
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-destructive 3D exploration of silicate glass corrosion: a combined multiscale approach from the macro to the nanoscale†
Giulia
Franceschin
 *a,
Roberta
Zanini
a,
Gianluca
Iori
b,
Elena
Longo
c,
Giorgio
Divitini
d,
Giuliana
Tromba
c and
Arianna
Traviglia
a
*a,
Roberta
Zanini
a,
Gianluca
Iori
b,
Elena
Longo
c,
Giorgio
Divitini
d,
Giuliana
Tromba
c and
Arianna
Traviglia
a
aCenter for Cultural Heritage Technologies (CCHT), Istituto Italiano di Tecnologia, Epsilon building, via Torino 155, Venice, 30170, Italy. E-mail: giulia.franceschin@iit.it
bSESAME – Synchrotron-light for Experimental Science and Applications in the Middle East, Allan 19252, Jordan
cElettra – Sincrotrone Trieste S.C.p.A., AREA Science Park, 34149 Basovizza, Trieste, 34149, Italy
dElectron Spectroscopy and Nanoscopy, Istituto Italiano di Tecnologia, Via Morego 30, Genoa, 16163, Italy
First published on 12th March 2024
Abstract
In this research, a comprehensive multi-technique analysis, including synchrotron-based X-ray micro-computed tomography, is used to visualize the microstructure of alteration in a very particular Roman glass fragment, in which millennia of corrosion history have not significantly impacted the integrity of the fragment itself. This exceptionally rare occurrence has allowed for the maximization of meaningful data acquisition, by examining the alteration structures from the macro to the nanoscale. This study elucidates the intricate mechanisms underlying glass corrosion when in contact with soil, providing quantitative data and phase correlations in the alteration structures. These findings validate and refine existing predictive corrosion models.
1. Introduction
Glass corrosion is a fascinating and complex process that occurs when environmental factors react with the surface of glass, leading to changes in its appearance and properties over time. This corrosion can result in both constructive and destructive surface defects, which create visually distinct (and sometimes aesthetically pleasing) marks on silicate glass.1,2 The alterations are often manifested as iridescence and a loss of the glass’ original clarity, which are caused by construction defects like the formation of a thick alteration patina on the surface.3–5 In some instances, destructive defects like micro- and meso-pits may also appear, which can be further exacerbated by biological activity.6 This occurs when the material degrades in conditions where it is not shielded from environmental elements or external influences.7Glass plays a pivotal role in numerous technological applications. Understanding the ageing process in glass samples with a known initial composition can unveil mechanisms that are not only technologically significant but also relevant to modern society. In the literature, studies on glass alteration from various fields have shown that constructive defects may develop on a glass surface as a silica-rich gel layer8,9 or in a multi-layered lamellar structure.10–14 This lamellar structure is explained as the trace left behind by the advancing alteration front, propagating from the surface towards the inner bulk areas of unaltered glass.15,16 Experiments aimed at reproducing the lamellar multi-layer structure of altered glass15,17 have provided significant evidence for this model, especially in nuclear waste glass studies. However, defining conditions to model the mechanism of lamellar multi-layer formation within experiments of realistic duration remains a challenge. Moreover, the formation mechanism of this layered structure, as well as the relationship between cracks, secondary crystal phases, and lamellae distribution, are still under study.15
The presence of laminated, altered glass surfaces on archaeological objects has only been observed in a few works.13,18,19 With the exception of rare examples (mainly relating to objects that have aged in protected conditions, like those that were part of undisturbed funerary archaeological contexts, for instance),18 preserved multiple layers of alteration patina in archaeological glass are primarily found in limited areas of an object.19 These areas have been normally safeguarded from external mechanical factors, thus preventing the detachment of alteration products due to their brittle nature. These specimens are highly valuable both from the perspective of archaeology and material science, as they provide direct evidence of the structure of the alteration patina that has remarkably endured over extremely long periods of time. However, characterising these complex and heterogeneous samples in a non-invasive manner, across large areas, and with sufficient contrast and spatial resolution to visualise their structural and physicochemical heterogeneity14 remains challenging.
This work analyses the exceptionally preserved multi-layer patina formed on a fragment of archaeological glass (I sec. B.C – I sec. A.D.) found in the archaeologically rich surroundings of ancient Aquileia (northeast Italy). The study of the laminated structure has provided new insight into the temporal progression of glass alteration. The combination of surface and 3D analytical techniques has enabled access to the structure and composition of the altered layer on the sample surface. Synchrotron radiation-based X-ray computed microtomography (SX-μCT) with phase contrast has offered direct evidence of the 3D structure of the alteration patina within a 1 cm-long pit on the surface of Roman glass, which could not be obtained through surface techniques alone. Although the origin of the studied pit remained unclear, the organisation of the corrosion products filling its volume yields valuable insight into the mechanisms of alteration that the glass has undergone over the centuries, as well as the reorganisation of the corrosion products in the alteration layers. It represents one example of those limited areas of samples where the alteration patina was safeguarded from external mechanical factors and endured over time. The preservation of the exceptionally thick stack of corrosion layers is attributed to the depth of the pit itself, which shielded the patina from external stresses and prevented detachment, thus maintaining it in a perfectly uncompromised state for centuries or even millennia. Phase contrast SX-μCT is confirmed as a breakthrough tool that can visualise the alteration history of ancient glass without damaging precious samples.
Our observations led us to interpret the multi-layer patina inside the pit as the result of a series of processes, including solubilisation and reprecipitation of glass constituents, transport of solutes from the environment, and slow allomorphic reorganisation of the secondary phases within the alteration layers. The loss of transparency and clarity of the original glass is likely due to a long-range organisation of the alteration products, creating a discontinuity with the original structure. The colour effects due to the growth of alteration products may be explained by light diffraction from overlapping silica-based layers with varying degrees of porosity.
2. Experimental
2.1. Material
The analysed sample (Italian National Inventory number: 581681 – Museo Archeologico Nazionale di Aquileia) was collected in 2012 in the vicinity of Aquileia (Italy), during archaeological field-walking survey activities. It is a highly degraded dark green glass fragment that was discovered on the topsoil of an agricultural field near the Grado lagoon. Additional information about the archaeological and geological context, can be found in the ESI.†In a previous study,20 the sample was identified as a fragment of a Roman glass object using laser ablation – inductively coupled plasma – mass spectroscopy (LA-ICP-MS). This analysis revealed that the unaltered core of the glass had a silica-soda-lime high-magnesium (HMG) composition. The presence of a high concentration of Fe, as indicated by the analysis, suggested that the original colour of the glass was dark green. It is likely that Fe-rich sand and plant ashes were added to the glass batch as a reducing agent to achieve this colour. The same study also reported on the composition and the distribution of elements within the alteration patina of the sample, which was correlated to the specific soil composition in the area where it was found.
