
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H2 formation from the E2–E4 states of nitrogenase†
Hao
Jiang
 and
Ulf
Ryde
*
and
Ulf
Ryde
*
Department of Computational Chemistry, Lund University, Chemical Centre, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: Ulf.Ryde@compchem.lu.se; Fax: +46 2228648; Tel: +46 2224502
First published on 8th December 2023
Abstract
Nitrogenase is the only enzyme that can cleave the strong triple bond in N2, making nitrogen available for biological lifeforms. The active site is a MoFe7S9C cluster (the FeMo cluster) that binds eight electrons and protons during one catalytic cycle, giving rise to eight intermediate states E0–E7. It is experimentally known that N2 binds to the E4 state and that H2 is a compulsory byproduct of the reaction. However, formation of H2 is also an unproductive side reaction that should be avoided, especially in the early steps of the reaction mechanism (E2 and E3). Here, we study the formation of H2 for various structural interpretations of the E2–E4 states using combined quantum mechanical and molecular mechanical (QM/MM) calculations and four different density-functional theory methods. We find large differences in the predictions of the different methods. B3LYP strongly favours protonation of the central carbide ion and H2 cannot form from such structures. On the other hand, with TPSS, r2SCAN and TPSSh, H2 formation is strongly exothermic for all structures and En and therefore need strict kinetic control to be avoided. For the E2 state, the kinetic barriers for the low-energy structures are high enough to avoid H2 formation. However, for both the E3 and E4 states, all three methods predict that the best structure has two hydride ions bridging the same pair of Fe ions (Fe2 and Fe6) and these two ions can combine to form H2 with an activation barrier of only 29–57 kJ mol−1, corresponding to rates of 7 × 102 to 5 × 107 s−1, i.e. much faster than the turnover rate of the enzyme (1–5 s−1). We have also studied H-atom movements within the FeMo cluster, showing that the various protonation states can quite freely be interconverted (activation barriers of 12–69 kJ mol−1).
Introduction
Nitrogenase (EC 1.18/19.6.1) is the only enzyme that can cleave the strong triple bond in N2, making atmospheric nitrogen available to plant life.1–4 Crystallographic studies have shown that the most active type of nitrogenase contains a MoFe7S9C(homocitrate) cofactor in the active site, called the FeMo cluster.5–9Nitrogenase catalyses the chemical reaction3,4
| N2 + 8e− + 8H+ + 16ATP → 2NH3 + H2 + 16ADP + 16Pi | (1) |
In spite of numerous spectroscopic, kinetic, biochemical and computational studies,1–9,16,17 many details of the reaction mechanism of nitrogenase are still unknown.4,17 An important reason for this is that different density-functional theory (DFT) methods give widely different predictions of the relative stability of various models of the active site of nitrogenase, e.g. different protonation states of the En states.17–20
