
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct spectroscopic evidence for the high-spin state of dioxidomanganese(V)†
Olesya S.
Ablyasova
 ab,
Vicente
Zamudio-Bayer
a,
Max
Flach
ab,
Mayara
da Silva Santos
ab,
J. Tobias
Lau
ab and
Konstantin
Hirsch
*a
ab,
Vicente
Zamudio-Bayer
a,
Max
Flach
ab,
Mayara
da Silva Santos
ab,
J. Tobias
Lau
ab and
Konstantin
Hirsch
*a
aAbteilung für Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, Berlin 12489, Germany. E-mail: Konstantin.Hirsch@helmholtz-berlin.de
bPhysikalisches Institut, Albert-Ludwigs-Universität Freiburg, Hermann-Herder-Str. 3, Freiburg 79104, Germany
First published on 29th January 2024
Abstract
The spin state of metal centers in many catalytic reactions has been demonstrated to be a rate limiting factor when high-valent metal centers such as manganese are involved. Although numerous manganese(V) complexes, including a few manganese(V) oxo complexes, have been identified, thus far only one of these, [MnVH3 buea(O)], has been directly confirmed to exist in a high spin state. Such a high-spin manganese(V) center may play a crucial role in the dioxygen formation process in the elusive S4 state of the Kok cycle in photosystem II. In this study, we provide direct experimental evidence, using X-ray magnetic circular dichroism (XMCD) and X-ray absorption spectroscopy (XAS), of gas phase [OMnO]+ as the second known high-spin manganese(V) oxo complex. We conclusively assign the ground state as 3B1 (C2v). Additionally, we provide fingerprint spectra not only for [OMnV O]+, but also for the high-spin hydroxidooxidomanganese(IV) ion [OMnIV OH]+ in its 4A′′ (Cs) ground state that is expected to exhibit similar XAS and XMCD spectral signatures to neutral dioxidomanganese(IV).
1 Introduction
High valent metal-oxo intermediates are believed to be key in facilitating numerous biological and abiotic catalytic oxidation reactions.1 There is also evidence that the spin state of these high-valent metal-oxo complexes determines their reactivity.1–5 For example, a combined theoretical and experimental study suggests that the catalytic hydrocarbon oxidation is only facilitated via the triplet state of a manganese(V)-oxo complex while the singlet state of that same species is inactive.6 Moreover, high-spin manganese(V) was also proposed to play a key role in water splitting as the main active site of the oxygen-evolving complex (OEC) of photosystem II.7–11 Despite the apparent importance of high-valent–high-spin manganese centers, only very few manganese(V)-oxo complexes are known,6,12–14 most of which are of tetragonal symmetry resulting in a quenched spin magnetic moment. To the best of our knowledge, only one manganese(V)-oxo complex, [MnVH3buea(O)], that exists in a high-spin (triplet) state has been reported thus far.15,16 This triplet state has been achieved by tailoring a trigonal ligand symmetry that results in a finite spin at the manganese site.Previously, we reported on cationic manganese oxide clusters, including [OMnO]+ for which we experimentally and theoretically determined the +5 oxidation state.13 The comparison of the spectral shape with the computational X-ray absorption spectroscopy (XAS) signature of [OMnVO]+ hinted at a high-spin 3B1 ground state, in line with a calculated high-spin ground state. Here, we present X-ray magnetic circular dichroism (XMCD) spectroscopy of [OMnO]+ that allows direct probing of the magnetic moments of gas-phase ions.17 By this, we now show conclusively that [OMnO]+ is indeed the second reported high-spin manganese(V)oxo species. Because manganese in oxidation states +5 and +4 might play a pivotal role in the oxygen evolution reaction mediated by the OEC,7,8,10,11 we provide fingerprint XAS and XMCD spectra, as well as median excitation energies of [OMnVO]+ and [OMnIVOH]+ whose oxidation states are determined by chemical shifts. Locally at the manganese site, the latter exhibits almost the same symmetry as the neutral OMnIV O complex.
