
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFörster resonance energy transfer within the neomycin aptamer†
Florian
Hurter
a,
Anna-Lena J.
Halbritter
bc,
Iram M.
Ahmad
b,
Markus
Braun
 a,
Snorri Th.
Sigurdsson
b and
Josef
Wachtveitl
*a
a,
Snorri Th.
Sigurdsson
b and
Josef
Wachtveitl
*a
aInstitute of Physical and Theoretical Chemistry, Goethe University Frankfurt, Main, Max-von-Laue-Str. 7, Frankfurt 60438, Germany. E-mail: wveitl@theochem.uni-frankfurt.de
bScience Institute, University of Iceland, Dunhaga 3, Reykjavik 107, Iceland
cDepartment of Chemistry, Ludwig-Maximilians-University (LMU), Munich, Germany
First published on 18th January 2024
Abstract
Förster resonance energy transfer (FRET) measurements between two dyes is a powerful method to interrogate both structure and dynamics of biopolymers. The intensity of a fluorescence signal in a FRET measurement is dependent on both the distance and the relative orientation of the dyes. The latter can at the same time both complicate the analysis and give more detailed information. Here we present a detailed spectroscopic study of the energy transfer between the rigid FRET labels Çmf (donor) and tCnitro (quencher/acceptor) within the neomycin aptamer N1. The energy transfer originates from multiple emitting states of the donor and occurs on a low picosecond to nanosecond time-scale. To fully characterize the energy transfer, ultrafast transient absorption measurements were performed in conjunction with static fluorescence and time-correlated single photon counting (TCSPC) measurements, showing a clear distance dependence of both signal intensity and lifetime. Using a known NMR structure of the ligand-bound neomycin aptamer, the distance between the two labels was used to estimate κ2 and, therefore, make qualitative statements about the change in orientation after ligand binding with unprecedented temporal and spatial resolution. The advantages and potential applications of absorption-based methods using rigid labels for the characterization of FRET processes are discussed.
Introduction
Ribonucleic acids (RNAs) play a central role in cellular regulation such as transcription, translation and gene expression.1 This includes functional non-coding RNAs, such as riboswitches that often target the 5′-untranslated regions of bacterial mRNA.2 A riboswitch contains two interacting domains, an aptamer region that binds with high affinity and specificity to a metabolite and the expression platform.3–5 These two domains act in concert to activate or repress gene expression through a conformational change that is triggered by ligand binding.2,6–9RNA aptamers can be readily generated by the technique of systematic evolution of ligands by exponential enrichment (SELEX)10–13 and have found use as diagnostic agents,14,15 biosensors,16,17 and therapeutics.18–20 It is of great interest to create artificial riboswitches for regulation of gene expression using ligands of choice, but only a few RNA aptamers have been utilized for creating non-natural riboswitches.6,7,21–23 Increased understanding of the structure–function relationships of aptamers and riboswitches is required to be able to purposefully design new functional riboswitches.24–26
Of the several methods that have been established for studying the structure and dynamics of RNA, fluorescence spectroscopy is particularly valuable.27,28 It has the advantages of being highly sensitive, nondestructive for the sample of interest and offers the ability to conduct experiments under native conditions. It is a very useful tool to detect the dynamics of biomolecules through various approaches. In addition to unraveling detailed information about RNA dynamics, fluorescence spectroscopy is a powerful method to study structure, even of single molecules, through Förster resonance energy transfer (FRET). This high-resolution fluorescence technique can obtain information about both distance and relative orientation of FRET pairs.29–33
The relative orientation between the donor and the acceptor is described with κ2, which is not easy to determine. For free moving labels a κ2 of 2/3 is usually assumed, which is valid in the dynamic isotropic regime.34 However, this assumption is in many cases insufficient,30,35–39 especially for rigid FRET pairs, where this assumption is considered invalid.35,40 On the flip side, rigid FRET labels can provide more structural details through interrogation of the relative orientation of the dyes.
The structural elements of nucleic acids provide a good framework for rigid labels, in particular when the labels are nucleobase analogues. When embedded in helical regions, they provide accurate distance measurements and the possibility to study relative orientations.41
We have previously prepared and evaluated the fluorescent nucleobase analogue Çmf (Fig. 1b), which forms a base pair with guanine, as a fluorescent probe for RNA.42 It was shown that Çmf does not perturb the structure of RNA duplexes and that it is highly sensitive to its microenvironment. Moreover, it was used to study ligand binding by the neomycin aptamer.
