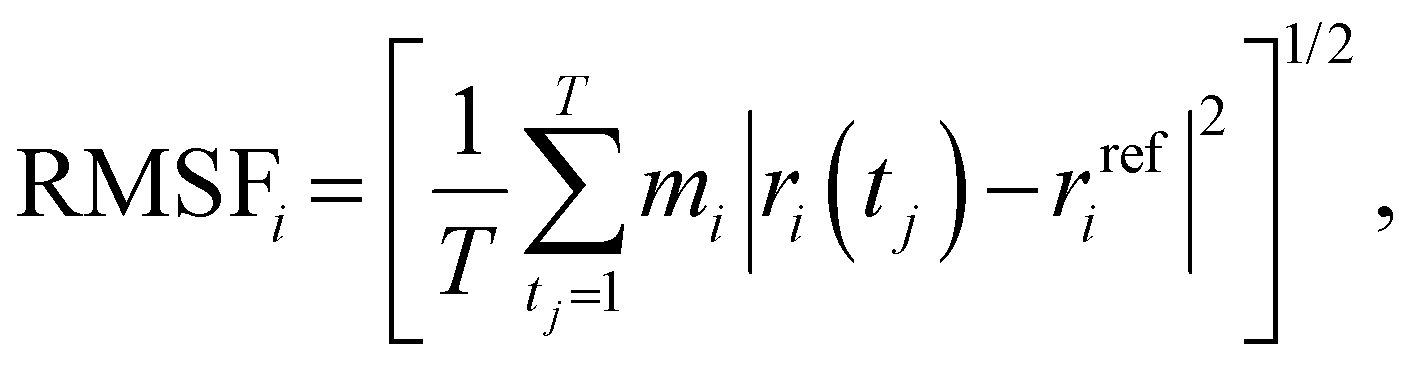 | (2) |
R g was obtained using eqn (3):
 | (3) |
Proteins alter their form to regulate their functions. The collective movements of its atoms govern the overall conformational change of a protein. PCA was used to examine the relative conformational dynamics and atomic fluctuations of the functionally significant substructures in their native and ligand-bound forms.36,37 For PCA analysis, the trajectories produced by the 500 ns MD simulations were utilized. After eliminating the translational and rotational movements, the relative Cα-backbone atomic fluctuations were observed, and a cross-correlation matrix was created. The direction of motion was represented by the eigenvectors, and the resulting eigenvalues indicate the energetic contribution from the respective principal components (PCs). The total flexibility of the protein was ascertained by analyzing the projected eigenvalues and eigenvectors. The formula provided in eqn (4) was used to perform PCA.
| ciJ = (xi − xi)(xJ − xJ), | (4) |
The MM-PBSA42,43 and MM-GBSA44,45 methods were used to calculate the binding free energy of the protein–ligand complexes. The studies suggest the goodness of the agreement between the experimentally determined binding affinities and those computed with MM-PBSA.46 While MM-PBSA has demonstrated efficacy in numerous drug design and biomacromolecular investigations, inherent limitations in the method have been identified through various studies. These limitations encompass challenges in accurately predicting solute entropies, and estimating solvation free energies for charged and buried groups, addressing issues related to conformational sampling, and making critical decisions in parameter selection, including the determination of radii for calculating solvation free energy and the choice of dielectric constant for the solute.47,48 We have also performed an enhanced sampling of both complexes to demonstrate the efficacy of Duvelisib and Eganelisib unambiguously. The MD configurations obtained at 500 ns were subjected to Umbrella sampling.49 Umbrella sampling focuses on specific collective variables (CVs) that define the progress of a rare event, such as a conformational change or a binding/unbinding process. In standard MD simulations, systems may get trapped in local energy minima, making it challenging to explore higher energy states separated by energy barriers. Umbrella sampling helps overcome these barriers by applying biasing potentials that push the system toward regions it would not normally explore, allowing for more efficient sampling. Umbrella sampling provides a means to calculate free energy profiles along the chosen reaction coordinate.49–51 Here, we have used the pull code to get the force required for releasing the drug molecules from the active site of Mpro. Beforehand, we followed all MD steps, such as energy minimization and NPT equilibration. Position restraint was applied to the protein and drug molecules. We used a dragging force of 1000 kJ mol−1 nm2 along the z-axis to snatch the molecules at the rate of 10 nm per sec. Pulling continued for 500 ps for both systems.
To calculate the mechanical stiffness of the pair of residues in both complexes, we used the ProDy python script. The anisotropic network model (ANM) was used to construct a complete map of the mechanical response of all residue pairs in a given protein to uniaxial deformation. The main application of ANM is to provide information on the relative mobilities of residues or the relative responses of various residue pairs under strain/tension rather than to predict the absolute sizes/strengths of deformation. Using ANM, we can readily construct a complete map of mechanical resistance in response to all possible pulling directions. Efficient assessment of such maps is an advantage of analytical models such as ANM over numerical approaches, such as steered MD or even coarse-grained simulations.52,53
We obtained minimum energy structures and Gibb's free energy landscape from MD trajectories using gmx_sham for both complexes.