2.2. Methods
The experiments were performed using focal spot size of ≈7 μm, X-ray tube voltage of 90 kV and current of ≈110 μA, integration time of 3 seconds to fully exploit the approximately 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 grey levels of the detector. The analyses were performed with fixed distances between source-detector, source-object, and object-detector. The distances used were respectively 650 mm, 100 mm and 550 mm. With the chosen magnification of ×6.5, the equivalent pixel size was determined to be 7.6 μm. During the acquisition, the sample was placed in a polystyrene support, with low attenuation effect at the energies used for the analysis, and rotated over 360° around the vertical axis, whit angular increments of 0.15°.
000 grey levels of the detector. The analyses were performed with fixed distances between source-detector, source-object, and object-detector. The distances used were respectively 650 mm, 100 mm and 550 mm. With the chosen magnification of ×6.5, the equivalent pixel size was determined to be 7.6 μm. During the acquisition, the sample was placed in a polystyrene support, with low attenuation effect at the energies used for the analysis, and rotated over 360° around the vertical axis, whit angular increments of 0.15°.
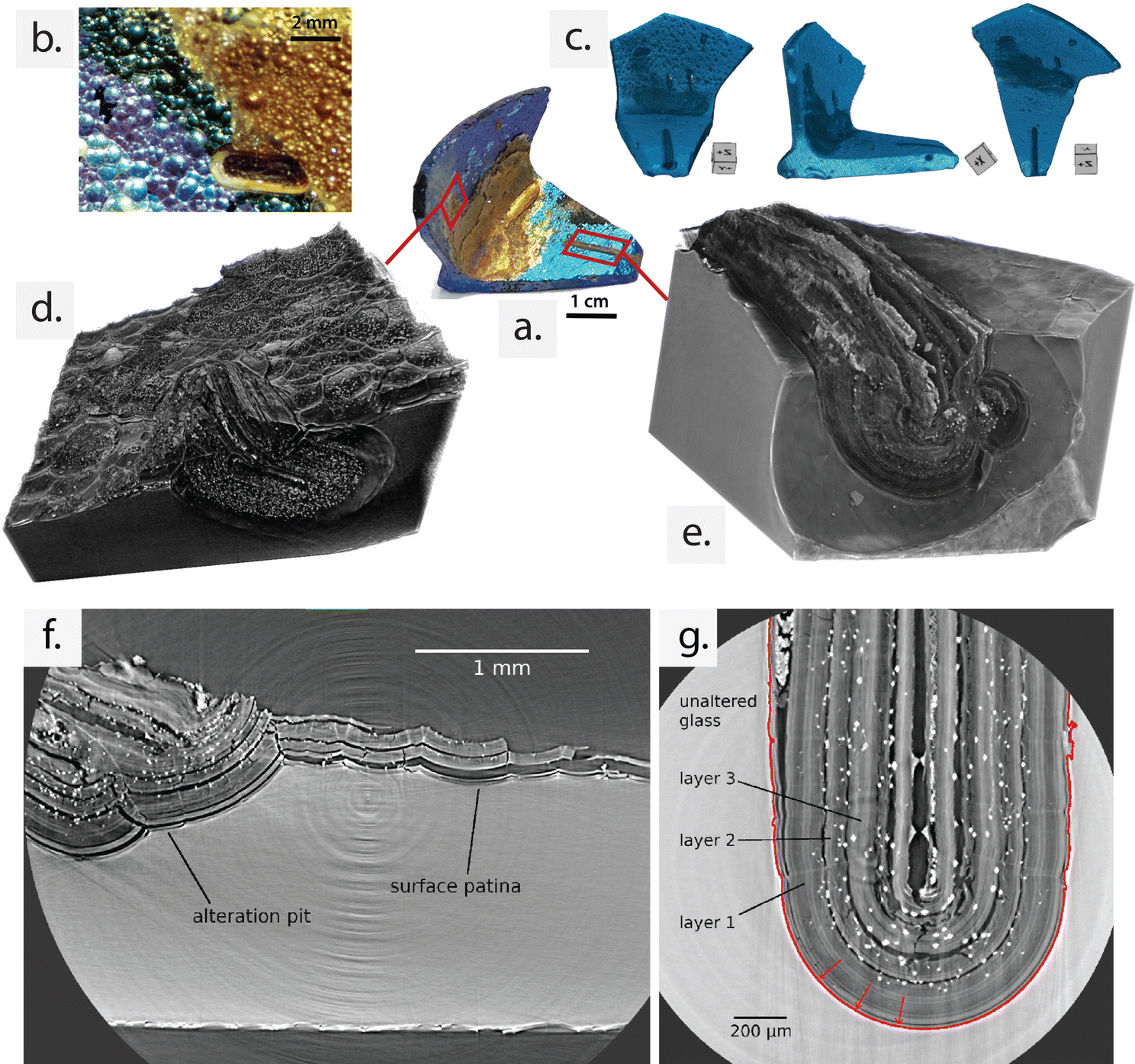 | ||
| Fig. 1 (a) Analysed archaeological sample with its characteristic L shape and (b) detail of the multi-layered alteration patina on surface of the sample captured by OM. (c) 3D rendering obtained from a large field of view laboratory X-ray computer tomography (CT) scan of the sample reported in front, lateral view, and top view from left to right. Different pits with different extensions are visible. Layered formations are visible in the 3D renderings of SX-μCT scans of (d) a second pit surrounded by the surface patina, and (e) the major longitudinal pit. The phase-contrast SX-μCT slice in (f) shows a portion of the sample covered with multi-layer surface patina and a pit containing altered glass layered structure adjacent to it. In (g), the high-resolution (1 μm 3 voxel size) phase-contrast SX-μCT slice shows the interior of the main U-shaped alteration pit. The alteration front and the direction of progression are shown as a red continuous line and arrows, respectively. (f) and (g) are selected SX-μCT slices from scans (d) and (e) of the imaged surface layers and pits, respectively. | ||
SX-μCT was performed at SYRMEP (Synchrotron Radiation MEdical Physics) beamline of Elettra Sincrotrone Trieste S.C.p.A., Italy.22 The experiment was carried out in parallel beam geometry using polychromatic X-ray beam (white/pink beam) filtered with 1.5 mm of Si and 3.5 mm of Al. Thus, a mean energy of 26.9 keV was achieved. Phase-contrast was obtained with free space propagation setting the sample-to-detector distance equal to 150 mm. The scan was accomplished collecting 1800 projections (i.e. sample radiographies) over 180 degrees. In addition, 20 flat fields (i.e. background images) and 20 dark-fields (i.e. dark images) were collected before and after each scan. The images were acquired with 2 s exposure time using ORCA Flash 4.0 Hamamatsu sCMOS detector (2048 × 2048 pixels, physical pixel size 6.5 μm) with a 17 μm thick GGG scintillator screen. The detector, equipped of a zoom system, enabled to collect images with an effective pixel size of 2 × 2 μm or 1 × 1 μm for some higher resolution details. The high-resolution scan of the alteration pit comprised four vertical scans with an overlap of approx. 100 μm allowing further 3D image registration and stitching. Four high-resolution scans were stitched together to extend the investigated volume of interest, which is approximately 7.9 mm × 2.0 mm × 2.0 mm. Four videos of 3D renderings of the investigated volume are available on Zenodo at the following link: https://doi.org/10.5281/zenodo.8169072. Tomographic reconstructions were computed using the open-source software SYRMEP Tomo-Project.23 Phase retrieval was obtained using Paganin's method24 and a delta/beta ratio between 50 and 100 depending on scan.
