
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the photophysical properties of naphthalimide derivatives using ultrafast spectroscopy†
Wei
Zhang
 a,
Yalei
Ma
b,
Hongwei
Song
c,
Rong
Miao
*b,
Jie
Kong
*a and
Meng
Zhou
*a
a,
Yalei
Ma
b,
Hongwei
Song
c,
Rong
Miao
*b,
Jie
Kong
*a and
Meng
Zhou
*a
aHefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Anhui 230026, China. E-mail: kongjie2021@ustc.edu.cn; mzhou88@ustc.edu.cn
bLaboratory of Applied Surface and Colloids Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, P. R. China. E-mail: miaorong2015@snnu.edu.cn
cDepartment of Chemistry–Angstrom Laboratory, Uppsala University, Box 523, Uppsala 75120, Sweden
First published on 4th January 2024
Abstract
Naphthalimide derivatives composed of donor–acceptor type structures hold significant promise across a wide range of applications. Here, the solvent polarity and viscosity controlled excited-state dynamics of a naphthalimide derivative with a donor–acceptor structure were studied using multiple spectroscopic techniques. From the stationary spectroscopic investigations, large Stokes shift and low fluorescence quantum yield were observed with increasing the solvent polarity, suggesting a more polar excited state relative to the ground state, which is evidenced by the Lippert–Mataga relationship. We also observe an enhanced fluorescence with a prolonged lifetime in a more viscous solution due to the restriction of excited-state molecular rearrangement. These observations result from the emerged twisted intramolecular charge transfer (TICT) state. The ultrafast spectroscopy studies further unravel a solvent polarity dependent excited state evolution from the intramolecular charge transfer state to the TICT state, revealing that the TICT state can be populated only in strong polar solvents. Control experiments by tuning the solvent viscosity in ultrafast experiments were employed to verify the excited state molecular rearrangement subsequently. These observations collectively emphasize how fine-tuning the photophysical properties of naphthalimide derivatives can be achieved through strategic manipulation of solvent polarity and viscosity.
Introduction
Organic functional molecules that consist of electron-donor (D) and -acceptor (A) moieties have attracted wide attention in the past few decades because of their potential applications in organic light emitting diodes, aggregation induced emission materials, organic photovoltaics, fluorescent probes,1–11etc. Normally, the molecules consisting of D and A subunits exhibit intramolecular charge transfer (ICT) characteristics, which can be easily modulated by the environmental polarity, viscosity, and D/A variations.12–14 The pump-initiated excited-state conformation relaxes to a new state characterized by the conformational change entailing twisted or planar intramolecular charge transfer (TICT/PICT) when there is an intramolecular rotation.14–23 The excited-state conformational change can thus in turn determine the unique photophysical and photochemical properties.24Naphthalimide is considered as one of the most important building blocks for efficient fluorescent materials owing to its unique photophysical properties, including high electron affinity, oxidation stability, good thermal stability, and biocompatibility,7,14,25–30 which have been explored extensively. These advantages make naphthalimide derivatives good candidates for biosensors, fluorescent cell imaging agents, and optoelectronic devices. Besides, the naphthalimide core is an electron acceptor in nature with redox potential of −1.8 V (vs. Ag/Ag+),31 and many of the above-mentioned applications cover the functionalization of naphthalimide with different electron-donor moieties to have the ICT character with marked solvatochromic effect.14,30,32–34 Thus, the local environment, such as solvent polarity and viscosity, plays a crucial role in modulating the excited-state behavior of such D–A type naphthalimide derivatives because a significant dipole moment enhancement in the excited state is obtained upon photon excitation.35–37 Previous research by Kubinyi et al. proposed a two state model, corresponding to the ICT and TICT, to describe the excited state dynamics of naphthalimide derivatives by applying theoretical calculations, stationary and time-resolved fluorescence spectra, whereas direct visual evidence of the conversion from ICT to TICT is absent.32 Therefore, investigating the photophysical properties of the long-term studied naphthalimide derivatives will be conducive to designing high-performance molecules with ICT character.