 | ||
| Fig. 1 (a) Secondary structure of the N1-aptamer in its free state (left) and when bound to its ligand (right).44 (b) Structure of the rigid labels Çmf and tCnitro. | ||
Interestingly, the fluorescent label Çmf has multiple emitting states. This makes Çmf even more interesting as a donor in FRET-studies, since the energy transfer can take place from multiple states.43
Here we present a detailed spectroscopic study of the energy transfer between the rigid labels Çmf (donor) and tCnitro (quencher/acceptor) (Fig. 1b) inside the neomycin aptamer N1 (Fig. 1a). The neomycin aptamer was chosen because both its structure and dynamics have been well studied and because this aptamer is one of the few aptamers that functions as an active riboswitch.6,26,42,44–46 We found that the energy transfer between Çmf and tCnitro extends over several orders of magnitude, from nanoseconds to the low picosecond range. This required application of different methods, to adequately describe the energy transfer over this large time-range. In general, static fluorescence measurements can be used to determine whether FRET occurs and on what time scale. Once this is known, it can be decided whether TCSPC or TAS is suitable to obtain temporal information about the energy transfer, depending on how fast the process proceeds.
By combining different spectroscopic and data evaluation methods, we provide new approaches to gain a deeper understanding of the distance and orientation dependence of rigid labels in RNA, specifically to precisely determine structural changes with unprecedented temporal and spatial resolution, even at donor–acceptor distances below 2 nm.
Results & discussion
Preparation of oligonucleotides/labeling strategy
We have previously labeled the N1-aptamer at positions C6, U8 and U15 with Çmf to study the kinetics of ligand binding.42 Since the label was well accommodated in these positions, we used the same labeling strategy for Çmf in this FRET study and chose the C3 position for tCnitro. Thus, this labeling strategy yielded three different samples, N1-tCnitro3-Çfm6, N1-tCnitro3-Çfm8 and N1-tCnitro3-Çfm15 (N-N3C6, N-N3C8 and N-N3C15, respectively) (Fig. 2), with increasing distance between donor and acceptor in this order. | ||
| Fig. 2 Positions of the donor (Çmf) and acceptor (tCnitro) within the N1-aptamer, outlined in PDBID:2KXM(NDB/PDB).44Çmf is highlighted in blue and tCnitro in red. The ligand neomycin is shown in black. Donor acceptor distances (center to center) from the modified NMR structure indicated on the side of the samples and the KD values are shown below. | ||
To investigate the binding capabilities of the doubly labeled aptamers, we initially performed ITC experiments (ESI,† Fig. S1). The dissociation constants of the doubly labeled aptamers are generally larger than the KDs of the unlabeled aptamer (ESI,† Table S1). However, the KDs of the doubly- and singly-labeled aptamers are in the same order of magnitude.42 This demonstrates that the incorporation of tCnitro as a second label does not significantly affect the binding capabilities to the ligand.
FRET: data collection and analysis. The donor and the acceptor used in this study were covalently linked to the backbone of the N1-aptamer and thus, both the distance (r) and the relative orientation of the transition dipole moments determine the efficiency of the FRET process. Since tCnitro acts as an efficient quencher, the fluorescence signal originates exclusively from the Çmf chromophore, which greatly simplifies the evaluation of the FRET data. We began by investigating the static fluorescence behavior.
Static FRET characterization
The singly Çmf-labeled N1-aptamers N-C6, N-C8 and N-C15,42 have previously been shown to have slightly different fluorescence intensity, due to the different flanking bases of Çmf (Fig. 3a).42 Upon ligand binding, the microenvironment of the Çmf changed, leading to an increased fluorescence signal for N-C6, N-C8 and to a decrease for N-C15.42 For the doubly-labeled aptamers (N-N3C6, N-N3C8 and N-N3C15), all signal intensities were significantly lower than the singly Çmf-labeled aptamers (Fig. 3b). Moreover, the fluorescence signals show a clear correlation with the distance between donor and acceptor: It is almost completely quenched for the shortest distance (N-N3C6), but the signals get stronger as the chromophores are further apart. | ||
| Fig. 3 Time-integrated fluorescence spectra of (a) Çmf labeled neomycin aptamers published by Gustman et al.42 (b) Çmf and tCnitro labeled neomycin aptamers. The fluorescence spectra of the aptamers without neomycin (− Neo) are shown as a segmented line. | ||
Ligand binding only resulted in a minor change in fluorescence for both N-N3C6 and N-N3C15. In contrast a large relative change was observed for N-N3C8, which was even larger than for the singly labeled aptamer N-C8.
