
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVarying the degree of oxidation of graphite: effect of oxidation time and oxidant mass†
Ioannis
Karnis
 ab,
Fanourios
Krasanakis
a,
Labrini
Sygellou
c,
Anastassia N.
Rissanou
ad,
Konstantinos
Karatasos
e and
Kiriaki
Chrissopoulou
*a
ab,
Fanourios
Krasanakis
a,
Labrini
Sygellou
c,
Anastassia N.
Rissanou
ad,
Konstantinos
Karatasos
e and
Kiriaki
Chrissopoulou
*a
aInstitute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas, P.O. Box 1527, Heraklion Crete 711 10, Greece. E-mail: kiki@iesl.forth.gr
bDepartment of Chemistry, University of Crete, Heraklion Crete, Greece
cInstitute of Chemical Engineering Studies, Foundation for Research and Technology-Hellas, Stadiou Str., 26504 Patras, Greece
dInstitute of Theoretical and Physical Chemistry, National Hellenic Research Foundation, 48 Vassileos Konstantinou Ave, Athens 11635, Greece
eDepartment of Chemical Engineering, Aristotle University of Thessaloniki, Thessaloniki, Greece
First published on 27th February 2024
Abstract
In this work, we employ a fast and less toxic modified Hummers’ method to develop graphene oxide (GO) with varying degrees of oxidation and investigate the effect of the latter on the structure and the thermal properties of the synthesized materials. Two different key parameters, the time of the oxidation reaction and the mass of the oxidation agent, were systematically altered in order to fine tune the oxidation degree. All graphene oxides were characterized by a plethora of experimental techniques, like X-ray diffraction (XRD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) as well as infrared spectroscopy (IR) and X-ray photoelectron spectroscopy (XPS) for their structural, thermal and chemical identification. The results revealed that for a certain amount of oxidant, the time does not affect the final degree of oxidation of the materials, at least for the examined reaction times, because very similar structural patterns and thermal properties were obtained. At the same time, the oxygen-containing functional groups were found very similar. On the other hand, the degree of oxidation was found highly dependent on the mass of the oxidizing agent. XRD analysis showed a systematic increase of the interlayer distance of the synthesized GOs with the increase of the oxidant mass, whereas both the enthalpy of reduction and the % weight loss were increased. Moreover, XPS measurements provided a quantitative evaluation of the amount of carbon and oxygen in the materials; the increase of the oxidant mass led to a decrease of the total carbon content with the concurrent increase of the total oxygen amount.
1. Introduction
In recent years, graphitic materials have garnered considerable attention from the scientific community and have undergone thorough study. They consist of sp2 carbon and exist in various forms such as graphite, carbon nanotubes (CNTs), graphene and activated carbon.1–4 Among them, the (multi)layered ones like graphite itself, graphene oxide as well as graphene have been intensely investigated; in particular, graphene is considered a very promising material for many applications due to its unique characteristics.1,2,4–6 It has a large theoretical specific surface area (∼2600 m2 g−1), high intrinsic mobility (∼200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1), high Young's modulus (∼1.0 TPa), thermal conductivity (∼5000 W m−1 K−1), optical transmittance (∼97.7%), and good electrical conductivity,7–11 whereas it can be utilized for the development of polymer nanocomposites with superior properties that can be used in many different applications.12,13 However, its poor solubility in a wide range of solvents14,15 limits its further applications in many fields. On the other hand, the graphene's surface can be easily modified to graphene oxide by adding hydrophilic functional groups such as oxygen groups. Graphene oxide, GO, is a multi-layered material that consists of graphene layers that are functionalized with different oxygen species (hydroxyl, carboxyl, epoxy groups) either on the surface or in the perimeter of the individual sheets.16–18 The sheets exist in a parallel configuration due to weak van der Waals forces, π–π interactions and hydrogen bonds formed between the hydrophilic groups, forming interlayer galleries with distances of ∼8 Å. Water molecules, other polar moieties as well as polar hydrophilic polymers can interact favorably with the surfaces due to their hydrophilicity and reside within the galleries19 giving rise to the formation of hydrophilic polymer nanocomposites.20–23
000 cm2 V−1 s−1), high Young's modulus (∼1.0 TPa), thermal conductivity (∼5000 W m−1 K−1), optical transmittance (∼97.7%), and good electrical conductivity,7–11 whereas it can be utilized for the development of polymer nanocomposites with superior properties that can be used in many different applications.12,13 However, its poor solubility in a wide range of solvents14,15 limits its further applications in many fields. On the other hand, the graphene's surface can be easily modified to graphene oxide by adding hydrophilic functional groups such as oxygen groups. Graphene oxide, GO, is a multi-layered material that consists of graphene layers that are functionalized with different oxygen species (hydroxyl, carboxyl, epoxy groups) either on the surface or in the perimeter of the individual sheets.16–18 The sheets exist in a parallel configuration due to weak van der Waals forces, π–π interactions and hydrogen bonds formed between the hydrophilic groups, forming interlayer galleries with distances of ∼8 Å. Water molecules, other polar moieties as well as polar hydrophilic polymers can interact favorably with the surfaces due to their hydrophilicity and reside within the galleries19 giving rise to the formation of hydrophilic polymer nanocomposites.20–23
The main method to fabricate graphene oxide is the oxidation of the natural graphite by utilizing strong oxidation agents. The first methods used were performed initially by Brodie24 and then by Staudenmaier25 using nitric acid; however, sodium oxide was formed as a by-product and a relatively long time was necessary to complete the reaction. A solution to these problems was given by Hummers, replacing the nitric acid by sodium nitrate.26 Nowadays, the Hummers' method is the most widely used method for the oxidation of graphite in large scale and with relatively low cost; moreover, equally or even more important is that the specific oxidation method involves less toxic reagents.
Graphene oxide can form dispersions with long term stability in water as well as in many organic solvents27 due to its high hydrophilic and polar character28 making them suitable for a broad range of applications. Critical for the formation of a fine dispersion in either aqueous or organic media is the overall degree of oxidation, the distribution of the oxygen groups and the functionalization of the material.29–33 Atomistic molecular dynamics simulations predict that indeed both the oxidation pattern and the amount of water present are critical for the structure and properties of the obtained graphene oxide; the former affects the spatial arrangement and the relative orientation of the flakes as well as the interlayer distance and the distribution of particle size, whereas the latter influences both the diffusion of GO flakes and the counterion dynamics.33
On the other hand, the significant hydrophilicity of graphene oxide limits its dispersibility in non-polar solvents and its interactions with hydrophobic materials. In order to enhance its dispersibility, GO can be covalently functionalized with different molecules and polymers,34 however, the presence of such stabilizers is not desirable for certain applications.35 To avoid using such molecules, one can fine tune the hydrophilicity of the GO sheets by controlling their oxidation degree, therefore, resulting in adjustable interactions with both solvent molecules as well as other additive materials.36 Hummers’ method is a reaction that can be easily modified,2,37–39 thus offering a great potential for the improvement of the final oxidation product.
The oxidation behavior of graphite can be influenced by many factors, including porosity, anisotropy, granularity, degree of graphitization, irradiation dose, micropores, presence of impurities, temperature, pressure, shape, and oxidizing gas. For graphite of different grades, due to different manufacturing processes and/or different raw materials, the oxidation behavior has been found different.40 At the same time, the graphite particle size was found to influence significantly both the degree of oxidation as well as the yield of the obtained GO with the smaller particles being more effective.41 In general, the degree of oxidation of GO inevitably influences further beyond the residual functional groups, impacting the defects on the basal plane of the prepared graphene as well, which in turn affects its final physical and chemical properties.2,42 Therefore, it is considered essential to investigate systematically all factors that influence the degree of oxidation of GO and clarify its effect on the final structure and properties of GO for preparing high-quality graphene with desired characteristics.
