
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Infrared spectra of SinH4n−1+ ions (n = 2–8): inorganic H–(Si–H)n−1 hydride wires of penta-coordinated Si in 3c–2e and charge-inverted hydrogen bonds†
Martin Andreas Robert
George
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, Berlin 10623, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 6th February 2024
Abstract
SinHm+ cations are important constituents in silane plasmas and astrochemical environments. Protonated disilane (Si2H7+) was shown to have a symmetric three-centre two-electron (3c–2e) Si–H–Si bond that can also be considered as a strong ionic charge-inverted hydrogen bond with polarity Siδ+–Hδ−–Siδ+. Herein, we extend our previous work to larger SinH4n−1+ cations, formally resulting from adding SiH4 molecules to a SiH3+ core. Infrared spectra of size-selected SinH4n−1+ ions (n = 2–8) produced in a cold SiH4/H2/He plasma expansion are analysed in the SiH stretch range by complementary dispersion-corrected density functional theory calculations (B3LYP-D3/aug-cc-pVTZ) to reveal their bonding characteristics and cluster growth. The ions with n = 2–4 form a linear inorganic H–(Si–H)n hydride wire with adjacent Si–H–Si 3c–2e bridges, whose strength decreases with n, as evident from their characteristic and strongly IR active SiH stretch fundamentals in the range 1850–2100 cm−1. These 3c–2e bonds result from the lowest-energy valence orbitals, and their high stability arises from their delocalization along the whole hydride wire. For SinH4n−1+ with n ≥ 5, the added SiH4 ligands form weak van der Waals bonds to the Si4H19+ chain. Significantly, because the SinH4n−1+ hydride wires are based on penta-coordinated Si atoms leading to supersaturated hydrosilane ions, analogous wires cannot be formed by isovalent carbon.
Introduction
Hydrogen passivation converts reactive Sin clusters into stable polysilanes nanostructures (SinHm), with potential applications of their derivatives and ions in material science, inorganic chemistry, catalysis, plasma- and astrochemistry, and theoretical chemistry.1–25 For example, polysilane oligomers and their radical ions reveal interesting electronic, photophysical, and optical properties arising from substantial σ-delocalization of the bonding Si–Si electrons.1,26 Recently, higher-order silanes have been identified during monosilane pyrolysis by gas chromatography–mass spectrometry.21 From an astrochemical point of view, Si and H are amongst the ten most abundant elements in the universe, and thus SinHm molecules and their ions are expected to occur in the interstellar medium, although so far only SiH4 has been identified.22 In the laboratory, saturated SinH2n+2 polysilanes up to n = 19 have been produced from bombarding SiH4 ices with electrons and detected by mass spectrometry.27 The discussed production mechanism involves radical–radical recombination and insertion reactions, although their structures have remained elusive. The structure and reactivity of SinHm+ cations relevant for silane plasmas used in plasma-enhanced chemical vapor deposition for the industrial fabrication of Si-based microelectronic thin films and devices17–20 have mostly been characterized by mass spectrometry.15,16,28,29 However, firm identification of structure and bonding of SinHm+ ions in plasma and astrochemical environments requires spectroscopic characterization, which is still rather scarce for ions with n ≥ 2.30–33 In the condensed phase, silyl cations are highly reactive,8,11 and only recently a SiH3+ compound could be synthesized and characterized.14Although Si and C are both group IV elements, their bonding properties differ substantially,34 which partly arises from their different electronegativity with respect to H (EN = 1.90, 2.20, 2.55 for Si, H, C on the Pauling scale). Due to their weaker and less directional Si–Si and Si–H bonds, SinHm molecules exhibit a larger variety of bonding motifs as compared to the corresponding CnHm molecules. For example, different from CnHm, SinHm have more frequently Si–H–Si bridges, characterized by a three-center two-electron (3c–2e) bond.35,36 In amorphous and crystalline silicon (a-Si:H and c-Si), used for example in commercial solar cells, linear Si–H–Si bridges are typical binding motifs for proton impurities. In c-Si, they form well-defined linear and symmetric Si–H–Si bonds.37 The Si–H–Si 3c–2e bond was first detected in crystals of cyclic and linear silyl cations.38–40 Subsequently, infrared photodissociation (IRPD) spectroscopy has been used in our group to characterize such 3c–2e bonds in a variety of SinHm+ cations in the gas phase, i.e. free from any perturbation by matrix effects and counter ions. The studied ions include protonated disilane (Si2H7+),31 an isomer of trisilane (Si3H8+),32 and SinH4n−4+ ions (n = 4–8)33 which may formally be described as silane aducts of the unsaturated sylilene cation, Si2H4+(SiH4)n−2. The characteristic IR fingerprint of the Si–H–Si bond is its strongly IR active antisymmetric stretch fundamental (σSiHSi) occurring in the 1600–2100 cm−1 range, whose frequency strongly depends on the structural and energetic details of the Si–H–Si bridges.33 The strength of the Si–H–Si bonds can range from strong 3c–2e chemical bonds to very weak van der Waals contacts (5–150 kJ mol−1).33 In the covalent case, the molecular orbital describing the 3c–2e bond is the lowest-energy valence orbital, illustrating its high stability.31,32 Because the EN of H is higher than that of Si, these 3c–2e bonds can also be considered as strong ionic charge-inverted hydrogen bonds (CIHB),41,42 with polarity Siδ+–Hδ−–Siδ+ resulting in the limit of ionic bonding in a hydride (H−) bond. This is opposite to regular Aδ−–Hδ+–Bδ− hydrogen bonds (H-bonds) in which, according to the IUPAC definition,43 a positive H atom (proton) is located between two basic atoms A (proton donor) and B (proton acceptor) with higher EN.
Herein, we extend our previous IRPD studies on silane cations to the SinH4n−1+ series with n = 2–8 to probe their structure, bonding and cluster growth mechanism as a function of the cluster size, along with complementary calculations. The most stable structures of these supersaturated hydrosilane ions may formally be considered as SiH3+(SiH4)n−1 clusters and exhibit unprecedented inorganic silicon hydride wires, H–(Si–H)n with multiple 3c–2e Si–H–Si bonds of penta-coordinated Si atoms. These 3c–2e bonds get weaker as the chain grows and, starting from n = 5, the Si4H15+ wire is solvated by SiH4 ligands via weak van der Waals bonds. The SinH4n−1+ ions differ qualitatively from the previously studied SinH4n−4+ series33 because they do not contain any Si–Si and/or Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si bonds. While SiH3+ and Si2H7+ (n = 1 and 2) have been well studied by IR spectroscopy,31,44–46 mass spectrometry,15,31,47,48 and calculations,31,49,50 the only study available for n ≥ 3 utilizes high-pressure mass spectrometry to determine the binding enthalpies of SiH3+(SiH4)n−1 as −ΔH0 = >146, 37.7, and 13.8 kJ mol−1 for n = 2–4, respectively.15 Although these data are not sensitive to structure, the authors concluded symmetric bonding of two SiH4 ligands to the two sides of the 3pz orbital of the planar SiH3+ cation and thus closure of the first solvation shell at n = 3.
Si bonds. While SiH3+ and Si2H7+ (n = 1 and 2) have been well studied by IR spectroscopy,31,44–46 mass spectrometry,15,31,47,48 and calculations,31,49,50 the only study available for n ≥ 3 utilizes high-pressure mass spectrometry to determine the binding enthalpies of SiH3+(SiH4)n−1 as −ΔH0 = >146, 37.7, and 13.8 kJ mol−1 for n = 2–4, respectively.15 Although these data are not sensitive to structure, the authors concluded symmetric bonding of two SiH4 ligands to the two sides of the 3pz orbital of the planar SiH3+ cation and thus closure of the first solvation shell at n = 3.