2 Methods
XMCD spectroscopy was carried out at the ion trap end station,18–21 located at beamline UE52-PGM of the synchrotron facility BESSY II operated by Helmholtz-Zentrum Berlin. Dioxidomanganese and hydroxidooxidomanganese cations are produced by DC-sputtering of a manganese target in a helium and argon atmosphere while simultaneously introducing trace amounts of oxygen to the discharge in presence of residual water vapour. The ion beam is then guided via a hexapole ion guide and quadrupole mass filter, selecting the species of interest, to a liquid-helium cooled quadrupole ion trap for cooling of the clusters to a typical temperature of approximately 20 K. A superconducting solenoid creates a homogeneous magnetic field of μBH = 4.5 T along the trap axis to magnetize the sample. Ion yield (action) spectroscopy is performed by collecting product ions resulting from X-ray absorption of photons with helicity parallel and antiparallel to the magnetic field axis, respectively.17 Photon energy calibration was performed using the neon 1s excitation in the beamline ionization cell and checked at the oxygen K-edge, giving a photon energy uncertainty of ±0.1 eV. Crystal field and charge transfer multiplet (CF/CTM) simulations were carried out using the program package CTM4XAS (version 5.5),22 and multiplet ligand-field theory calculations were performed using the software package Xclaim.23 Density functional theory (DFT) calculations were carried out using ORCA version 4.2.124 and turbomole version 7.6.1 employing the B3LYP functional25,26 and def2-TZVP basis set.27 Search for the [OMnIVOH]+ ground state structure was performed starting from the reported [OMnVO]+ ground state structure13 adding a hydrogen atom either at the manganese or oxygen site, respectively. See the ESI† for details.3 Results and discussion
3.1 Spin ground state of dioxidomanganese(V) cation
The experimental X-ray absorption spectrum of the [OMnVO]+ complex at the manganese L2,3 edge is shown in the top panel of Fig. 1(a) reproducing the data presented earlier13 despite being collected on different ion yield channels, namely O+ here and Mn2 + in the earlier study.13 From the median L3 excitation energy of 642.6 ± 0.2 eV, also shown in Fig. 1(a) as a dashed black line, we can deduce the oxidation state28 of the manganese center in [OMnO]+ to be +5 in agreement with the earlier report,13cf. ESI,† Fig. S11. The oxidation state assignment is further substantiated by a DFT Mulliken 3d spin population of 2.2 which is known to correlate with the oxidation state in manganese.29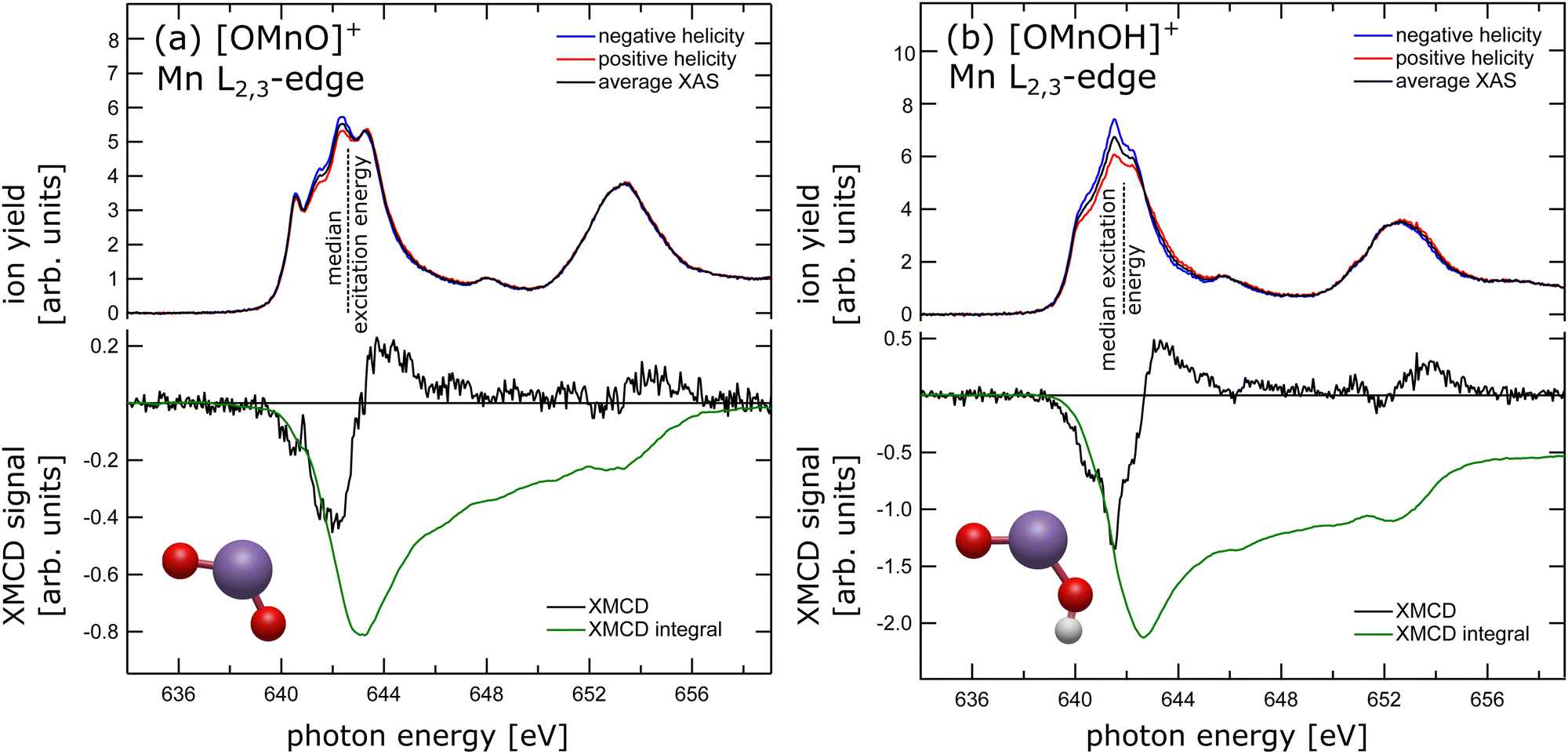 | ||
| Fig. 1 X-ray absorption spectra (upper panel) recorded with negative (blue trace) and positive (red trace) helicity with respect to the magnetic field axis, and average XAS (black trace) taken at an energy resolution of 170 meV of (a) [OMnVO]+ and (b) [OMnIVOH]+ complexes. The dashed lines indicate the median L3 excitation energies of 642.6 ± 0.2 eV and 641.9 ± 0.3 eV, respectively. Lower panel: Experimental XMCD signal (black trace) along with its integral (green trace) of (a) [OMnVO]+ and (b) [OMnIVOH]+ complexes. | ||
In the lower panel of Fig. 1(a) the XMCD signal is presented, which is the difference in X-ray absorption with helicity of the X-ray beam parallel and antiparallel to the magnetic field axis. Additionally, we present the XMCD integral in the lower panel of Fig. 1(a). The XMCD orbital sum rule30–32 directly links the XMCD integral over both, L3 and L2, edges to the expectation value of the orbital magnetic moment at the manganese center. As can be seen, the integral over both L2,3-edges approaches zero, indicating a vanishing orbital magnetic moment. A quantitative analysis using the orbital XMCD sum rule30 yields an orbital magnetization of μL = 0.01 ± 0.05μB per electron hole. This agrees with quenching of the orbital magnetic moment as is expected in C2v symmetry.33 Hence, the dichroic signal observed is of pure spin character. Since the manganese center has been shown to be in oxidation state +5, which is equivalent to a nominal local 3d2 electron configuration at the manganese site, the non-vanishing XMCD signal, arising from spin only, is evidence for a triplet state. We have therefore experimentally and unambiguously demonstrated that the ground state of the high-valent [OMnVO]+ complex is a high spin 3B1 (C2v) state.
3.2 XAS and XMCD spectral fingerprinting of [OMnVO]+ and [OMnIVOH]+