The structure of the resting E0 state is well-known from accurate crystal structures, and combined quantum mechanical and molecular mechanical (QM/MM) calculations.4,5,21–23 Moreover, quantum refinement has shown that this state does not contain any extra protons.20 For the singly reduced and protonated E1 state, DFT studies and EXAFS measurements indicate that one of the μ2 bridging sulfide ions, S2B (atom names are shown in Fig. 1), is protonated.20,24–26 However, spectroscopic studies of Fe-nitrogenase (in which the Mo ions is replaced by Fe), indicate that it instead should contain a dissociable hydride ions,27 although this is not supported by recent QM/MM calculations.28
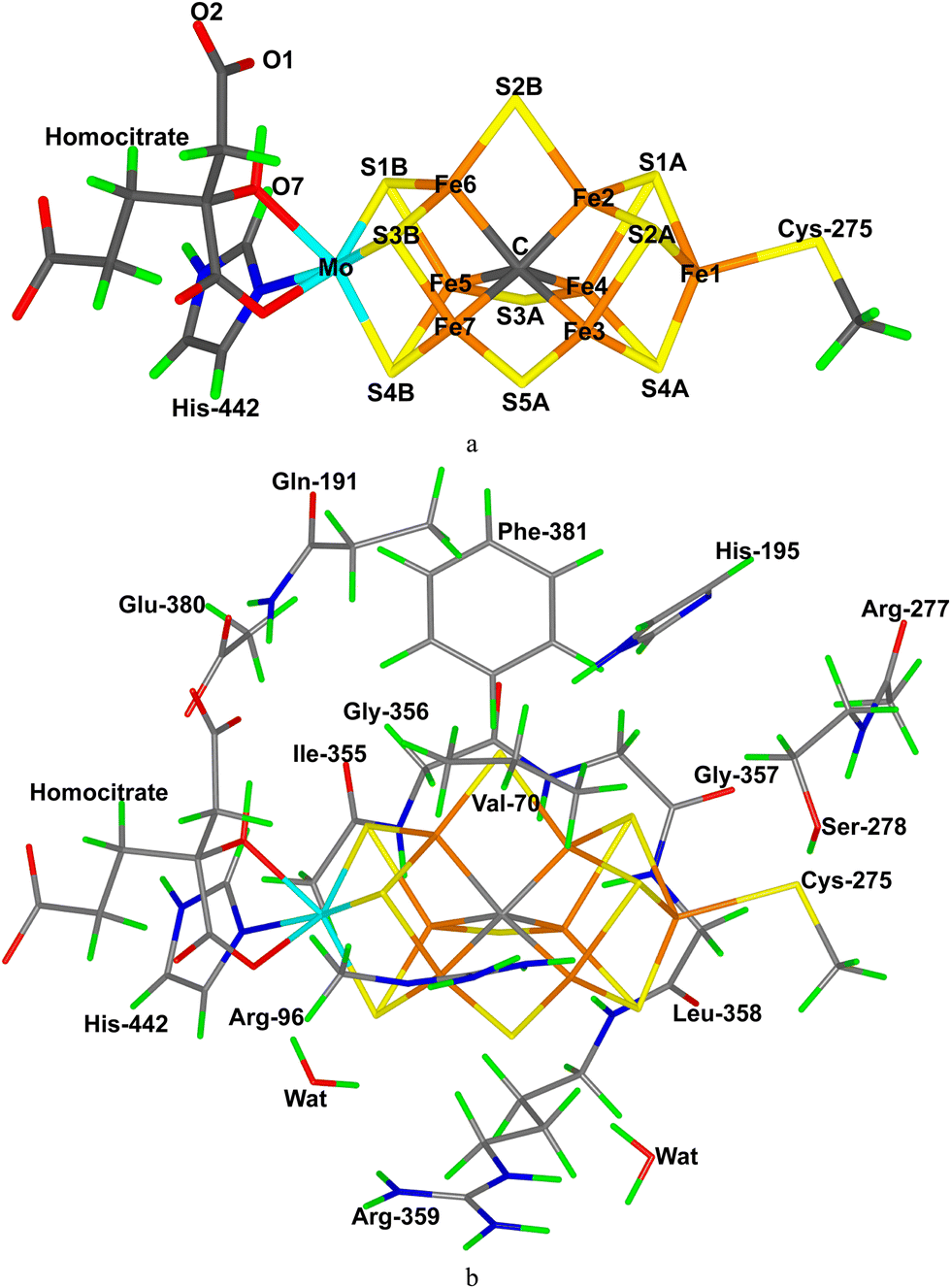 | ||
| Fig. 1 (a) The FeMo cluster of nitrogenase, showing also the atom names. (b) The QM system used in the calculations, showing the names of the surrounding residues. | ||
For the E2 state, the results with different DFT methods start to differ.20 Calculations with the pure meta generalised gradient approximation (mGGA) functional TPSS29 suggest that the best structure involves a proton on S2B and a hydride ion bridging the Fe2 and Fe6 ions.19 Other mGGA or hybrid functionals with a low amount of Hartree–Fock (HF) exchange (r2SCAN30 and TPSSh31) suggest a similar structure, but with the protonated S2B ion dissociated from one of the Fe ions.32 A structure with both S2B and S5A protonated is also competitive with TPSSh.33 The hybrid B3LYP functional (with 20% HF exchange)34–36 suggests instead that the central carbide ion is doubly protonated. Likewise, for the E3 state B3LYP suggests a structure with a triply protonated carbide ions, whereas the other functionals suggest a structure with S2B protonated and dissociated from one Fe ion, and two hydride ions both bridging between the Fe2 and Fe6 ions.32 Other structures with a terminal hydride ion or a bound H2 molecule have also been proposed.37
For the key E4 intermediate, many different structures have been suggested.17 ENDOR experiments show that E4 should contain two bridging hydride ions12,13,15 and based on this and computational studies, Hoffman and coworkers have suggested a structure in which S2B and S5A are protonated and there are two hydride ions bridging the Fe2/6 and the Fe3/7 pairs of ions, with all H atoms pointing towards the same face of the FeMo cluster.38 We made a systematic search of structures with two bridging hydride ions and showed that a structure with the protons and the hydride ions at the same positions is most stable, but three of them were pointing towards to other faces of the FeMo cluster.39 On the other hand, Bjornsson and coworkers have advocated for structures in which the two hydride ions both bridge Fe2/6, S2B is protonated and dissociated from one Fe and the second proton is on S5A.40 A thorough study of many of these structures indicated that with TPSS, the Hoffman and Bjornsson-type structures are nearly degenerate, whereas the latter are most stable with r2SCAN and TPSSh.32 A structure with a triply protonated carbide ion and the fourth proton on S2B is by far best by B3LYP (and competitive with TPSSh).41,42 It has also been suggested that S2B may fully dissociate from the FeMo cluster,43–46 inspired by crystallographic studies showing lability of this ligand.8,47,48