In this work, we have employed a modified Hummers’ method to develop graphene oxides with varying degrees of oxidation; to achieve this, two parameters, the oxidation time and the amount of the oxidizing agent, were systematically changed. The effect of the different degrees of oxidation on the interlayer distance and the overall structure of the obtained graphene oxides as well as on their thermal properties and their thermal stability in terms of the reduction temperature and enthalpy and the % weight loss was investigated and discussed for the first time in the literature. A series of experimental techniques like X-ray diffraction (XRD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) and Raman spectroscopies were used to systematically verify the obtained results. In addition, atomistically resolved molecular dynamics (MD) simulations were employed to provide details on the organization of the GO flakes at the nanoscale, under a microscopic environment similar to the experimental conditions. Our study provides new insights on the duration of the oxidation process and the mass of the oxidant needed to achieve all the intermediate degrees of oxidation, up to a fully oxidized GO sample and on the structural features of the resulted materials.
2. Materials and methods
2.1. Reagents and consumables
Graphite powder, sodium nitrate (NaNO3), sulfuric acid 98% (H2SO4), potassium permanganate (KMnO4) and hydrogen peroxide (H2O2) 30% were purchased from Sigma-Aldrich. According to its datasheet, the graphite used is a powder of synthetic graphite with particles size less than 20 μm and density of 2.26 g cm−3.2.2. GO synthesis
The graphene oxide samples were prepared using a modified Hummers’ method.2 In a typical procedure, to obtain a fully oxidized material, the mass of potassium permanganate, KMnO4, usually utilized as the oxidizing agent, is 6 g and the oxidation time is 24 hours. In the present work, the time of oxidation as well as the mass of the oxidant were individually and systematically changed in order to achieve varying degrees of oxidation. More specifically, different oxidant masses ranging from 0.5 g to 6 g and oxidation times varying from 15 minutes to 24 hours were utilized. These parameters cover the whole range of reaction times and mass of the oxidant agent from excess to scarce while ensuring the prerequisite of the homogeneous mixing of the reactants. The synthesized materials and the different reaction parameters are shown in Table 1 where the code of each material denotes the time used for the oxidation reaction and the mass of the oxidant that was utilized.| Code | Oxidation time (h) | Oxidant mass (g) |
|---|---|---|
| Graphite | — | — |
| GO 0.25 h 1 g | 0.25 | 1 |
| GO 0.25 h 2 g | 0.25 | 2 |
| GO 0.5 h 2 g | 0.5 | 2 |
| GO 1 h 0.5 g | 1 | 0.5 |
| GO 1 h 1 g | 1 | 1 |
| GO 1 h 1.5 g | 1 | 1.5 |
| GO 1 h 2 g | 1 | 2 |
| GO 1 h 4 g | 1 | 4 |
| GO 1 h 6 g | 1 | 6 |
| GO 2 h 2 g | 2 | 2 |
| GO 2 h 4 g | 2 | 4 |
| GO 2 h 6 g | 2 | 6 |
| GO 3 h 2 g | 3 | 2 |
| GO 3 h 4 g | 3 | 4 |
| GO 3 h 6 g | 3 | 6 |
| GO 12 h 6 g | 12 | 6 |
| GO 24 h 6 g | 24 | 6 |
The procedure used was as follows: 1 g of graphite was dispersed in 48 ml of concentrated sulfuric acid, H2SO4, and the dispersion was left under constant stirring in an ice-bath for 20 minutes. Subsequently, 1 g of NaNO3 was added and the mixture was stirred for another 1 hour. Thereafter, the specific amount of KMnO4 (between 0.5 g to 6 g) was added and the suspension was left stirring for a certain amount of time (15 minutes to 24 hours). Following the stirring, the dispersion was removed from the ice-bath and heated to 35 °C for 100 minutes. Next, 80 ml of nanopure water was added and the mixture was heated to 90 °C for 40 more minutes. Subsequently, the reaction beaker was removed from the heat, and 200 ml of water and 20 ml of H2O2 were added and the mixture was left to cool. Finally, the mixture was centrifuged several times at 4500 rpm for 10 minutes, in order the material to precipitate, and the sediment was washed thoroughly with water until the supernatant reached a pH value of 7. The synthesized GOs were then left to dry for 48 hours at 60 °C under vacuum. When the samples were completely dried, they were pulverized using a pestle and a mortar until a fine powder was formed.
2.3. Characterization methods
The structural characterization of all graphene oxides was performed by X-ray diffraction, XRD, on a Brucker (Billerica, MA, USA) D8 Advance X-ray diffractometer, using Cu Kα radiation (λ = 1.5406 Å). Measurements were performed for diffraction angles 2θ from 1.5° to 50°, with a scanning step of 0.01° and measuring time 1 s per step, whereas the voltage and current used were 40 kV and 40 mA, respectively.The thermal properties of the GOs were analyzed using differential scanning calorimetry, DSC, with a TA Instruments Discovery DSC250 calorimeter. All measurements were performed in a temperature range between 30 °C and 300 °C with a heating/cooling rate of 10 °C min−1 under nitrogen flow to create inert conditions. The cooling was performed using a Refrigerated Cooling System 120.
The thermal stability of the materials was carried out using thermogravimetric analysis, TGA, with a SDT600 TGA/DTA of TA Instruments under argon flow between 25 °C and 700 °C with a heating rate of 10 °C min−1.
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) measurements were performed using a Bruker Vertex 70v FTIR vacuum spectrometer equipped with an A225/Q platinum ATR unit with a single reflection diamond crystal, which allows the infrared analysis of unevenly shaped solid samples and liquids through total reflection measurements, over the spectral range of 4000–700 cm−1.
X-ray photoelectron spectroscopy (XPS) measurements were performed using a UHV chamber (P ∼ 5 × 10−10 mbar) equipped with a SPECS Phoibos 100-1D-DLD hemispherical electron analyzer and a non-monochromatized dual-anode Mg/Al X-ray source for XPS. The XP spectra were recorded using MgKa at 1253.6 eV photon energy and an analyzer pass energy of 10 eV. The analyzed area was a spot of 3 mm diameter. The samples were measured in powder form, and for XPS measurements, they were pressed on indium foil.
All XRD, XPS, ATR-FTIR and Raman spectroscopy measurements were performed at room temperature.
2.4. Simulation method and system details
All-atom molecular dynamics (MD) simulations were performed using GROMACS software in the isobaric–isothermal (NPT) statistical ensemble.43 The model simulated system was an oxidized graphene sheet with carboxyl, hydroxyl and epoxy groups distributed on both surfaces as well as around the edges, according to the Lerf–Klinowski model;44 the lateral dimension of the graphene sheet was 3.2 × 4 nm2. Moreover, all the flakes that were utilized to constitute the investigated systems were identical.45 To resemble experimental systems that contain a certain amount of water, depending on their hydrophilicity, the simulated system includes 10 wt% water, as well. The carboxyl groups of the oxidized graphene flakes were taken to be deprotonated with a degree of deprotonation close to neutral pH conditions to resemble the corresponding experimental system.The OPLS-AA forcefield46 was used for the description of the energetic parameters for all graphene molecules and the TIP3P explicit solvent model47–49 for water molecules. After energy minimization, a production run followed for 100 ns, whereas the last 40 ns was used for the statistical data analysis. Because of the limited motion of the graphene sheets in such low water concentration systems, three runs starting from different initial configurations were performed in order to enrich data statistics. Each configuration comprised of 56 GO flakes, a number of water molecules necessary to achieve the desired water weight fraction and an appropriate number of counterions to secure the overall electrical neutrality of the system. More details concerning the composition of the system, and the simulation protocol, can be found in a previous publication.33
3. Results and discussion
3.1. Varying oxidation time
Different series of materials were synthesized utilizing a constant mass of the oxidation medium and varying the time of the oxidation reaction. Reaction times from 0.25 to 24 h were utilized; it is noted that reaction times shorter than 15 min (0.25 h) were not attempted because adequate mixing of the reaction constituents and thus a homogeneous mixture at this small interval would not be assured.Fig. 1 shows X-ray diffraction (XRD) measurements of two series of materials synthesized utilizing 2 g (Fig. 1(a)) and 6 g (Fig. 1(b)) of KMnO4 and varying reaction times. The corresponding measurement of the original graphite material is included for comparison in both cases, in which a very sharp peak at 2θ = 26.5° is observed; this corresponds to an interlayer distance of d001 = 3.3 Å, calculated based on Bragg's law, in accordance with what is well known from the literature.9,50
 | ||
| Fig. 1 XRD measurements of GO samples with (a) 2 g and (b) 6 g of oxidant mass for various oxidation times. The corresponding measurement for graphite is included for comparison. All curves have been shifted vertically for clarity. | ||
From the XRD measurements shown in Fig. 1(a), it is evident that when 2 g of oxidant is utilized, a successful oxidation of the graphite material has been achieved for all the materials synthesized for reaction periods between 0.25 h (15 min) and 3 h. This is attested by the appearance of a new peak corresponding to the layered periodic structure of graphene oxide in all diffractograms and for all the reaction periods. In all cases, the main diffraction peak appears at an angle of 2θ ∼ 12.0° ± 0.2° that corresponds to an interlayer distance of d001 ∼ 7.4 Å ± 0.1 Å. This increase of the interlayer distance is attributed to the existence of oxygen-containing functional groups functionalizing the graphene sheets, whereas the presence of a small quantity of water is also anticipated due to the increased hydrophilicity of the obtained GO. At the same time, there is only a trace of the graphite peak at 2θ = 26.5° indicating that there is still a small number of sheets that have not been oxygen-functionalized when 2 g of KMnO4 is used, even after 3 h of reaction.