In our special case of [OMnVO]+ there is no need to further analyse the spectral shape of the L-edge absorption in order to extract information on the electronic ground state. However, fingerprinting of the L2,3-edge multiplet structure is often successfully employed to determine the electronic configuration of 3d metals and their compounds.34–38 Additionally, in case of early or high-valent transition metals, the XMCD spin sum rule breaks down39 and fingerprinting of the XMCD signal is the only fallback option to extract information on the spin state from helicity dependent L-edge absorption. Furthermore, modelling the spectral shape also allows to extract semi-empirical parameters such as the crystal field strength induced by the coordinating ligands.38We therefore employed crystal field and charge transfer multiplet modelling of both XAS and XMCD spectral signatures of the [OMnO]+ complex. However, despite searching a wide parameter space starting from parameters reported for other 3d2 systems, namely VI340,41 and FeV2O4,42 the agreement between experiment and simulation is rather poor as can be seen from ESI,† Fig. S1–S6. It is worthwhile mentioning that even wave-function based theoretical models struggle to reproduce all the features of systems exhibiting a 3d2 electron configuration.43 Moreover, the agreement between XAS and XMCD signatures of [OMnVO]+ with the reported experimental XAS and XMCD signatures of 3d2 systems40–42 is subpar. For a thorough overview of the CF/CTM simulations and a comparison of [OMnVO]+ XAS and XMCD to reports for VI340,41 or FeV2O442 see the ESI.† Interestingly, we find a similarly satisfying agreement of the [OMnVO]+ XMCD signal with that of CrI3, as can be seen from ESI,† Fig. S8, where the metal center, however, exhibits a 3d3 local electronic configuration.44,45 This seems to indicate that spectral fingerprinting might not be robust for high-valent manganese systems.46
In order to test this hypothesis we have prepared manganese in oxidation state +4 in a similar local symmetry by reducing the [OMnVO]+ complex using residual water and forming the hydroxidooxido manganese(IV) cation, [OMnIVOH]+. Its manganese L2,3-edge X-ray absorption spectrum is shown in the upper panel of Fig. 1(b) alongside the median L3 excitation energy indicated by the dashed line and its ground state geometry as identified by DFT. For related manganese oxide species the shift of the median L3 excitation energy has been shown to be 0.95 ± 0.02 eV per unit oxidation state.13 Here, we report the median L3 excitation energy of [OMnIVOH]+ as 641.9 ± 0.3 eV, i.e., 1.1 eV below and 0.9 eV above the median L3 excitation energy of MnV and MnIII centers, respectively.13 This reveals an oxidation state of the manganese center in [OMnIVOH]+ of +4 or, put differently, a local 3d3 electron configuration. The spins of these three 3d electrons can couple to give either doublet or quartet spin states. Although application of the XMCD spin sum rule in this instance would result in a significant error,39 comparison of the XMCD intensity of [OMnIVOH]+ as presented in the lower panel of Fig. 1(b) to that of [OMnVO]+ allows for exclusion of one of the potential spin states of [OMnIVOH]+. Since the XMCD signal of [OMnIVOH]+ is larger by a factor of 3 compared to that of 3B1 (C2v) [OMnVO]+ this is evidence for [OMnIVOH]+ to be in a quartet spin state. Furthermore, the XMCD integral over both L2,3 edges indicates a small positive orbital magnetization that accounts to 0.011 to 0.014μB per hole, showcasing an almost quenched orbital magnetic moment. This can be expected as the local symmetry of the metal center is only slightly disturbed by the addition of hydrogen atom with respect to the [OMnVO]+ complex. The quartet ground state identified from the experimental data is also in agreement with our ground state as determined by DFT, for details see the ESI.†
Despite the significantly different XMCD intensity in [OMnVO]+ and [OMnIVOH]+ complexes, there are only marginal differences in the XMCD spectral signatures of both molecules as can be seen from a direct comparison shown in ESI,† Fig. S9. Consequently, the fingerprint of the XMCD signal of high-valent manganese is less suitable for determining the respective electronic configuration and spin state. However, there is a distinct difference in their XAS spectral signature (see ESI,† Fig. S9) that is even more pronounced than in manganese with a local 3d3 and 3d4 electron configuration, respectively,46 and might be used as a spectral fingerprint.