In this work, we report a comprehensive investigation of a naphthalimide derivative, 2-(2-ethylhexyl)-6-(1H-pyrrol-2-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (HP-NAP, the molecular structure is shown in Scheme 1),8 that is composed of electron-acceptor naphthalimide and electron-donor pyrrole. We found that the Stokes shifts and fluorescence quantum yields of HP-NAP are sensitive to the environmental polarity, and viscosity-dependent measurements suggest that restricting molecular rotation is beneficial for obtaining stronger fluorescence intensity and longer lifetime. Ultrafast spectroscopy provides the main insights and allows visualization of the dynamic process of ICT converting to the TICT state, which is governed by the environmental polarity and viscosity. These results are important for deeply understanding the photophysical properties of naphthalimide derivatives.
 | ||
| Scheme 1 The molecular structure of HP-NAP. | ||
Materials and methods
Materials
The synthesis and characterization of HP-NAP were reported previously.8 All solvents used in this work, including hexane (Hex), dichloromethane (CH2Cl2), tetrahydrofuran (THF), acetonitrile (ACN), and cyclohexane (CHX), were purchased from Sinopharm Chemical Reagent Co., Ltd, and all solvents were used without further purification. 9,10-Diphenylanthracene (9,10-DPA) and polymethyl methacrylate (PMMA) were purchased from Sinopharm Chemical Reagent Co., Ltd and Shanghai Macklin Biochemical Technology Co., Ltd, respectively.Quantum chemical calculations
Theoretical calculations were performed based on density functional theory (DFT) and time-dependent DFT (TD-DFT) by employing the Gaussian 16, Revision A.03 package.38 Optimization of the ground state geometry was obtained using the B3LYP39 functional at the basis set 6-311G** level. The alkyl chain on the N-atom is reduced to a methyl group for calculation simplicity.Stationary spectroscopy
The steady-state spectra (including UV-vis absorption and fluorescence spectra) were collected using Agilent Cary 60 UV-Vis and Agilent Cary Eclipse fluorescence spectrometers, respectively. The fluorescence quantum yields were measured using a comparative method, where the reference is a solution of 9,10-DPA in CHX solvent (Φf = 0.955, excited at 366 nm).40 The stationary data was obtained with sample concentrations of 10−5 M.Fluorescence lifetime and ultrafast spectroscopy measurements
The fluorescence lifetime was acquired on a time-correlated single photon counting spectrometer (FluoTime 300, PicoQuant, Germany) under 375 nm picosecond laser (PDL 820, PicoQuant diode laser) excitation.The femtosecond transient absorption (fs-TA) spectroscopy measurements were performed on a home-built pump–probe set-up. The laser pulse (800 nm, 35 fs pulse width, 1 kHz repetition rate) was generated by a regeneratively amplified Ti:sapphire laser (Coherent Astrella-Tunalbe-USP, USA). The output of the pulse is divided into two beams with a beam splitter. The 400 nm pump beam was produced by doubling the 800 nm pulse with a beta barium borate crystal (type I, 0.5 mm thickness); the power of the pump pulse was set as 0.1 μJ. The probe pulse was delayed by a computer-controlled optical delay line and then focused on a thin sapphire plate to generate the white light supercontinuum, which split into two beams by using a broadband 50/50 beam splitter as the signal and reference beams (450–800 nm). The focused pump and probe pulses overlapped into a sample cuvette. The mutual polarization between the pump and probe pulses was set to 54.7° by placing a half-wave plate in the pump beam. The solution concentrations were set to around 10−5 M, and there was no photodegradation after fs-TA experiments by checking the stationary absorption spectra.