As far as the series of GOs synthesized with 6 g of oxidant is concerned, their XRD measurements (Fig. 1(b)) show that there is practically no dependence of the observed structure on the reaction time for periods between 1 and 24 h. The main diffraction peak is observed at the same diffraction angle, however in this case, this angle is lower than the one measured for the materials that had been oxidized with lower amount (i.e., 2 g) of oxidant. The diffraction angle in this case is 2θ = 11.0° + 0.5° corresponding to an interlayer distance of d001 = 8.1 ± 0.3 Å.22,23,51,52 Furthermore, there is no trace of a peak at the diffraction angle corresponding to the existence of graphite; the latter in conjunction with the increased interlayer distance indicates that the materials have been fully oxidized and that all sheets possess the maximum amount of oxygen groups. Aside from the interlayer distance of the periodic structure obtained from the position of the main diffraction peak, one can calculate the average size of the graphene oxide assembly and therefore the number of layers in each crystallite. The size of such a crystallite, τ, can be calculated according to the Scherrer equation,  , where K is a shape factor equal to 0.9, λ is the wavelength of the X-rays and θ and β are the position and half width at half maximum of the main diffraction peak, respectively. The so-calculated size of the crystallite in conjunction with the interlayer distance estimated by the position of the corresponding diffraction peak results in the number of oxidized sheets per GO particle, that is ∼11 ± 1 and ∼13 ± 1 in the case where 2 g and 6 g of KMnO4 were utilized, respectively. It should be noted that in the case of non-oxidized graphite, each crystallite consists on the average of ∼70 sheets, indicating that the oxidation reaction leads to a significant reduction in the coherence of the layered periodic structure. From the discussion above, it follows that for a constant amount of oxidant, the oxidation time does not play an important role in the overall degree of oxidation because the final structure of the synthesized materials is very similar.
, where K is a shape factor equal to 0.9, λ is the wavelength of the X-rays and θ and β are the position and half width at half maximum of the main diffraction peak, respectively. The so-calculated size of the crystallite in conjunction with the interlayer distance estimated by the position of the corresponding diffraction peak results in the number of oxidized sheets per GO particle, that is ∼11 ± 1 and ∼13 ± 1 in the case where 2 g and 6 g of KMnO4 were utilized, respectively. It should be noted that in the case of non-oxidized graphite, each crystallite consists on the average of ∼70 sheets, indicating that the oxidation reaction leads to a significant reduction in the coherence of the layered periodic structure. From the discussion above, it follows that for a constant amount of oxidant, the oxidation time does not play an important role in the overall degree of oxidation because the final structure of the synthesized materials is very similar.
Fig. 2 shows differential scanning calorimetry (DSC) measurements of graphene oxides that have been oxidized during different reaction times when 6 g of oxidant is utilized, within the temperature range wherein the reduction of GO takes place.
 | ||
| Fig. 2 DSC measurements expressed in terms of heat capacity for graphene oxides oxidized with 6 g of oxidant during varying reaction times. | ||
All samples demonstrate a strong exothermal peak that corresponds to their reduction, in agreement with previous literature results.22,23,52,53 The maximum of each peak can provide the corresponding reduction temperature, Tr, whereas the integral under each peak represents the respective reduction enthalpy, ΔH. In analogy with the discussion concerning the periodic structure of these materials, at a constant amount of oxidant, both the reduction temperature and the reduction enthalpy do not show any appreciable dependence on the oxidation time, because they can be determined to be Tr = 212 ± 4 °C and ΔH = 337 ± 15 cal g−1, respectively. Moreover, the results are qualitatively very similar showing no dependence on the reaction time for the GOs oxidized utilizing different masses of the oxidation agent indicating that the oxidation time does not influence the degree of oxidation.
In addition to the thermal properties of the materials, information on the degree of oxidation of the different samples can be obtained via the estimation of the % weight loss of the oxygen containing groups as well as via the differences in the decomposition steps obtained by thermogravimetric analysis, TGA. Fig. 3 shows TGA measurements for graphene oxide samples obtained after different reaction times utilizing 2 g (Fig. 3(a)) and 6 g (Fig. 3(b)) of oxidant, respectively. All samples exhibit a similar thermal degradation pattern consisting of three major steps. The first weight reduction is completed up to T ∼ 100 °C and is attributed to the loss of water molecules, which via hydrogen bonding reside between the galleries of the oxidized materials due to their hydrophilic character. For temperatures T ∼ 150–220 °C, a second step in the weight loss is observed, which can be ascribed to the decomposition of labile oxygen-containing functional groups, like hydroxyl groups (OH). In the region between 220 and 300 °C, the weight loss is associated with the removal of the more stable oxygen moieties such as the carboxyl (COOH) and epoxy groups (C–O–C).23,51,52,54,55 Finally, at higher temperatures, a weak weight loss is initiated, corresponding to the pyrolysis of the carbon skeleton that is completed at temperatures outside the range of the performed measurements.54,55
 | ||
| Fig. 3 TGA measurements of GO samples oxidized during different reaction times utilizing (a) 2 g and (b) 6 g of oxidant mass. The measurement of graphite is shown for comparison. | ||
A common feature that can be observed in all samples prepared with 2 g of oxidant is that they display almost the same % weight loss with only minor deviations. For oxidation times varying between 15 minutes and 3 hours, the water content approaches an average value of about 9.3% with a standard deviation ±1%, while the organic content amounts to 49% with a standard deviation of ±2%, indicating that all materials possess very similar degree of oxidation. If this was not the case, a different amount of both water and organic content would be anticipated. An increased amount of water and an increased organic content are observed for the series of materials synthesized utilizing 6 g; however, no effect of the different reaction time (ranging from 1 to 24 hours) can be observed regarding either the water content or the amount of oxygen groups, in this case as well. Therefore, TGA provides a supplementary support to the conclusion that the oxidation time within the examined limits does not significantly influence the degree of oxidation.
ATR-FTIR measurements were performed to provide information regarding the oxygen functional groups of the materials synthesized during different reaction times for the samples prepared with different oxidant mass. Fig. 4 shows the corresponding measurements for the graphene oxide samples prepared with 6 g of oxidant, for reaction times ranging between 1 and 24 h. The corresponding measurement of graphite is included in the figure for comparison, however, it does not display any peaks, as expected, because the dipole moment of the carbon atoms is zero. In contrast, the spectra of all GOs show the same peaks which correspond to the vibrations of different carbon–oxygen bonds; more specifically, the peaks at 1045 cm−1, 1164 cm−1, 1384 cm−1, 1729 cm−1 and 3418 cm−1 are related to the C–O, epoxy (C–O–C), carboxyl (C–OH), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and O–H vibrations, respectively.51,56–58 The presence of these bonds between carbon and oxygen atoms attests to the full oxidation of the materials because in all cases not only hydroxyl but also carboxyl and even epoxy groups, which possess high binding energy, have been formed. At the same time, it is observed that oxidation time has no effect on the formation of the functional groups, because they appear the same for all samples for all oxidation times examined.