For a quantitative analysis, the FRET efficiencies can be calculated. The reduction of the FRET efficiency (E) is dominated by the donor–acceptor distance, but also the relative orientation of the transition dipole moments (eqn (1)):
 | (1) |
Here, R0 (eqn (2)) corresponds to the Förster radius, r to the distance between donor and acceptor, τD to the fluorescence lifetime of the donor alone and kFRET(r) (eqn (3)) to the transfer rate.
 | (2) |
 | (3) |
The relative orientation between the transition dipole moments of the donor and acceptor is described by κ2 (eqn (4)). Here, JDA is the overlap integral between the emission spectrum of the donor and the absorption spectrum of the acceptor, n corresponds to the refractive index of the solvent and φD to the fluorescence quantum yield of the donor.30
κ2 = (sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θDsinθAcosϕ − 2cosθDcosθA)2 θDsinθAcosϕ − 2cosθDcosθA)2 | (4) |
The static FRET efficiencies (Esta) can be determined using the integrated donor fluorescence signal of the donor only (ID) and the doubly labeled (IDA) aptamers (eqn (5), Table 1). The Förster radii R0,sta for the FRET pair labeled aptamers were determined by use of these estimated distances between donor and acceptor (Fig. 2, eqn 1 and Table 1).
 | (5) |
| Neo | E sta [%] | r DA [Å] | R 0,sta [Å] | |
|---|---|---|---|---|
| N-N3C6 | − | 99.1 | 12.3 | 26.8 |
| + | 98.2 | 23.8 | ||
| N-N3C8 | − | 97.1 | 20.1 | 36.1 |
| + | 83.5 | 26.3 | ||
| N-N3C15 | − | 61.9 | 24.6 | 26.6 |
| + | 64.0 | 27.0 |
Overall, high static FRET efficiencies (Esta) were obtained for all samples (Table 1). The most drastic change of Esta upon ligand binding can be seen for N-N3C8 (14%). For N-N3C6 and N-N3C15, the FRET efficiencies change by only 1% and 2%, respectively. The absolute FRET efficiencies, as well as their change due to ligand binding, are strongly dependent on the distance and κ2 (see below). Since the largest flexibility is expected at position 8, it is reasonable that the actual distance between donor and acceptor for the aptamer without ligand is different from the modelled one. Moreover, the comparatively large Förster radius for N-N3C8 (− Neo) could also suggest a more favorable orientation between donor and acceptor for the energy transfer (eqn (1) and (2)).
Time-resolved FRET characterization
In conjunction with the static experiments, it is essential to characterize the energy transfer by time-resolved fluorescence measurements. Time-resolved FRET efficiencies (Etrf) can be obtained by measuring the respective amplitude weighted averaged fluorescence lifetimes τD and τDA (eqn (6), Table 2).30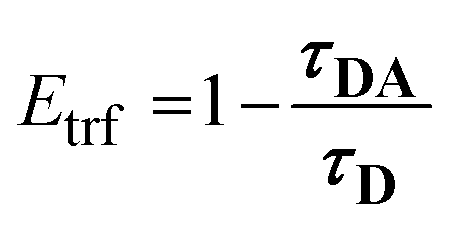 | (6) |
| Neo | τ D [ns]42 | τ DA [ns]a | E trf [%] | R 0,trf [Å] | |
|---|---|---|---|---|---|
| a Amplitude weighted averaged fluorescence lifetime for the time-resolved fluorescence signals from Fig. 4. | |||||
| N-N3C6 | − | 4.06 | 0.02 | 99.5 | 29.2 |
| + | 4.97 | 0.12 | 97.5 | 22.6 | |
| N-N3C8 | − | 4.14 | 0.30 | 92.8 | 30.7 |
| + | 3.73 | 1.09 | 70.7 | 23.3 | |
| N-N3C15 | − | 4.35 | 1.42 | 67.3 | 27.7 |
| + | 5.96 | 1.64 | 72.6 | 28.9 | |
Time-correlated single photon counting (TCSPC) and Etrf
We performed TCSPC measurements to monitor the fluorescence decay of the doubly labeled aptamers and to determine the corresponding fluorescence lifetimes.Similarly to the static fluorescence measurements, the time-resolved fluorescence data (Fig. 4) show a faster decay of the fluorescent state compared to the singly labeled aptamers.42
 | ||
| Fig. 4 Time-resolved fluorescence signals for N-N3C6, N-N3C8 and N-N3C15 (−/+ Neo). Data points are shown as dots, the fits are shown as solid lines. | ||
Fitting the data with two respectively three exponential functions results in the averaged fluorescence lifetimes shown in Table 2 (τD, τDA, for further information see ESI,† Table S2).42 It should be noted that the fast decays from N-N3C6 (−/+ Neo) and N-N3C8 (− Neo) were deconvoluted from the IRF (Instrument response function) of the used spectrometer.
Using the averaged fluorescence lifetimes of the donor in presence of the acceptor (τDA) and the donor alone (τD),42 the time-resolved FRET efficiencies Etrf can be derived mathematically using eqn (6) (Table 2).30,47 The efficiencies from the static measurements (Esta, Table 1) are in good agreement with those from the time-resolved measurements (Etrf), within an error of roughly 10%.