Recently, we studied the binding of N2 to the E0–E4 states of nitrogenase and showed that only the TPSS29 functional gave binding energies in accordance with experiments (i.e. a favourable binding to E3 and E4), but not to E0–E2.32 The other tested functionals gave too weak binding to E3 or E4. Other studies have suggested stronger binding of N2, but this depends on how the binding energy is defined and how the entropy loss of the N2 ligand is treated.40,49–51
A related question is the formation and dissociation of H2 from the FeMo cluster. This is a very important reaction. If H2 forms from the E2 and E3 states of nitrogenase, the enzyme goes back to the E0 and E1 states, respectively, and two electrons and protons have been lost on an unproductive side reaction.4,33 On the other hand, formation of H2 is desirable for the E4 state, but only if it is directly coupled to the binding of N2.
Khadka and coworkers have studied H2 evolution from the E2 state with mediated bioelectrocatalysis and DFT calculations.52 The experiments showed that the rate-limiting step for H2 formation is hydride protonation. The calculations were performed with a minimal QM model of the active site in a continuum solvent. They were started from a structure with S2B protonated and a hydride ion bridging Fe2 and Fe6. A free-energy barrier of only 29 kJ mol−1 was obtained.
Thorhallsson and Bjornsson have studied the formation of H2 from the E2 state of nitrogenase by QM/MM calculations.33 They found activation barriers of 86–95 kJ mol−1 from a structure with S2B dissociated from one of the Fe ions, depending on the protonation state of the nearby His-195 residue. Interestingly, the barrier was much lower (48 kJ mol−1) when calculated with a small QM-cluster model, indicating that the surrounding protein counteracts this side reaction.
Here, we extend these investigations by studying the formation and dissociation of H2 from the FeMo cluster in the E2–E4 states. The aim of this investigation is three-fold: The first is to study the H2-formation reaction for many different structures: combining either two hydride ions or one proton and one hydride ion, and studying reactions for different positions in the cluster, e.g. both terminal and bridging hydride ions, located on different Fe ions or on the same Fe ion. The second is to study how easily different protonation states can be interconverted. This may answer the question whether the enzyme can avoid the formation of H2 by posing the proton and hydride ions far from each other. The third is to compare the results of four different DFT functionals. We know that the enzyme needs to avoid the loss of H2 from the E2 and E3 states, whereas H2 formation is necessary for the E4 state, but only in conjunction with the binding of N2. If the H2 formation is too facile for the E2 and E3 states, it is an indication of problems with the DFT method used or that the proper structure is still not found.