3.3 Energy level diagram and oxygen K-edge X-ray absorption spectra
Complementary to manganese L2,3-edge X-ray absorption we also recorded oxygen K-edge X-ray absorption spectra, which are presented in the right panels of Fig. 2. In the absence of multiplet effects, X-ray absorption is a measure of the unoccupied density of states. Hence, we also show the oxygen p-projected density of states in Fig. 2 in comparison to the experimental oxygen K-edge absorption spectrum. The good agreement between the calculated density of states and the experimental data allows us to directly extract energy level diagrams as presented in the left panels of the same figure.38 This has the advantage that we do not have to rely on simulations of oxygen K-edge spectra capitalizing on multiconfigurational self-consistent field wavefunction methods as recently reported.47 The crystal field induced by the oxygen atoms lifts the degeneracy of the manganese 3d states and groups the molecular orbitals with significant 3d character.48 In the [OMnVO]+ complex the aforementioned grouping nicely correlates with the orientation of the 3d-derived states relative to the ligand orbitals. Therefore, the two occupied α-spin molecular orbitals (MOs) 10a1 and 4b1 are grouped together and are separated from the 11a1 and 2a2 MOs by about 3.8 eV. The molecular orbital 7b2 with significant 3d electron density along the Mn–O axes experiences an additional energy penalty pushing it up in energy by another 1.5 eV. We in turn used these parameters as a starting point for further CF simulations of the manganese L2,3-edge XAS and XMCD spectra. Again, the agreement between the resulting simulations and the experimental spectra remains poor as can be seen from ESI,† Fig. S7.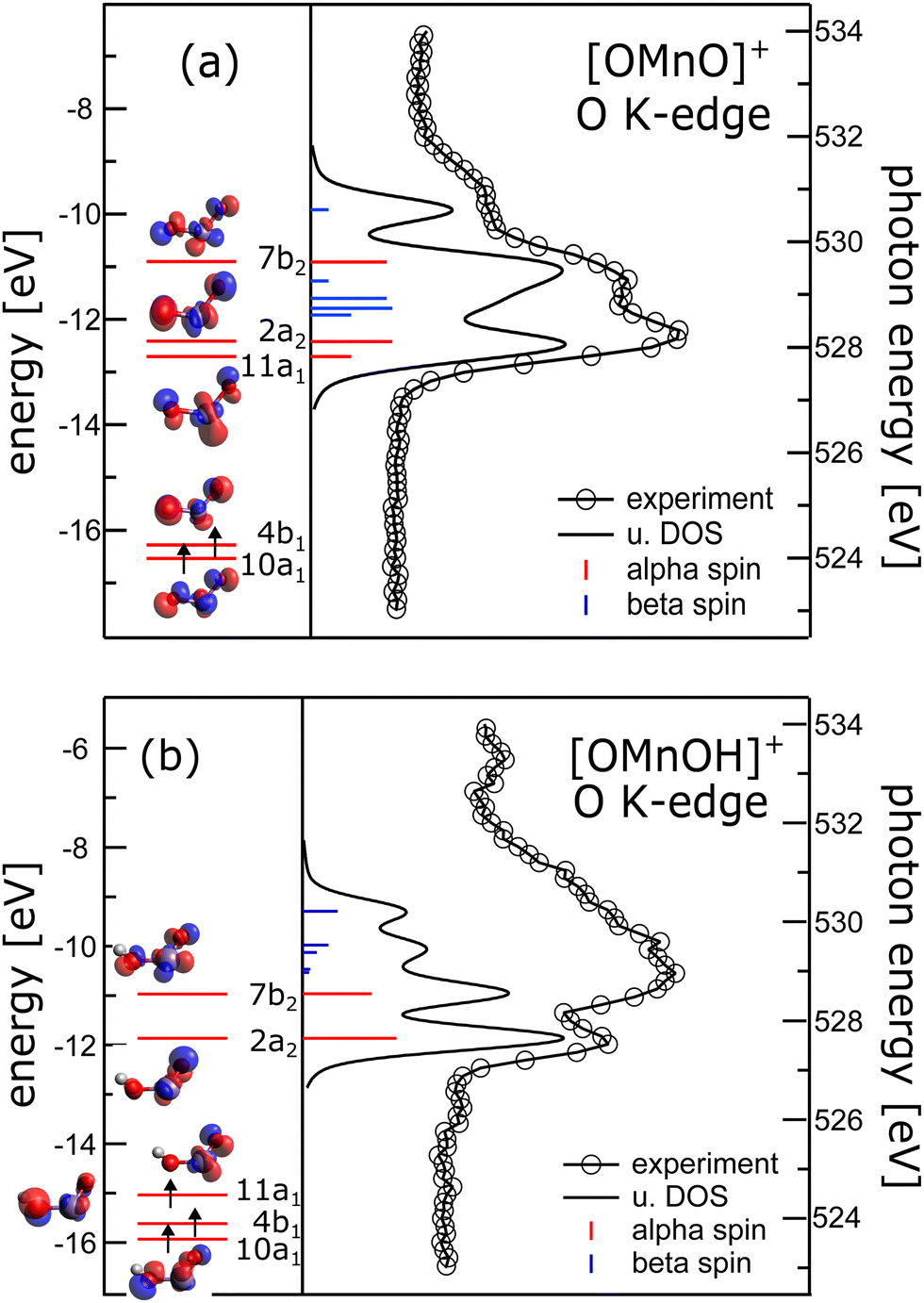 | ||
| Fig. 2 Left panels: Energy level diagram deduced from α spins along isosurface plots of the molecular orbitals of (a) [OMnVO]+ and (b) [OMnIVOH]+ complexes. Arrows indicate occupied molecular orbitals. Right panel: Comparison of oxygen K-edge absorption (340 meV energy resolution) and oxygen p-projected density of states (broadened with a Gaussian of 0.3 eV full width half maximum and a 0.16 eV Lorentzian49 to account for experimental resolution and core hole lifetime, respectively) of (a) [OMnVO]+ and (b) [OMnIVOH]+ complexes. Blue and red sticks show α and β spin states, respectively. For [OMnIVOH]+ we use the molecular orbital nomenclature in C2v symmetry as the local symmetry at the metal is only slightly disturbed by the addition of the hydrogen atom. This allows for a more direct comparison to [OMnVO]+. | ||