The energy transfer rate kFRET,sta(r) was subsequently calculated from the donor fluorescence lifetime τD and the static FRET efficiencies Esta, using eqn (1). The reciprocal of this transfer rate, τFRET,sta (in ns), indicates the time scale in which the energy transfer predominantly takes place. Likewise, the FRET efficiencies (Etrf), determined by TCSPC, yielded kFRET,trf(r) and τFRET,trf. The transfer-rates and -times from the static and the time-resolved measurements are in good agreement (Table 3).
| Neo | k FRET,sta(r) [ns−1]a | τFRET,sta [ns]b | k FRET,trf(r) [ns−1]a | τ FRET,trf [ns]b | |
|---|---|---|---|---|---|
| a Determined transfer rates for the energy transfer using eqn (1) and the FRET efficiencies Esta from Table 1 or Etrf from Table 2. b Reciprocal of kFRET,sta(r) and kFRET,trf(r) correspond to the FRET time given in ns. | |||||
| N-N3C6 | − | 27.06 | 0.04 | 45.63 | 0.02 |
| + | 10.94 | 0.09 | 7.85 | 0.13 | |
| N-N3C8 | − | 8.20 | 0.16 | 3.11 | 0.32 |
| + | 1.36 | 0.74 | 0.65 | 1.54 | |
| N-N3C15 | − | 0.37 | 2.68 | 0.47 | 2.11 |
| + | 0.30 | 3.35 | 0.44 | 2.25 | |
For high static FRET efficiencies (Esta), when the fluorescence of Çmf in the doubly labeled samples is strongly quenched, it is possible that the weak signals can be distorted by signals from impurities or detector noise. For high time-resolved FRET efficiencies (Etrf, determined by TCSPC), the fluorescence lifetimes were so short that they had to be deconvoluted from the IRF. The fact that FRET takes place in the picosecond time-range (e.g. for N-N3C6, Table 3), shows that both experimental methods are at their limits, although the derived FRET times are similar with both methods. The dependence of the FRET times on the distance between the FRET pairs is also evident. However, we decided to examine the energy transfer more closely with femtosecond transient absorption experiments in order to resolve the FRET process for the high FRET rates.
Ultrafast FRET dynamics
Ultrafast measurements were performed to characterize energy transfer processes that are at the edge of the time-resolution limit of the TCSPC setup. This is especially important for the N-N3C6 and N-N3C8 samples, both of which have high FRET efficiencies and thus ultrafast FRET dynamics. The transient absorption data (Fig. 5) were fitted by global lifetime analysis (GLA) to obtain the lifetimes of the observed states.48 | ||
| Fig. 5 Transient absorptions maps (left) and decay associated spectra (DAS) (right), top to bottom N-N3, N-N3C6, N-N3C8 and N-N3C15: left column without neomycin, right column with neomycin. τ2 for the decay of the directly excited tCnitro is shown in violet, τ3 for one emitting state of Çmf is shown in orange, τ4 for later emitting states is shown in red. For the samples N-N3 (−/+ Neo), N-N3C6 (−/+ Neo), and N-N3C8 (− Neo) an additional residual lifetime (inf.) is needed to fit the data properly (shown in brown). | ||
The aptamer that was singly labeled with tCnitro (N-N3) served as a control. tCnitro shows a ground state absorption in the range of 350 to 550 nm.41 Therefore, a fraction of the acceptor is always directly excited at the same time as Çmf, when irradiating at 388 nm. Upon excitation, tCnitro shows an excited state difference absorption signal (ESA1A) between 450 and 530 nm with a maximum at 505 nm and a second signal (ESA2A) above 600 nm. The directly excited tCnitro (−/+ Neo) decays with a lifetime of 5.1 and 3.8 ps, respectively. Due to the co-excitation, the dynamics of the directly excited acceptor are visible in all the measurements. The Çmf donor shows similar excited state dynamics as the free chromophore, with several emitting states that decay with different lifetimes (Fig. 5).43
Next to the transient absorption measurements (Fig. 5) are the corresponding decay associated spectra (DAS), along with the most importantant decay times. A positive signal for ESA1D, between 410 to 490 nm, partially overlaps the signal of the directly excited tCnitro. A second positive signal, between 590 and 650 nm, also overlaps the signal of the directly excited tCnitro (ESA2A/D).
For Çmf, a negative signal is expected between the two ESA bands at 490 to 590 nm, which is assigned to the stimulated emission difference signal (SED). Due to the high FRET efficiency for N-N3C6 (− Neo) and the simultaneous overlap by the dynamics of the directly excited acceptor (ESA1A & ESA2A), the SED signal only contributes with a small amplitude to this measurement. However, for the other measurements, the stimulated emission is clearly identifiable.