Results and discussion
IRPD spectra of size-selected SinH4n−1+ ions with n = 2–8 in the SiH stretch range (1400–2300 cm−1, Fig. 1, Table 1) were obtained in a tandem quadrupole mass spectrometer coupled to an electron ionization source.51,52 The ions were produced in a pulsed supersonic plasma expansion of a SiH4/H2/He gas mixture (ratio 1/1/38) at 3 bar stagnation pressure. This setup was used previously to record IRPD spectra of bare SinH4n−4+ ions and Ar/Ne tagged Si2H6+,30 Si3H8+,32 SiH3OH2+,53 and Si2H7+.31 Chemical ionisation of this gas mixture causes silane polymerization reactions forming SinH4n−1+ up to n = 8. They form in barrierless aggregation reactions of SiH4 units to SiH3+,15,31,32 which is the major fragment of ionizing SiH4. A typical mass spectrum of the ion source is shown in Fig. S1 in the ESI.† SinH4n−1+ ions selected by the first quadrupole were irradiated with a tuneable IR laser pulse of an optical parametric oscillator (νIR, 2–5 mJ, 10 Hz repetition rate, 1 cm−1 bandwidth). Resonant vibrational excitation by single-photon absorption resulted in the evaporation of either one (n = 3–4) or two (n ≥ 5) SiH4 molecules, indicating a formal cluster composition of SiH3+(SiH4)n−1 with decreasing SiH4 binding energy as n increases. IRPD spectra were then obtained as a function of νIR by monitoring the fragment ions selected by the second quadrupole and normalization for laser intensity. Because the SiH4 binding energy of Si2H7+ (ca. 150 kJ mol−1) is much higher than the employed IR photon energy (<25 kJ mol−1), its spectrum could only be obtained by Ne/Ar tagging.31 This spectrum was reported and analysed previously31 and is included in Fig. 1 only for comparison.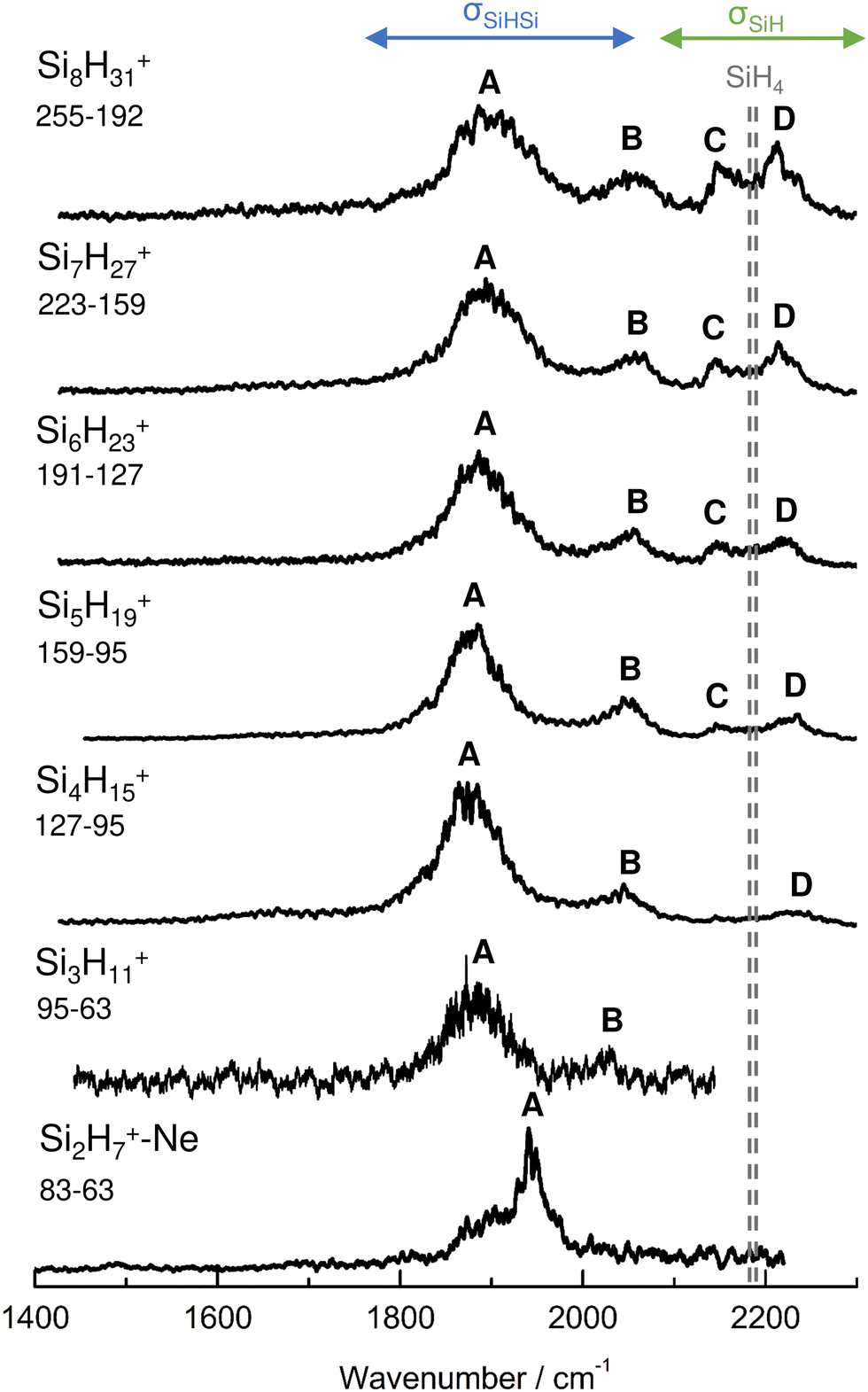 | ||
| Fig. 1 IRPD spectra of SinH4n−1+ (n = 3–8) in the SiH stretch range recorded in the SiH4 or 2SiH4 loss channel as indicated by the m/z values of parent and daughter ions. The Si2H7+Ne spectrum (n = 2) recorded in the Ne loss channel is included for comparison.31 The probed spectral range (1400–2300 cm−1) covers σSiH fundamentals (C/D) of free Si–H bonds and σSiHSi modes (A/B) of 3c–2e bonds (Table 1). The dashed lines indicate the positions of the SiH stretch modes of bare SiH4 at ν1(a1) = 2187 and ν3(t2) = 2191 cm−1.54 | ||
| Peak | Mode | SiH4a | Si2H7+Neb | Si3H11+ | Si4H15+ | Si5H19+ | Si6H23+ | Si7H27+ | Si8H31+ |
|---|---|---|---|---|---|---|---|---|---|
| a Ref. 54. b Ref. 31. | |||||||||
| A | σ SiHSi(a) | 1941 (30) | 1886 (65) | 1873 (70) | 1877 (65) | 1885 (70) | 1893 (80) | 1903 (100) | |
| B | σ SiHSi(s) | 2031 (15) | 2045 (50) | 2051 (45) | 2055 (60) | 2058 (60) | 2060 (60) | ||
| C | σ SiH | 2187 | 2144 (25) | 2147 (30) | 2144 (25) | 2147 (35) | |||
| D | σ SiH | 2191 | 2236 (45) | 2235 (40) | 2225 (40) | 2216 (40) | 2211 (45) | ||
The four major peaks A–D observed in the IRPD spectra are readily assigned to the strongly IR active Si–H–Si stretch modes of the 3c–2e bond(s) between 1800 and 2100 cm−1 (σSiHSi, A and B) and the much weaker SiH stretch modes of the free Si–H bonds between 2100 and 2300 cm−1 (σSiH, C and D). For comparison, Fig. 1 also indicates the symmetric and triply degenerate antisymmetric SiH stretch frequencies of bare SiH4 by dashed lines, ν1(a1)/ν3(t2) = 2187/2191 cm−1.54 Significantly, while ν1 is IR forbidden in bare SiH4, it becomes slightly allowed by the presence of the nearby positive charge and experiences shifts and splittings. The IRPD spectra are dominated by peak A, assigned to antisymmetric σSiHSi(a) modes of the Si–H–Si 3c–2e bonds by comparison to the previous analysis of the Si2H7+ and SinH4n−4+ spectra. This band experiences a substantial monotonic red shift from 1941 to 1873 cm−1 for n = 2–4 (by −55 to −13 cm−1), while it shows smaller incremental blue shifts of +4, +8, +8, and +10 cm−1 for n = 5–8, indicating a