3. Results and discussion
3.1. Evidence of multilayer patina at macroscale
The shimmery patina covering the surface of the investigated sample has revealed the presence of products deriving from the natural deterioration of silicate glass,1,13 which leads to an extraordinary and pronounced colour variation.Laboratory (Fig. 1c) and synchrotron (Fig. 1d–g) μCT scans (isotropic voxel sizes 7 μm and 2 μm, respectively) confirmed the presence of highly heterogeneous features across the entire surface of the analysed sample, consistent with its highly corroded state. Both constructive (i.e., multi-layered patina) and destructive defects (i.e., pits) were observed. The multi-layered patina was found on all sides of the object, with varying thickness. The inner edge, in particular, exhibited a thicker and more prominent patina, characterised by a gold-yellow colour (Fig. 1a and d–g). The variation in thickness indicates that in this area the patina has been less affected by the detachment and loss of alteration products due to external mechanical factors. Detailed observations using optical microscopy (OM) allowed for the distinction of different colour shades, ranging from metallic blue (where the patina is less thick) to gold-brownish (in the inner edge where the patina is thicker) (Fig. 1a and b).
The rendering in Fig. 1c reveals several mm-scale pits that extend from the sample surface into the bulk material. From the μCT 3D reconstructions, it is possible to visualise that all pits are filled with material that exhibits a longitudinal, multi-layered structure. This structure is oriented parallel to the interface with the unaltered core, mirroring the arrangement observed in the iridescent patina on the sample's surface. To capture high-resolution details of the alteration patina, phase-contrast SX-μCT was performed on selected portions of the sample. The results revealed that both the surface and the pit-confined spaces consistently display multiple overlapping lamellae, which embed secondary globular phases in between (white spots in CT slices). The structure of the alteration, characterised by alternating lamellae with void and polygonal grains, is more extensively preserved inside the confined space of the pits and in the internal edge of the L-shaped fragment. These areas correspond to regions that are less exposed to external agents. Consequently, the high-resolution, phase-contrast SX-μCT analysis focused on a single pit of exceptional size, the one depicted Fig. 1e.
This pit, characterised by a regular U-shaped interface between the altered and unaltered glass, extends along the entire scan length (Fig. 2a) and exhibits the typical multi-layer organisation, exceptionally preserved in its entirety. Remarkably, the laminated layers inside the pit volume have been well preserved, with a notably high thickness, retaining the historical record of the alteration process for centuries or even millennia within the immobilised patina.
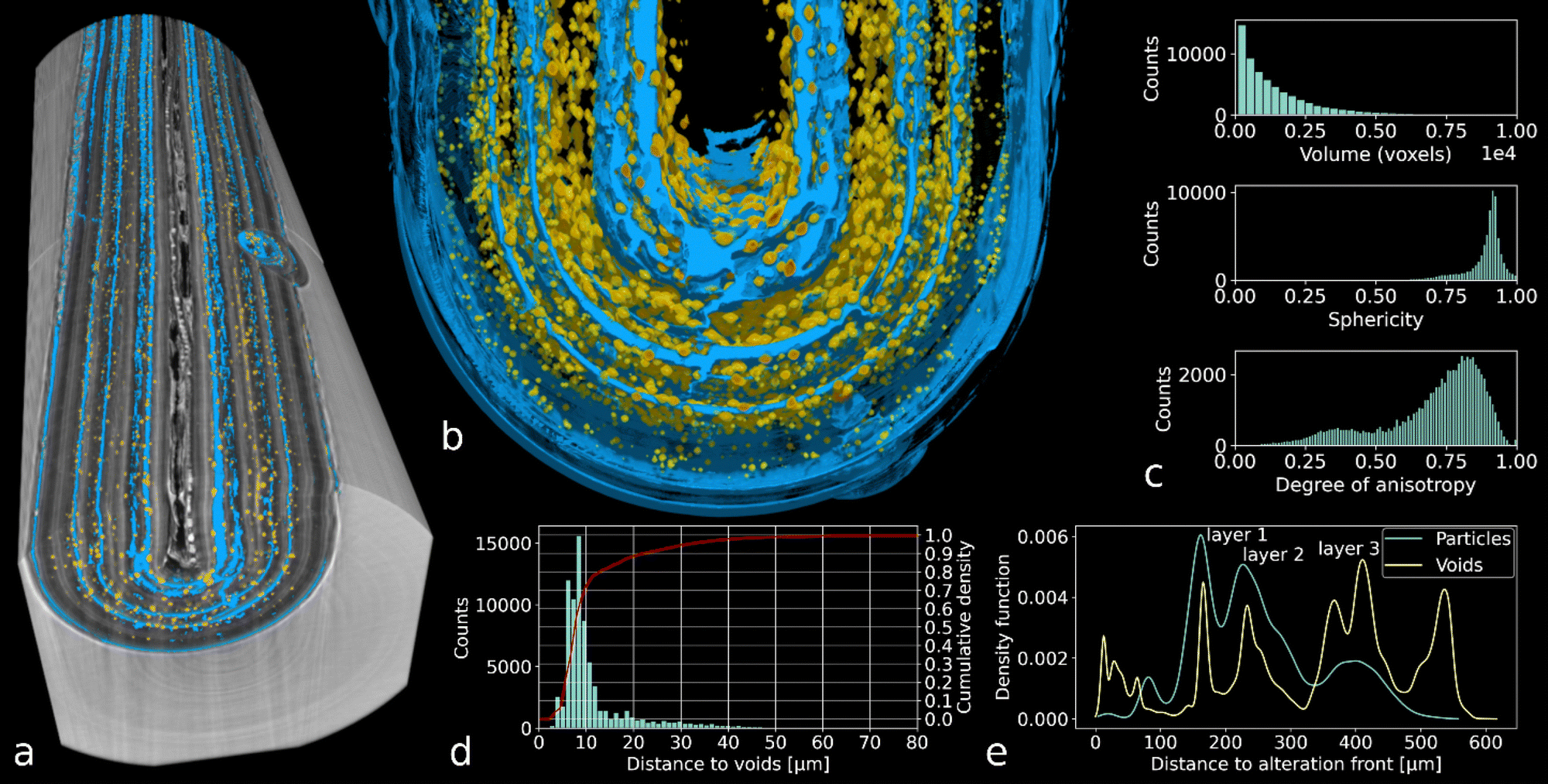 | ||
| Fig. 2 (a) 3D rendering showing the main pit filled with alteration layers, alternating with voids (in blue) and grains of secondary phases (in yellow). (b) Detail view of the pit section showing the distribution of voids and secondary inclusions. (c) Volume, sphericity, and anisotropy distribution of the grains inside the pit volume. (d) Histogram of the grain distance from a void surface: 90% of the analysed grains is located within 20 μm from a void space. (e) Void spaces and secondary grains occur at the same distances from the alteration front. | ||
The investigated pit was scanned at the maximum magnification achievable, leading to 1 μm voxel size.
The primary component of the inner pit volume (80.7%) consists of layers of alteration products that resemble bundles of multiple thinner lamellae that faithfully replicate the geometry of the alteration front. In Fig. 1g, three macroscopic layers can be observed within the altered glass material (dark grey), with void spaces (black) separating them. Layer 1 corresponds to the layer in contact with the unaltered bulk, while layer 3 is the one closest to the centre of the pit. The thickness of the three layers ranges between 70 and 160 μm. However, the precise thickness of each individual thin lamella composing the layers and the number of units in each bundle of layers cannot be determined at the spatial resolution of the SX-μCT acquisitions.