The decay of the excited state of Çmf in the FRET system follows an identical path for each sample; only the corresponding lifetimes are different (Table 4 and ESI,† Fig. S3). The decay occurs in a multi-exponential fashion. The smallest lifetime (τ1) corresponds to an internal relaxation process after excitation, which leads to the formation of several emitting states.43 The formation of the emitting states is described with τ3 and τ4, while the lifetime τ2 describes the decay of the directly excited tCnitro.
| Neo | τ 1 [ps] | τ 2 [ps] | τ 3 [ps] | τ 4 [ps] | |
|---|---|---|---|---|---|
| N-N3C6 | − | 0.5 | 3.1 | 14 | 380 |
| + | 0.4 | 5.6 | 52 | 290 | |
| N-N3C8 | − | 0.7 | 4.7 | 110 | 1300 |
| + | 0.3 | 3.9 | 160 | 1500 | |
| N-N3C15 | − | 0.3 | 5.2 | 840 | 3000 |
| + | 0.3 | 4.3 | 350 | 1800 |
Comparing the lifetimes of the emitting states of Çmf with kFRET,trf(r), and consequently τFRET,trf (Table 3), shows that the τFRET,trf value is either closer to the lifetime τ3 or τ4. It can thus be deduced that, depending on the efficiency, FRET tends to occur predominantly from one of the first or the later emerging emitting states of Çmf. However, it must be kept in mind that in all cases, energy transfer starts from the first emitting state, but since this state decays rapidly, the transfer progressively shifts towards a second state at lower efficiencies.
In the DAS of τ3 and τ4 of the individual transient absorption measurements (Fig. 5), a shoulder at 505 nm (ESA1A) can be observed. When tCnitro was directly excited, the excited state decayed within approximately 5 ps, indicating that the decay of tCnitro in the FRET system occurs after the energy transfer. This shoulder is more recognizable in the normalized DAS of the emitting states (ESI,† Fig. S4) further supporting a FRET process from several emitting states.
To further analyze the ultrafast measurements, the transients at 428 nm (ESA1D) and 505 nm (ESA1A) were plotted (Fig. 6). The transients at 428 nm correspond to the decay of the excited state of the Çmf. It is evident that the excited state of Çmf is longer-lived at relative greater DA distances and therefore at lower FRET efficiencies.
 | ||
| Fig. 6 Transients of the samples N-N3 (black), N-N3C6 (blue), N-N3C8 (yellow) and N-N3C15 (pink) without (− Neo) and with neomycin (+ Neo). 428 nm corresponds to the wavelength of the excited state of Çmf. 505 nm corresponds to the wavelength where tCnitro shows its strongest signal. | ||
The transients at 505 nm, associated with the decay of the excited state of tCnitro, also include the SED signal of Çmf. The transient of the directly excited tCnitro (− Neo) (black) is almost identical to the transient of N-N3C8 (− Neo) (yellow). This also applies to the transients of the directly excited tCnitro (+ Neo) and N-N3C6 (+ Neo) (blue). For these labeling positions, the FRET occurs after 50 ps, so the transient reflects the decay of the directly excited acceptor up to approximately 10 ps. The transient of N-N3C6 (− Neo) (blue) decays later than for N-N3 (− Neo), because of the superposition of the signal for both the directly excited tCnitro and the excited tCnitro after FRET, due to the high efficiency of the energy transfer. The transients of N-N3C8 (+ Neo) (yellow) and N-N3C15 (−/+ Neo) (pink) show a superposition of the positive signal of the directly excited tCnitro and the negative signal of the stimulated emission of Çmf. Thus, it appears that the signal of the directly excited acceptor decays faster. Taken together, the transients clearly elucidate the distance dependence of the energy transfer within the aptamer.
An averaged lifetime τDA(TAS) can also be derived from the transient absorption measurements, specifically using the amplitudes of the two DAS spectra (associated with τ3 and τ4) for the emitting states of Çmf. The respective lifetimes were weighted by percentage according to their amplitudes to obtain τDA(TAS) (Table 5 and Table S3, ESI†). Despite small deviations, the averaged lifetimes are in good agreement with those obtained by the TCSPC.
| Neo | τ DA(TAS) [ps] | E TAS [%] | E trf [%] | E sta [%] | |
|---|---|---|---|---|---|
| a Averaged fluorescence lifetime obtained using the amplitudes in the respective DAS of the two observed emitting states for Çmf (Fig. 5). | |||||
| N-N3C6 | − | 110 | 97.3 | 99.5 | 99.1 |
| + | 224 | 95.5 | 97.5 | 98.2 | |
| N-N3C8 | − | 411 | 90.1 | 92.8 | 97.1 |
| + | 1032 | 72.3 | 70.7 | 83.5 | |
| N-N3C15 | − | 1427 | 67.2 | 67.3 | 61.9 |
| + | 1475 | 75.3 | 72.6 | 64.0 | |
With τDA(TAS) (Table 5) and τD (Table 2) the FRET efficiency can now be derived from the transient absorption measurements (ETAS, Table 5). When comparing the FRET efficiencies derived from the three different methods, it can be seen that similar energy transfer efficiencies are obtained for all three.