change in bonding at n = 5. Starting from n = 3, band B assigned to the corresponding symmetric σSiHSi(s) modes of the 3c–2e bonds grow in intensity. This mode exists only for n ≥ 3, indicating the appearance of a second Si–H–Si 3c–2e bond in Si3H11+. Band B experiences gradual monotonic blue shifts by +14, +6, +4, +3, and +2 cm−1 for n = 4–8. The magnitude of the shifts drops for n ≥ 5, again indicating a change in bonding in this size regime. Bands C and D occur in the range of the free SiH stretch modes and grow roughly linearly in intensity starting from n = 5, again indicating a change in bonding at this size range. Their positions at around 2145 and 2225 cm−1 hardly change as a function of n, although their widths get somewhat broader. Hence, the first rough analysis of the number and position of the IRPD bands, and their incremental shifts and changes in IR activity suggests a SinH4n−1+ cluster growth with the formation of chemical 3c–2e bonds up to n = 4 and subsequent attachment of weakly bonded SiH4 ligands attached to a Si4H15+ core ion by induction and dispersion forces. The weaker SiH4 binding energy of the larger ions is also visible in the photofragmentation branching ratios measured at band A (Table S1 in the ESI†). While SinH4n−1+ ions with n ≤ 4 evaporate only one SiH4 ligand, those with n ≥ 5 can dissociate two ligands at an IR photon energy of around 2000 cm−1. Neglecting kinetic energy release and assuming that the absorbed photon energy is merely used for ligand dissociation (and not for changes in internal energy) and all SiH4 ligands have the same binding energy for n = 5–8, one can bracket their dissociation energy as D0 = 850 ± 150 cm−1 or 10.2 ± 1.8 kJ mol−1. This value is consistent with the SiH4 binding enthalpy of the somewhat stronger bonded n = 4 ion reported as −ΔH0 = 13.8 ± 1.3 kJ mol−1.15
For a more detailed structural and vibrational assignment and the analysis of chemical bonding (charge distributions, binding energies, molecular orbitals), quantum chemical calculations were performed at the dispersion-corrected B3LYP-D3/aug-cc-pVTZ level for SinH4n−1+ ions with n ≤ 5 (Fig. 2, Fig. S2–S7 and Tables S2–S5 in ESI†). Reported relative energies (E0) and binding energies (D0) are corrected for harmonic zero-point vibrational energies. All calculated frequencies are scaled by a factor of 0.97878 to optimize the agreement between the calculated and measured νSiH frequencies of SiH3 and SiH4. The atomic charge distribution is evaluated using the natural bond orbital (NBO) analysis. We consider herein mainly the most stable structure for each cluster size, while information for less stable local minima is provided in the ESI.†
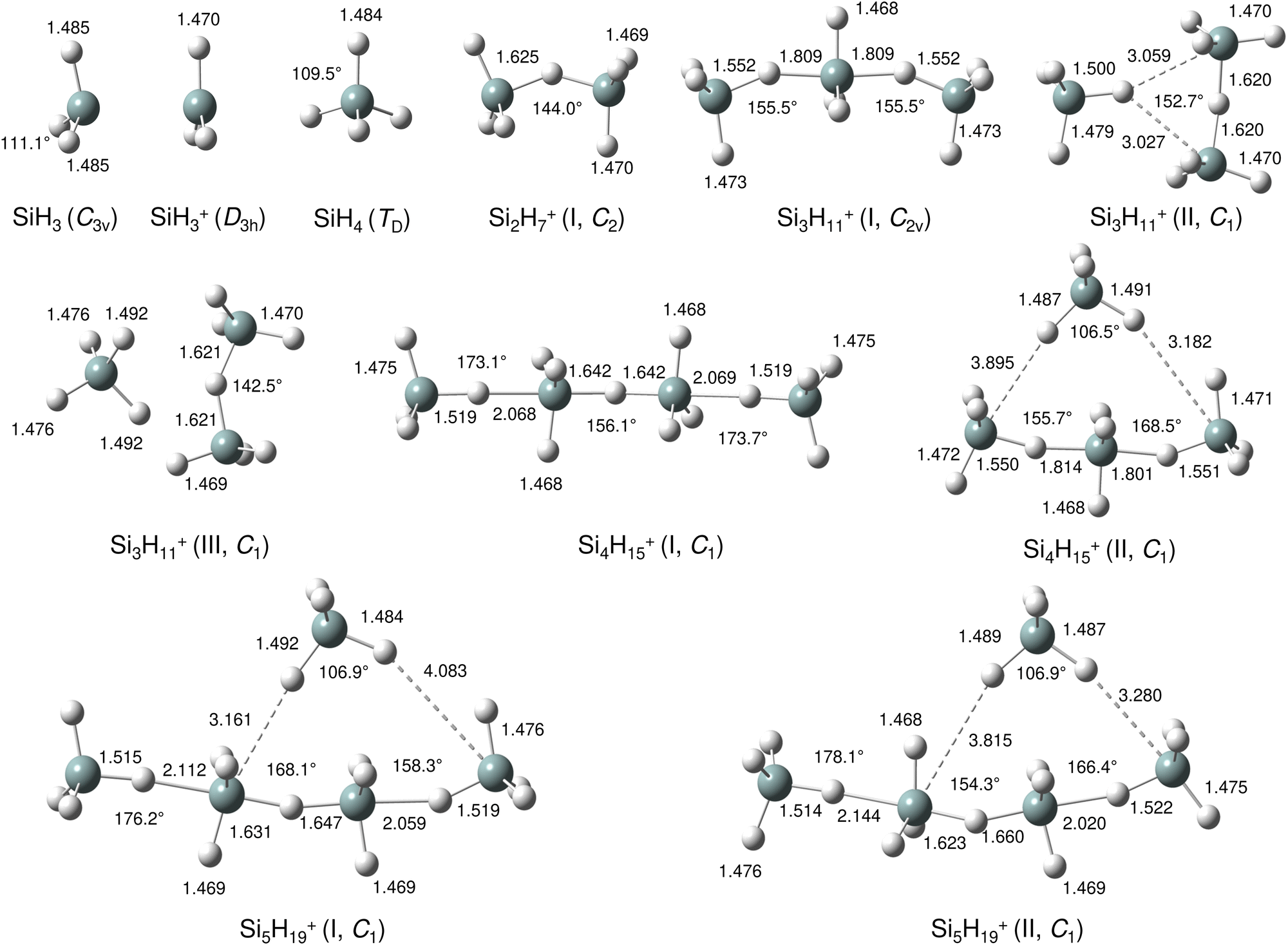 | ||
| Fig. 2 Lowest energy structures of SiH3, SiH3+, SiH4, Si2H7+, Si3H11+(I–III), Si4H15+(I,II) and Si5H19+(I,II) in their ground electronic states calculated at the B3LYP-D3/aug-cc-pVTZ level. Selected bond lengths and bond angles are given in Å and degree, respectively. | ||
Our calculations yield a planar structure for SiH3+ (D3h) with a Si–H bond length and stretch frequencies (re = 1.470 Å, ν1/3 = 2218/2295 cm−1) in good agreement with previous experimental and computational data.55,56 In line with the EN of Si and H, the partial charges are qH = −124 and qSi = +1371 me. The closed-shell SiH3+ cation (1A1) has a vacant and thus rather electrophilic 3pz orbital, which serves as attractive binding sites for neutral ligands, such as rare gas atoms,57 H2,45,46 or H2O.53 Barrierless attachment of SiH4 to this orbital results in protonated disilane, Si2H7+, forming a strong symmetric Si–H–Si 3c–2e bond characterized in detail previously.31 Our computed binding energy for SiH4 loss (D0 = 150.1 kJ mol−1) agrees well with the measured bond enthalpy (−ΔH0 > 146 kJ mol−1)15 and our previous MP2 and CBS-QB3 