574 individual particles were identified and segmented based on their high grey level in the reconstructed images. They were then labelled in 3D within the analysed pit region (details of the particle analysis can be found in the ESI†).
The particles identified as secondary phases within the pit region have a volume of 1.4 ± 4.3 × 10−6 mm3 (min: 1 μm3; max: 0.7 mm3), a sphericity of 0.88 ± 0.08, and a broad anisotropy distribution (DA: 0.70 ± 0.18), as reported in Fig. 2c. Studying the distance between secondary grains and voids within the pit region reveals that secondary phases have grown adjacent to air spaces between layers of the pit (Fig. 2d and e). This confirms that the polyhedral phases are located at the interface between each layer of corrosion products and empty space, as observed for the patina on surface (Fig. 1f). In these inter-layer spaces, the polyhedral objects find the best condition to grow with the minimal energy requirements.
Once reconstructed in 3D (Fig. 3b), the external shape of secondary grains shows a clear axis of symmetry and appears rhomboidal when looked perpendicular to this axis. Furthermore, the orientation of individual secondary grains can be quantified in 3D on SX-μCT images thanks to their anisotropic shape (Fig. 3). This reveals that dense, secondary alteration objects have an axis of symmetry which is preferentially oriented along the lamination direction of the patina, whereas they grow with a more isotropic shape in the layer plane.
 | ||
| Fig. 3 3D orientation of secondary crystalline phases. (a) Orientation distribution plot of the grain principal axis. (b) 3D rendering of one secondary phase grain, with principal axis (eigenvector) shown in green. (c)–(e) Orthogonal sections through the analysed pit; principal axes of secondary grains shown as line overlay coloured according to their orientation with respect to the image X-axis. Tick bars: 500 μm. | ||
3.2. Evidence at the microscale of the preserved alteration patina on surface and within the major corrosion pit
Examples of altered silicate glass, as reported in the literature, indicate that the multi-layer patina on the surface consists of a random arrangement of amorphous silica nanoparticles (NPs) with a varying level of packing density.13,16,17 The initial surface state of the glass significantly influences the formation of the alteration layer.29 Additionally, other than silica, crystalline and amorphous phases are frequently detected in the spaces between silica-rich layers. The formation of these phases is a consequence of adventitious elements diffusing from the environment and interacting with the dissolved elements of the altered glass surface.13,26To gain a more comprehensive understanding of the structure and chemical properties of the alternating layers observed by SX-μCT analysis, a combination of surface and subsurface analytical techniques was employed. The techniques offered valuable insight into the composition, morphology, and defects within the material, including the grains of secondary phases detected with SX-μCT and OM analyses (Fig. S1, ESI†). SEM and STEM analysis of debris of surface patina confirmed that each block of superimposed layers is in fact formed by multiple thinner lamellae of aggregated NPs with alternating packing density (Fig. S2, ESI†). SEM analysis showed the presence of aggregated packed NPs within the stacked lamellae as well as the thickness of each lamella unit composing the alteration layers observed with SX-μCT analysis both on the surface and within pits (Fig. 1f and g). The Experimental section provides a detailed description of the ultramicrotomy procedure that enabled to prepare the 3.5 μm (for SEM) and 100 nm-thin (for STEM) samples used for the study in cross-section of the surface patina.