O) and O–H vibrations, respectively.51,56–58 The presence of these bonds between carbon and oxygen atoms attests to the full oxidation of the materials because in all cases not only hydroxyl but also carboxyl and even epoxy groups, which possess high binding energy, have been formed. At the same time, it is observed that oxidation time has no effect on the formation of the functional groups, because they appear the same for all samples for all oxidation times examined.
 | ||
| Fig. 4 ATR-FTIR measurements of GO samples synthesized with 6 g of oxidant during different reaction times. The measurement of graphite is shown for comparison as well. The curves have been shifted for clarity. | ||
Lastly, a quantitative characterization of the degree of oxidation of the series of GOs which were synthesized during different reaction times was performed using X-ray photoelectron spectroscopy, XPS, in order to quantify the oxygen functional groups on the graphene sheets as well as the carbon (C) to oxygen (O) ratio. Fig. 5 shows the XPS measurements of the graphene oxides synthesized using 2 g of oxidant during 15 min, 1 h and 3 h.
 | ||
| Fig. 5 XPS measurements of GOs synthesized with 2 g of oxidant during different reaction times. | ||
Table 2 lists the % amount of carbon and oxygen that resulted from the peak areas of the detailed C1s and O1s spectra divided by the corresponding relative sensitivity factors given by the photoemission cross-sections, the electrons kinetic energy and the analyser transmission for the GOs that were oxidized for different reaction times. It is obvious that these concentrations remain constant for the three materials and independent of the reaction time in agreement with the results of all techniques discussed so far; the analysis results in an average C:O atomic ratio of 69:31%.
| Code | C–C sp2 | C–C sp3 | C–O(H) | CO |
COOH | %C at | %O at | C/O |
|---|---|---|---|---|---|---|---|---|
| GO 0.25 h 2 g | 37.5 | 14.2 | 37.1 | 8.1 | 3.1 | 69.2 ± 0.2 | 30.8 ± 0.2 | 2.2 |
| GO 1 h 2 g | 41.1 | 6.3 | 43.4 | 6.8 | 2.4 | 68.9 ± 0.2 | 31.1 ± 0.2 | 2.2 |
| GO 3 h 2 g | 45.2 | 7.2 | 37.1 | 8.5 | 2.0 | 69.4 ± 0.2 | 30.6 ± 0.2 | 2.3 |
Thereupon, deconvolution of the C1s peak for each measurement was performed, to quantify the presence of the oxygen functional groups. Fig. 6 shows such an analysis for the graphene oxides that were synthesized using 2 g of oxidant for three different reaction times.
 | ||
| Fig. 6 C1s peak deconvolution for the determination of O functional groups in GO oxidized with 2 g of oxidant during (a) 15 min, (b) 1 h and (c) 3 h of oxidation reaction. | ||
As shown from the analysis, the C peak can be deconvoluted to five peaks corresponding to the C–C (sp2), C–C (sp3), C–OH, CO και COOH bonds at 284.2 eV, 284.9 eV, 286.2 eV, 287.4 eV and 288.7 eV, respectively.59–61 The percentage of each functional group is listed in Table 2, as well. It is observed that the ratios are almost the same for the graphene oxides oxidized during different reaction times, with some non-systematic deviations. In a similar manner, the O1s peak deconvolution can be performed. The O1s peak can be analyzed into three components attributed to the C–O single bonds (epoxides/hydroxides) with a binding energy of 532.25 ± 0.05 eV to CO double bonds (carbonyls/carboxyls) at 530.60 ± 0.05 eV and to adsorbed H2O at ∼534.5 eV.61 The percentage of each component is 90 ± 1 for C–O, 5 ± 1 for CO and 5 ± 1 for adsorbed water across all samples (adsorbed water for 15 min, 1 and 3 h), indicating that the oxidation time longer than 15 min has no effect on the oxidation of graphene. The corresponding analysis is shown in Fig. S1 of the ESI.†
Consequently, from the overall investigation of the effect of the reaction time on the degree of oxidation of GO, it can be derived that for a certain amount of oxidant mass, the oxidation time does not show any important effect, because no significant changes were observed in either the obtained structure and the chemical constitution of the materials or in their thermal properties and their thermal decomposition. However, it should be noted that this conclusion applies for oxidation times longer than 15 min because shorter time intervals could not ensure the homogeneity of the synthesized graphene oxides.
3.2. Varying oxidant mass
Following the result that the oxidation time has no significant effect on the degree of oxidation of the synthesized GOs, the possible influence of the mass of the oxidant for a certain reaction time was investigated; this is anticipated to affect the degree of oxidation and therefore the overall structure of the prepared materials and the spatial arrangement of the GO sheets.33 To elaborate on this, six materials were synthesized utilizing a varying amount of oxidant ranging from 0.5 g to 6 g keeping the oxidation time constant at 1 h. The code names of all GOs together with the parameters of the synthesis are presented in Table 1.Fig. 7(a) shows the XRD measurements for all the synthesized graphene oxides together with the respective measurement of graphite that is shown for comparison. As discussed above, the graphite shows a very sharp peak at 2θ = 26.5° corresponding to its well-known interlayer distance of d002 = 3.3 Å. The measurement of the graphene oxide that has been synthesized utilizing 0.5 g of KMnO4 (GO 1 h 0.5 g) shows a diffraction peak at the exact same angle with that of graphite indicating that this amount of oxidant is not enough to cause a significant oxidation of the material. However, there is a strong broadening of this peak which suggests that the coherence of the graphite structure is significantly reduced. Moreover, another very broad diffraction peak exists in the diffractogram at lower angle, 2θ = 14.2°. This corresponds to an interlayer distance of d001 = 6.2 Å and is attributed to the presence of a low percentage of oxidized sheets. Additionally, the low intensity of the GO peak in comparison with that of the graphite corroborates the fact that GO 1 h 0.5 g possesses a low degree of oxidation. When the structure of the materials that have been synthesized using a higher amount of oxidant is investigated, two characteristic findings can be derived from the diffractograms of Fig. 7(a). As the mass of the oxidant increases, there is a systematic decrease of the ‘graphite’ peak, which is accompanied by the concurrent increase of the intensity of the second ‘graphene oxide’ peak. The latter is located at a lower scattering angle, it eventually becomes the dominant peak and it systematically shifts towards lower diffraction angles. This peak is located at 2θ = 13.4° corresponding to an interlayer distance of d001 = 6.6 Å for the GO 1 h 1 g sample and reaches 2θ = 10.9° (d001 = 8.1 Å) for GO 1 h 6 g, which is the sample synthesized with the highest amount of oxidant.
 | ||
| Fig. 7 (a) XRD measurements of GO samples oxidized with varying oxidant masses during 1 h of reaction time. The graphite peak is included for comparison. (b) Dependence of the interlayer distance on the mass of the oxidant used for the oxidation reaction. | ||
All diffraction angles together with the corresponding interlayer distances of the synthesized graphene oxide samples are reported in Table 3. It can be noted that for all GOs synthesized using up to 2 g of oxidant, the graphite peak remains discernible in the diffractogram, indicating that for those materials, the oxidation is incomplete. On the other hand, as already discussed, the interlayer distance of the different GOs increases upon increasing the mass of the oxidant due to the increasing number of oxygen-containing functional groups attached to the sheets; the interlayer dependence on the mass of the oxidant used is shown in Fig. 7(b).
| Code | 2θ (°) | d (Å) | Enthalpy (cal g−1) | %Water | %Weight loss |
|---|---|---|---|---|---|
| GO 1 h 0.5 g | 14.2 ± 0.6 | 6.2 ± 0.3 | 68.9 ± 2 | 2 | 16 |
| GO 1 h 1 g | 13.4 ± 0.3 | 6.6 ± 0.2 | 152.5 ± 3 | 2 | 21 |
| GO 1 h 1.5 g | 12.1 ± 0.2 | 7.3 ± 0.1 | 198.9 ± 3 | 4 | 32 |
| GO 1 h 2 g | 11.8 ± 0.1 | 7.5 ± 0.1 | 282.2 ± 2 | 9 | 38 |
| GO 1 h 4 g | 11.2 ± 0.2 | 7.9 ± 0.1 | 290.6 ± 2 | 11 | 52 |
| GO 1 h 6 g | 10.9 ± 0.1 | 8.1 ± 0.1 | 337.5 ± 2 | 12 | 51 |
It is evident that the interlayer distance, d001, increases with the mass of the oxidant used, reaching a plateau for the materials that have been oxidized using 4 and 6 g of oxidant which are the ones for which the ‘graphite’ peak has vanished, indicating that the oxidation is complete. Furthermore, aside the shift of the peak towards lower diffraction angles, a narrowing of its width can be observed when the mass of the oxidant is increased, indicating that completion of the oxidation reaction leads to a better coherence of the GO structure; an estimation of the number of layers per crystallite, as discussed earlier, yields on average ∼4 sheets for the barely oxidized GO 1 h 0.5 g, and ∼13 for the fully oxidized GO 1 h 6 g sample.