calculations (148.2 and 150.1 kJ mol−1).31 The SiH4 ligand attacks the 3pz orbital with one of the negative H atoms of SiH4 (qH = −161 and qSi = +643 me) in a bent configuration with C2 symmetry (θ = 144°), which is slightly more stable than the linear Si–H–Si structure (D3h/3d) by only a few kJ mol−1 (<5 kJ mol−1),31 indicating a rather flat and anharmonic double minimum potential for H motion perpendicular to the Si–Si axis. The SiH3 groups become slightly nonplanar (intermediate between sp2 and sp3 hybridisation) and the barriers for hindered internal rotation are rather low (<3 kJ mol−1).31 The corresponding molecular orbital (MO) describing the rather strong 3c–2e bond (1.625 Å) is delocalized along the Si–H–Si bridge and is the lowest valence MO (HOMO-6, Fig. 3). Although formally a proton is added to Si2H6, the bridging H atom has a negative charge of qH = −304 me, while the two Si atoms carry most of the excess positive charge (qSi = +954 me), and are thus attractive binding sites for both rare gas31 and further SiH4 ligands. The alternating charge distribution in Siδ+–Hδ−–Siδ+ qualifies Si2H7+ as prototypical cationic example of a CIHB, and its formation causes charge transfer of 348 me from SiH3+ to SiH4. The strongly IR active antisymmetric SiH stretch mode is computed as σSiHSi = 1975 cm−1 (I = 1314 km mol−1), in good agreement with the value of the Ne-tagged complex (1941 cm−1).
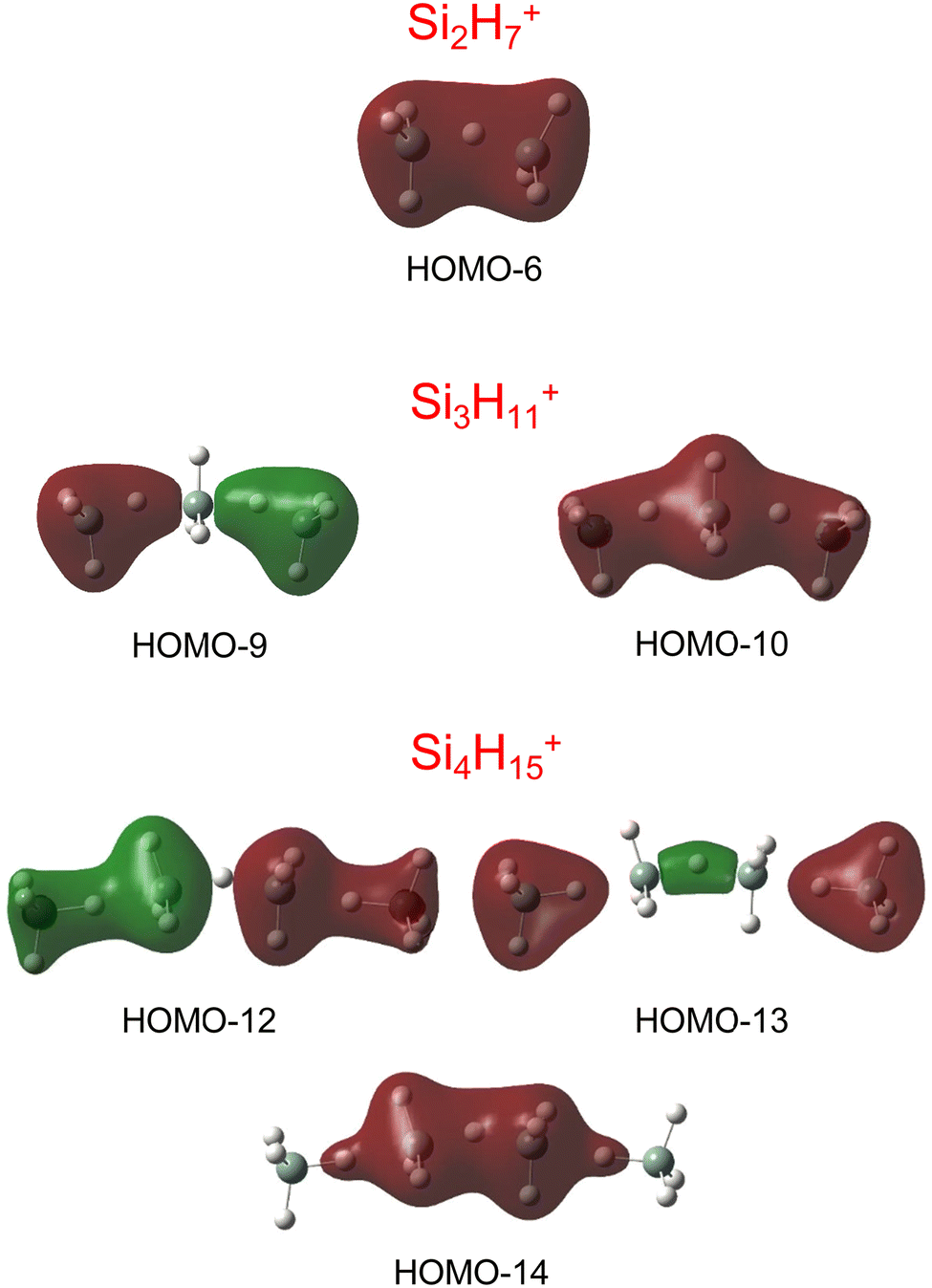 | ||
| Fig. 3 Visualisation of the lowest valence MOs of the most stable structures of Si2H7+, Si3H11+, and Si4H15+, representing the 3c–2e bonds. | ||
By far, the most stable structure of Si3H11+ is obtained by barrierless attachment of SiH4 to a Si atom of Si2H7+ (isomer I, Fig. 2). In this C2 symmetric structure, two equivalent SiH4 ligands bind to the 3pz orbital of a central SiH3+ cation forming two strongly asymmetric and bent 3c–2e bonds (1.809 and 1.552 Å, 155.5°). According to the much longer Si–H bond in the bridge (1.809 vs. 1.625 Å), the SiH4 binding energy is computed to be much weaker than in Si2H7+ (40.8 vs. 150.0 kJ mol−1) but again in excellent agreement with the measured bond enthalpy of −ΔH0 = 37.7 ± 1.3 kJ mol−1.15 The lower binding energy enables the IRPD spectrum of Si3H11+ to be recorded without tagging. This SN2 like complex features one penta-coordinated Si atom and can be considered as a supersaturated hydrosilane molecule, because even protonated aliphatic trisilane would only have nine H atoms for three Si atoms (Si3H9+). Formation of the two CIHBs causes charge transfer of 433 me from SiH3+ to the two SiH4 ligands. The two negative H atoms in Si3H11+ carry a slightly more negative charge than in Si2H7+ (−306 vs. −304 me). The two MOs describing the two 3c–2e bonds are completely delocalized along the Si–H–Si–H–Si bridge and are again the two lowest valence MOs (HOMO-9/10, Fig. 3). The coupling of the two σSiHSi modes of the two 3c–2e bonds results in a splitting of the σSiHSi mode of 1975 cm−1 of n = 2 in a rather weak symmetric and a rather strong antisymmetric normal mode at σSiHSi(s) = 2031 cm−1 and σSiHSi(a) = 1904 cm−1 for n = 3, respectively. These bands are in excellent agreement with the observed bands B and A predicted at 2031 and 1886 cm−1 with respect to their absolute frequency, the frequency shifts with respect to band A of Si2H7+, and their relative IR activities (Fig. 4). We also calculated two further less stable Si3H11+ isomers (II and III), in which SiH4 ligands are attached to the side of a Si2H7+ core ion by much weaker induction and dispersion forces (D0 = 13.8 and 12.3 kJ mol−1) and these feature only one 3c–2e bond. As a consequence, their predicted IR spectra are rather similar to that of Si2H7+ and in strong contradiction with the experiment (Fig. S3 in ESI†). Hence, we do not discuss them further here.