Results and discussion
Optimized ground state (S0) geometry and charge density distribution on frontier molecular orbitals (FMOs) of HP-NAP are depicted in Fig. 1a–c. The highest occupied molecular orbital (HOMO) comprises the charge distribution between the naphthalimide and pyrrole moieties, while the charge density on the lowest unoccupied molecular orbital (LUMO) is mainly localized on the naphthalimide core, making the vertical transition a local excited (LE) dominated character. More discussion about the theoretically calculated excited state (S1) of HP-NAP, as well as a short comment, can be found in the S1 part of the ESI.† | ||
| Fig. 1 (a) The optimized geometry of HP-NAP at the S0 state. The (b) HOMO and (c) LUMO of HP-NAP at the optimized S0 geometry. (d) The normalized stationary absorption and fluorescence spectra of HP-NAP in different solvents. (e) Stokes shifts of HP-NAP relative to the solvent orientation polarizability. (f) The fluorescence decay profiles of HP-NAP in different solvents. | ||
Fig. 1d shows the stationary absorption and fluorescence spectra of HP-NAP in different polar solvents. These solvent polarity-independent absorption bands with well-resolved vibronic peaks at ∼345 nm can be ascribed to the LE transition. In terms of the lower-energy absorption bands centered at ∼400 nm, the marginal solvatochromic shifts are observed, accounting for the LE dominated vertical transition from S0 to S1. This is also evidenced by the shoulder peak at ∼400 nm in Hex that collaborates with the absorption bands in THF and ACN. In contrast, the fluorescence spectra show a significant red shift with an enhancement of dielectric environment, where we observe the structured band in Hex evolve into broader and Gaussian-like bands in THF and ACN. The dynamic evolution of absorption and fluorescence spectra of HP-NAP suggests the increase of the dipole moment under photon excitation, accounting for a pump-initiated S1 state with LE character, while the relaxed S1 state with LE character in Hex evolves into the ICT character in polar solvents.
To further clarify the ICT character caused by the dipole moment difference between the S0 and S1 states (Δμge), the Lippert–Mataga equation41–44 is applied to evaluate the Δμge by plotting the Stokes shift against the solvent orientation polarizability (Δf) as shown in Fig. 1e, the detailed data are provided in Table S1 (ESI†). The relationship between Stokes shift and Δf is as follows,
 | (1) |
 | (2) |
The fluorescence quantum yields and lifetime (Fig. 1f) were also measured, and the radiative and nonradiative rate constants were calculated by the following equations:
| kr = Φ/τ | (3) |
| 1/τ = kr + knr | (4) |
| Solvent | Abs/nm | Em/nm | Stokes shift/cm−1 | τ/ns | Φ | k r/ns−1 | k nr/ns−1 |
|---|---|---|---|---|---|---|---|
| Hex | 395 | 460 | 3577 | 3.36 | 1.00 | 0.30 | 0.00 |
| THF | 419 | 528 | 4927 | 6.11 | 0.54 | 0.09 | 0.08 |
| ACN | 412 | 550 | 6090 | 2.69 | 0.18 | 0.07 | 0.31 |
The viscosity-dependent measurements of HP-NAP are shown in Fig. 2, where the different mass concentrations of PMMA are used to provide a conformational change limiting environment. It is found that the increased concentration of PMMA results in fluorescence enhancement (Fig. 2a), and one can observe a gradual blue-shift of fluorescence spectra (Fig. S2a, ESI†). To show a clearer presentation of the PMMA concentration controlled blueshift of the fluorescence spectra, the differential spectrum between the normalized fluorescence spectra with PMMA concentration of 150 mg mL−1 and of 0 mg mL−1 is provided in Fig. S2b (ESI†), where the positive signal at the blue side (∼ 515 nm) clearly shows the blueshift of the fluorescence spectrum at a PMMA concentration of 150 mg mL−1 relative to that of 0 mg mL−1. In addition, it is also noted that the increase of the viscosity can boost the fluorescence lifetime (Fig. 2b and Table S2, ESI†). These results should be considered as an outcome of restricting the nonradiative decay channel that enhances the fluorescence, while restricting the excited-state bond rotation will blue-shift the fluorescence.
 | ||
| Fig. 2 The viscosity-dependent measurements of HP-NAP in the ACN solvent dissolved with different mass concentrations of PMMA. The viscosity-dependent (a) absorption and fluorescence spectra and (b) fluorescence decay profiles of HP-NAP. | ||
So far, it is evident that the excited state relaxation pathways of HP-NAP are strongly dependent on the environment polarity and viscosity. The efficient nonradiative decay channel and pronounced decrease of fluorescence quantum yield upon enhancement of solvent polarity correlate well with the relatively stronger excited-state dipole moment μe. The longer fluorescence lifetime, and stronger and blue-shifted fluorescence with increasing solvent viscosity result from the suppression of excited-state conformation change process. To gain a deeper understanding of the excited state dynamics of HP-NAP, we then performed fs-TA spectroscopy under different dielectric constant conditions.