3. Results and discussion
3.1. Quantum parameter calculations and screening
Tables S1 and S2 (ESI†) summarize all electronic descriptors and DFT-optimized coordinates for Duvelisib and Eganelisib, respectively. Fig. S1 (ESI†) pictorially displays the energies of the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and band gaps. The large dipole moment of Eganelisib compared to Duvelisib indicates its significant polarization. The band gap values of 0.12 eV and 0.16 eV, respectively, for Eganelisib and Duvelisib, imply that the former is more chemically reactive. The HOMO–LUMO energy levels are related to the photostability and chemical reactivity of the molecules.54–56 In protein–ligand complexes, the HOMO of a molecule interacts with the LUMO of a protein and vice versa.57–60 Ionization potential is the energy required to remove a single electron from the HOMO; Eganelisib and Duvelisib have ionization potentials very close to each other, at 0.20 eV and 0.22 eV, respectively. Chemical softness and hardness are related to the polarizability of a molecule. More polarization means more softness, which is inversely related to chemical hardness. The electronegativity and electrophilicity index indicate the ability of a molecule to attract electrons. Eganelisib has a larger electrophilicity index than Duvelisib, while both have the same electronegativity and chemical potential values. Dipole moments below 10 Debye indicate better oral availability and membrane permeability. Compared to Eganelisib, Duvelisib has a higher band gap because of its higher ionization potential and lower electron affinity.3.2. Interaction analysis
Table S3 (ESI†) furnishes the docking scores of Duvelisib against 13 different targets. The binding affinity of Duvelisib with the Mpro and helicase NCB site (Nsp13) is higher than with other targets. Similarly, we have also considered another potential PI3Kγ inhibitor, Eganelisib, and docked it in the active site of Mpro and helicase protein. With Mpro being a crucial target involved in the proteolytic cleavage of the polyprotein and replication of the virus, we investigated its complexes with the drugs. The binding affinity of Duvelisib and Eganelisib to Mpro is −8.2 and −8.3 kcal mol−1, respectively. The unwinding of double-stranded DNA or RNA is catalyzed by the 67 kDa helicase NSP13 protein.61 The replication-transcription complex (NSP7/NSP8/NSP12) functions in conjunction with NSP13, which has been demonstrated to interact with the viral RNA-dependent RNA polymerase.62 Similarly, the binding affinity of Duvelisib and Eganelisib against the helicase NCB site is −8.2 kcal mol−1 and −8.0 kcal mol−1, respectively. For reference, we have also performed molecular docking studies against the natural receptor of these two drugs i.e., the PI3Kγ receptor. The binding affinity of Duvelisib and Eganelisib against PI3Kγ is −8.5 kcal mol−1 and −9.2 kcal mol−1, respectively.Fig. S2, ESI† displays the 2D interaction plots of the Mpro complexes. Mpro and Duvelisib have the following: H-bonds with Gly143, Asn142, Thr26, and Glu166; a π–sulfur interaction with Cys145; a π–π stacking interaction with His41; and a π–alkyl interaction with Met165. Similarly, Mpro and Eganelisib exhibit: H-bonds with Phe140, Glu166 (2), and His163; a π–sulfur interaction with Cys145; an amide–π interaction with Leu141; and a π–alkyl interaction with Met165 and Met49. Between Mpro and α-Ketoamide 13b, His41, Gly143, Cys145, Ser144, His163, Glu166, His163, and Asn142 exhibit a H-bond interaction and His41, Met165, and Pro168 exhibit a hydrophobic interaction. Both Duvelisib and Eganelisib interact with some of the most important Mpro residues, including Glu166, Met49, Cys145, and His41.63,64 Similarly, Duvelisib shows a H-bond interaction with Ser310, a π–cation interaction with Arg178, and hydrophobic interactions with Arg178, Ala312, Ala313, and Ala316 of the helicase protein. The Eganelisib-helicase complex has H-bond interactions with Lys202 and Asn179, including a hydrophobic interaction with Ala520. It was noted that the hydrophobic, π–alkyl, and π–π interactions predominate over the other interactions in all of the complexes. Table 1 summarizes the various interactions observed.
| Complex | Time (ns) | Interactions | |||||
|---|---|---|---|---|---|---|---|
| Conventional hydrogen bond | Carbon hydrogen bond | π–Lone pair/cation | π–Sulfur | π–π T shaped/amide–π stacked | Alkyl/π–alkyl | ||
| Mpro-Duvelisib | 0 | Glu166 (2.3), Phe140 (2.62) | Glu166 (2.99), His163 (2.83) | Cys145 (5.13) | Leu141 (3.42 & 4.01) | Met165 (3.45), Met49 (5.19) | |
| 500 | Glu166 (2.62) | Ser144 (2.91), Met165 (2.82) | Asn142 (2.94) | His41 (4.91) | Met165 (4.01, 3.79, & 4.44), Met49 (4.64 & 4.69) | ||
| Mpro-Eganelisib | 0 | Gly143 (2.60), Asn142 (2.61) | Thr26 (2.18), Glu166 (2.74) | Cys145 (3.98) | His41 (4.69) | Met165 (4.48, 4.71, & 4.77) | |
| 500 | Thr190 (3.02), Asn142 (2.35), Gln189 (2.64) | Glu166 (2.67), Pro168 (2.74), Thr26 (2.99) | Met49 (5.43) | His41 (4.93), Asn142 (4.55) | Leu167 (4.48), Ala191 (4.27), Leu27 (4.59), Met165 (4.07, 4.74, & 4.88), & Met49 (5.21) | ||