Discussion of the label movement and orientation in the aptamer
The FRET efficiency (E) for the labeled aptamers can be experimentally determined, with steady state fluorescence, time-resolved fluorescence (TCSPC) or transient absorption experiments (Table 5). From eqn (1) and (2), it is obvious that E depends on the donor–acceptor distance (rDA) and the relative orientation of the transition dipole moments (κ2). Therefore, it is not possible to determine this relative orientation, without independent knowledge about the distance rDA and vice versa.38,49Nevertheless, the donor–acceptor distance rDA can be extracted with reasonable accuracy (see Table 6) from the NMR structure of the ligand-bound N1-aptamer.44 Thus the Förster radius R0 and the orientation factor κ2 can be calculated from E (Table 6; the time-resolved FRET efficiencies Etrf from Table 5 are used for the calculation).
| Neo | r DA [Å] | R 0,trf [Å] | κ 2 | |
|---|---|---|---|---|
| N-N3C6 | − | 12.3 | 29.2 | 0.40 |
| + | 22.6 | 0.07 | ||
| N-N3C8 | − | 20.1 | 30.7 | 0.49 |
| + | 23.3 | 0.06 | ||
| N-N3C15 | − | 24.6 | 27.7 | 0.29 |
| + | 28.9 | 0.36 |
The three-dimensional structure of N1 without the ligand is not available and, therefore, it is not possible to calculate κ2 from the distance rDA for the FRET pair in this case. However, using eqn (1) and (2) and keeping Etrf constant, the distance rDA can be plotted as a function of orientation κ2 (Fig. 7). These value pairs result in the corresponding FRET efficiency (here Etrf, Table 5). For the ligand-bound aptamer the data from Table 6 is marked in Fig. 7 with crosses.
 | ||
| Fig. 7 Dependence of the distance between donor and acceptor (rDA) on the orientation κ2 for a given FRET efficiency (Etrf), areas marked in grey indicate the region of value pairs between rDA and κ2. | ||
For N-N3C15, it is obvious that the resulting curves from the combinations between rDA and κ2 are almost identical in the absence and presence of the ligand (Fig. 7). This is in agreement with previous a EPR study in which the distance between positions 4 and 15 did not change upon ligand binding.50 This indicates that the small changes in FRET efficiency could be due to a slight change in the relative orientation between donor and acceptor in the ligand-bound state.
Assuming the same behavior for N-N3C6 and N-N3C8, the corresponding values for κ2 in the unbound state can be calculated (Table 6). Although the structure in the unbound state is unknown, the value pairs (rDA and κ2) and the FRET curves (Fig. 7) for both forms of the aptamer can be used to construct areas (gray regions in Fig. 7) that reflect the range of motion of the labels relative to each other. The flexibility of the RNA is accounted for by the construction of the gray areas. This flexibility and the resulting slight differences in κ2 lead to changes in the FRET efficiency. The gray areas take this into account, with the measured FRET efficiencies corresponding to the average FRET efficiencies in the ensemble.
Based on this analysis, neither distance nor orientation changes significantly for N-N3C15 upon ligand binding. For N-N3C6, a more significant change in both distance and orientation can be observed, which is in agreement with a previous NMR study.46 The greatest range of changes is seen for N-N3C8, which is to be expected, because the Çmf is located at the edge of the binding pocket where ligand binding should result the most pronounced movement. Therefore, our results are consistent with previous studies that have indicated that the N1-aptamer is already prefolded to a significant extent; the binding pocket conformation primarily adapts to the ligand, with little change in the rest of the aptamer.42
Conclusions
We have presented a detailed spectroscopic study of the energy transfer process between Çmf and tCnitro using experimental methods that can address a range of time-scales. The FRET efficiency was determined for FRET times from about 20 ps to 3 ns (Table 3). While methods using steady state fluorescence (Esta) and TCSPC (Etrf) are commonly used, the third method, transient absorption spectroscopy (ETAS), allowed determination of fast FRET processes (e.g.N-N3C6) with appropriate time-resolution; even small changes at very high FRET efficiencies (and therefore very small distances) can be determined, for which TCSPC is not sensitive enough.We were able to evaluate several emitting states for Çmf and showed that the resulting FRET efficiencies, from the three methods that we used, Esta, Etrf and ETAS are identical within a range of 10%. It should be noticed that for the transient absorption method it is not necessary for the donor to be a strong-emitting fluorophore with high quantum yield, as for the evaluation, exclusively the absorption data is used. A lower quantum yield results in lower FRET efficiency and also makes TCSPC experiments difficult to perform, because the method depends on emitted photons. In the case presented here, TAS can be used to study the ultrafast (excited state) dynamics of the donor and the change in dynamics due to energy transfer.This extends the toolbox for the experimentalist.
We have shown that the FRET pair Çmf and tCnitro is highly efficient whereby the fluorescence of the donor can be almost completely quenched. However, as with other pairs, it is not possible to determine the exact distance and orientation for this pair from the FRET efficiency E alone. Using a known NMR structure of the ligand-bound N1-aptamer, a distance can be approximated to determine κ2 and, therefore, make qualitative statements about the change in orientation after ligand binding.