The obtained fs-TA data (Fig. 3) in different polar solvents illustrate the competition between the electronic excited states upon photon excitation at the lowest absorption band (400 nm). The initial recorded spectra of HP-NAP in nonpolar solvent Hex consist of a negative peak centered at 450 nm and a broad positive band across from 475 nm to 750 nm with several absorption peaks (Fig. 3(a) and (b)). The former negative peak can be ascribed to the stimulated emission (SE) by comparing it with the stationary fluorescence spectrum in Hex (Fig. 1d), confirming that the pump-populated state is a LE state; the latter positive peaks are the excited state absorption (ESA) from S1 to the higher singlet excited states. No pronounced dielectric-controlled dynamics is observed during the detection window. In the medium polar THF, the dynamic spectral evolution is different. The initial fs-TA spectrum of HP-NAP in THF is different from that in Hex, as the initial SE signal in THF is located at ∼525 nm, which can be ascribed to the ICT signal compared to the stationary fluorescence spectrum in Fig. 1d. This is because of the strong solvation effect making a quick transformation from LE to ICT, which cannot be captured in our fs-TA set-up. As the time delay continues, the band at 525 nm shows a slight red shift and intensity decrease, which is accompanied by the generation of a new SE band at 625 nm. This spectral evolution is attributed to the excited-state conversion from the ICT to TICT state. However, we should highlight that the SE intensity at 525 nm is stronger than the newly formed SE signal at 625 nm from the initial time to the end time. The spectral evolution in ACN shows a faster TICT generation than in THF because a stronger dielectric environment accelerates the conversion from ICT to TICT, and the ICT SE signal disappears within ∼1 ps (Fig. 3e).
 | ||
| Fig. 3 The fs-TA maps (up panel) and spectra (low panel) of HP-NAP in different polar solvents. | ||
To reveal the relaxation mechanism, the global analysis45 was conducted to extract the transient species. Fig. 4 shows the evolution-associated spectra (EAS) and the representative kinetics at the characteristic wavelengths, and the detailed fitting data is displayed in Table S3 (ESI†). It is obvious that there is no new transient species generated in Hex, and two EAS are obtained by global analysis (Fig. 4a), corresponding to LE and vibrational cooling LE (LE’). Being in such a regime, the vibrational cooling process of excited state species toward ambient temperature leads to band narrowing (Fig. S3a, ESI†),46,47i.e., the band narrowing process is due to the vibrational cooling (Fig. S3b, ESI†). For HP-NAP in THF, the first transient component (EAS1) extracted from the fs-TA data is the ICT state, whose spectral profile is similar to the initial spectral profile from fs-TA data (Fig. 3d). While the second transient component contains two SE signals which belong to the ICT and TICT states, thus we assign the EAS2 to the equilibrium state between the ICT and TICT. This is also confirmed by the kinetic comparison in Fig. 4d, which shows that though the kinetic traces at 625 nm exhibit the generation of the TICT state, there is no obvious intensity decrease of the ICT state even at 300 ps. Then we turn our attention to HP-NAP in ACN, and there is an isosbestic point located at ∼560 nm (Fig. 3f), suggesting the state-state conversion process and that the fs-TA can be well decomposed into two species (Fig. 4e), corresponding to the ICT and TICT states. The kinetic traces shown in Fig. 4f disclose that the decay of the ICT state (505 nm) generates the TICT state (605 nm) rather than the S0 state. One could argue that the TICT signals in THF (625 nm) and ACN (605 nm) are not at the same position because the ICT signal in THF (525 nm) is always much stronger than the TICT signal, which shifts the TICT signal to a red position. Compared with previous literature,14 the equilibrium state between the ICT and TICT of the naphthalimide derivative is firstly reported in our work.