| Mpro-α-Ketoamide 13b | 0 | His41 (2.37), Gly143 (2.09), Cys145 (2.11, 1.74, & 2.89), Ser144 (2.50), His163 (2.26), Glu166 (1.84 & 2.39) | His163 (2.99), His164 (2.83), Met165 (2.42), Asn142 (3.06) | His41 (4.28), Met165 (5.47), His163 (5.29) | |||
| 500 | Ser144 (2.32 & 2.03) | His41 (4.85), Cys145 (5.23), Met49 (5.22), Met165 (5.45), His163 (5.18) | |||||
| PI3Kγ-Duvelisib | 0 | Lys690 (2.37), Asp821 (2.19) | Lys690 (2.79), Asp80 (2.46) | Asp821 (2.94) | Met661 (4.02), Met166 (4.02) | Tyr24 (4.82) | Val739 (4.42), Met661 (4.44), Pro667 (3.9), Ile688 (4.38), Phe818 (5.01), Ile688 (4.96), Met810 (5.37), Ile820 (4.26), Ile736 (4.71), Ile820 (3.94), Met661 (5.12), Lys665 (5.44), Pro667 (5.08) |
| 500 | Thr744 (3.01), Lys747 (2.12), Asp807 (2.92) | Ala742 (2.83), | Met810 (4.02 & 4.48) | Trp669 (5.39), Tyr724 (5.05) | Ile738 (5.09), Met661 (4.07 & 5.48), Pro667 (4.46), Ile688 (4.46), Phe818 (4.74), Ile688 (4.69 & 5.28), Ile736 (4.83), Met810 (5.29), Ile820 (5.49), Ala662 (4.83 & 4.00), Ala742 (5.49) | ||
| PI3Kγ-Eganelisib | 0 | Glu737 (3.35 Å), Val739 (2.01 Å) | Glu737 (2.81 Å), | Lys747 (4.15 Å) | Met661 (4.18 Å) | Trp669 (5.42 Å, 5.10 Å, & 4.92 Å), Tyr724 (4.91 Å) | Trp669 (2.45 Å), Met661 (5.17 Å, 4.26 Å), Ile738 (5.31 Å), Val739 (5.41 Å), Met810 (4.83 Å), Ile688 (5.34 Å), Ile736 (5.16 Å), Met810 (5.37 Å), Ile820 (4.45 Å), Lys747 (5.03 Å), Lys659 (4.72 Å) |
| 500 | Lys659 (2.37 Å), Val739 (1.98 Å) | Lys659 (2.67 Å), Glu737 (2.84 Å), Val660 (2.76 Å) | Lys747 (4.22 Å) | Met661 (4.49 Å), Met810 (4.03 Å), | Trp669 (5.30 Å & 5.42 Å), Tyr724 (5.01 Å) | Ile820 (5.39 Å & 4.57 Å), Met661 (5.11 Å), Lys659 (5.00 Å), Pro667 (5.21 Å), Ile688 (5.36 Å, 4.90 Å, & 4.70 Å), Ile736 (5.21 Å), Lys747 (5.34 Å) | |
3.3. MD trajectory analysis
First, the protein conformational dynamics and stability of the complexes were examined by evaluating various post-MD parameters, such as RMSD, RMSF, and Rg, and performing principal component analysis (PCA), based on 500 ns trajectories, as discussed in the succeeding sections.RMSD, defined as the average distance between atoms over the simulation period, is a crucial term to judge conformational stability. Mpro-Duvelisib shows less stable binding until 400 ns, ascertained by Fig. S3B (ESI†) with large RMSD deviations that converge and become smooth upon the attainment of the stable conformation of the complex afterward. Based on the simulation snapshots (see Fig. S4, ESI†) taken at various points in time, Duvelisib gradually moves into the catalytic pocket and binds strongly, accompanied by protein conformational changes. On the contrary, the smooth RMSD plots in Fig. S3C (ESI†) indicate Mpro-Eganelisib's stable binding over the entire MD simulation period without any conformational changes (Fig. S5, ESI†). Despite the RMSD plot of the protein being smooth, the α-Ketoamide 13b peptide inhibitor has large fluctuations in RMSD. This suggests that the inhibitor is unable to bind to the Mpro active site strongly enough, indicating instability of the complex (Fig. S6, ESI†). While calculating RMSD for the binding domains of all three complexes, interestingly we observe the following order: Mpro-Duvelisib < Mpro-Eganelisib < Mpro-α-Ketoamide 13b (see Fig. 1(A)). Calculation of the average RMSD and RMSF of the three Mpro complexes shows more stability of Duvelisib and Eganelisib compared to α-Ketoamide 13b (Fig. 1(B)). The average RMSF also exhibits a similar trend with values of 0.11 nm, 0.1 nm, and 0.12 nm, respectively, for Mpro-Duvelisib, Mpro-Eganelisib, and Mpro-α-Ketoamide 13b. In contrast, we notice pronounced fluctuations of RMSD for Helicase NCB site-bound Duvelisib and Eganelisib with an average RMSD value of 0.47 nm and 0.53 nm, respectively (Fig. S7, ESI†). This implies the sheer instability of the corresponding complexes. We also do not observe any distinct change in the binding location of the drug molecules. Therefore, these two systems are excluded from further investigation. Moreover, the catalytic role of the NCB site is not appealing for drug discovery. Furthermore, drug discovery is unlikely to benefit from the catalytic role of the NCB site.
 | ||
| Fig. 1 (A) RMSD plots of Mpro's receptor binding domain (RBD) when bonded to Duvelisib, Eganelisib, and α-Ketoamide 13b and (B) average values with standard deviations of RMSD and RMSF for RBD in the three complexes. | ||
Similarly, the MD simulation snapshots of Mpro-Eganelisib taken at various points in time (Fig. S5, ESI†) show no remarkable change in the conformation of the protein or ligand, signifying strong binding with Eganelisib. Again, comparing the 0 ns and 500 ns superimposed structures of the complexes in Fig. S8 (ESI†), very little conformational change is observed in Mpro-Eganelisib compared to Mpro-Duvelisib, further suggesting a strong interaction of Eganelisib. However, Duvelisib benefits from conformational changes that strengthen binding to the active site pocket. There is an inward movement of Duvelisib during the simulation (see Fig. S4, ESI†). Poor overlap is noticeable in the Mpro-α-Ketoamide 13b structures, specifically for α-Ketoamide 13b, as shown in Fig. S8 (ESI†). The MD snapshots in Fig. S6 (ESI†) at various points in time guide us to striking conformational changes of the compound, while there is no discernible alteration of the Mpro conformation.