Bundles of multiple lamellae of slightly varying thickness (of about 200–300 nm) are stacked, producing the distinct thicker layers (70–160 μm-thick) detected with the SX-μCT analysis. STEM analysis confirmed the presence of aggregated NPs building the internal structure of the lamellae and showed that they have an amorphous nature (Fig. S2b, ESI†). These high-resolution observations complement those by SX-μCT, where the detection limit is driven by the coarser voxel size of 1 μm. Additionally, the stacks of multiple lamellae with varied thickness may also explain the wide range of colour shades macroscopically visible on the sample surface (Fig. 1b), as the local optical properties interact with incoming light.
Confirmation about the correlation between the Fe-rich aggregates found in the surface patina and inside the pits, as well as the bright particles observed with SX-μCT, was obtained through STEM coupled with energy-dispersive X-ray spectroscopy (STEM-EDXS) and micro-Raman analyses (Fig. S3, ESI†).
The SX-μCT reconstructions had already demonstrated the presence of polyhedral inclusions of comparable size and shape both between the lamellae of the surface patina, and within the confined space inside the pits (Fig. 2f and g). OM observations of the thin section and the surface patina debris corroborated the results of the SX-μCT. In the plane containing the lamination direction, the cross-section of secondary phases showed a rhomboidal shape, with the major axis aligned with the direction of lamination (Fig. 4a). When sectioned along a plane parallel to the alteration front, the same objects presented a more regular circular shape (Fig. 4b). However, a minority of particles developed with a random orientation. These results align with the observations previously made on the 3D reconstructions obtained from SX-μCT scans on grains located within the main pit (Fig. 3b).
 | ||
| Fig. 4 OM images of polyhedral grains of the surface patina (a) in cross section from the ultramicrotome thin sample, and (b) parallel to the surface from a fragment of surface patina. (c) HAADF STEM image of an area of patina cross-section containing a polyhedral grain oriented in the direction of lamination. (d) Chemical distribution of Fe (yellow) and Si (blue) of the area reported in c. (e) HAADF STEM image of an area of an area of patina cross-section containing a polyhedral grain oriented along a different principal axis. (f) Overlay of Fe (yellow), Si (blue) chemical distribution and HAADF image of the area in e. | ||
STEM images collected in specific areas of the patina thin section, where secondary phase grains were previously selected using OM (Fig. 4a), provided additional information on the nature of these secondary phases. Electron diffraction analysis (ED) performed in situ during the STEM observation confirmed the amorphous nature of the polyhedral inclusions, while chemical analysis confirmed the prevalence of Fe in their composition (Fig. 4c and d). Unexpectedly, the polyhedral grains were found to be homogeneous in phase with the silica layers and devoid of any signature of crystallinity. They were identified as Fe-rich inclusions embedded in the amorphous silica lamellae. The fast phase transformation of the iron compound under the influence of the laser, obtained during the Raman analysis of the grains inside the pit (reported in ESI†), confirmed the presence of a low degree of crystallinity in the analysed material30 and the transformation from amorphous to crystalline phase of the analysed particles.
3.3. Insight into glass alteration mechanisms
The thorough analysis of the tomographic images has enabled us to identify a correlation between the volume of particles and their distribution within the alteration layer. As illustrated in Fig. 5, larger particles (depicted in yellow) tend to concentrate in the outer layers of the patina, while smaller particles (depicted in purple) dominate the deeper layers near the boundary with unaltered glass. By combining the insights gained from the tomographic, microstructural, and chemical analysis, three distinct stages in the formation of this long-term alteration patina have emerged (Fig. 6). | ||
| Fig. 5 Statistical analysis of the Fe-based (bright, X-ray opaque) particles distribution inside the pit in function of their volume in voxels. Colour ranges from purple for smaller to yellow for larger particles. Within layers adjacent to unaltered glass, almost no particle is captured at the 3D image resolution of SX-μCT. This is likely because the small size of the particles in these strata makes them undetectable by the tomographic scans. | ||
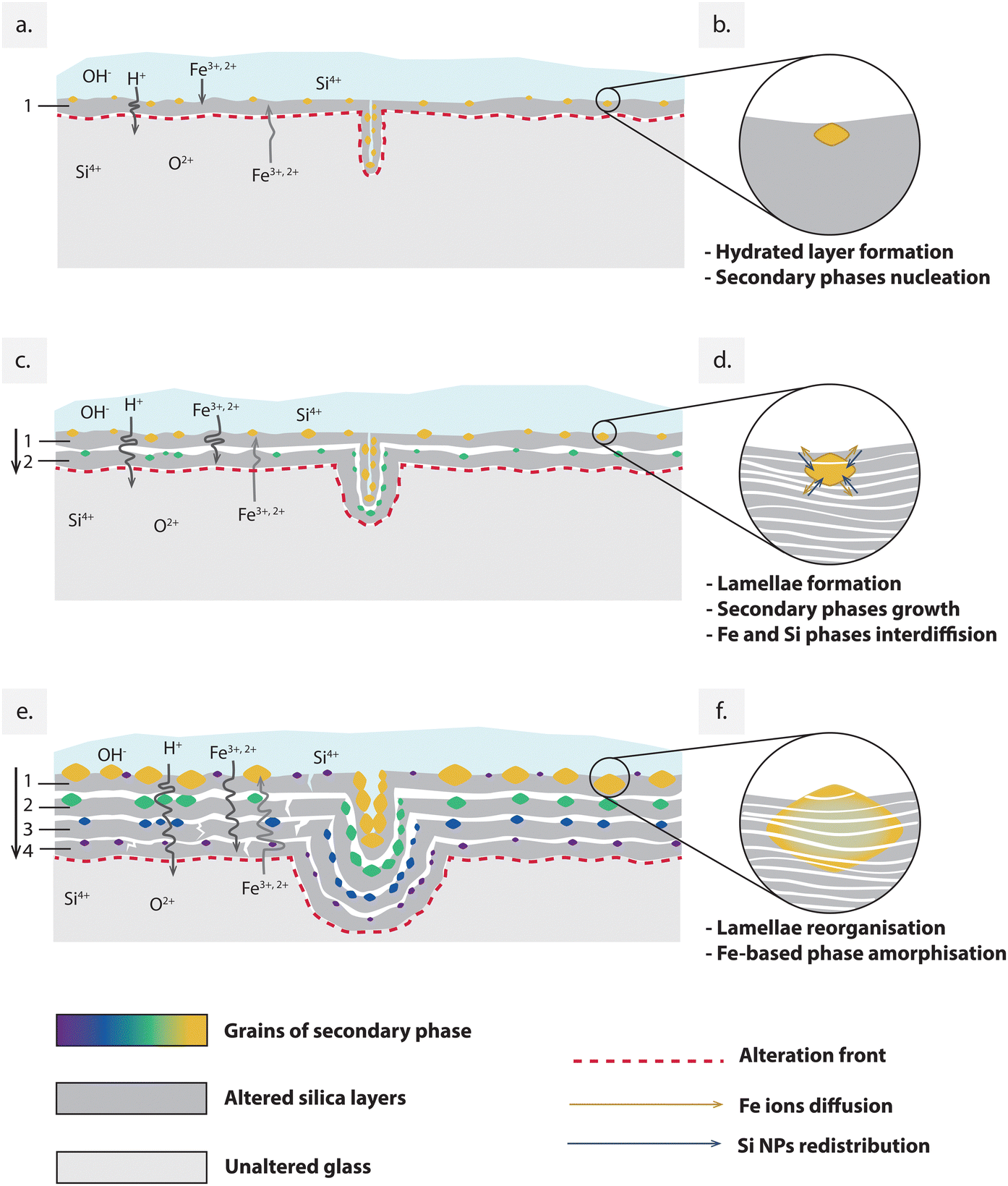 | ||
| Fig. 6 (a), (c) and (e) Schematic representation of the progressive formation of the alteration layers’ structure including the embedding of the amorphous crystalline phase. The temporary evolution of the secondary phase's structure is shown in the detail's circles (b), (d) and (f) on the left. | ||
The mineral and chemical composition of the clay-rich soil that surrounded the analysed fragment during the alteration process played a key role in determining the composition of the first alteration layer. The presence of Fe in the original glass composition, combined with the mineral content of the soil, possibly led to a high availability of Fe ions for the nucleation of secondary Fe-based phases at the interface between glass and water (Fig. 6b). In alkaline environments, silicic solutions may form silica particles rather than a homogenous gel, depending on the silica saturation conditions determined by the glass composition at the surface.8,38 We could hypothesize that during this initial stage (Stage 1), amorphous silica NPs and the other hydrated species precipitated from the water film, coexisting in the first hydrated layer while maintaining a high degree of freedom to reorganise into a more ordered and dense structure.