Methods
The protein
The calculations were based on the 1.0 Å crystal structure of Mo nitrogenase from Azotobacter vinelandii (PDB code 3U7Q).5 The setup of the protein is identical to that of our previous studies.18,32,39,53 The entire heterotetramer was included in the calculations and the quantum mechanical (QM) calculations were concentrated on the FeMo clusters in the C subunit because there is a buried imidazole molecule rather close to the active site (∼11 Å) in the A subunit. The two P clusters and the FeMo cluster in subunit A were modelled by MM in the fully reduced and resting states, respectively, using a QM charge model.53 The protonation states of all residues were the same as before,53 and the homocitrate ligand was modelled in the singly protonated state with a proton shared between the hydroxyl group (O7 that coordinates to Mo) and the O1 carboxylate atom.23,53 The protein was solvated in a sphere with a radius of 65 Å around the geometrical centre of the protein. Cl− and Na+ ions were added to an ionic strength of 0.2 M.54 The final system contained 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 915 atoms. For the protein, we used the Amber ff14SB force field55 and water molecules were described by the TIP3P model.56 The metal sites were treated by a non-bonded model57 and charges were obtained with the restrained electrostatic potential method.58
915 atoms. For the protein, we used the Amber ff14SB force field55 and water molecules were described by the TIP3P model.56 The metal sites were treated by a non-bonded model57 and charges were obtained with the restrained electrostatic potential method.58
The FeMo cluster was modelled by MoFe7S9C(homocitrate)(CH3S)(imidazole), where the two last groups are models of Cys-275 and His-442. In addition, all groups that form hydrogen bonds to the FeMo cluster were also included in the QM model, viz. Arg-96, Gln-191 and His-195 (sidechains), Ser-278 and Arg-359 (both backbone and sidechain, including the CA and C and O atoms from Arg-277), Gly-356, Gly-357 and Leu-358 (backbone, including the CA and C and O atoms from Ile-355), as well as two water molecules. Finally, the sidechain of Glu-380 was included because it forms hydrogen bonds to Gln191 and His-442, as well as the sidechains of Val-70 and Phe-381 because they are close to S2B, Fe2 and Fe6, i.e. the expected reactive site. The QM system contained 191–193 atoms depending on the En state and is shown in Fig. 1. The net charge of QM region was −4 e.
QM calculations
All QM calculations were performed with the Turbomole software (versions 7.5 and 7.6).59 All structures were studied with the TPSS,29 r2SCAN,30 TPSSh31 and B3LYP34–36 functionals, whereas reactions were primarily studied with the TPSS and TPSSh functionals. TPSS and r2SCAN are mGGA functionals, whereas TPSSh and B3LYP are hybrid functionals with 10 and 20% HF exchange, respectively. TPSS has been used in most of our previous studies of nitrogenase20,39,53,60 and gives the best N2 binding energies.32 r2SCAN and TPSSh have been shown to give accurate structures of nitrogenase models.61 All calculations involved the def2-SV(P) basis set.62 The calculations were sped up by expanding the Coulomb interactions in an auxiliary basis set, the resolution-of-identity (RI) approximation.63,64 Empirical dispersion corrections were included with the DFT-D4 approach,65 as implemented in Turbomole.In this investigation we study the E0–E4 states of the FeMo cluster. The resting E0 state has the formal MoIIIFeII3FeIII4 oxidation state21,23,66 and is a quartet state according to experiments.4 The other four states were obtained by successively adding one electron and one proton to the previous state. Several positions of the added protons were tested, based on previous investigations,32 as will be discussed below. E2 was studied in the quartet spin state and E4 in the doublet state, in agreement with experiments1–4,67–69 For E1 and E3, no experimental data are available and we assumed the quintet and triplet states, respectively (previous studies have shown that different spin states are close in energy).19,20
The electronic structure of all QM calculations was obtained with the broken-symmetry (BS) approach:70 Each of the seven Fe ions was modelled in the high-spin state, with either a surplus of α (four Fe ions) or β (three Fe ions) spin. Such a state can be selected in 35 different ways.71 The various BS states were obtained either by swapping the coordinates of the Fe ions72 or with the fragment approach by Szilagyi and Winslow.73 The BS states are named by listing the numbers of the three Fe ions with minority spin, e.g. BS-235. The selection of the BS states was based on our previous experience with the similar systems.19,20,32,39,71,74
For the H2 dissociation energies (ΔEH2), H2 was optimised in a conductor-like screening model (COSMO)75,76 continuum solvent with a dielectric constant of 80. These calculations employed the default optimised COSMO atomic radius for H (1.30 Å) and a water solvent radius of 1.3 Å.77 The COSMO solvation energy of H2 is small, 1–3 kJ mol−1, making this energy correction insignificant.