To delineate the dominating interactions leading to the final conformations, Fig. S9 (ESI†) presents an interaction analysis of the three complexes at 500 ns. It is evident that the π–alkyl and π–π stacking interactions have a dominant role in binding the three molecules with Mpro. There happen to be numerous interactions between Mpro-Duvelisib, such as H-bond interactions with Ser144, Met165, and Glu166; a π–lone pair interaction with Asn142; a π–π stacking interaction with His41; and π–alkyl interactions with Met49 and Met165. Here, the Met49, Met165, and Glu166 residues retain their interaction until the end of the simulation, indicating that they may be critical for the catalytic activity of the active site. An intramolecular π–π interaction is observed in Duvelisib, stabilizing the molecules and the aromatic group and providing a site for interacting with Met49, Met165, and His41.
Similarly, at 500 ns, the prominent interactions between Mpro-Eganelisib are H-bonds with Thr190, Asn142, Gln189, Glu166, Pro168, and Thr26; a π–sulfur interaction with Met49; a π–π interaction with His41; a π–amide interaction with Asn142; and a π–alkyl interaction with Leu167, Ala191, Leu27, Met165, and Met49. Markedly, Thr26, His41, Asn142, Met165, and Glu166 keep intact their interaction with Mpro until the end of the simulation. In the case of Mpro-α-Ketoamide 13b, however, the number of interactions is drastically reduced at 500 ns (see Table 1).
Based on the interaction table (Table 1) and residue contribution energy plots presented in Fig. 3(B) and (D), it is evident that hydrophobic interactions exert a pivotal influence throughout the simulations in all examined complexes. Notably, both drug molecules feature an indole group within their respective structures. Following the findings by Atukuri Dorababu et al., this indole group holds significant relevance to the antiviral and anticancer realms.65 The indole moiety plays a crucial role in various hydrophobic and π–π stacking interactions across all complexes. Consequently, the indole moiety presents promising avenues for further exploration in drug design targeting Mpro.
RMSF quantifies the average flexibility of the protein's Cα atoms over the simulated time. Compared to Mpro-Duvelisib and Mpro-α-Ketoamide 13b, Mpro-Eganelisib has a lower average RMSF (see Fig. 1) similar to RMSD, with fewer fluctuations of the Cα atoms of the residues (see Fig. 2(A)). Once again, Mpro-Eganelisib is proven to be more stable than Mpro-Duvelisib. Mpro in Mpro-α-Ketoamide 13b fluctuates somewhat more than in Mpro-Eganelisib but less than in Mpro-Duvelisib.
 | ||
| Fig. 2 RMSF plots of the Cα-atoms of the Mpro-Duvelisib, Mpro-Eganelisib, and Mpro-α-Ketoamide 13b complexes considering (A) all and (B) only active-site residues. (C) and (D) Show radial distribution function (RDF) plots of the drug molecules with respect to the His41 and Cys145 catalytic-dyad residues in the three complexes. | ||
Concerning the active site residues (see Fig. 2(B)), the magnitude of RMSF fluctuations is still lower for Mpro-Eganelisib than Mpro-Duvelisib and Mpro-α-Ketoamide 13b, demonstrating that Mpro is more stable with Eganelisib than Duvelisib and α-Ketoamide 13b. In addition, we analyze Rg, which indicates the compactness of the protein–ligand complexes. Fig. S10 (ESI†) depicts the Rg plots of the complexes. The Rg plots of Mpro-Eganelisib and Mpro-α-Ketoamide 13b are stable and smoothly varying, and have lower values than Mpro-Duvelisib. The latter shows a striking deviation from smoothness, indicating protein conformational changes that promote stronger binding to Duvelisib (Fig. S4, ESI†). A closer investigation reveals that Duvelisib's binding is dominated by π–π and π–alkyl/sulfur interactions rather than H-bonds (see Fig. S2, ESI†). Compared to Eganelisib, Duvelisib gives rise to very few H-bonds with Mpro.
Fig. 2(C) and (D) portray the radial distribution function (RDF) of the three molecules (Duvelisib, Eganelisib, and α-Ketoamide 13b) with respect to the catalytic dyad residues (His41 and Cys145). RDF measures a drug molecule's propensity to be near the Mpro active site. Concerning the distribution, Eganelisib has a higher probability of being found in close proximity to the His41 catalytic dyad residue than Duvelisib, ascertained by its larger peak height at a shorter distance (r). On the contrary, even though their most probable distances from the catalytic residue are virtually the same, Duvelisib has a much higher probability of being near Cys145 than Eganelisib. The likelihood distances of Duvelisib and Eganelisib are 0.64 and 0.40 nm from His41 and 0.72 and 0.73 nm from Cys145, respectively. Thus, it is evident that Eganelisib is closer to His41, reflecting a stronger interaction between them.
Fig. 3 displays the MM-PBSA binding free energy with its various components and the per-residue energy contribution in panels A and B for the three Mpro complexes, computed using the last 20 ns MD trajectories. The total binding free energy of Mpro-Eganelisib (Mpro-Duvelisib) is −68.8 kJ mol−1 (−65.8 kJ mol−1), in which the van der Waals, electrostatic, polar solvation, and solvent accessible surface area (SASA) contributions are −143.2 (−151.4), −17.1 (−27.9), 104.4 (128.7), and −13.1 (−14.4) kJ mol−1, respectively. The total binding free energy of Mpro-α-Ketoamide 13b is −66.1 kJ mol−1, with the van der Waals, electrostatic, polar solvation, and SASA contributions being −137.9, −32.1, 118.4, and −15.1 kJ mol−1, respectively. According to Fig. 3(B), the His41, Met49, Leu141, Asn142, Cys145, Met165, and Gln189 residues in Mpro-Eganelisib, the Leu27, Met49, Met165, Gln189, and Ala191 residues in Mpro-Duvelisib, and the His41, Met49, Asn142, Ser144, Met165, Pro168, and Gln189 residues in Mpro-α-Ketoamide 13b are the major contributors to stable binding. The residues that do not favor binding and consequently have positive energy contributions to the total binding energy are Glu166 (common in all three complexes), Gln192 in Mpro-Eganelisib, Asn142 and Asp187 in Mpro-Duvelisib, and Lys137, Arg188, and Ala191 in Mpro-α-Ketoamide 13b. Based on energetics, Met49 and Met165 have a major role in drug binding to the active site of Mpro, driven by π–alkyl interactions in the Mpro-Duvelisib and Mpro-Eganelisib complexes. Earlier, we reinforced that Duvelisib and Eganelisib do not form stable complexes with the Helicase NCB site due to the large RMSD. This is also strongly supported by their meagre total binding free energies of −34.8 kJ mol−1 and −22 kJ mol−1, respectively (see Fig. S11, ESI†).