It is clear that the normal approximation of κ2 having the value of 2/3 is insufficient when rigid labels are incorporated. A distinct advantage of rigid labels is the possibility to reveal the dynamic binding behaviour of aptamers.
This study also highlights the importance of the photophysical properties of both the donor and the acceptor in a FRET pair. Ideally, the acceptor should fluoresce spectrally separately from the donor. This would enable measurements of the angle between donor and acceptor using anisotropy measurements.51 With the procedure described here, in particular using transient absorption experiments, the determination of the angle between donor and acceptor is also possible without fluorescence. This requires a different acceptor than tCnitro, whose excited state lives longer and is spectrally separated from the signal of the excited state of the donor.
Author contributions
Florian Hurter conducted all experiments, data analysis and interpretation and wrote the manuscript. Anna-Lena J. Halbritter and Iram M. Ahmad prepared the labeled RNA aptamers. Markus Braun helped with data interpretation. All experiments were performed under the supervision of Snorri Th. Sigurdsson and Josef Wachtveitl. All authors read and agreed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. J. Wöhnert for access to his ITC and J. Vögele for the help during the measurements. This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG) through the Collaborative Research Center (CRC) 902; ‘Molecular Principles of RNA-based Regulation’ sub-project A7.References
- R. R. Breaker and G. F. Joyce, Chem. Biol., 2014, 21, 1059–1065 CrossRef CAS PubMed.
- N. Pavlova, D. Kaloudas and R. Penchovsky, Gene, 2019, 708, 38–48 CrossRef CAS PubMed.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 1–14 CAS.
- R. J. Trachman, A. Autour, S. C. Y. Jeng, A. Abdolahzadeh, A. Andreoni, R. Cojocaru, R. Garipov, E. V. Dolgosheina, J. R. Knutson, M. Ryckelynck, P. J. Unrau and A. R. Ferré-D’Amaré, Nat. Chem. Biol., 2019, 15, 472–479 CrossRef PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- M. Etzel and M. Mörl, Biochemistry, 2017, 56, 1181–1198 CrossRef CAS PubMed.
- F. Groher and B. Suess, Biochim. Biophys. Acta, Gene Regul. Mech., 2014, 1839, 964–973 CrossRef CAS PubMed.
- A.-S. V. Bédard, E. D. M. Hien and D. A. Lafontaine, Biochim. Biophys. Acta, Gene Regul. Mech., 1863, 2020, 194501 Search PubMed.
- A. V. Sherwood and T. M. Henkin, Annu. Rev. Microbiol., 2016, 70, 361–374 CrossRef CAS.
- R. D. Jenison, S. C. Gill, A. Pardi and B. Polisky, Science, 1994, 263, 1425–1429 CrossRef CAS PubMed.
- S. C. B. Gopinath, Anal. Bioanal. Chem., 2006, 387, 171–182 CrossRef PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- Y. Zhang, B. S. Lai and M. Juhas, Molecules, 2019, 24, 941 CrossRef PubMed.
- P. Kumar Kulabhusan, B. Hussain and M. Yüce, Pharmaceutics, 2020, 12, 646 CrossRef PubMed.
- A. K. Deisingh, in RNA Towards Medicine, ed. V. Erdmann, J. Barciszewski and J. Brosius, Springer, 2006 Search PubMed.
- W. Zhou, P.-J. J. Huang, J. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- S. Ni, Z. Zhuo, Y. Pan, Y. Yu, F. Li, J. Liu, L. Wang, X. Wu, D. Li, Y. Wan, L. Zhang, Z. Yang, B.-T. Zhang, A. Lu and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 9500–9519 CrossRef CAS PubMed.
- H. Kaur, J. G. Bruno, A. Kumar and T. K. Sharma, Theranostics, 2018, 8, 4016–4032 CrossRef CAS PubMed.
- K.-N. Kang and Y.-S. Lee, in Future Trends in Biotechnology, ed. J.-J. Zhong, Springer, Berlin, Heidelberg, 2013, pp. 153–169 Search PubMed.
- C. Berens, F. Groher and B. Suess, Biotechnol. J., 2015, 10, 246–257 CrossRef CAS PubMed.
- A. Wittmann and B. Suess, FEBS Lett., 2012, 586, 2076–2083 CrossRef CAS PubMed.
- J. E. Weigand, S. R. Schmidtke, T. J. Will, E. Duchardt-Ferner, C. Hammann, J. Wöhnert and B. Suess, Nucleic Acids Res., 2011, 39, 3363–3372 CrossRef CAS PubMed.
- J. Hoetzel and B. Suess, J. Mol. Biol., 2022, 434, 167631 CrossRef CAS PubMed.
- C. Berens and B. Suess, Curr. Opin. Biotechnol, 2015, 31, 10–15 CrossRef CAS PubMed.