During this second stage of glass alteration, progression occurred inward from the surface, as illustrated in Fig. 6. Chemical elements dissolved from the surrounding environment and came into contact with the glass, primarily interacting with the pre-altered surface, thus leading to the development of secondary phases atop it.49
In the literature, the formation of the layered structure is described as a recurring process involving the formation and precipitation of silica nanoparticles at the interface between the unaltered glass and inner alteration layer. Simultaneously, secondary phases with varying degrees of crystallinity may precipitate on the surface of each newly formed silica layer.50 In the studied sample, these dissolution and reprecipitation cycles have occurred repeatedly over an extended period, leading to the formation of multiple stacked layers. Embedding grains of Fe-rich secondary phases distributed within the interlayer spaces. Observing Fig. 6, the nucleation and growth of such Fe-rich grains seem to have been influenced by their distance from the surface. This was possibly due to the decreasing availability of Fe species with depth, as they primarily interacted with the more superficial layers, thus contributing to the growth of the pre-nucleated grains.
In Fig. 2a and b, smaller grains of secondary phases are visible in the layers close to the pristine glass boundary, while their size increases as we approach the surface layers. The size-depth dependence of these secondary phases can be explained by two parallel factors. On one side, the diffusion of Fe ions from the outside to the internal layers may have contributed to the formation of larger particles in the layers closer to the external environment, where the concentration of adventitious elements was higher. On the other hand, the grains progressively increased in size over time. In this scenario, if we assume that the nucleation of secondary phases coincided with the formation of the gel layer of amorphous silica NPs, we can determine the order of formation of different stacking layers based on the particles size. Deeper layer may have formed more recently compared to surface layers, where the secondary particles exhibit the largest size.
Furthermore, our study suggests that secondary grains also undergo structural reorganisation over more extended periods (Fig. 6d and f). While the presence of Fe-rich amorphous phases in the altered layers has been previously observed in glass corroded underground, our study unveiled a distinctive organisation of these phases within the nanostructured silica lamellae, which has never been described before. In the case of nuclear glass altered in an anoxic clayey environment, the presence of amorphous Fe-rich phases was reported as distinct nanophases entrapped within the porous gel network. These were attributed to the diffusion of dissolved Fe through the porous network of the silica gel and the subsequent co-precipitation of nanosized Fe-silicate entrapped within the porous silica gel layer.51
This observation aligns with similar findings of amorphous structures in alteration layers formed on Fe-rich basaltic glass under aerated conditions,52 as well as in presence of microbial species from the environment. It has been demonstrated that microorganisms indeed have an impact on glass alteration,27 and one of their effects may be the conversion of both native and newly formed crystalline Fe oxides into an amorphous form.53 However, as observed in TEM analysis (Fig. 4), the amorphous Fe-rich particles found in our sample are today completely homogenised within the lamellae of packed silica NPs, while maintaining a polygonal shape that may reflect the one of the minerals originally formed and grown during the alteration process (Fig. 6f).
Given the amorphous atomic structure of silica NPs, which restricts dislocation movement and renders the material brittle,54 it can be inferred that the reorganisation of Fe-rich grains occurred through solid-state diffusion of Fe ions within the structure of silica NPs, thereby resulting in a reorganisation of the packing order of the NPs. The solid-state interdiffusion of Fe in silica NPs can be interpreted as a Kirkendall effect,55 where Fe has a higher diffusion coefficient compared to Si NPs. This effect is facilitated by the extended duration given for the transformation, which takes place under conditions of quasi-equilibrium and slow kinetics.
4. Conclusions
The analysis of this archaeological glass sample that has undergone alteration for multiple centuries offers a rare opportunity to observe exceptionally preserved alteration symptoms that cannot be replicated in laboratory-based experiments.The characteristic alteration pattern observed, with multiple stacking lamellae composed of silica NPs, aligns with previous studies of naturally and experimentally corroded silica glass. However, the long-range ordered distribution of the Fe-based secondary phase within the lamellae is unprecedented and the examination of their structure and morphology has allowed for a better understanding of the successive stages of formation and evolution involved in the glass alteration process. Moreover, the presence of amorphous secondary phases within the alteration layers, as observed in the literature, may be hypothesized to contribute to the slowing down the diffusion rate of water and solutes through the layers.