 | ||
| Fig. 4 The global analysis results (up panel) and representative kinetics (low panel) of HP-NAP in different polar solvents. | ||
The observed changes in fs-TA spectra in ACN are attributed to the population conversion from ICT to TICT accompanied by the excited-state molecular rearrangement from a quasi-planar structure to a perpendicular conformation as we discussed above. To verify the excited state molecular rearrangement, we conducted the viscosity-dependent fs-TA measurements of HP-NAP (Fig. 5). At the initial time delay of 0.1 ps, the SE signals that belong to the ICT state are almost the same for all samples; while we observe gradual blue shifted SE signals at a time delay of 10 ps (Fig. 5b) with an increase in the solvent viscosity, which is due to the limited molecular rearrangement environment. The TA differential spectrum at a time delay of 10 ps between the spectra with PMMA concentration of 150 mg mL−1 and PMMA concentration of 0 mg mL−1 is also provided in Fig. S4 (ESI†), the negative signal at 520 nm (at the SE region) confirms the blueshift of the SE signal upon increasing the solvent viscosity. In addition, the kinetic trace comparison at 600 nm (Fig. 5c) shows a slower generation and slower relaxation of the TICT state with 150 mg mL−1 PMMA in ACN, further verifying that the high viscosity environment can limit the excited state molecular rotation, thus slowing the generation and deactivation of TICT. The detailed fitting time constants from viscosity-dependent fs-TA spectra can be found in Table S4 (ESI†).
 | ||
| Fig. 5 The viscosity-dependent fs-TA spectra of HP-NAP in ACN solvents that dissolved different mass concentrations of PMMA. The normalized fs-TA spectra at a time delay of (a) 0.1 ps and (b) 10 ps, and (c) the normalized viscosity-dependent kinetics comparison at 600 nm. | ||
Based on the current results, the fs-TA measurements in Hex represent the excitation relaxation from the LE state with quasi-planar geometry, while the result from medium polar THF show a competition between the ICT and TICT state, and finally, the fs-TA data in ACN corresponds to the TICT dominated relaxation. The results from the fs-TA spectra nicely match the earlier discussion obtained from the solvent polarity- and viscosity-dependent stationary absorption and fluorescence spectra, as well as the time-resolved fluorescence experiments. As seen from Scheme 2, we provide the schematic diagram of the solvent polarity controlled excited-state relaxation pathways of HP-NAP, which presents that the dynamic solvation determines whether the TICT generates, and more polar solvent induces more pronounced fluorescence quenching.
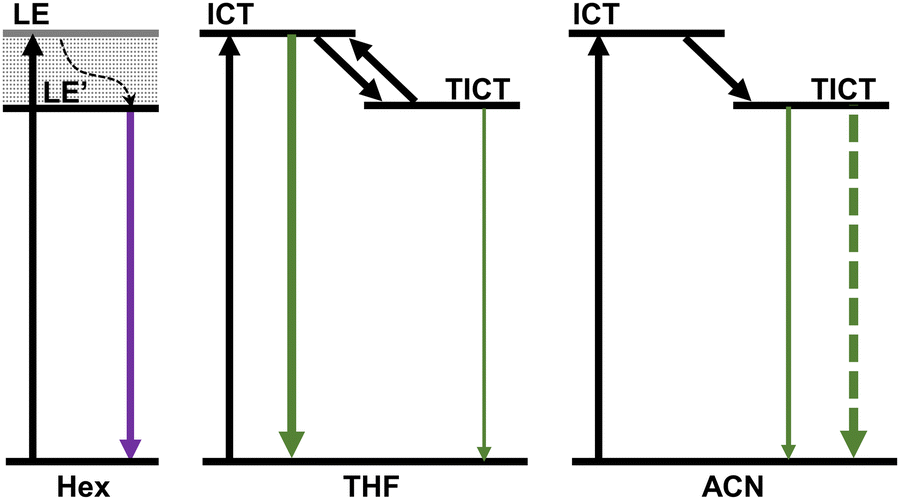 | ||
| Scheme 2 The schematic diagram of the solvent polarity controlled excited-state relaxation pathways of HP-NAP. | ||
Conclusions
In summary, the photophysical properties of naphthalimide derivative HP-NAP were thoroughly investigated by employing solvent polarity- and viscosity-dependent stationary and ultrafast spectroscopies. HP-NAP shows LE character in nonpolar Hex and a high fluorescence quantum yield (∼100%) is achieved. However, an increase in solvent polarity gives rise to the formation of the TICT state, and efficient nonradiative pathways are enabled, which strongly quenched the fluorescence. The fs-TA spectra confirm the results from stationary measurements and reveal the detailed excited-state evolution pathways: the vibrational cooling process from the pump-initiated LE state in Hex, the competition of ICT and TICT in THF, and the TICT-oriented relaxation in ACN. Furthermore, the viscosity-controlled experiments of similar polarity with ACN show enhancement of fluorescence as viscosity increases, as well as a blue-shifted SE signal and slower generation of the TICT state, confirming the excited state molecular rotation process. These results are helpful for understanding the photophysics of naphthalimide derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSFC (22203085) and Chinese Academy of Sciences (YSBR-007).References
- J. Kong, W. Zhang, X. Zhang, B. Liu, Y. Li and A. Xia, J. Chem. Phys., 2022, 156, 174902 CrossRef CAS PubMed.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- Y.-K. Qu, Q. Zheng, J. Fan, L.-S. Liao and Z.-Q. Jiang, Acc. Mater. Res., 2021, 2, 1261–1271 CrossRef CAS.