- J. E. Weigand, M. Sanchez, E.-B. Gunnesch, S. Zeiher, R. Schroeder and B. Suess, RNA, 2008, 14, 89–97 CrossRef CAS PubMed.
- P. St-Pierre, K. McCluskey, E. Shaw, J. C. Penedo and D. A. Lafontaine, Biochim. Biophys. Acta, Gene Regul. Mech., 2014, 1839, 1005–1019 CrossRef CAS PubMed.
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Frontiers in Chemistry Search PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- P. G. Wu and L. Brand, Anal. Biochem., 1994, 218, 1–13 CrossRef CAS PubMed.
- A. Y. Kobitski, M. Hengesbach, M. Helm and G. U. Nienhaus, Angew. Chem., Int. Ed., 2008, 47, 4326–4330 CrossRef CAS PubMed.
- H. Li, S. Cao, S. Zhang, J. Chen, J. Xu and J. R. Knutson, Phys. Chem. Chem. Phys., 2023, 25, 7239–7250 RSC.
- S. S. Vogel, B. W. Van der Meer and P. S. Blank, Methods, 2014, 66, 131–138 CrossRef CAS PubMed.
- S. Ranjit, K. Gurunathan and M. Levitus, J. Phys. Chem. B, 2009, 113, 7861–7866 CrossRef CAS PubMed.
- L. Loura, Int. J. Mol. Sci., 2012, 13, 15252–15270 CrossRef CAS PubMed.
- S. S. Vogel, T. A. Nguyen, B. W. van der Meer and P. S. Blank, PLOS ONE Search PubMed.
- C. Steinmetzger, C. Bäuerlein and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 6760–6764 CrossRef CAS PubMed.
- S. C. Y. Jeng, R. J. Trachman, F. Weissenboeck, L. Truong, K. A. Link, M. D. E. Jepsen, J. R. Knutson, E. S. Andersen, A. R. Ferré-D’Amaré and P. J. Unrau, RNA, 2021, 27, 433–444 CrossRef CAS PubMed.
- A. F. Füchtbauer, M. S. Wranne, M. Bood, E. Weis, P. Pfeiffer, J. R. Nilsson, A. Dahlén, M. Grøtli and L. M. Wilhelmsson, Nucleic Acids Res., 2019, 47, 9990–9997 CrossRef PubMed.
- K. Börjesson, S. Preus, A. H. El-Sagheer, T. Brown, B. Albinsson and L. M. Wilhelmsson, J. Am. Chem. Soc., 2009, 131, 4288–4293 CrossRef PubMed.
- H. Gustmann, A.-L. J. Segler, D. B. Gophane, A. J. Reuss, C. Grünewald, M. Braun, J. E. Weigand, S. T. Sigurdsson and J. Wachtveitl, Nucleic Acids Res., 2019, 47, 15–28 CrossRef CAS PubMed.
- H. Gustmann, D. Lefrancois, A. J. Reuss, D. B. Gophane, M. Braun, A. Dreuw, S. T. Sigurdsson and J. Wachtveitl, Phys. Chem. Chem. Phys., 2017, 19, 26255–26264 RSC.
- E. Duchardt-Ferner, J. E. Weigand, O. Ohlenschläger, S. R. Schmidtke, B. Suess and J. Wöhnert, Angew. Chem., Int. Ed., 2010, 49, 6216–6219 CrossRef CAS PubMed.
- S. Stampfl, A. Lempradl, G. Koehler and R. Schroeder, ChemBioChem, 2007, 8, 1137–1145 CrossRef CAS PubMed.
- E. Duchardt-Ferner, S. R. Gottstein-Schmidtke, J. E. Weigand, O. Ohlenschläger, J.-P. Wurm, C. Hammann, B. Suess and J. Wöhnert, Angew. Chem., Int. Ed., 2016, 55, 1527–1530 CrossRef CAS PubMed.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CrossRef CAS.
- C. Slavov, H. Hartmann and J. Wachtveitl, Anal. Chem., 2015, 87, 2328–2336 CrossRef CAS PubMed.
- M. Khrenova, I. Topol, J. Collins and A. Nemukhin, Biophys. J., 2015, 108, 126–132 CrossRef CAS PubMed.
- I. Krstić, O. Frolow, D. Sezer, B. Endeward, J. E. Weigand, B. Suess, J. W. Engels and T. F. Prisner, J. Am. Chem. Soc., 2010, 132, 1454–1455 CrossRef PubMed.
- D. Laskaratou, G. S. Fernández, Q. Coucke, E. Fron, S. Rocha, J. Hofkens, J. Hendrix and H. Mizuno, Nat. Commun., 2021, 12, 1–12 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, Fig. S1–S4 and Tables S1–S3. See DOI: https://doi.org/10.1039/d3cp05728c |
| This journal is © the Owner Societies 2024 |