 | ||
| Fig. 3 The MM-PBSA binding free energy of Duvelisib, Eganelisib, and α-Ketoamide 13b with (A) Mpro and (C) PI3Kγ, including its various components. The per-residue energy contribution to the corresponding complexes of (B) Mpro and (D) PI3Kγ. All energies are in kJ mol−1. | ||
For a comparative analysis, we also performed 500 ns MD simulations of both drugs with the PI3Kγ cancer target. In the complex form, the PI3Kγ protein appears quite stable, as evidenced by its smooth RMSD plots; the same is true for Eganelisib (Fig. S12C and D, ESI†). Interestingly, the RMSD plot of Duvelisib shows two sets of values, implying that it encounters two different conformations during the simulation. At ∼70 ns, it adopts a different conformation and changes back to the initial conformation after 330 ns. In the interim period (70–330 ns), certain interactions may become predominant, which may explain the conformational change. Conversely, the Cα atomic fluctuations of PI3Kγ are also more in PI3Kγ-Eganelisib than PI3Kγ-Duvelisib, demonstrated by the large values of RMSD and RMSF shown in Fig. S12A and B (ESI†). However, it does not affect the stability of the complex since fluctuations mostly originate from residues in the non-binding domain (1–480) rather than in the molecular-binding domain (480–948), cf. Fig. S12B (ESI†). The average values of RMSD and RMSF for PI3Kγ-Duvelisib (PI3Kγ-Eganelisib) are 0.35 nm (0.41 nm) and 0.15 nm (0.17 nm), respectively. Graphically, Fig. 3(C) illustrates the binding free energies and their various components. PI3Kγ amounts to −76.7 kJ mol−1 (−71 kJ mol−1) energy with Duvelisib (Eganelisib), in which the van der Waals, electrostatic, polar solvation, and SASA contributions are −123.3 (−140.8), −18 (−36.3), 75.2 (117.2), and −11.6 (−12.8) kJ mol−1, respectively. According to the per-residue contributions, in Fig. 3(D), the Met661, Pro667, Ile668, Ile738, Met810, and Ile820 residues or Met661, Trp669, Ile738, Th744, Met810, and Ile820 residues have significant binding contributions to PI3Kγ-Eganelisib or PI3Kγ-Duvelisib, respectively. Noticeably, both complexes contain several interactions that significantly contribute to binding, such as Met661, Ile738, Met810, and Ile820. Thus, these residues could be considered hotspot residues for the future design of more potent drugs against the PI3Kγ receptor. Based on the above investigations, both compounds exhibit stable complex formation and binding with the PI3Kγ receptor protein.
The computed MM-GBSA binding free energies for the various complexes exhibit consistent trends with those obtained through the MM-PBSA analysis. Specifically, the total binding free energies for the Mpro-Eganelisib, Mpro-Duvelisib, Mpro-α-Ketoamide 13b, PI3Kγ-Eganelisib, and PI3Kγ-Duvelisib complexes are determined to be −9.1, −7.66, −8.5, −8.36, and −9.59 kcal mol−1, respectively (see Fig. S13, ESI†). In addition to providing valuable insights into the energetics of ligand binding to the target proteins, these values represent the thermodynamic stability of the respective complex systems.
We also employed ten-replica enhanced sampling simulations to determine the average force required for dissociating the drug molecules from their respective Mpro-drug complexes for each complex. Fig. 4 reveals that three molecules need nearly the same force to detach from Mpro's active site. The averaged maximum forces required to dissociate Mpro-Duvelisib, Mpro-Eganelisib, and Mpro-α-Ketoamide 13b are 396.2, 387, and 388, kJ mol−1 nm−1, respectively. As is obvious, there is a sudden decrease in force for Mpro-Duvelisib, whereas the Mpro-Eganelisib force decreases gradually over time. This implies that the disintegration of strong interactions in Mpro-Eganelisib is slower than in Mpro-Duvelisib. Mpro-α-Ketoamide 13b has a force profile akin to Mpro-Duvelisib. In the former, some interactions are retained up to 200 ps, giving rise to a peak of the largest dissociating forces. Afterward, there is a rapid fall, indicating that there is no longer any firm contact with the Mpro residues. Fig. S14–S16 (ESI†) display 2D-projected interactions of Mpro-Duvelisib, Mpro-Eganelisib, and Mpro-α-Ketoamide 13b at different points in time during enhanced sampling, while gradually pulling the drugs off the active site. To help understand better, we also provide three movies in the ESI.†
 | ||
| Fig. 4 Plots depicting the pulling forces required to dissociate Duvelisib, Eganelisib, and α-Ketoamide 13b from the active site of Mpro in the three complexes. | ||
Additionally, mechanical stiffness maps of the residues in Fig. 5 and Fig. S17 (ESI†) provide insights into the observed force profiles. The residue pairs marked in blue have less deformation and, in turn, high mechanical stiffness. The maps show that the residues that belong to the secondary structure tend to exhibit relatively strong resistance to deformation. It is evident from the 3D diagram of both complexes that most active site residues (His41, Met49, Leu141, Asn142, Cys145, Met165, and Gln189) show strong resistance to external tension (see Fig. 5). Usually, most parts of the secondary structure remain stiff. The loop regions in both complexes are more deformable and less stiff. This indicates that drug binding to the active site residues makes them stiffer than the other regions. Some secondary structures outside the active site, marked in red, are less stiff, eventually turning into loops as observed during dynamics.