QM/MM calculations
QM/MM calculations were performed with the COMQUM software.78,79 In this approach, the protein and solvent are split into three subsystems: system 1 (the QM region) was relaxed by QM methods. System 2 was kept fixed at the original coordinates (equilibrated crystal structure), to avoid the risk that different calculations end up in different local minima.In the QM calculations, system 1 was represented by a wavefunction, whereas all the other atoms were represented by an array of partial point charges, one for each atom, taken from the MM setup. Thereby, the polarisation of the QM system by the surroundings is included in a self-consistent manner. When there is a bond between systems 1 and 2 (a junction), the hydrogen link-atom approach was employed: The QM system was capped with hydrogen atoms, the positions of which are linearly related to the corresponding carbon atoms (carbon link atoms, CL) in the full system.78,80 All atoms were included in the point-charge model, except the CL atoms.81 ComQum employs a subtractive scheme with van der Waals link-atom corrections.82 No cut-off is used for the QM and QM–MM interactions. The geometry optimisations were continued until the energy change between two iterations was less than 2.6 J mol−1 (10−6 a.u.) and the maximum norm of the Cartesian gradients was below 10−3 a.u. Approximate transition states for the formation of H2 were obtained by performing relaxed scans of H–H distances.
Result and discussion
We have studied the formation of H2 in nitrogenase. This is not possible until the E2 state when two protons have accumulated on the cluster. Therefore, we have studied the E2–E4 states. The results at each reduction level are discussed in separate sections. We also study proton transfers within the FeMo cluster, connecting the various protonated structures.H2 formation in the E2 state
We first studied the formation and dissociation of H2 from the E2 state. As discussed in the Introduction, different DFT functionals give different predictions of what is the most stable structure (protonation) of the E2 state. Based on previous investigations,19,32 we have studied H2 formation and proton transfers within seven structural candidates for the E2 state. These and some additional low-energy structural candidates are shown in Fig. 2. Relative energies calculated with four DFT functionals are shown in Table 1. | ||
| Fig. 2 The eight considered E2 structures, showing the structure obtained with TPSS (B3LYP for C2). The labels are explained in the text. H–H distances in Å are marked in the figures. | ||
| Struct. | H1 | H2 | BS | TPSS | r2SCAN | TPSSh | B3LYP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H–H | ΔE | ΔEH2 | ΔE‡ | ΔE | ΔEH2 | H–H | ΔE | ΔEH2 | ΔE‡ | ΔE | ΔEH2 | ||||
| a BS-346. b BS-235. c ΔE‡ > 220 kJ mol−1. | |||||||||||||||
| B55 | S2B(5) | Fe2/6(5) | 235 | 2.23 | 33 | −133 | 51 | 63 | −166 | 2.21 | 42 | −146 | 59 | 171 | −158 |
| B53 | S2B(5) | Fe2/6(3) | 235 | 3.41 | 4 | −104 | 46 | −150 | 3.45 | 21 | −125 | 62 | 152 | −139 | |
| B33 | S2B(3) | Fe2/6(3) | 235 | 2.32 | 0 | −100 | 86 | 41 | −145 | 2.33 | 14 | −119 | 91 | 142a | −130 |
| B35 | S2B(3) | Fe2/6(5) | 235 | 3.47 | 17 | −117 | 17 | −120 | 3.55 | 9 | −114 | 133 | −120 | ||
| T53 | S2B(3) | Fe5 | 235 | 5.82 | 19 | −119 | 160 | 25 | −129 | 5.82 | 16 | −120 | 115 | −102 | |
| H6 | S2B(H6) | Fe2/6 | 235 | 2.43 | 13 | −113 | 79 | 0 | −104 | 2.48 | 0 | −104 | 104 | 88a | −75 |
| H2 | S2B(H2) | Fe2/6 | 235 | 3.66 | 75 | −175 | 80 | −183 | 3.78 | 71 | −175 | 186 | −173 | ||
| C2 | C2367 | C3457 | 345 | 1.87 | 155b | −255 | 113 | −217 | 1.77 | 35 | −139 | 0c | 13 | ||
| C1 | S2B(3) | C2456 | 346 | 3.90 | 32 | −18 | |||||||||
| E0 + H2 from B55 | 235 | 0.75 | −57 | −43 | −60 | −44 | 0.75 | −65 | −40 | −32 | −43 | ||||
| E0 + H2 from B33 | 235 | 0.75 | −44 | −56 | −40 | −64 | 0.76 | −48 | −57 | −9 | −66 | ||||
| E0 + H2 from H6S | 235 | 0.76 | −105 | 5 | −109 | 5 | 0.76 | −109 | 5 | −82 | 6 | ||||
With the TPSS functional, the most stable structures have one proton bound to S2B (which bridges Fe2 and Fe6, cf.Fig. 1) and one hydride ion also bridging Fe2 and Fe6. There are four structural isomers of this structure depending on the direction of the two H atoms. The best one has the hydride ion on the same side of S2B as S3B (called Fe2/6(3)) and the proton on S2B also points towards S3A (called S2B(3)). This is structure B33 in Fig. 2 (“B” because both S2B and the hydride ion bridge Fe2 and Fe6). The other three isomers are called B35, B53 and B55, depending on whether the proton on S2B points towards S3A or S5A (first number) and whether the hydride ion is on the same side of S2B as S3A or S5A (second number). They are 4–33 kJ mol−1 less stable than the B33 structure with TPSS.