The energy separation of 10a1 and 4b1 of 0.2 eV is considerably smaller than the 3d exchange interaction of 0.45 eV (CASPT2/RASPT2.13) and 0.55 eV (DFT, B3LYP/def2-TZVP), respectively, as deduced from the singlet–triplet gap of [OMnVO]+, which is also consistent with the exchange splitting of 0.53 eV in atomic Ti2 + with 3d2 configuration.50 This results in the stabilization of the triplet ground state, also present in other isoelectronic high-valent transition metal dioxide molecules.51–60
In [OMnIVOH]+ with an overall Cs symmetry, the local symmetry at the manganese site deviates only slightly from C2v symmetry. We therefore use the same nomenclature for the molecular orbitals as in [OMnVO]+. The crystal field splitting in [OMnIVOH]+ is less pronounced than in [OMnVO]+, but the molecular orbitals 10a1 and 4b1 remain grouped relatively close (0.3 eV) in energy, and 4b1 is separated from 11a1 by 0.58 eV. This splitting is still considerably smaller than the high-spin–low-spin transition energy of manganese with a 3d3 electronic configuration46 resulting in the observed quartet spin state. As expected the molecular orbital 7b2 suffers an additional energy penalty relative to 2a2, which amounts to 0.9 eV. From the electron configuration of the [OMnIVOH]+ complex we deduce a 4A′′ (Cs) ground state, which is identical to the reported ground state of isoelectronic [OVOH]−.61
At the manganese site the local symmetry resembles neutral OMnO with similar Mn–O bond distances and angle of 1.603/1.699 Å, 123.5° in [OMnIVOH]+ compared to 1.639 Å, 118.4°13 and 1.581 Å, 131°62 in neutral OMnO. In C2v symmetry the ground state of the [OMnIVOH]+ complex could be described as a 4B1 state with (10a1)1 (4b1)1 (11a1)1 configuration, in agreement with the ground state reported for neutral OMnO by DFT,52,53,56,62 but distinct from a 4B2 ground state found by employing multiconfigurational wavefunction methods.13 However, the latter differs from the former only by swapping the energetic ordering of the nearly degenerate 11a1 and 2a2 molecular orbitals. Furthermore, the leading line in the oxygen K-edge absorption of [OMnVO]+ was predicted to be reduced in neutral OMnO.13 Indeed we observe a reduction in intensity of the low energy line when comparing [OMnVO]+ and [OMnIVOH]+ complexes further substantiating the similarity of [OMnIVOH]+ and neutral OMnIVO. Note, that this reduction is not present in the oxygen p-projected density of states, which may also not be expected as this approach neglects any variation of the oscillator strength due to changing overlap of the 2p wavefunction and the involved molecular orbitals.