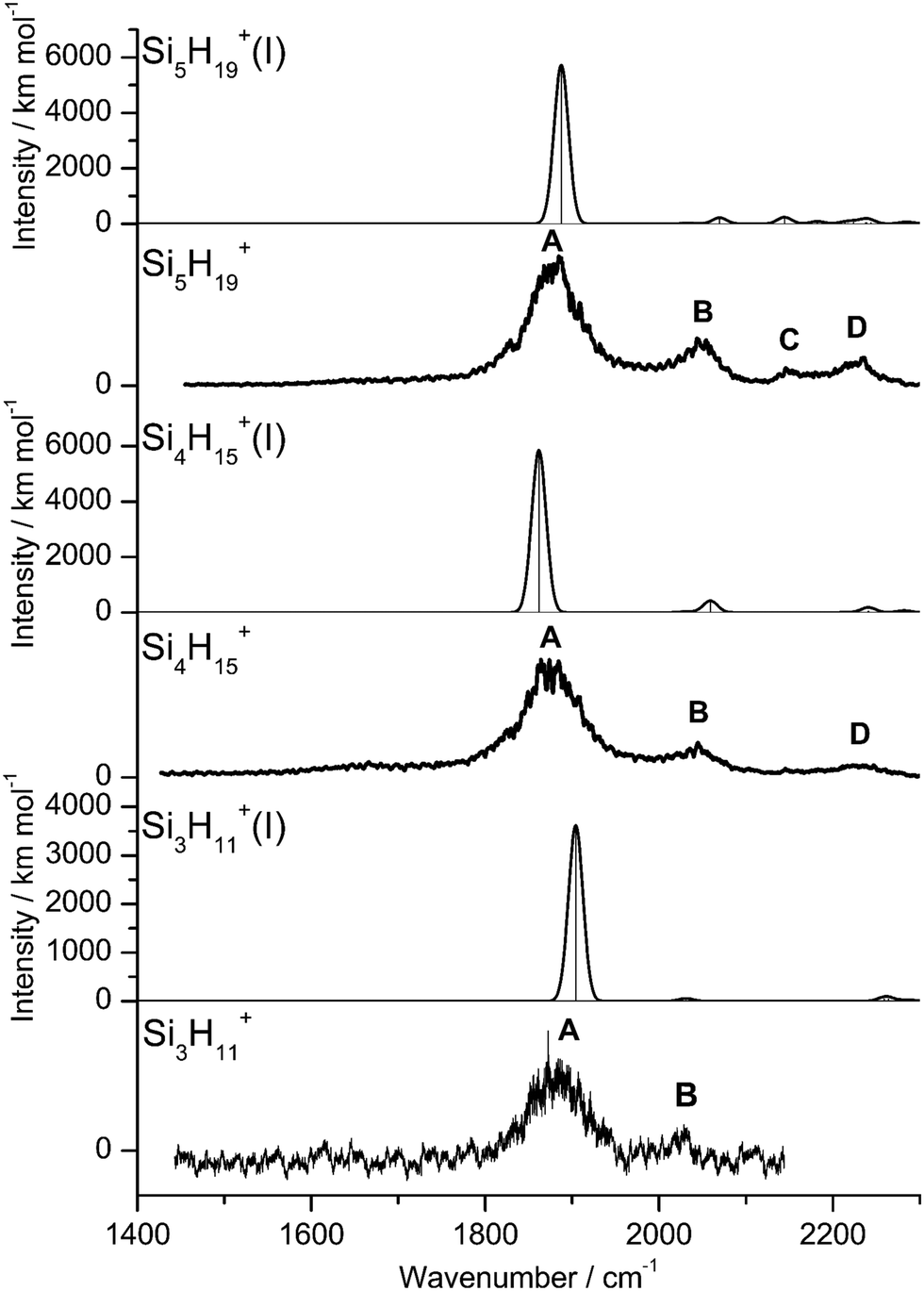 | ||
| Fig. 4 Experimental IRPD spectra of Si3H11+, Si4H15+, and Si5H19+ compared to linear IR absorption spectra of the most stable isomers calculated at the B3LYP-D3/aug-cc-pVTZ level (Table S2 in ESI†). | ||
The most stable structure of Si4H15+ (isomer I) is derived by barrierless addition of a further SiH4 ligand to a terminal Si atom of the linear Si3H11+ wire. In this nearly symmetric structure, two almost equivalent SiH4 ligands bind to the highly positive Si atoms of a central Si2H7+ cation forming a rather strong symmetric 3c–2e bond in the middle of the wire (1.642 Å, 156°) and two nearly equivalent and weaker asymmetric 3c–2e bonds at both ends (2.069 and 1.519 Å, 173°). (The C2 symmetric structure is predicted as a transition state.) The MOs describing the three 3c–2e bonds are again completely delocalized along the Si–H–Si–H–Si bridge and these are again the three lowest valence MOs (HOMO-12/13/14, Fig. 3). The binding energy of the terminal SiH4 units drops further down to D0 = 14.6 kJ mol−1, again in excellent agreement with the measured bond enthalpy of −ΔH0 = 13.8 ± 1.3 kJ mol−1.15 The lower binding energy is consistent with the smaller charge transfer from Si3H11+ to the added SiH4 ligand (110 me). This linear inorganic hydride wire features two penta-coordinated Si atoms and is an even more supersaturated silane molecule than Si3H11+, because protonated aliphatic tetrasilane has eleven H atoms for four Si atoms (Si4H11+), i.e. four H atoms less than Si4H15+. Attachment of the two SiH4 ligands at opposite ends of Si2H7+ changes its parameters with respect to both bond lengths and angle of the 3c–2e bridge (1.642 Å and 156° vs. 1.625 Å and 144°) and the negative charge on the central H atom (qH = −317 vs. −304 me). The three 3c–2e bonds have their characteristic σSiHSi modes predicted at 1862 (very strong), 2032 (very weak), and 2059 (weak) cm−1, with I = 5844, 34, and 423 km mol−1, respectively. These predicted frequencies show again good agreement with the corresponding bands A and B at 1873 and 2045 cm−1, where the latter band covers both weak σSiHSi modes (Fig. 4). Finally, the weak band D at 2236 cm−1 can also be explained by unresolved σSiH modes with low IR activity in this spectral range. We also computed further less stable Si4H15+ isomers, in which either one SiH4 ligand is attached to the side of Si3H11+ or two SiH4 ligands attached to the side of Si2H7+, all with D0 < 12 kJ mol−1. As their predicted IR spectra do not match the measured IRPD spectrum recorded for Si4H15+, they are not considered further here (Fig. S4 in ESI†).