For the B33 structure, the proton and the hydride ion are both on the same side of the cluster, with a distance of 2.32–2.33 Å in the optimised structures, and therefore the formation of H2 from these two atoms is quite straight forward. The transition state is late, at a H–H distance of 1.0–1.1 Å, and the barrier is 86–91 kJ mol−1 (calculated with TPSS or TPSSh). For the B55 structure, the hydride ion and the proton are also on the same side of the cluster, at a distance of 2.21–2.23 Å. However, for this structure, the activation energy for H2 formation is lower, 51–55 kJ mol−1.
For the other two structures (B35 and B53), the proton and the hydride ion are on different sides of the cluster, with initial distances of 3.4–3.5 Å. Therefore, the reaction has to be performed in two steps. For example, B35 could first be isomerised to B33 by a rotation of the proton on S2B to the other side and then the transfer of the proton to the Fe2/6 hydride ion (which we have already studied). The rotation has a barrier of 55–62 kJ mol−1.
With TPSSh and r2SCAN, the best structure still has a proton on S2B and a hydride ion bridging Fe2/6, but S2B has dissociated from Fe2, but not from Fe6. This structure is called H6 in Fig. 2, indicating that S2B is half-dissociated, still binding to Fe6. The corresponding structure with S2B dissociated from Fe6 instead (H2 in Fig. 2) is 71–97 kJ mol−1 higher in energy. With TPSS, B33 is 13 kJ mol−1 more stable than H6, whereas the opposite is true with r2SCAN and TPSSh by 41 and 14 kJ mol−1, respectively.
For the H6 structure, the two H atoms are also on the same side of the cluster, with a distance of 2.43–2.48 Å. However, the activation barrier for H2 formation is quite high, 79 kJ mol−1 with TPSS and 104 kJ mol−1 with TPSSh. The transition state has a H–H distance of 1.0–1.1 Å. For this structure, we also studied how easily the proton on the half-dissociated S2B ion may rotate 360° around the Fe6–S2B axis. We found two local minima. The lowest is that shown in Fig. 2, with the proton pointing between the hydride ion and S1B. In the second, which is 2–3 kJ mol−1 less stable, it instead points towards S3B. The two minima are separated by barriers of only 12–17 kJ mol−1. Thus, the rotation of this proton is essentially free and it is enough to study the most stable structure.
Moreover, we studied the formation of H6 from B33 by cleavage of the S2B–Fe2 bond. This cleavage turned out to be quite facile, with a barrier of 35–39 kJ mol−1. Similar reactions could also be used twice to move hydride ion in the B-type structures.
With B3LYP, the most stable structure instead has a doubly protonated central carbide ion. This structure is called C2 (two protons on the carbide ion). It is 88 kJ mol−1 more stable than the H6 state with B3LYP, whereas the opposite is true by 66–155 kJ mol−1 with the other functionals. We tried to form H2 in the C2 structure using the B3LYP functional. Interestingly, even if the two protons on the carbide ion are quite close (1.71 Å), no reaction could be obtained; instead the energy increased monotonically to over 200 kJ mol−1 when the H–H distance was decreased. We also tried to move the Fe2/6 hydride ion in the H6S structure to the central carbide ion. However, the barrier for such a movement was quite high, 106 kJ mol−1 with B3LYP. The product, which has protons on the carbide ion and on S2B (which rebound to Fe2 during the reaction) is 57 kJ mol−1 more stable than the reactant and therefore only 32 kJ mol−1 less stable than the C2 structure (i.e. the second-best structure with B3LYP; called C1 in Table 1).
Finally, we considered also H2 formation from a structure with a hydride ion on Fe5 and a proton on S2B(3) (called T53, highlighting the terminal hydride ion). This structure is 16–25 kJ mol−1 less stable than the best one for TPSS, r2SCAN and TPSSh (but 115 kJ mol−1 with B3LYP). The distance between the two H atoms is 5.82 Å. Therefore, the hydride ion on Fe5, first has to move from the exo position (trans to C), to an endo position, where it almost bridges to Fe4. The barrier for this is quite low, only 32 kJ mol−1, and the intermediate is only 13 kJ mol−1 less stable than the starting T53 structure. However, the reaction from this intermediate with a H–H distance of 3.6 Å has a barrier of 160 kJ mol−1, making the reaction prohibitive.