The XMCD and XAS spectra of [OMnIVOH]+ should be very similar to those of the neutral OMnO complex given their similar local electronic structure and symmetry. Therefore the XMCD and XAS spectra of [OMnIVOH]+ might be used as a spectral fingerprint for neutral OMnO.
Furthermore, we want to emphasize that neither in the oxygen K-edge spectrum of [OMnVO]+ nor in [OMnIVOH]+ the molecular orbital contributing to the energetically lowest lying excitation of the spectrum has a prominent 3d character. This is in contrast to the case of oxygen centered radicals,63 which can therefore be excluded here.
4 Conclusion and outlook
Using XMCD spectroscopy we conclusively demonstrate that the ground state of the dioxidomanganese(V) cation is the high-spin state 3B1 as foreshadowed by an earlier study employing experimental and computational XAS only.13 Hence, we fully establish [OMnVO]+ as the second known manganese(V)-oxo complex to exhibit a high-spin ground state.15,16 This is an important piece of information when considering reaction pathways involving high-valent species such as [OMnVO]+ that might well be intermediate species for which the spin state could be rate determining.Furthermore, we present the first XMCD spectrum of a high-spin manganese(V) oxo complex, i.e., of a high-spin manganese center in a local 3d2 electronic configuration. Reproducing the spectral signature properly is a challenge for both semi-empirical models – as demonstrated here but also for other 3d2 systems40–42 – as well as for wavefunction-based ab initio electronic structure theory.43 Nonetheless, considerable progress has been made by wavefunction-based ab initio methods in reproducing both XAS and XMCD signatures of transition metal compounds and will become invaluable in interpreting experimental data.
Moreover, we also provide the XAS and XMCD spectrum for the high-spin hydroxidooxidomanganese(IV) cation with almost identical local symmetry at the manganese site as in [OMnO]+,0. Manganese centers in both, oxidation states +4 and +5, might play a significant role in the oxygen evolving reaction of photosystem II. Hence, the reported spectra can serve as a benchmark not only for studying the OEC but also for studying other multinuclear manganese systems64,65 and to further refine quantum chemical methods.
However, what remains elusive is the existence of a high-valent–high-spin species in a multinuclear manganese complex, which is an issue closely intertwined with important oxidation reactions such as the Kok cycle.66 We hope that this study will encourage future studies into such systems.
Author contributions
JTL, OSA, and VZB devised the project, designed the experimental study, and prepared the beamtime proposal. JTL, KH, and VZB designed the experimental setup. MF, MSS, OSA and VZB performed the experiments, and validated their reproducibility. OSA analyzed the data under guidance of JTL, KH, and VZB. OSA, KH and VZB performed the calculations. JTL, KH, and VZB supervised the project. OSA wrote the original draft, with reviewing and editing by JTL, KH, and VZB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Beamtime for this project was granted at the Ion Trap endstation of BESSY II, beamline UE52-PGM, operated by Helmholtz Zentrum Berlin. This project has received funding from the German Federal Ministry of Education and Research through Grant No. BMBF-05K16VF1. JTL, MF, MSS and OSA acknowledge support by the DFG funded Research Training Group RTG 2717, “Dynamics of Controlled Atomic and Molecular Systems”.Notes and references
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS PubMed.
- X. P. Zhang, A. Chandra, Y. M. Lee, R. Cao, K. Ray and W. Nam, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- T. Yang, M. G. Quesne, H. M. Neu, F. G. Cantú Reinhard, D. P. Goldberg and S. P. de Visser, J. Am. Chem. Soc., 2016, 138, 12375–12386 CrossRef CAS PubMed.
- Z. Chen and G. Yin, Chem. Soc. Rev., 2015, 44, 1083–1100 RSC.
- V. Krewald, M. Retegan, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Inorg. Chem., 2016, 55, 488–501 CrossRef CAS PubMed.
- X.-X. Li, M. Guo, B. Qiu, K.-B. Cho, W. Sun and W. Nam, Inorg. Chem., 2019, 58, 14842–14852 CrossRef CAS PubMed.
- J. Barber, Nat. Plants, 2017, 3, 17041 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis and W. Lubitz, Annu. Rev. Biochem., 2020, 89, 795–820 CrossRef CAS PubMed.
- S. H. Kim, H. Park, M. S. Seo, M. Kubo, T. Ogura, J. Klajn, D. T. Gryko, J. S. Valentine and W. Nam, J. Am. Chem. Soc., 2010, 132, 14030–14032 CrossRef CAS PubMed.