For the Si5H19+ ions, our calculations yield six low-energy isomers, all within an energy range of 4 kJ mol−1. The binding energy for adding SiH4 ligands to the SinH4n−1+ wire drops with the length of the chain, and at n = 5 it is no longer favourable to extend the Si4H15+ chain further in a linear fashion. Indeed, we could not find a stable minimum for such a n = 5 chain, because the added SiH4 ligand prefers to move closer to the central Si2H7+ unit, which carries a substantial amount of the positive excess charge. To this end, the global minimum identified for Si5H19+ has a SiH4 unit attached to the side of the Si4H15+ wire, with D0 = 10.6 kJ mol−1 and very long SiH⋯Si contacts (3.161 and 4.083 Å). Further low-energy minima can be obtained for other isomers, with one SiH4 attached to Si4H15+ or two SiH4 ligands attached to the Si3H11+ core via induction and dispersion forces. The IR spectra predicted for Si5H19+ clusters with one or two SiH4 ligands are quite different, because the core ions, Si4H15+ or Si3H11+, determine the coarse structure in the strongly IR active σSiHSi range. Comparison of these spectra with the measured IRPD spectrum suggests that the core in the observed Si5H19+ ions is the Si4H15+ wire. Depending on the exact position of the last weakly-bonded SiH4 ligand in Si5H19+, the symmetry, structure, charge distribution, and IR spectrum changes slightly. Taking the computed global minimum, the charge transfer from Si4H15+ to SiH4 is rather small (7 me). The intense σSiHSi transition predicted at 1887 cm−1 agrees well with band A observed at 1877 cm−1, while the two lower-intensity σSiHSi modes computed at 2033 and 2070 cm−1 match with peak B at 2051 cm−1. In the higher frequency range, the σSiH modes of the SiH4 ligand and SiH3 units predicted at 2144, 2182, 2214, 2224, 2238, 2239, and 2244 cm−1 can be attributed to the weak broad and unresolved peaks C and D at 2144 and 2235 cm−1, respectively.
No calculations have been performed for SinH4n−1+ clusters with n ≥ 6, because of their multiple low-energy local minima, which differ from their global minimum merely by the different attachment of weakly-bonded SiH4 ligands to the linear Si4H15+ core. The excellent agreement of the measured σSiHSi frequencies with those calculated for the global minima obtained for n ≤ 5 in Fig. 4 confirms the deduced cluster growth. The intense band A drops significantly in frequency for n = 2–4 and increases the monotonically for n ≥ 4 with a smaller slope, confirming the change in bonding mechanism from chemical to van der Waals between n = 4 and 5 (Fig. 5).
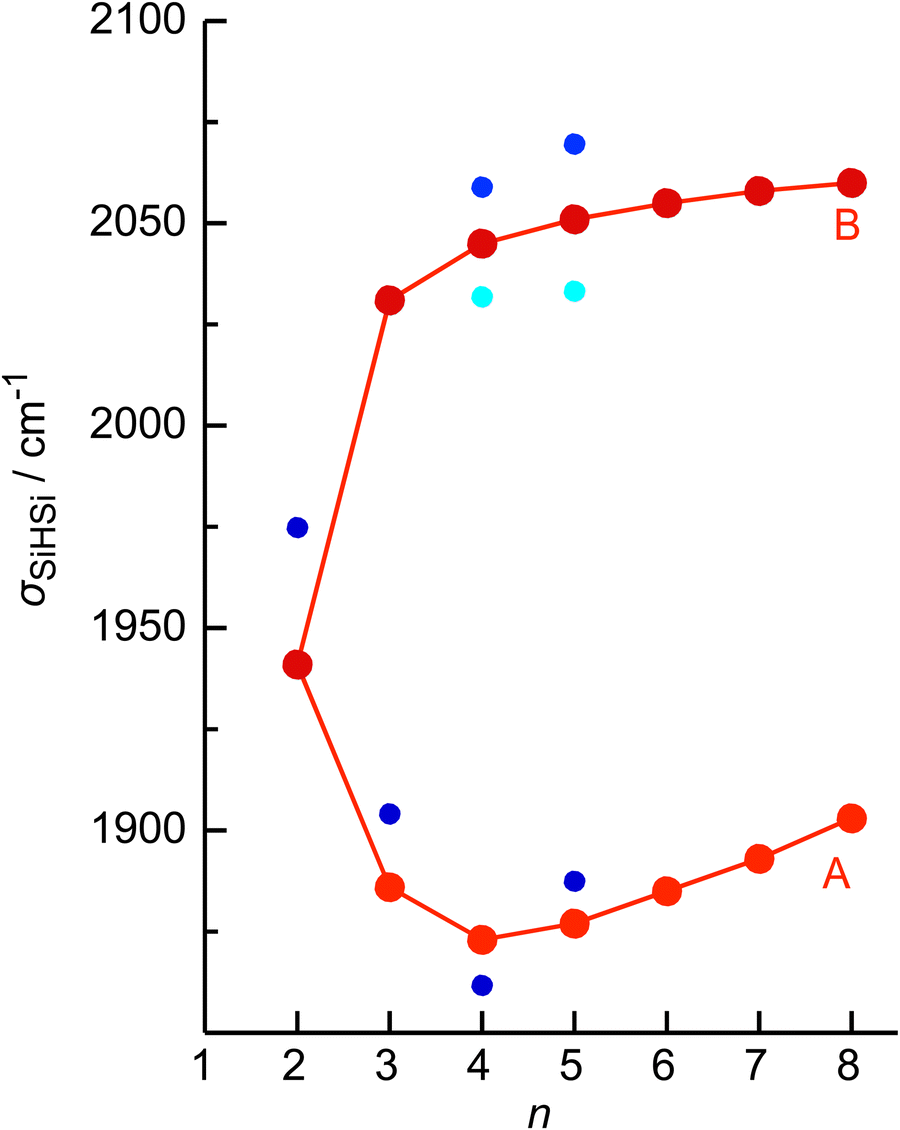 | ||
| Fig. 5 Experimental σSiHSi frequencies of SinH4n−1+ (bands A and B, red) as a function of n compared to computed values for the strongest (blue) and weaker (cyan) component, illustrating the change from strong chemical to weak van der Waals bonding between n = 4 and 5. | ||
In the following, we analyse the binding properties in the identified H–(Si–H)n wire in more detail. In general, Si–H–Si bridges are often formed by a 3c–2e bond in which two electrons in a bonding orbital establish two stable chemical bonds. These Si–H⋯Si H-bonds in SinHm+ cations feature a cationic CIHB, because the EN of Si is lower than that of H, leading to a charge configuration (Siδ+–Hδ−⋯Siδ+) opposite from that of a conventional σ-type H-bond between a proton donor A and an acceptor B (Aδ−–Hδ+⋯Bδ−). However, in many aspects these CIHBs show similar features as the regular H-bonds, including the redshifts of the proton donor stretch frequency (σAH) observed in the IR spectrum. The adjacent Si–H–Si bridges observed in SinH4n−1+ form an interesting inorganic hydride wire of the type H–(Si–H)n in which the H atoms carry negative partial charges. A systematic characterization of individual Si–H–Si bridges as a function of the two Si–H bond lengths (R1 and R2) has been presented in our previous study of SinH4n−4+.33 In general, the more symmetric the Si–H–Si geometry, the stronger the 3c–2e bond. This can be observed in the H–(Si–H)n wires with n = 2 and 4, in which the (inner) 3c–2e bond is rather strong and short with R1 = R2 = 1.625 and 1.642 Å, respectively, and D0(n = 2) = 150 kJ mol−1. On the other hand, the (outer) 3c–2e bonds for n = 3 and 4 are more asymmetric (R1 = 1.552/1.519 Å and R2 = 1.809/2.069 Å), reducing the bond strength to D0(n = 3/4) = 41/15 kJ mol−1, respectively. The correlation of the Si–H–Si bond angle with the 3c–2e bond strength is less strong, although it appears that they are more linear for weaker bonds. The observed trend is fully consistent with the analysis of Wiberg bond indices for the two bonds in the Si–H–Si bridge, yielding 0.44/0.44 for n = 2, 0.60/0.27 for n = 3, and 0.43/0.43 and 0.75/0.14 for the inner and outer bridges of n = 4. Clearly, for the weakly-bonded SiH4 ligand in n = 5, the indices of 0.92/0.00 illustrate the situation of a bare van der Waals bond. In general, the H atoms in the 3c–2e bridges carry a substantially more negative partial charge (qH = −290 to −320 me) than the free H atoms (−90 to −135 me). At the same time, the Si atoms carry nearly a full positive charge (qSi = +800 to +960 me), justifying the view of a hydride bride in the limit of ionic bonding (e.g., Si+H−Si+).