- M. Mamada, H. Katagiri, C.-Y. Chan, Y.-T. Lee, K. Goushi, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Funct. Mater., 2022, 32, 2204352 CrossRef CAS.
- W. Zhang, L. Zhao, B. W. Laursen and J. Chen, Phys. Chem. Chem. Phys., 2022, 24, 26731–26737 RSC.
- H.-Q. Dong, T.-B. Wei, X.-Q. Ma, Q.-Y. Yang, Y.-F. Zhang, Y.-J. Sun, B.-B. Shi, H. Yao, Y.-M. Zhang and Q. Lin, J. Mater. Chem. C, 2020, 8, 13501–13529 RSC.
- R. Miao, J. Li, C. Wang, X. Jiang, Y. Gao, X. Liu, D. Wang, X. Li, X. Liu and Y. Fang, Adv. Sci., 2022, 9, 2104609 CrossRef CAS PubMed.
- G. Yang, Y. Ran, Y. Wu, M. Chen, Z. Bin and J. You, Aggregate, 2022, 3, e127 CrossRef CAS.
- N. Masimukku, D. Gudeika, D. Volyniuk, O. Bezvikonnyi, J. Simokaitiene, V. Matulis, D. Lyakhov, V. Azovskyi and J. V. Gražulevičius, Phys. Chem. Chem. Phys., 2022, 24, 5070–5082 RSC.
- C. Rizzo, P. Cancemi, L. Mattiello, S. Marullo and F. D’Anna, ACS Appl. Mater. Interfaces, 2020, 12, 48442–48457 CrossRef CAS PubMed.
- W. Zhang, J. Kong, R. Z. An, J. Zhang, Y. Zhou, L.-S. Cui and M. Zhou, Aggregate, 2023, e416 CrossRef.
- X. Niu, P. Gautam, Z. Kuang, C. P. Yu, Y. Guo, H. Song, Q. Guo, J. M. W. Chan and A. Xia, Phys. Chem. Chem. Phys., 2019, 21, 17323–17331 RSC.
- J. Jovaišaitė, P. Baronas, G. Jonusauskas, D. Gudeika, A. Gruodis, J. V. Gražulevičius and S. Juršėnas, Phys. Chem. Chem. Phys., 2023, 25, 2411–2419 RSC.
- K. Hanaoka, S. Iwaki, K. Yagi, T. Myochin, T. Ikeno, H. Ohno, E. Sasaki, T. Komatsu, T. Ueno, M. Uchigashima, T. Mikuni, K. Tainaka, S. Tahara, S. Takeuchi, T. Tahara, M. Uchiyama, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2022, 144, 19778–19790 CrossRef CAS PubMed.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656–12678 RSC.
- R. Grotjahn, J. Chem. Phys., 2023, 159, 174102 CrossRef CAS PubMed.
- A. Iagatti, B. Patrizi, A. Basagni, A. Marcelli, A. Alessi, S. Zanardi, R. Fusco, M. Salvalaggio, L. Bussotti and P. Foggi, Phys. Chem. Chem. Phys., 2017, 19, 13604–13613 RSC.