The use of SX-μCT is demonstrated to be a powerful method to visualise microscale structural features over macroscopical distances. By combining insights from SR-μCT and microstructural analyses, a comprehensive view of the intricate processes underlying the formation of the observed structure over extended periods has been achieved. This has led to the identification of three distinct stages in the formation mechanism, shedding new light on the underlying dynamics of this long-term alteration process.
The obtained results highlight the contribution of this multi-technique analysis in deciphering the mechanisms of silicate glass alteration. The preservation of the multi-layer structure of the alteration further strengthens the interest in studying ancient samples to assist in the development of long-term models of alteration, not limited to glass samples. Various avenues can be explored to identify the molecular nature of the alteration products, including the iron-based secondary phase, by employing different non-invasive spectroscopic techniques. These techniques will be crucial to identify minor spectral features in the complex silica-based compound, thereby deepening our understanding of the specific mechanisms of formation and reorganisation within the altered patina.
Author contributions
Giulia Franceschin: conceptualisation, methodology, writing – original draft preparation, investigation, funding acquisition Roberta Zanini: investigations, methodology, writing – review & editing Gianluca Iori investigation, visualisation, data curation, writing – review & editing Elena Longo investigation, writing – review & editing Giorgio Divitini investigation, writing – review & editing Giuliana Tromba writing – review & editing, methodology Arianna Traviglia writing – review & editing, resources, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the CERIC-ERIC Consortium for access to experimental facilities and financial support. The authors are grateful to the SYRMEP beamline scientists and coordinators for the support before and during the synchrotron measurements, to Dr Victoria Beltran (University of Antwerp) for preparing the thin sections from the analysed sample, and to Prof. Alessandro Re (Physics Department of the University of Torino and INFN) and his team for supporting us during the laboratory CT scanning performed in the framework of the OpenAIAr project. The glass fragment studied in this article is identified with the Italian National ID 581681. The image of the glass fragment in Fig. 1a is published by permission of the Ministry of Culture, Regional Directorate of Museum of Friuli Venezia Giulia (Museo Archeologico Nazionale di Aquileia – Direzione Regionale Musei del Friuli Venezia Giulia, Ministero della Cultura). The use of these images is regulated by current legislation (art. 108, co. 3 del D. Lgs 42/2004s.m.i. – DM 161/23). Any reproduction, duplication by any mean, downloading and manipulation is strictly prohibited.References
- S. Fiorentino, T. Chinni, D. Galusková, S. Mantellini, A. Silvestri, A. E. Berdimuradov and M. Vandini, Minerals, 2021, 11, 1364 CrossRef CAS.
- N. A. R. van Giffen and S. P. Koob, The Encyclopedia of Archaeological Sciences, John Wiley & Sons, Ltd, 2018, pp. 1–4 Search PubMed.
- G. Barbera, G. Barone, V. Crupi, F. Longo, D. Majolino, P. Mazzoleni, G. Sabatino, D. Tanasi and V. Venuti, J. Non-Cryst. Solids, 2012, 358, 1554–1561 CrossRef CAS.
- D. Brewster, Earth Environ. Sci. Trans. R. Soc. Edinburgh, 1863, 23, 193–204 Search PubMed.
- C. Macquet and J. H. Thomassin, Appl. Clay Sci., 1992, 7, 17–31 CrossRef CAS.
- A. A. Gorbushina and K. A. Palinska, Aerobiologia, 1999, 15, 183–192 CrossRef.
- M. F. Macedo, M. G. Vilarigues and M. L. Coutinho, Appl. Sci., 2021, 11, 9552 CrossRef.
- S. Gin, A. H. Mir, A. Jan, J. M. Delaye, E. Chauvet, Y. De Puydt, A. Gourgiotis and S. Kerisit, J. Phys. Chem. C, 2020, 124, 5132–5144 CrossRef CAS.
- A. Verney-Carron, S. Gin and G. Libourel, Geochim. Cosmochim. Acta, 2008, 72, 5372–5385 CrossRef CAS.
- W. Huang, D. E. Day, K. Kittiratanapiboon and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2006, 17, 583–596 CrossRef CAS PubMed.
- W. Huang, M. N. Rahaman, D. E. Day and Y. Li, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2006, 47, 647–658 CAS.
- Y. Wang, C. F. Jove-Colon, C. Lenting, J. Icenhower and K. L. Kuhlman, npj Mater. Degrad., 2018, 2, 1–10 CrossRef.
- O. Schalm and W. Anaf, J. Non-Cryst. Solids, 2016, 442, 1–16 CrossRef CAS.
- R. Zanini, G. Franceschin, E. Cattaruzza and A. Traviglia, npj Mater. Degrad., 2023, 7, 1–17 CrossRef.
- L. Dohmen, C. Lenting, R. O. C. Fonseca, T. Nagel, A. Heuser, T. Geisler and R. Denkler, Int. J. Appl. Glass Sci., 2013, 4, 357–370 CrossRef CAS.
- O. Schalm, G. Nuyts and K. Janssens, J. Non-Cryst. Solids, 2021, 569, 120984 CrossRef CAS.
- M. I. Ojovan, MRS Adv., 2020, 5, 111–120 CrossRef CAS.
- M. Emami, S. Nekouei, H. Ahmadi, C. Pritzel and R. Trettin, Int. J. Appl. Glass Sci., 2016, 7, 59–68 CrossRef CAS.
- A. Silvestri, G. Molin and G. Salviulo, J. Non-Cryst. Solids, 2005, 351, 1338–1349 CrossRef CAS.
- G. Guidetti, R. Zanini, G. Franceschin, M. Moglianetti, K. Taehoon, N. Cohan, L. Chan, J. Treadgold and A. Traviglia, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(39), e2311583120 CrossRef CAS PubMed.
- V. Beltran, A. Marchetti, G. Nuyts, M. Leeuwestein, C. Sandt, F. Borondics and K. De Wael, Angew. Chem., 2021, 133, 22935–22942 CrossRef.
- C. Dullin, F. di Lillo, A. Svetlove, J. Albers, W. Wagner, A. Markus, N. Sodini, D. Dreossi, F. Alves and G. Tromba, Phys. Open, 2021, 6, 100050 CrossRef.