In the following, we briefly compare the properties of the SinH4n−1+ hydride wires to those of related systems. Overall, due to higher symmetry and the lack of any Si–Si/SiSi bonds, the IRPD spectra of SinH4n−1+ are much simpler and feature less bands than those of the previously studied SinH4n−4+ ions,33 which cannot form a regular hydride wire and lack supersaturation. The bonding of rare gas (Rg) atoms and H2 ligands to the SiH3+ cation is much weaker than that of SiH4 ligands. Thus, while in SiH3+Rg1–2 and SiH3+(H2)1–2 the ligands form weak van der Waals bonds to the 3pz orbital of SiH3+,15,45,46,57 SiH4 ligands form stronger covalent 3c–2e bonds in the SiH3+(SiH4)1–2 hydride wires. The interaction of Rg atoms and H2 with the related carbon analogue CH3+ is much stronger than with SiH3+, leading to covalent bonds in CH3+Rg dimers with heavy Rg atoms and in CH5+.51,58–61 As (i) the EN of C is higher than that of H and (ii) C does not like to be penta-coordinated, the CnH4n−1+ ions do not form supersaturated hydride wires but instead form weakly-bonded van der Waals clusters of CH4 ligands with a covalently-bonded C2H7+ core (Fig. S8–S10 in ESI†).62,63 The linear H–(Si–H)n hydride wires in SinH4n−1+ differ in several aspects from the corresponding proton wires and networks frequently observed, for example in protonated water and ammonia clusters.64–67 First, the H atoms in SinH4n−1+ are negatively charged because EN(Si) < EN(H), while the H atoms in O–H–O and N–H–N bridges are positive because EN(O/N) > EN(H). Moreover, the H–(Si–H)n hydride wires are essentially one-dimensional (1D), while H+(H2O)n and H+(NH3)n form 2D to 3D networks exhibiting proton transport via the Grotthuss mechanism.68–70 Linear proton wires can be formed for example in protonated imidazole clusters, in which however a heterocyclic spacer is involved.71–73
Conclusions
The interpretation of IRPD spectra of SinH4n−1+ ions with n = 2–8 by complementary quantum chemical calculations reveals the formation of an inorganic H–(Si–H)n hydride wire in the size range n = 2–4, with up three adjacent nonlinear covalent 3c–2e bonds involving penta-coordinated Si atoms. The binding energy with respect to SiH4 elimination becomes weaker as the chain length (n) increases. As a result, starting from n = 5 further SiH4 ligands bind weakly to the side of the linear Si4H15+ chain by induction and dispersion forces. The formation of such a wire is not favourable for the carbon analogue because penta-coordination is highly unfavourable and the EN order (Si < H < C), as illustrated by a comparison of the most stable structures of CnH4n−1+ and SinH4n−1+ (Fig. S8–S9 in ESI†). As a result, the SinH4n−1+ clusters are considered to be supersaturated with hydrogen when compared to carbon analogues. The adjacent 3c–2e bonds are associated with the lowest-lying valence MOs, due to their complete delocalization over the whole wire. Due to the relative EN values of Si and H, the SinH4n−1+ hydride wires with negatively charged H atoms represent prototypical cationic examples for charge-inverted H-bonds (CIHB)31–33,41 not yet covered by the IUPAC definition of H-bonds,43 which thus should be revised for this aspect.42 Similar to SiH3+ and Si2H7+, longer SinH4n−1+ wires may be synthesized and stabilized in the condensed phase. Moreover, such SinH4n−1+ ions may be observed in the future in SiH4-rich interstellar regions. Finally, it may be interesting to investigate the effects of substitution of H atoms by functional groups on the properties of such hydride wires.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (project DO 729/9). The authors thank J. Langer, A. Lagutschenkov, and M. Savoca for support in the acquisition of the experimental data.References
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS PubMed.
- P. D. Lickiss, in Adv. Inorg. Chem., ed. A. G. Sykes, Academic Press, 1995, vol. 42, pp. 147–262 Search PubMed.
- J. Fischer, J. Baumgartner and C. Marschner, Science, 2005, 310, 825 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2003, 90, 055502 CrossRef PubMed.
- F. Pichierri, V. Kumar and Y. Kawazoe, Chem. Phys. Lett., 2004, 383, 544–548 CrossRef CAS.
- R. Singh, J. Phys.: Condens. Matter, 2008, 20, 045226 CrossRef.
- J. B. Lambert, L. Kania and S. Z. Zhang, Chem. Rev., 1995, 95, 1191–1201 CrossRef CAS.
- K. C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. J. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5898–5985 CrossRef PubMed.
- V. Y. Lee, Russ. Chem. Rev., 2019, 88, 351–369 CrossRef CAS.
- J. C. L. Walker, H. F. T. Klare and M. Oestreich, Nat. Rev. Chem., 2020, 4, 54–62 CrossRef CAS.
- Q. Wu, E. Irran, R. Müller, M. Kaupp, H. F. T. Klare and M. Oestreich, Science, 2019, 365, 168 CrossRef CAS PubMed.
- K. Hiraoka, J. Katsuragawa and A. Minamitsu, Chem. Phys. Lett., 1997, 267, 580 CrossRef CAS.
- T. P. Martin and H. Schaber, J. Chem. Phys., 1985, 83, 855–858 CrossRef CAS.
- H. Chatham and A. Gallagher, J. Appl. Phys., 1985, 58, 159–169 CrossRef CAS.