It is thus evident that the mass of the oxidant has a significant effect on the interlayer distance between the GO sheets and therefore to the formed structure.
To obtain a more detailed view on the spatial arrangement of the oxidized flakes beyond the information provided by the experimental techniques, fully atomistic molecular dynamics simulations were employed. Our goal was to visualize generic features of the microscopic morphology developed in a dispersion of oxidized flakes bearing oxygen groups similar to those detected in the experimental systems (i.e., carboxylates, hydroxyls and epoxides). A system with a carbon to oxygen ratio (C/O) of 4.94/1 was examined that was close to the 4.7/1 ratio detected by the XPS measurements for GO 1 h 0.5 g prepared by using 0.5 g of oxidant (see below). The total water content in the simulated system was 10 wt%; however, it can be calculated that 1.5 wt% of the water molecules reside between the GO galleries. At that low water content, the system is practically in a dynamically arrested state in which the structural features of the formed GO clusters remain essentially unchanged.33 To check the consistency between the simulated model and the corresponding synthesized material, the tendency for the formation of clusters with parallel-oriented sheets has been examined and the inter-flake separation has been compared to that obtained from the analysis of the corresponding XRD spectra. Fig. 8(a) shows the distribution of the centers of mass of the GO flakes in a direction normal to a GO plane that is taken as a reference. For the calculation of this profile, only the neighboring GO flakes have been taken into account, for which the projection of their centers of mass vector onto the plane of a reference GO falls within the area covered by the reference flake. Evidently, the most probable distance for the detection of neighboring GO centers of mass is close to 6 Å ambilateral from a GO flake.
 | ||
| Fig. 8 (a) Distribution of the GO centers of mass as a function of their distance from the plane of a reference GO flake, in a direction normal to the latter. The coordinate 0 in the x-axis corresponds to the position of the center of mass of a reference flake. The negative and positive values in this axis denote the presence of GO flakes at the left and at the right of central plane, respectively. (b) Orientational parameter of the GO flakes as a function of their separation. | ||
To verify that the GO flakes separated by that distance, assuming parallel orientation, the orientational order parameter, P2(cosθ(r)), of the GO flakes has been calculated; it is given by  , where θ(r) is the angle between the planes of two flakes whose centers of mass are separated by distance r. The orientational parameter assumes a value of 1 if the examined planes are parallel, −0.5 if the planes are perpendicular to each other and 0 if there is no preferred relative orientation between the planes. Fig. 8(b) shows that the flakes separated by a distance close to 6 Å, indeed assume parallel orientations, that is, they form stacked configurations. This value of the interlayer distance in stacked GO arrangements is in good agreement with the separation detected experimentally by XRD measurements, i.e. 6.2 Å (see Fig. 7(b) and Table 3). At larger separations, the tendency of GO flakes to assume a parallel arrangement diminishes, indicating the absence of GO clusters with a longer orientational order between their flakes.
, where θ(r) is the angle between the planes of two flakes whose centers of mass are separated by distance r. The orientational parameter assumes a value of 1 if the examined planes are parallel, −0.5 if the planes are perpendicular to each other and 0 if there is no preferred relative orientation between the planes. Fig. 8(b) shows that the flakes separated by a distance close to 6 Å, indeed assume parallel orientations, that is, they form stacked configurations. This value of the interlayer distance in stacked GO arrangements is in good agreement with the separation detected experimentally by XRD measurements, i.e. 6.2 Å (see Fig. 7(b) and Table 3). At larger separations, the tendency of GO flakes to assume a parallel arrangement diminishes, indicating the absence of GO clusters with a longer orientational order between their flakes.
Based on this validation of the simulated model, the spatial arrangement of the flakes in the simulated system has been examined in more detail. Fig. 9(a) shows a snapshot, where both low-occupancy stacked configurations coexist with GO assemblies of greater size. To better understand the reasons for a GO spatial arrangement with such characteristics, we must note that although there is a tendency of the GO flakes to form stacked configurations (e.g., through hydrogen-bond formation),33 the presence of the oxidized groups disrupts the π–π interactions responsible for the stacking in pristine graphene, while the presence of entrapped water molecules between the hydrophilic flakes even at relatively low water contents may also affect the structural characteristics of the clusters.62,63 These factors combined with kinetic reasons (due to the low water content) essentially prohibit the formation of extended crystallites, resulting in the coexistence of low-populated clusters of stacked flakes together with larger in size clusters consisting of non-parallel flakes. Such configurations could play a significant role in key physical properties of the dispersions, such as their mechanical and electrical response.63,64
 | ||
| Fig. 9 (a) Representative configuration of a system where different clusters of GO flakes are observed. The flakes are shown in dark cyan, water molecules are shown in blue and the oxygen atoms are shown in red. (b) Probability distribution h(Ns) of the number of GO flakes within a cluster, Ns, characterizing a system with an overall 10 wt% water content. | ||
Based on such snapshots, Fig. 9(b) portrays the probability distribution of the clusters formed by the GO flakes as a function of the number of flakes they contain, irrespective of their relative orientation. The implementation of the clustering detection was based on a previously described algorithm.65 Evidently, the most frequently met clusters are comprised of 2–4 flakes in good agreement with the XRD results. However, clusters with larger size (not necessarily comprised of parallel flakes as argued above) are also present.
The morphology of the synthesized GOs was imaged utilizing scanning electron microscopy and the images are shown in Fig. S2 of the ESI† together with the image of the original graphite material. It is obvious that the structure of graphite is relatively compact and as the oxidant mass increases, the layers start getting separated from one another. In the case of the GO 1 h 0.5 g sample, the stack of layers is larger than the GO 1 h 1.5 g sample, indicating the presence of more parallel layers. On the other hand, in GO 1 h 6 g, a more ‘exfoliated’ structure is observed. Moreover, the presence of cracks and a crumpled structure becomes more prominent as the oxidation degree increases, indicating an enhanced disorder of the GO sample.
The thermal properties and more specifically the thermal reduction of all graphene oxides that were synthesized using different amounts of oxidant were investigated utilizing DSC. Fig. 10 shows the thermograms of all materials oxidized for 1 h in terms of the specific heat. In all curves, an exothermic peak is evident from which both the reduction temperature and the heat of reduction can be calculated. It is clear that the reduction temperature of the samples is constant with an average value at 210 ± 3 °C. On the other hand, the reduction enthalpy that can be calculated by the integral under each peak gradually increases with increasing oxidant mass and for the larger amount of oxidant, it seems to reach a plateau, in contrast to the case of varying oxidation time; all values are reported in Table 3 as well. This increase indicates an increase of the overall degree of oxidation of the materials. Complete oxidation is achieved for oxidant mass between 4 and 6 g according to DSC measurements in agreement with the findings of the structural investigations of the materials based on XRD that was discussed above.
 | ||
| Fig. 10 (a) DSC thermograms, expressed in units of heat capacity, for GOs oxidized with different oxidant masses during 1 h of reaction. (b) Heat of reduction as a function of the oxidant mass calculated based on the DSC measurements in (a). | ||
Fig. 11 shows TGA measurements of all GO materials. In all cases, three steps of thermal decomposition (removal of the oxygen groups) can be observed, with different amplitudes. As the oxidant mass increases, both the overall weight loss and the percentage of the first weight loss that corresponds to the amount of water that is adsorbed on the GO sheets increase (they are both reported in Table 3); the latter is attributed to the increased hydrophilicity of the materials due to the increased degree of oxidation. It is noted that the amount of water that resides within the galleries of GO 1 h 0.5 g is measured to be ∼2 wt%, a value that is very close to the one predicted by atomistic molecular dynamic simulations for a very similar system, as described above. The thermal decomposition traces of GO 1 h 4 g and GO 1 h 6 g are almost identical which indicates that these materials possess a similar number of functional groups and therefore a very similar degree of oxidation in accordance with the results obtained with all the characterization techniques discussed.