- Y. Guo, J. Messinger, L. Kloo and L. Sun, J. Am. Chem. Soc., 2023, 145, 4129–4141 CrossRef CAS PubMed.
- V. Krewald, F. Neese and D. A. Pantazis, J. Inorg. Biochem., 2019, 199, 110797 CrossRef CAS PubMed.
- H. M. Neu, R. A. Baglia and D. P. Goldberg, Acc. Chem. Res., 2015, 48, 2754–2764 CrossRef CAS PubMed.
- M. G. Delcey, R. Lindblad, M. Timm, C. Bülow, V. Zamudio-Bayer, B. von Issendorff, J. T. Lau and M. Lundberg, Phys. Chem. Chem. Phys., 2022, 24, 3598–3610 RSC.
- D. Feichtinger and D. A. Plattner, Angew. Chem., Int. Ed. Engl., 1997, 36, 1718–1719 CrossRef CAS.
- T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- M. Niemeyer, K. Hirsch, V. Zamudio-Bayer, A. Langenberg, M. Vogel, M. Kossick, C. Ebrecht, K. Egashira, A. Terasaki, T. Möller, B. V. Issendorff and J. T. Lau, Phys. Rev. Lett., 2012, 108, 57201 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Kossick, A. Ławicki, A. Terasaki, B. V. Issendorff and J. T. Lau, J. Chem. Phys., 2015, 142, 234301 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Niemeyer, M. Vogel, A. Ławicki, A. Terasaki, J. T. Lau and B. von Issendorff, Angew. Chem., Int. Ed., 2015, 54, 4498–4501 CrossRef CAS PubMed.
- A. Langenberg, K. Hirsch, A. Ławicki, V. Zamudio-Bayer, M. Niemeyer, P. Chmiela, B. Langbehn, A. Terasaki, B. V. Issendorff and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 184420 CrossRef.
- K. Hirsch, J. T. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. von Issendorff, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154029 CrossRef.
- E. Stavitski and F. M. F. de Groot, Micron, 2010, 41, 687–694 CrossRef CAS PubMed.
- J. Fernandez-Rodriguez, B. Toby and M. Van Veenendaal, J. Electron Spectrosc. Relat. Phenom., 2015, 202, 81–88 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- M. Flach, K. Hirsch, M. Timm, O. S. Ablyasova, M. D. S. Santos, M. Kubin, C. Bülow, T. Gitzinger, B. von Issendorff, J. T. Lau and V. Zamudio-Bayer, Phys. Chem. Chem. Phys., 2022, 24, 19890–19894 RSC.
- M. R. A. Blomberg and P. E. M. Siegbahn, Theor. Chem. Acc., 1997, 97, 72–80 Search PubMed.
- B. T. Thole, P. Carra, F. Sette and G. van der Laan, Phys. Rev. Lett., 1992, 68, 1943 CrossRef CAS PubMed.
- P. Carra, B. T. Thole, M. Altarelli and X. Wang, Phys. Rev. Lett., 1993, 70, 694 CrossRef CAS PubMed.
- C. T. Chen, Y. U. Idzerda, H.-J. Lin, N. V. Smith, G. Meigs, E. Chaban, G. H. Ho, E. Pellegrin and F. Sette, Phys. Rev. Lett., 1995, 75, 152 CrossRef CAS PubMed.
- G. F. Dionne, Magnetic Oxides, Springer US, Boston, MA, 2009 Search PubMed.
- F. M. F. de Groot, Coord. Chem. Rev., 2005, 249, 31–63 CrossRef CAS.
- F. M. F. de Groot, J. Electron Spectrosc. Relat. Phenom., 1994, 67, 529–622 CrossRef CAS.
- S. P. Cramer, F. M. F. DeGroot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, M. K. Chan, W. H. Armstrong, E. Libby, G. Christou, S. Brooker, V. McKee, O. C. Mullins and J. C. Fuggle, J. Am. Chem. Soc., 1991, 113, 7937–7940 CrossRef CAS.
- M. Risch, K. A. Stoerzinger, B. Han, T. Z. Regier, D. Peak, S. Y. Sayed, C. Wei, Z. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2017, 121, 17682–17692 CrossRef CAS.
- O. S. Ablyasova, M. Guo, V. Zamudio-Bayer, M. Kubin, T. Gitzinger, M. da Silva Santos, M. Flach, M. Timm, M. Lundberg, J. T. Lau and K. Hirsch, J. Phys. Chem. A, 2023, 127, 7121–7131 CrossRef CAS PubMed.
- C. Piamonteze, P. Miedema and F. M. F. de Groot, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 184410 CrossRef.
- D. Hovančík, J. Pospíšil, K. Carva, V. Sechovský and C. Piamonteze, Nano Lett., 2023, 23, 1175–1180 CrossRef PubMed.
- R. Sant, A. D. Vita, V. Polewczyk, G. M. Pierantozzi, F. Mazzola, G. Vinai, G. van der Laan, G. Panaccione and N. B. Brookes, J. Phys.: Condens. Matter, 2023, 35, 405601 CrossRef PubMed.