- M. L. Mandich and W. D. Reents, J. Chem. Phys., 1989, 90, 3121–3135 CrossRef CAS.
- M. J. Kushner, J. Appl. Phys., 1993, 74, 6538–6553 CrossRef CAS.
- G. Turban, Y. Catherine and B. Grolleau, Plasma Chem. Plasma Process., 1982, 2, 61–80 CrossRef CAS.
- G. M. Wyller, T. J. Preston, H. Klette, T. Mongstad and E. S. Marstein, J. Cryst. Growth, 2018, 498, 315–327 CrossRef CAS.
- D. M. Goldhaber and A. L. Betz, Astrophys. J., 1984, 279, L55–L58 CrossRef CAS.
- B. A. McGuire, Astrophys. J., Suppl. Ser., 2022, 259, 30 CrossRef.
- M. C. McCarthy, C. A. Gottlieb and P. Thaddeus, Mol. Phys., 2003, 101, 697–704 CrossRef CAS.
- E. Herbst, T. J. Millar, S. Wlodek and D. K. Bohme, Astron. Astrophys., 1989, 222, 205–210 Search PubMed.
- F. Fang, Q. Jiang and R. S. Klausen, J. Am. Chem. Soc., 2022, 144, 7834–7843 CrossRef CAS PubMed.
- G. Tarczay, M. Förstel, P. Maksyutenko and R. I. Kaiser, Inorg. Chem., 2016, 55, 8776–8785 CrossRef CAS PubMed.
- N. Goldberg and H. Schwarz, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, 1998, ch. 18, vol. 2, pp. 1105–1142 Search PubMed.
- S. Fornarini, The Chemistry of Organic Silicon Compounds, 2001, ch. 18, pp. 1027–1057 Search PubMed.
- M. Savoca, M. A. R. George, J. Langer and O. Dopfer, Phys. Chem. Chem. Phys., 2013, 15, 2774–2781 RSC.
- M. Savoca, J. Langer and O. Dopfer, Angew. Chem., Int. Ed., 2013, 52, 1568–1571 CrossRef CAS PubMed.
- M. A. R. George, M. Savoca and O. Dopfer, Chem. – Eur. J., 2013, 19, 15315–15328 CrossRef CAS PubMed.
- M. A. R. George and O. Dopfer, Int. J. Mass Spectrom., 2019, 435, 51–60 CrossRef CAS.
- S. Patai and Z. Rappoport, The Chemistry of Organic Silicon Compounds, Wiley, Chichester, 1989 Search PubMed.
- R. L. DeKock and W. B. Bosma, J. Chem. Educ., 1988, 65, 194–197 CrossRef CAS.
- J. E. McMurry and T. Lectka, Acc. Chem. Res., 1992, 25, 47–53 CrossRef CAS.
- S. K. Estreicher, Mater. Sci. Eng., R, 1995, 14, 319–412 CrossRef.
- T. Müller, Angew. Chem., Int. Ed., 2001, 40, 3033–3036 CrossRef.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed.
- S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767–769 RSC.
- M. Jablonski, Chem. Phys. Lett., 2009, 477, 374–376 CrossRef CAS.
- S. Civiš, M. Lamanec, V. Špirko, J. Kubišta, M. Špet’ko and P. Hobza, J. Am. Chem. Soc., 2023, 145, 8550–8559 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- D. M. Smith, P. M. Martineau and P. B. Davies, J. Chem. Phys., 1992, 96, 1741 CrossRef CAS.
- Y. B. Cao, J. H. Choi, B. M. Haas, M. S. Johnson and M. Okumura, J. Phys. Chem., 1993, 97, 5215–5217 CrossRef CAS.
- D. W. Boo and Y. T. Lee, J. Chem. Phys., 1995, 103, 514–519 CrossRef CAS.
- T. Y. Yu, T. M. H. Cheng, F. W. Lampe and V. Kempter, J. Phys. Chem., 1972, 76, 3321–3330 CrossRef CAS.
- A. Ding, R. A. Cassidy, L. S. Cordis and F. W. Lampe, J. Chem. Phys., 1985, 83, 3426–3432 CrossRef CAS.
- K. Raghavachari, J. Chem. Phys., 1990, 92, 452–465 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. W. Deutsch and J. A. Pople, J. Chem. Phys., 1991, 95, 2433–2444 CrossRef CAS.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- M. A. R. George, N. X. Truong, M. Savoca and O. Dopfer, Angew. Chem., Int. Ed., 2018, 57, 2919–2923 CrossRef CAS PubMed.
- T. Shimanouchi, Tables of Molecular Vibrational Frequencies, Consolidated Volume I, NSRDS-NBS, 1972 Search PubMed.
- P. Botschwina and M. Oswald, J. Chem. Phys., 1992, 96, 4044–4045 CrossRef CAS.
- J. Kapp, P. R. Schreiner and P. v R. Schleyer, J. Am. Chem. Soc., 1996, 118, 12154–12158 CrossRef CAS.
- S. Pan, D. Moreno, G. Merino and P. Chattaraj, Chem. Phys. Chem., 2014, 15, 3554–3564 CrossRef CAS PubMed.
- R. V. Olkhov, S. A. Nizkorodov and O. Dopfer, J. Chem. Phys., 1998, 108, 10046–10060 CrossRef CAS.
- D. W. Boo, Z. F. Liu, A. G. Suits, J. S. Tse and Y. T. Lee, Science, 1995, 269, 57–59 CrossRef CAS PubMed.
- S. P. Gnanasekar and E. Arunan, J. Phys. Chem. A, 2019, 123, 1168–1176 CrossRef CAS PubMed.
- O. Asvany, K. M. T. Yamada, S. Brunken, A. Potapov and S. Schlemmer, Science, 2015, 347, 1346–1349 CrossRef CAS PubMed.
- K. Hiraoka and P. Kebarle, J. Am. Chem. Soc., 1976, 98, 6119–6125 CrossRef CAS.
- L. I. Yeh, J. M. Price and Y. T. Lee, J. Am. Chem. Soc., 1989, 111, 5597 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Science, 2004, 304, 1134–1137 CrossRef CAS PubMed.
- J.-W. Shin, N. I. Hammer, E. G. Diken, M. A. Johnson, R. S. Walters, T. D. Jaeger, M. A. Duncan, R. A. Christie and K. D. Jordan, Science, 2004, 304, 1137–1140 CrossRef CAS PubMed.
- M. Park, I. Shin, N. J. Singh and K. S. Kim, J. Phys. Chem., 2007, 111, 10692–10702 CrossRef CAS PubMed.
- M. Katada, R. Shishido and A. Fujii, Phys. Chem. Chem. Phys., 2014, 16, 7595–7601 RSC.
- C. J. D. Grotthuss, Ann. Chim. Phys., 1806, 58, 54–57 Search PubMed.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456 CrossRef CAS.
- D. Marx, Chem. Phys. Chem., 2006, 7, 1848–1870 CrossRef CAS PubMed.
- W. Münch, K. D. Kreuer, W. Silvestri, J. Maier and G. Seifert, Solid State Ionics, 2001, 145, 437–443 CrossRef.
- H. K. Gerardi, G. H. Gardenier, U. Viswanathan, S. M. Auerbach and M. A. Johnson, Chem. Phys. Lett., 2011, 501, 172–178 CrossRef CAS.
- A. A. Adesokan, G. M. Chaban, O. Dopfer and R. B. Gerber, J. Phys. Chem. A, 2007, 111, 7374–7381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05918a |
| This journal is © the Owner Societies 2024 |