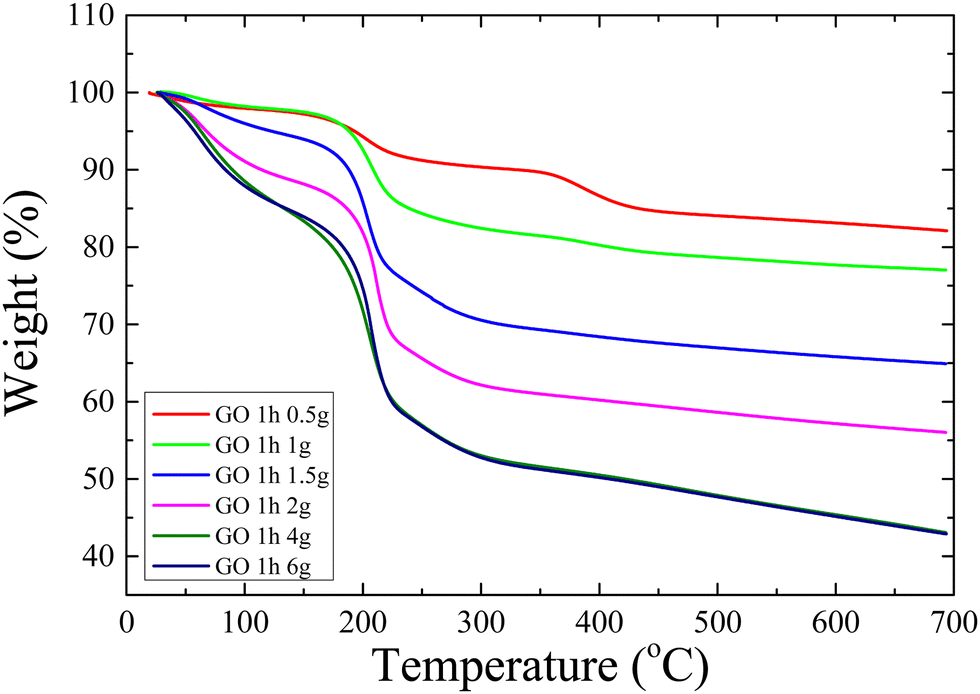 | ||
| Fig. 11 TGA measurements of GO samples oxidized with varying oxidant masses during 1 h of reaction. | ||
A widely accepted technique to provide the quality of graphene oxides is Raman spectroscopy; in such measurements, the ratio of D and G bands (ID/IG) can be an indication on how the uniformity of the carbon lattice changes during the progress of the oxidation reaction and the formation of the oxygen groups. Such changes are considered as defects of the carbon lattice and ID/IG determines their density. The Raman spectroscopy measurements for all synthesized GOs as well as of the original graphite are shown in Fig. S3 of the ESI.† The D and G bands can be observed for all materials and the ratio ID/IG equals 0.64 for the GO synthesized with 0.5 g of oxidant, whereas for all the other materials, its value varies from 0.96 to 1.06. It is noted that the respective value for graphite is ∼0.07. Therefore, it seems that apart from the sample synthesized with a small amount of oxidant mass, in all other cases, the effective oxidation of graphite to GO can be confirmed. Moreover, all GOs seem to be of similar quality and with the same density of defects.
All the results discussed so far are confirmed using XPS in order to quantitatively determine the C and O content as well as the percentage of the different functional groups of the synthesized materials. Fig. 12 shows the XPS wide scans and Fig. 13 shows the deconvolution of the C1s peaks. It is observed that as the amount of the oxidant mass increases, the intensity of the O peak increases from 18 to 40% and concurrently the intensity of the C peak decreases from 82 to 60%. The percentages for all materials are shown in Table 4 from which it is noted that GO 1 h 4 g and GO 1 h 6 g possess a very similar C/O ratio.
 | ||
| Fig. 12 XPS measurements of GOs synthesized with different oxidant masses during 1 h of reaction time. | ||
 | ||
| Fig. 13 C1s peak deconvolution for the determination of O functional groups of GOs synthesized with different oxidant masses during 1 h of reaction time, (a) G) 1h 0.5g, (b) GO 1h 1g, (c) GO 1h 1.5g, (d) GO 1h 2g, (e) GO 1h 4g and (f) GO 1h 6g. | ||
| Code | C–C sp2 | C–C sp3 | C–O(H) | CO |
COOH | %C at | %O at | C/O |
|---|---|---|---|---|---|---|---|---|
| GO 1 h 0.5 g | 62.4 | 5.9 | 20.0 | 6.8 | 4.8 | 82.4 ± 0.2 | 17.6 ± 0.2 | 4.7 |
| GO 1 h 1g | 52.4 | 17.7 | 22.0 | 5.6 | 2.2 | 80.3 ± 0.2 | 19.7 ± 0.2 | 4.1 |
| GO 1 h 1.5g | 41.8 | 9.0 | 40.2 | 6.0 | 3.0 | 71.9 ± 0.2 | 28.1 ± 0.2 | 2.6 |
| GO 1 h 2g | 41.0 | 6.3 | 43.4 | 6.8 | 2.4 | 68.9 ± 0.2 | 31.1 ± 0.2 | 2.2 |
| GO 1 h 4 g | 42.2 | 8.9 | 35.4 | 8.6 | 4.9 | 62.1 ± 0.2 | 37.9 ± 0.2 | 1.6 |
| GO 1 h 6 g | 31.5 | 10.5 | 45.9 | 9.1 | 3.0 | 60.4 ± 0.2 | 39.6 ± 0.2 | 1.5 |
From the C1s peak deconvolution (Fig. 13), it is evident that the intensity of the peak at 284.2 eV corresponding to the sp2 C–C peak is reduced, whereas there is a simultaneous and significant increase of the C–OH peak at 286.2 eV, as the oxidant mass increases (Table 4). The hydroxide and O content increase with the simultaneous decrease of the C content and the CC ratio of the carbon plane proves that the oxidizing agent mass directly affects the oxidation degree of the samples. Moreover, the less notable change of the GO 1 h 4 g and GO 1 h 6 g samples probably means that they have the same oxidation degrees. A deconvolution of the O1s core level spectra has been performed as well. The peak shapes of the O1s spectra for the different oxidant masses are similar; however, there are significant variations in the peak intensities, with a systematic increase in intensity as the oxidant mass increases due to the enhanced oxidation of carbon atoms.
It is evident that for a certain period of oxidation, the mass of the oxidizing agent significantly affects the degree of oxidation and therefore the structure and the thermal properties of the samples. By increasing the oxidant mass, the interlayer distance increases and reaches a plateau for oxidant mass between 4 and 6 g. The same behavior was observed from DSC, TGA and XPS measurements. This increase is attributed to the different number of oxygen groups that each sample contains, as was quantitatively shown by XPS. Overall, our results prove that at oxidation periods longer than 15 min, the GO degree of oxidation can only be affected by the mass of the oxidizing agent with 4 g being sufficient to yield fully oxidized graphene oxide materials.
4. Conclusions
In this work, the effect of the oxidation time and of the mass of the oxidation agent on the final degree of oxidation of synthesized graphene oxides was investigated. All materials were synthesized utilizing a modified Hummers' method which is considered as the most efficient, fast and less toxic method for the oxidation of graphite. A plethora of experimental techniques in conjunction with atomistic molecular dynamics simulations were employed to examine the structural and thermal properties of the materials.As far as the role of oxidation time at constant mass of the oxidant is concerned, XRD measurements showed that the reaction time does not affect the interlayer distance and consequently the structure of the synthesized materials. In addition to the structural investigation, thermal analysis (DSC and TGA) revealed that the enthalpy of reduction, the reduction temperature as well as the %weight loss were not affected by the time of the oxidation. Additionally, ATR-FTIR and XPS analysis showed that the oxygen-containing functional groups and the % content of C and O do not change with increasing time of oxidation. Consequently, the oxidation time does not seem to impart any significant effect on the final oxidation degree of the GO samples.