 | ||
| Fig. 5 The 3D diagrams displaying the mechanical stiffness of the Mpro residues while bonded to (A) Eganelisib and (B) Duvelisib. | ||
To examine protein conformational motions, PCA was performed using molecular dynamics trajectories. The covariance matrix of Cα atomic fluctuations was diagonalized against the equivalent eigenvector (EV) indices to generate eigenvalues utilized later to determine the magnitude of the conformational changes. An exponentially decaying curve of the eigenvalues against the EVs is formed (Fig. S18A, ESI†) when the first 12 modes are considered in the study of the essential subspace since they account for around 95% of the protein's motion.
Moreover, while plotting the first two PCs, Fig. S18B (ESI†) shows a smaller subspace dimension of Mpro-α-Ketoamide 13b and Mpro-Eganelisib than Mpro-Duvelisib as the trend of their eigenvalues. As a result of reduced collective movements, Mpro-Eganelisib and Mpro-α-Ketoamide 13b have high stability and fewer conformational changes consistent with other dynamical parameters.
Fig. S19 (ESI†) shows the Gibb's free energy landscape. The red contours represent different energy-minimized configurations of the protein-drug complexes in the PC1-PC2 space. Compared with Mpro-Duvelisib, most conformations of Mpro-Eganelisib have reached an energy minimum. The energy-minimized configurations of Mpro-Eganelisib sprawl over a low subspace dimension compared to Mpro-Duvelisib, indicating that the latter suffers more fluctuations and has lower stability. One of the minimum energy structures of both complexes is also shown in the 2D interaction plots. In both complexes, the π–alkyl interaction with Met165 is typical. Besides, there is a π–alkyl interaction with Met49 in Mpro-Eganelisib and a π–π stacking interaction with His41 in Mpro-Duvelisib. In addition, there are H-bond interactions with Thr190, Gln189, and Asn142 and one hydrophobic interaction with Ala191 in Mpro-Eganelisib. The above analysis shows that the hydrophobic interaction, such as π–alkyl interaction, is more dominant in both complexes.
We conducted MD simulations for all systems employing the TIP3P water model to validate the findings derived from using the SPC water model. Our observations indicate congruence in average RMSD and RMSF trends for both the binding domain and the entire Mpro protein, affirming consistency between outcomes obtained with the two water models (Fig. S20, ESI†). Furthermore, our observations reveal analogous binding energy profiles for Mpro-Duvelisib and Mpro-Eganelisib compared to Mpro-α-Ketoamide 13b (Fig. S21, ESI†). Regarding the PI3K-γ complexed with Duvelisib, the absolute RMSD and RMSF values remain consistent. However, in the case of PI3K-γ complexed with Eganelisib, a notable reduction in these values is evident when utilizing the TIP3P water model compared to the SPC model. Upon examination of the binding free energy, it is evident that the trend remains consistent when transitioning from the SPC water model to TIP3P. Specifically, the binding free energy for PI3K-γ in complex with Duvelisib is consistently higher than that observed for Eganelisib. Upon comparing the binding free energy values for the Mpro complexes with those of the PI3K-γ complexes, it is notable that all values have undergone simultaneous negative increments while maintaining a consistent trend.
Based on the aforementioned molecular dynamics (MD) analyses encompassing dissociation force, mechanical stiffness, and free energy calculations, it has been deduced that the interaction between Duvelisib and Eganelisib with the Mpro and PI3K receptors results in the formation of a robust and enduring complex. This observation underscores the multitarget affinity attributes inherent in both molecules. Notably, both Duvelisib and Eganelisib exhibit characteristics indicative of stable complexation, as evidenced by consistently smooth RMSD plots, minimal fluctuations in RMSF, sustained lower and stable Rg, diminished conformational movements in the PCA calculations, heightened dissociation forces, and reduced binding free energy.
Conclusions
SARS-CoV-2 has spread worldwide. The appearance of new variants has instilled fear due to their stellar immune escape nature and infectivity. Since the spike undergoes mutations frequently, a less mutation-prone target could be a better choice. A cytokine storm is a common trait in both cancer and COVID-19. Here, Duvelisib, a potent PI3Kγ inhibitor helping in the cytokine storm, was screened against different SARS-CoV-2 targets along with Eganelisib. Duvelisib and Eganelisib show strong binding with Mpro, a key protein target involved in SARS-CoV-2 replication with reportedly no mutation in its active site thus far. Long-term MD simulations ascertain that Mpro-Duvelisib and Mpro-Eganelisib are pretty stable (compared to the natural inhibitor, α-Ketoamide). Comparatively, Mpro-Eganelisib is more stable with fewer fluctuations, smoother RMSD, and a lower binding free energy than the others. As per RDF calculations, they are more likely to remain in proximity to the catalytic dyad. Additionally, both complexes exhibit significant mechanical stiffness concerning the active site residues. The Gibb's free energy landscape demonstrates that most conformations attain an energy minimum in Mpro-Eganelisib, occupying a lower subspace dimension than Mpro-Duvelisib. The minimum energy structures of both complexes have π–π and π–alkyl as dominant interactions. Simultaneously, computational studies demonstrate that, as anticipated, Duvelisib and Eganelisib could inhibit PI3Kγ owing to stable complex formation. Hence, the present work deciphers that both drugs could help treat cancer and COVID-19 with multitarget capability. Moreover, as the active site of Mpro remains conserved across all variants, they could also be effective against the latest variants of SARS-CoV-2, such as the Omicron, BA.2, and BA.5 lineages, etc.Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
The authors acknowledge IISER Berhampur for computational support. SKP is also grateful for the Institute PhD fellowship provided by IISER Berhampur to carry out this work. The supercomputing facility vested for carrying out the major work by AMD and CSIR under the Covid Care Network (CCN) is also much appreciated. A fraction of computation was performed using the C-DAC PARAM Siddhi-AI supercomputer under NSM, India.References
- https://www.who.int/news/item/29-06-2020-covidtimeline .