- J.-S. Kang, J. Hwang, D. H. Kim, E. Lee, W. C. Kim, C. S. Kim, S. Kwon, S. Lee, J.-Y. Kim, T. Ueno, M. Sawada, B. Kim, B. H. Kim and B. I. Min, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165136 CrossRef.
- D. Maganas, J. K. Kowalska, C. Van Stappen, S. DeBeer and F. Neese, J. Chem. Phys., 2020, 152, 114107 CrossRef CAS PubMed.
- A. Frisk, L. B. Duffy, S. Zhang, G. van der Laan and T. Hesjedal, Mater. Lett., 2018, 232, 5–7 CrossRef CAS.
- A. Ghosh, H. J. M. Jönsson, D. J. Mukkattukavil, Y. Kvashnin, D. Phuyal, P. Thunström, M. Agåker, A. Nicolaou, M. Jonak, R. Klingeler, M. V. Kamalakar, T. Sarkar, A. N. Vasiliev, S. M. Butorin, O. Eriksson and M. Abdel-Hafiez, Phys. Rev. B: Condens. Matter Mater. Phys., 2023, 107, 115148 CrossRef CAS.
- G. van der Laan and B. T. Thole, J. Phys.: Condens. Matter, 1992, 4, 4181 CrossRef CAS.
- M. Scott and M. G. Delcey, J. Chem. Theory Comput., 2023, 19, 5924–5937 CrossRef CAS PubMed.
- F. Habib, O. R. Luca, V. Vieru, M. Shiddiq, I. Korobkov, S. I. Gorelsky, M. K. Takase, L. F. Chibotaru, S. Hill, R. H. Crabtree and M. Murugesu, Angew. Chem., Int. Ed., 2013, 52, 11290–11293 CrossRef CAS PubMed.
- M. Coreno, M. de Simone, K. C. Prince, R. Richter, M. Vondráček, L. Avaldi and R. Camilloni, Chem. Phys. Lett., 1999, 306, 269–274 CrossRef CAS.
- E. B. Saloman, J. Phys. Chem. Ref. Data, 2012, 41, 013101 CrossRef.
- G. V. Chertihin, W. D. Bare and L. Andrews, J. Phys. Chem. A, 1997, 101, 5090–5096 CrossRef CAS.
- G. L. Gutsev, B. K. Rao and P. Jena, J. Phys. Chem. A, 2000, 104, 11961–11971 CrossRef CAS.
- E. L. Uzunova, H. Mikosch and G. S. Nikolov, J. Chem. Phys., 2008, 128, 094307 CrossRef PubMed.
- F. Grein, Chem. Phys., 2008, 343, 231–240 CrossRef CAS.
- Y. Wang, X. Gong and J. Wang, Phys. Chem. Chem. Phys., 2010, 12, 2471–2477 RSC.
- Y. Gong, M. Zhou and L. Andrews, Chem. Rev., 2009, 109, 6765–6808 CrossRef CAS PubMed.
- G. L. Gutsev, P. Jena, H.-J. Zhai and L.-S. Wang, J. Chem. Phys., 2001, 115, 7935–7944 CrossRef CAS.
- P. B. Armentrout, J. Chem. Phys., 2013, 139, 084305 CrossRef CAS PubMed.
- M. Zhou and L. Andrews, J. Chem. Phys., 1999, 111, 4230–4238 CrossRef CAS.
- J. Gancheff, C. Kremer, E. Kremer and O. N. Ventura, J. Mol. Struct. THEOCHEM, 2002, 580, 107–116 CrossRef CAS.
- Y. Wang, C. Han, J. Hong, Z. Fei, C. Dong, H. Liu and X. Xiong, RSC Adv., 2021, 11, 18729–18736 RSC.
- G. V. Chertihin and L. Andrews, J. Phys. Chem. A, 1997, 101, 8547–8553 CrossRef CAS.
- M. da Silva Santos, R. Medel, M. Flach, O. S. Ablyasova, M. Timm, B. von Issendorff, K. Hirsch, V. Zamudio-Bayer, S. Riedel and J. T. Lau, Chem. Phys. Chem., 2023, e202300390 CrossRef CAS PubMed.
- M. Mannini, P. Sainctavit, R. Sessoli, C. Cartier dit Moulin, F. Pineider, M.-A. Arrio, A. Cornia and D. Gatteschi, Chem. – Eur. J., 2008, 14, 7530–7535 CrossRef CAS PubMed.
- M. Tombers, J. Meyer, J. Meyer, A. Lawicki, V. Zamudio-Bayer, K. Hirsch, J. T. Lau, B. von Issendorff, A. Terasaki, T. A. Schlathölter, R. A. Hoekstra, S. Schmidt, A. K. Powell, E. Kessler, M. H. Prosenc, C. van Wüllen and G. Niedner-Schatteburg, Chem. – Eur. J., 2022, 28, e202102592 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05468c |
| This journal is © the Owner Societies 2024 |