Regarding the effect of the oxidant mass at constant oxidation time, it was found that its increase leads to larger interlayer spacings and to a more coherent structure (XRD), accompanied by an increase in the reduction enthalpy (DSC), as well as in the weight loss and the hydrophilicity (TGA) of the materials. At the same time, the C/O ratio decreases upon the increase of the mass of the oxidant, as revealed by XPS measurements. Four (4) grams of oxidant seems to be sufficient to achieve a complete oxidation of graphite, indicating that the commonly utilized 6 grams are more than necessary for the complete oxidation of the samples and they are utilized in excess. Therefore, changing the mass of the oxidant offers a way of modifying the degree of oxidation and thus the hydrophilicity of graphene oxide in a controllable manner, and thus provides a way to alter the interactions with other materials opening new routes in their applications.
The structural information obtained experimentally, regarding the interlayer distance and the average size of the crystallites comprised of stacked GO sheets, is in qualitative and quantitative agreement with findings from atomistic molecular simulations. Moreover, the simulation results indicate the coexistence of clusters of a few stacked GO flakes with orientationally disordered ones, and a certain percentage of entrapped water molecules within the galleries, the presence of which should be taken into account when a bottom-up design of such materials with desired thermal, mechanical and electrical properties is to be performed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund – ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning 2014–2020” in the context of the project “POLYGRAPH” (MIS: 5050562).” This work received a computational support from the Greek Research & Technology Network (GRNET) in the National HPC facility – ARIS – under project ID CONGRESS. The authors also acknowledge the use of computational resources of the High-Performance Computing Infrastructure of the Aristotle University of Thessaloniki (AUTh). The authors acknowledge the access to the XPS facility obtained through the ‘Innovation-EL’ National Research Infrastructure on nanotechnology, advanced materials and micro/nanoelectronics” (MIS 5002772).References
- A. K. Geim and K. S. Novoselov, The Rise of Graphene, Nat. Mater., 2007, 6(3), 183–191 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and Graphene Oxide: Synthesis, Properties, and Applications, Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Carbon Nanotubes: Present and Future Commercial Applications, Science, 2013, 339(6119), 535–539 CrossRef CAS PubMed.
- A. C. Ferrari, Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects, Solid State Commun., 2017, 143(1–2), 47–57 Search PubMed.
- C. Backes, A. M. Abdelkader, C. Alonso, A. Andrieux-Ledier, R. Arenal, J. Azpeitia, N. Balakrishnan, L. Banszerus, J. Barjon and R. Bartali, et al., Production and processing of graphene and related materials, 2D Mater., 2020, 7, 022001 CrossRef CAS.
- R. Ikram, B. M. Jan and W. Ahmad, Advances in synthesis of graphene derivatives using industrial wastes precursors; prospects and challenges, J. Mater. Res. Technol., 2020, 9, 15924–15951 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Fine structure constant defines visual transparency of graphene, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Large-Scale Pattern Growth of Graphene Films for Stretchable Transparent Electrodes, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- F. T. Johra, J.-W. Lee and W.-G. Jung, Facile and safe graphene preparation on solution based platform, J. Ind. Eng. Chem., 2014, 20, 2883–2887, DOI:10.1016/j.jiec.2013.11.022.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Mechanical properties of graphene and graphene-based nanocomposites, Prog. Mater. Sci., 2017, 90, 75–127 CrossRef CAS.
- H. Li and K. Leifer, Determining the Elasticity of Graphene, Image Microsc., 2019, 21, 32–34 Search PubMed.
- A. Koutsioukis, V. Georgakilas, V. Belessi and R. Zboril, Highly Conductive Water-Based Polymer/Graphene Nanocomposites for Printed Electronics, Chem. – Eur. J., 2017, 23, 8268–8274 CrossRef CAS PubMed.
- W. Chen, G. Lv, J. Shen, D. Li, X. Liu and Z. Dai, The preparation and application of polymer/graphene nanocomposites, Emerg. Mater. Res., 2020, 9(3), 943–959 Search PubMed.
- M. Ayán-Varela, J. I. Paredes, S. Villar-Rodil, R. Rozada, A. Martínez-Alonso and J. M. D. Tascón, A quantitative analysis of the dispersion behavior of reduced graphene oxide in solvents, Carbon, 2014, 75, 390–400, DOI:10.1016/j.carbon.2014.04.018.
- K. Anagnostou, M. M. Stylianakis, G. Atsalakis, D. M. Kosmidis, A. Skouras, I. J. Stavrou, K. Petridis and E. Kymakis, An extensive case study on the dispersion parameters of HI-assisted reduced graphene oxide and its graphene oxide precursor, J. Colloid Interface Sci., 2020, 580, 332–344, DOI:10.1016/j.jcis.2020.07.040.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The Chemistry of Graphene Oxide, Chem. Soc. Rev., 2010, 39(1), 228–240, 10.1039/b917103g.
- J. Zhao, L. Liu and F. Li, Graphene Oxide: Physics and Applications, Springer Berlin, Heidelberg, 2015 DOI:10.1007/978-3-662-44829-8.
- M. Ayrat and S. E. Dimiev, Graphene Oxide: Fundamentals and Applications, John Wiley & Sons, Ltd., 2017 Search PubMed.
- L. Liu, R. Zhang, Y. Liu, W. Tan and G. Zhu, Insight into Hydrogen Bonds and Characterization of Interlayer Spacing of Hydrated Graphene Oxide, J. Mol. Model., 2018, 24, 137, DOI:10.1007/s00894-018-3679-7.
- C. Y. Wan and B. Q. Chen, Reinforcement and interphase of polymer/graphene oxide nanocomposites, J. Mater. Chem., 2012, 22, 3637–3646 RSC.
- K. Pourzare, Y. Mansourpanah and S. Farhadi, Advanced Nanocomposite Membranes for Fuel Cell Applications: A Comprehensive Review, Biofuel Res. J., 2016, 3, 496–513 CrossRef CAS.
- K. Chrissopoulou, K. Androulaki, M. Labardi and S. H. Anastasiadis, Static and Dynamic Behavior of Polymer/Graphite Oxide Nanocomposites before and after Thermal Reduction, Polymers, 2021, 13, 1008 CrossRef CAS PubMed.
- K. Androulaki, K. Chrissopoulou, M. Labardi and S. H. Anastasiadis, Effect of Interfacial Interactions on Static and Dynamic Behavior of Hyperbranced Polymers: Comparison between Different Layered Nanoadditives, Polymer, 2021, 222, 123646, DOI:10.1016/j.polymer.2021.123646.
- B. C. Brodie XIII, On the Atomic Weight of Graphite, Philos. Trans. R. Soc. London, 1859, 149, 249–259, DOI:10.1098/rstl.1859.0013.
- L. Staudenmaier, Verfahren Zur Darstellung Der Graphitsäure, Ber. Dtsch. Chem. Ges., 1898, 31(2), 1481–1487, DOI:10.1002/cber.18980310237.
- W. S. Hummers Jr and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80(6), 1339, DOI:10.1021/ja01539a.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Graphene Oxide Dispersions in Organic Solvents, Langmuir, 2008, 24, 10560–10564, DOI:10.1021/la801744a.
- J.-A. Yan and M. Y. Chou, Oxidation functional groups on graphene: Structural and electronic properties, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125403, DOI:10.1103/PhysRevB.82.125403.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- H. Yan, X. Tao, Z. Yang, K. Li, H. Yang, A. Li and R. Cheng, Effects of the oxidation degree of graphene oxide on the adsorption of methylene blue, J. Hazard. Mater., 2014, 268, 191–198, DOI:10.1016/j.jhazmat.2014.01.015.
- M. Wang, Y. Niu, J. Zhou, H. Wen, Z. Zhang, D. Luo, D. Gao, J. Yang, D. Liang and Y. Li, The dispersion and aggregation of graphene oxide in aqueous media, Nanoscale, 2016, 8, 14587–14592 RSC.