- F. Brun, L. Massimi, M. Fratini, D. Dreossi, F. Billé, A. Accardo, R. Pugliese and A. Cedola, Adv. Struct. Chem. Imaging, 2017, 3, 4 CrossRef PubMed.
- D. Paganin, S. C. Mayo, T. E. Gureyev, P. R. Miller and S. W. Wilkins, J. Microsc., 2002, 206, 33–40 CrossRef CAS PubMed.
- O. Stamati, E. Andò, E. Roubin, R. Cailletaud, M. Wiebicke, G. Pinzon, C. Couture, R. C. Hurley, R. Caulk, D. Caillerie, T. Matsushima, P. Bésuelle, F. Bertoni, T. Arnaud, A. O. Laborin, R. Rorato, Y. Sun, A. Tengattini, O. Okubadejo, J.-B. Colliat, M. Saadatfar, F. E. Garcia, C. Papazoglou, I. Vego, S. Brisard, J. Dijkstra and G. Birmpilis, J. Open Source Softw., 2020, 5, 2286 CrossRef.
- T. Lombardo, L. Gentaz, A. Verney-Carron, A. Chabas, C. Loisel, D. Neff and E. Leroy, Corros. Sci., 2013, 72, 10–19 CrossRef CAS.
- S. Gin, J.-M. Delaye, F. Angeli and S. Schuller, npj Mater. Degrad., 2021, 5, 1–20 CrossRef.
- J. Sterpenich and G. Libourel, Chem. Geol., 2001, 174, 181–193 CrossRef CAS.
- L. Neill, S. Gin, T. Ducasse, T. De Echave, M. Fournier, P. Jollivet, A. Gourgiotis and N. A. Wall, npj Mater. Degrad., 2017, 1, 1–11 CrossRef CAS.
- M. Hanesch, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
- P. Frugier, S. Gin, Y. Minet, T. Chave, B. Bonin, N. Godon, J.-E. Lartigue, P. Jollivet, A. Ayral, L. De Windt and G. Santarini, J. Nucl. Mater., 2008, 380, 8–21 CrossRef CAS.
- R. Zanini, G. Franceschin, E. Cattaruzza, M. Prato, M. Barozzi and A. Traviglia, J. Non-Cryst. Solids, 2023, 612, 122356 CrossRef CAS.
- R. H. Brill, Stud. Conserv., 1975, 20, 121–134 CrossRef.
- O. Majérus, P. Lehuédé, I. Biron, F. Alloteau, S. Narayanasamy and D. Caurant, npj Mater. Degrad., 2020, 4, 1–16 CrossRef.
- J. K. Bates, M. G. Seitz and M. J. Steindler, Nucl. Chem. Waste Manage., 1984, 5, 63–73 CrossRef CAS.
- W. H. Casey and B. Bunker, Rev. Mineral. Geochem., 1990, 23, 397–426 CAS.
- M.-T. Doménech-Carbó, A. Doménech-Carbó, L. Osete-Cortina and M.-C. Saurí-Peris, Microchim. Acta, 2006, 154, 123–142 CrossRef.
- Y. Tardy, Sci. Geol., Bull., 1982, 35, 93 Search PubMed.
- A. Rodrigues, S. Fearn, T. Palomar and M. Vilarigues, Corros. Sci., 2018, 143, 362–375 CrossRef CAS.
- D. Rebiscoul, A. Van der Lee, F. Rieutord, F. Né, O. Spalla, A. El-Mansouri, P. Frugier, A. Ayral and S. Gin, J. Nucl. Mater., 2004, 326, 9–18 CrossRef CAS.
- R. H. Brill and H. P. Hood, Nature, 1961, 189, 12–14 CrossRef.
- T. Geisler, A. Janssen, D. Scheiter, T. Stephan, J. Berndt and A. Putnis, J. Non-Cryst. Solids, 2010, 356, 1458–1465 CrossRef CAS.
- W. Anaf, Study on the formation of heterogeneous structures in leached layers during the corrosion process of glass, CeROArt. Conservation, exposition, Restauration d’Objets d’Art, 2010, EGG 1 DOI:10.4000/ceroart.1561.
- B. Dal Bianco, R. Bertoncello, L. Milanese and S. Barison, J. Non-Cryst. Solids, 2004, 343, 91–100 CrossRef CAS.
- J. H. E. Cartwright, J. M. Garcia-Ruiz and A. I. Villacampa, Comput. Phys. Commun., 1999,(121–122), 411–413 CrossRef CAS.
- S. C. Müller, S. Kai and J. Ross, Science, 1982, 216, 635–637 CrossRef PubMed.
- H. K. Henisch, Crystals in Gels and Liesegang Rings, Cambridge University Press, Cambridge, 1988 Search PubMed.
- D. A. Scott, Stud. Conserv., 2005, 50, 179–189 CrossRef.
- J.-L. Crovisier, T. Advocat and J.-L. Dussossoy, J. Nucl. Mater., 2003, 321, 91–109 CrossRef CAS.
- H. Aréna, N. Godon, D. Rébiscoul, P. Frugier, R. Podor, E. Garcès, M. Cabie and J.-P. Mestre, Appl. Geochem., 2017, 82, 119–133 CrossRef.
- E. Burger, D. Rebiscoul, F. Bruguier, M. Jublot, J. E. Lartigue and S. Gin, Appl. Geochem., 2013, 31, 159–170 CrossRef CAS.
- L. L. Baker, D. G. Strawn, P. A. McDaniel, R. N. Nickerosn, J. L. Bishop, D. W. Ming and R. V. Morris, Poorly Crystalline Iron-Bearing Aluminosilicates and Their Importance on Mars, 42nd Lunar and Planetary Science Conference, The Woodlands, TX, 2011.
- P. A. Wahid and N. V. Kamalam, Biol. Fertil. Soils, 1993, 15, 144–148 CrossRef CAS.
- L. Aucott, R. Bamber, A. Lunev, T. Darby, P. Maquet, N. Gimbert, S. Pak, M. Walsh, V. Udintsev, G. Eaton and B. Conway, TMS 2020 149th Annual Meeting & Exhibition Supplemental Proceedings, Springer International Publishing, Cham, 2020, pp. 2085–2094 Search PubMed.
- H. Nakajima, JOM, 1997, 49, 15–19 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05221d |
| This journal is © the Owner Societies 2024 |