- D. Cucinotta and M. Vanelli, Acta Biomed. Atenei Parmensis, 2020, 91, 157–160 Search PubMed.
- M. Pal, G. Berhanu, C. Desalegn and V. Kandi, Cureus, 2020, 12, e7423 Search PubMed.
- M. Prajapat, P. Sarma, N. Shekhar, P. Avti, S. Sinha, H. Kaur, S. Kumar, A. Bhattacharyya, H. Kumar, S. Bansal and B. Medhi, Indian J. Pharmacol., 2020, 52, 56–65 CrossRef PubMed.
- P. Sarma, N. Shekhar, M. Prajapat, P. Avti, H. Kaur, S. Kumar, S. Singh, H. Kumar, A. Prakash, D. P. Dhibar and B. Medhi, J. Biomol. Struct. Dyn., 2021, 39, 2724–2732 CrossRef CAS PubMed.
- C.-k Chang, S.-C. Sue, T.-h Yu, C.-M. Hsieh, C.-K. Tsai, Y.-C. Chiang, S.-j Lee, H.-h Hsiao, W.-J. Wu, W.-L. Chang, C.-H. Lin and T.-h Huang, J. Biomed. Sci., 2006, 13, 59–72 CrossRef CAS PubMed.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao, W. Tan, I. China Novel Coronavirus and T. Research, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS PubMed.
- N. Muralidharan, R. Sakthivel, D. Velmurugan and M. M. Gromiha, J. Biomol. Struct. Dyn., 2021, 39, 2673–2678 CrossRef CAS PubMed.
- Y. Fan, X. Li, L. Zhang, S. Wan, L. Zhang and F. Zhou, Signal Transduction Targeted Ther., 2022, 7, 141 CrossRef CAS PubMed.
- R. Yadav, J. K. Chaudhary, N. Jain, P. K. Chaudhary, S. Khanra, P. Dhamija, A. Sharma, A. Kumar and S. Handu, Cells, 2021, 10(4), 821 CrossRef CAS PubMed.
- J. Yang, J. Nie, X. Ma, Y. Wei, Y. Peng and X. Wei, Mol. Cancer, 2019, 18, 26 CrossRef PubMed.
- S. Fattahi, Z. Khalifehzadeh-Esfahani, M. Mohammad-Rezaei, S. Mafi and M. Jafarinia, Immunol. Res., 2022, 70, 269–275 CrossRef CAS PubMed.
- https://clinicaltrials.gov/ct2/show/NCT04372602 .
- Y.-Y. Ke, T.-T. Peng, T.-K. Yeh, W.-Z. Huang, S.-E. Chang, S.-H. Wu, H.-C. Hung, T.-A. Hsu, S.-J. Lee, J.-S. Song, W.-H. Lin, T.-J. Chiang, J.-H. Lin, H.-K. Sytwu and C.-T. Chen, Biomed. J., 2020, 43, 355–362 CrossRef PubMed.
- K. El Bairi, D. Trapani, A. Petrillo, C. Le Page, H. Zbakh, B. Daniele, R. Belbaraka, G. Curigliano and S. Afqir, Eur. J. Cancer, 2020, 141, 40–61 CrossRef CAS PubMed.
- L. Jia, L. Wang, Y. Jiang, L. Xu, Y. Cai, Y. Chen, J. Jin, H. Sun and J. Zhu, Comput. Biol. Med., 2022, 147, 105642 CrossRef CAS PubMed.
- S. E. Coutré, J. C. Barrientos, J. R. Brown, S. de Vos, R. R. Furman, M. J. Keating, D. Li, S. M. O’Brien, J. M. Pagel, M. H. Poleski, J. P. Sharman, N.-S. Yao and A. D. Zelenetz, Leuk. Lymphoma, 2015, 56, 2779–2786 CrossRef PubMed.
- K. Khayer and T. Haque, ACS Omega, 2020, 5, 4507–4531 CrossRef CAS PubMed.
- F. Pereira and J. Aires-de-Sousa, J. Cheminf., 2018, 10, 43 Search PubMed.
- K. P. Safna Hussan, M. S. Thayyil, V. K. Rajan and K. Muraleedharan, Comput. Biol. Chem., 2018, 72, 113–121 CrossRef CAS PubMed.
- U. Bhattacharya, S. K. Panda, P. S. S. Gupta and M. K. Rana, J. Mol. Struct., 2022, 1253, 132258 CrossRef CAS.
- P. S. S. Gupta, H. R. Bhat, S. Biswal and M. K. Rana, J. Mol. Liq., 2020, 320, 114375 CrossRef CAS.
- N. Eswar, B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M.-Y. Shen, U. Pieper and A. Sali, Curr. Protoc. Bioinf., 2006, 15, 5.6.1–5.6.30 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- N. T. Nguyen, T. H. Nguyen, T. N. H. Pham, N. T. Huy, M. V. Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2020, 60, 204–211 CrossRef CAS PubMed.
- A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS PubMed.