- R. David, A. Tuladhar, L. Zhang, C. Arges and R. Kumar, Effect of Oxidation Level on the Interfacial Water at the Graphene Oxide–Water Interface: From Spectroscopic Signatures to Hydrogen-Bonding Environment, J. Phys. Chem. B, 2020, 124, 8167–8178 CrossRef CAS PubMed.
- A. N. Rissanou, I. Karnis, F. Krasanakis, K. Chrissopoulou and K. Karatasos, The Role of Oxidation Pattern and Water Content in the Spatial Arrangement and Dynamics of Oxidized Graphene-Based Aqueous Dispersions, Int. J. Mol. Sci., 2022, 23, 13459 CrossRef CAS PubMed.
- C. Xu, X. Wu, J. Zhu and X. Wang, Synthesis of amphiphilic graphite oxide, Carbon, 2008, 46, 386–389, DOI:10.1016/j.carbon.2007.11.045.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105, DOI:10.1038/nnano.2007.451.
- S. Pareek, D. Jain, R. Shrivastava, S. Dam, S. Hussain and D. Behera, Tunable degree of oxidation in graphene oxide: cost effective synthesis, characterization and process optimization, Mater. Res. Exp., 2019, 6, 85625, DOI:10.1088/2053-1591/ab243a.
- R. Muzyka, M. Kwoka, Ł. Smędowski, N. Díez and G. Gryglewicz, Oxidation of graphite by different modified Hummers methods, New Carbon Mater., 2017, 32, 15–20, DOI:10.1016/S1872-5805(17)60102-1.
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W.-W. Liu and C. H. Voon, Synthesis of Graphene Oxide using Modified Hummers Method: Solvent Influence, Procedia Eng., 2017, 184, 469–477, DOI:10.1016/j.proeng.2017.04.118.
- J. Chen, B. Yao, C. Li and G. Shi, An improved Hummers method for eco-friendly synthesis of graphene oxide, Carbon, 2013, 64, 225–229 CrossRef CAS.
- X. Luo, J.-C. Robin and S. Yu, Comparison of Oxidation Behaviors of Different Grades of Nuclear Graphite, Nuclear Sci. Eng., 2005, 151, 121–127 CrossRef CAS.
- L. Shen, L. Zhang, K. Wang, L. Miao, O. Lan, K. Jiang, H. Lu, M. Li, Y. Li, B. Shen and W. Zheng, Analysis of oxidation degree of graphite oxide and chemical structure of corresponding reduced graphite oxide by selecting different-sized original graphite, RSC Adv., 2018, 8, 17209–17217 RSC.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Atomic Structure of Reduced Graphene Oxide, Nano Lett., 2010, 10, 1144–1148, DOI:10.1021/nl9031617.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- A. Lerf, H. He, M. Forster and J. Klinowski, Structure of Graphite Oxide Revisited, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Rissanou, A. Konstantinou and K. Karatasos, Morphology and Dynamics in Hydrated Graphene Oxide/Branched Poly(ethyleneimine) Nanocomposites: An In Silico Investigation, Nanomaterials, 2023, 2, 1865, DOI:10.3390/nano13121865.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. Tang, Y. Zhao, X. N. Yang, D. M. Liu, S. J. Shan, F. Y. Cui and B. S. Xing, Understanding the pH-dependent adsorption of ionizable compounds on graphene oxide using molecular dynamics simulations, Environ. Sci.-Nano, 2017, 4, 1935–1943, 10.1039/c7en00585g.
- A. K. Giri, F. Teixeira and M. N. D. S. Cordeiro, Salt separation from water using graphene oxide nanochannels: A molecular dynamics simulation study, Desalination, 2019, 460, 1–14, DOI:10.1016/j.desal.2019.02.014.
- A. N. Popova, Crystallographic analysis of graphite by X-Ray diffraction, Coke Chem., 2017, 60, 361–365, DOI:10.3103/S1068364X17090058.
- I. Sengupta, S. Chakraborty, M. Talukdar, S. K. Pal and S. Chakraborty, Thermal reduction of graphene oxide: How temperature influences purity, J. Mater. Res., 2018, 33, 4113–4122, DOI:10.1557/jmr.2018.338.
- R. Fryczkowski, M. Gorczowska, C. Ślusarczyk, B. Fryczkowska and J. Janicki, The possibility of obtaining graphene/polymer composites from graphene oxide by a one step process, Comput. Sci. Technol., 2013, 80, 87–92, DOI:10.1016/j.compscitech.2013.03.012.
- V. Patil, R. V. Dennis, T. K. Rout, S. Banerjeeb and G. D. Yadav, Graphene oxide and functionalized multi walled carbon nanotubes as epoxy curing agents: a novel synthetic approach to nanocomposites containing active nanostructured fillers, RSC Adv., 2014, 4, 49264–49272, 10.1039/c4ra09693b.
- B. Y. S. Chang, N. M. Huang, M. N. An’amt, A. R. Marlinda, Y. Norazriena, M. R. Muhamad, I. Harrison, H. N. Lim and C. H. Chia, Facile hydrothermal preparation of titanium dioxide decorated reduced graphene oxide nanocomposite, Int. J. Nanomed., 2012, 7, 3379–3387, DOI:10.2147/IJN.S28189.
- F. Farivar, P. L. Yap, R. U. Karunagaran and D. Losic, Effect of Particle Size of Graphene, Graphene Oxide and Graphite on Thermal Parameters, J. Carbon Res., 2021, 7, 41, DOI:10.3390/c7020041.
- Sudesh, N. Kumar, S. Das, C. Bernhard and G. D. Varma, Effect of graphene oxide doping on superconducting properties of bulk MgB2, Supercond. Sci. Technol., 2013, 26, 095008, DOI:10.1088/0953-2048/26/9/095008.
- V. Sharma, Y. Jain, M. Kumari, R. Gupta, S. K. Sharma and K. Sachdev, Synthesis and Characterization of Graphene Oxide (GO) and Reduced Graphene Oxide (rGO) for Gas Sensing Application, Macromol. Symp., 2017, 376, 1–5, DOI:10.1002/masy.201700006.
- C. Valencia, C. H. Valencia, F. Zuluaga, M. E. Valencia, J. H. Mina and C. D. Grande-Tovar, Synthesis and application of scaffolds of chitosan-graphene oxide by the freeze-drying method for tissue regeneration, Molecules, 2018, 23, 2651, DOI:10.3390/molecules23102651.
- S. Yumitori, Correlation of C1s chemical state intensities with the O1s intensity in the XPS analysis of anodically oxidized glass-like carbon samples, J. Mater. Sci., 2000, 35, 139–146, DOI:10.1023/A:1004761103919.
- J. R. Rani, J. Lim, J. Oh, D. Kim, D. Lee, J. W. Kim, H. S. Shin, J. H. Kim and S. C. Jun, Substrate and buffer layer effect on the structural and optical properties of graphene oxide thin films, RSC Adv., 2013, 3, 5926–5936, 10.1039/c3ra00028a.
- L. Sygellou, G. Paterakis, C. Galiotis and D. Tasis, Work Function Tuning of Reduced Graphene Oxide Thin Films, J. Phys. Chem. C, 2016, 120, 281–290, DOI:10.1021/acs.jpcc.5b09234.
- A. Buchsteiner, A. Lerf and J. Pieper, Water Dynamics in Graphite Oxide Investigated with Neutron Scattering, J. Phys. Chem., 2006, 110, 22328–22338, DOI:10.1021/jp0641132.
- O. C. Compton, S. W. Cranfrond, K. W. Putz, Z. An, L. Catherine Brinson, M. J. Buehler and S. T. Nguyen, Tuning the Mechanical Properties of Graphene Oxide Paper and Its Associated Polymer Nanocomposites by Controlling Cooperative Intersheet Hydrogen Bonding, ACS Nano, 2012, 6, 2008–2019, DOI:10.1021/nn202928w.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, Hydrogen Bond Networks in Graphene Oxide Composite Paper: Structure and Mechanical Properties, ACS Nano, 2010, 4, 2300–2306, DOI:10.1021/nn901934u.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Peptide Folding: When Simulation Meets Experiment, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05268k |
| This journal is © the Owner Societies 2024 |