- A. Barak, N. Dhiman, F. Sturm, F. Rauch, Y. A. Lakshmanna, K. S. Findlay, A. Beeby, T. B. Marder and S. Umapathy, J. Phys. Chem. C, 2023, 127, 5855–5865 CrossRef CAS.
- I. Knysh, I. Duchemin, X. Blase and D. Jacquemin, J. Chem. Phys., 2022, 157, 194102 CrossRef CAS PubMed.
- J. Dobkowski, J. Wójcik, W. Koźmiński, R. Kołos, J. Waluk and J. Michl, J. Am. Chem. Soc., 2002, 124, 2406–2407 CrossRef CAS PubMed.
- C. Wang, W. Jiang, D. Tan, L. Huang, J. Li, Q. Qiao, P. Yadav, X. Liu and Z. Xu, Chem. Sci., 2023, 14, 4786–4795 RSC.
- K. Basuroy, J. d J. Velazquez-Garcia, D. Storozhuk, R. Henning, D. J. Gosztola, S. Thekku Veedu and S. Techert, J. Chem. Phys., 2023, 158, 054304 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- R. Tandon, V. Luxami, N. Tandon and K. Paul, Bioorg. Chem., 2022, 121, 105677 CrossRef CAS PubMed.
- A. S. Oshchepkov, M. S. Oshchepkov, M. V. Oshchepkova, A. Al-Hamry, O. Kanoun and E. A. Kataev, Adv. Opt. Mater., 2021, 9, 2001913 CrossRef CAS.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- H. Yu, Y. Guo, W. Zhu, K. Havener and X. Zheng, Coord. Chem. Rev., 2021, 444, 214019 CrossRef CAS.
- X. Tian, K. Shoyama, B. Mahlmeister, F. Brust, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2023, 145, 9886–9894 CrossRef CAS PubMed.
- J. R. Swartzfager, G. Chen, T. Francese, G. Galli and J. B. Asbury, Phys. Chem. Chem. Phys., 2023, 25, 3151–3159 RSC.
- D. Gudeika, R. R. Reghu, J. V. Grazulevicius, G. Buika, J. Simokaitiene, A. Miasojedovas, S. Jursenas and V. Jankauskas, Dyes Pigm., 2013, 99, 895–902 CrossRef CAS.
- Z. Szakács, S. Rousseva, M. Bojtár, D. Hessz, I. Bitter, M. Kállay, M. Hilbers, H. Zhang and M. Kubinyi, Phys. Chem. Chem. Phys., 2018, 20, 10155–10164 RSC.
- W. Zhang, J. Kong, R. Miao, H. Song, Y. Ma, M. Zhou and Y. Fang, Adv. Funct. Mater., 2023, 2311404 CrossRef.
- X. Xiao, Y. Yan, A. A. Sukhanov, S. Doria, A. Iagatti, L. Bussotti, J. Zhao, M. Di Donato and V. K. Voronkova, J. Phys. Chem. B, 2023, 127, 6982–6998 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- E. W. Castner Jr, M. Maroncelli and G. R. Fleming, J. Chem. Phys., 1987, 86, 1090–1097 CrossRef.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsujiet al., Gaussian, Revision C.02, Gaussian, Inc., Wallingford CT, 2019, vol. 16 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- N. Mataga, Bull. Chem. Soc. Jpn., 1963, 36, 654–662 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- E. Lippert, Z. Naturforschung A, 1955, 10, 541–545 CrossRef.
- J. Snellenburg, S. Laptenok, R. Seger, K. Mullen and I. Van Stokkum, J. Stat. Soft., 2012, 49, 1–22 Search PubMed.
- W. Zhang, J. Kong, D. Hu, M. Tao, X. Niu, S. Vdović, D. Aumiler, Y. Ma and A. Xia, J. Phys. Chem. C, 2020, 124, 5574–5582 CrossRef CAS.
- S. M. Aly, A. Usman, M. AlZayer, G. A. Hamdi, E. Alarousu and O. F. Mohammed, J. Phys. Chem. B, 2015, 119, 2596–2603 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05654f |
| This journal is © the Owner Societies 2024 |