- K. B. Santos, I. A. Guedes, A. L. M. Karl and L. E. Dardenne, J. Chem. Inf. Model., 2020, 60, 667–683 CrossRef CAS PubMed.
- P. S. Sen Gupta, S. Biswal, S. K. Panda, A. K. Ray and M. K. Rana, J. Biomol. Struct. Dyn., 2022, 40, 2217–2226 CrossRef CAS PubMed.
- P. S. Sen Gupta, S. Biswal, D. Singha and M. K. Rana, J. Biomol. Struct. Dyn., 2021, 39, 5327–5333 CrossRef CAS PubMed.
- S. K. Panda, S. Saxena and L. Guruprasad, J. Biomol. Struct. Dyn., 2020, 38, 1887–1902 CrossRef CAS PubMed.
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- S. A. Showalter and R. Brüschweiler, J. Chem. Theory Comput., 2007, 3, 961–975 CrossRef CAS PubMed.
- T. T. Nguyen, B. K. Mai and M. S. Li, J. Chem. Inf. Model., 2011, 51, 2266–2276 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- V. Tiwari, S. Garg and T. Karmakar, ACS Appl. Bio Mater., 2024, 7, 685–691 CrossRef CAS PubMed.
- C. Coppa, A. Bazzoli, M. Barkhordari and A. Contini, J. Chem. Inf. Model., 2023, 63, 3030–3042 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- S. K. Panda, P. S. Sen Gupta, S. Karmakar, S. Biswal, N. C. Mahanandia and M. K. Rana, J. Phys. Chem. Lett., 2023, 14, 10278–10284 CrossRef CAS PubMed.
- G. Weng, E. Wang, Z. Wang, H. Liu, F. Zhu, D. Li and T. Hou, Nucleic Acids Res., 2019, 47, W322–W330 CrossRef CAS PubMed.
- P. Kollman, Chem. Rev., 1993, 93, 2395–2417 CrossRef CAS.
- R. Kulandaisamy, T. Kushwaha, A. Dalal, V. Kumar, D. Singh, K. Baswal, P. Sharma, K. Praneeth, P. Jorwal, S. R. Kayampeta, T. Sharma, S. Maddur, M. Kumar, S. Kumar, A. Polamarasetty, A. Singh, D. Sehgal, S. L. Gholap, M. B. Appaiahgari, M. R. Katika and K. K. Inampudi, Front. Microbiol., 2022, 13, 877813 CrossRef PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- T. Yang, J. C. Wu, C. Yan, Y. Wang, R. Luo, M. B. Gonzales, K. N. Dalby and P. Ren, Proteins: Struct., Funct., Bioinf., 2011, 79, 1940–1951 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, J. Phys. Chem. B, 2010, 114, 1652–1660 CrossRef CAS PubMed.
- C. Matthews, J. Weare, A. Kravtsov and E. Jennings, Mon. Not. R. Astron. Soc., 2018, 480, 4069–4079 CrossRef CAS.
- H. Fu, H. Bian, X. Shao and W. Cai, J. Phys. Chem. Lett., 2024, 15, 1774–1783 CrossRef CAS PubMed.
- E. Eyal and I. Bahar, Biophys. J., 2008, 94, 3424–3435 CrossRef CAS PubMed.
- K. Mikulska-Ruminska, A. J. Kulik, C. Benadiba, I. Bahar, G. Dietler and W. Nowak, Sci. Rep., 2017, 7, 8852 CrossRef PubMed.
- S. Armaković, S. J. Armaković, J. P. Šetrajčić and I. J. Šetrajčić, J. Mol. Model., 2012, 18, 4491–4501 CrossRef PubMed.
- H. N. Banavath, O. P. Sharma, M. S. Kumar and R. Baskaran, Sci. Rep., 2014, 4, 6948 CrossRef PubMed.
- U. Soykan, Y. Sert and G. Yıldırım, J. Mol. Struct., 2021, 1244, 130940 CrossRef CAS.
- E. Margiotta, S. C. C. van der Lubbe, L. de Azevedo Santos, G. Paragi, S. Moro, F. M. Bickelhaupt and C. Fonseca Guerra, J. Chem. Inf. Model., 2020, 60, 1317–1328 CrossRef CAS PubMed.
- M. K. Rana, M. Sinha and S. Panda, Chem. Phys., 2018, 513, 23–34 CrossRef CAS.
- E. P. Jesudason, S. K. Sridhar, E. J. P. Malar, P. Shanmugapandiyan, M. Inayathullah, V. Arul, D. Selvaraj and R. Jayakumar, Eur. J. Med. Chem., 2009, 44, 2307–2312 CrossRef CAS PubMed.
- H. R. Bhat, P. S. S. Gupta, S. Biswal and M. K. Rana, ACS Omega, 2019, 4, 4505–4518 CrossRef CAS PubMed.
- J. A. Tanner, R. M. Watt, Y.-B. Chai, L.-Y. Lu, M. C. Lin, J. S. M. Peiris, L. L. M. Poon, H.-F. Kung and J.-D. Huang, J. Biol. Chem., 2003, 278, 39578–39582 CrossRef CAS PubMed.
- J. Chen, B. Malone, E. Llewellyn, M. Grasso, P. M. M. Shelton, P. D. B. Olinares, K. Maruthi, E. T. Eng, H. Vatandaslar, B. T. Chait, T. M. Kapoor, S. A. Darst and E. A. Campbell, Cell, 2020, 182, 1560–1573 CrossRef CAS PubMed.
- S.-C. Cheng, G.-G. Chang and C.-Y. Chou, Biophys. J., 2010, 98, 1327–1336 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- A. Dorababu, RSC Med. Chem., 2020, 11, 1335–1353 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05934k |
| This journal is © the Owner Societies 2024 |