In all product structures of these reactions, the formed H2 molecule has dissociated from the FeMo cluster. The structure of the cluster in these product states is similar because S2B binds back to Fe2 for the H6 structure, forming a normal E0 structure. However, H2 resides in different positions in the second coordination sphere. The most stable structure is the one starting from H6. In this structure, H2 is 3.8 Å from Fe2 and it interacts weakly with His-195, Arg-277 and Ser-278 (cf.Fig. 3). This structure is 105–109 kJ mol−1 more stable than the best E2 structure with the TPSS, r2SCAN and TPSSh functionals, but it is 6 kJ mol−1 less stable than the C2 structure with B3LYP. This shows that formation of H2 is strongly downhill with the former three functionals.
 | ||
| Fig. 3 The best E2 structure with H2 in the second coordination sphere. The H–H bond length and distances to the nearest residues are marked (in Å). | ||
Another way to quantify this is to calculate the energy difference between a certain structure and the E0 state plus H2 in water solution (optimised with the COSMO continuum-solvation model). This H2 dissociation energy (ΔEH2) is given for all structures in Table 1. It can be seen that formation and dissociation of H2 are strongly favourable for all structures considered, by 100–255 kJ mol−1 for TPSS, r2SCAN and TPSSh. For B3LYP, the energies are lower, and for the most stable C2 state, the H2 dissociation energy is actually unfavourable by 13 kJ mol−1. Yet, H2 dissociation would be further enhanced by translational and rotational entropy of the released H2 molecule (typically estimated to 17–45 kJ mol−1).40,51,83,84 For the three structures with H2 already formed and located in the second coordination sphere, ΔEH2 is smaller and actually unfavourable by 5–6 kJ mol−1 for the best structure with all four functionals (but again this will be reversed by the entropy term).
H2 dissociation from the E3 state
Next, we studied H2 formation from the E3 state. As was discussed in the Introduction, the best E3 structure with TPSS, r2SCAN and TPSSh has S2B protonated and dissociated from Fe2, but still binding to Fe6, and two hydride ions both bridging Fe2 and Fe6 (called structure S6, because the two hydride ions bind to the same pair of Fe ions and S2B binds to Fe6; cf.Fig. 4). A structure with the proton on S2B pointing to another direction (towards S3B rather than S1B) is only 2–9 kJ mol−1 less stable and since the results in the previous section indicated that the proton on the half-dissociated S2B may rotate quite freely around the Fe6–S2B axis, this structure is not further discussed. A structure with the proton and the hydride ions at the same positions, but with S2B binding to Fe2 instead (S2) is 52–85 kJ mol−1 less stable than S6. | ||
| Fig. 4 The eleven considered E3 structures, showing the structure obtained with TPSS (B3LYP for C3). The labels are explained in the text. | ||
On the other hand, with B3LYP, a structure with a triply protonated carbide ion, is 195 kJ mol−1 better (C3 in Fig. 4). With TPSS and r2SCAN, C3 is 141–189 kJ mol−1 less stable than S6, but with TPSSh, the difference is only 4 kJ mol−1. We have studied several other E3 structures, of two different types. One is related to Hoffman's suggestion for the E4 state (i.e. with protons on S2B and S5A and hydride ions bridging Fe2/6 and Fe3/7), but missing one of the H atoms for E3.38 They are named according to the direction of the four H atoms (towards, S3A, S2B or S5A), given by a number in the order S2B–Fe2/6–Fe3/7–S5A and with an underscore marking the missing H atom, e.g. 35_2, (cf.Fig. 4). The second type has a proton on S2B(3) (which bridges Fe2/6), a terminal hydride on Fe5 and either another terminal hydride on Fe4 (called 345) or a hydride ion bridging Fe2/6, pointing either towards S3A or S5A (called 335 or 355, respectively; cf.Fig. 4). The best structures of these two types are 21–46 and 44–58 kJ mol−1 less stable than the S6 structure (35_3 and 355 best), respectively, with TPSS, r2SCAN and TPSSh, but 178 kJ mol−1 worse than C3 with B3LYP (35_2 best).
For all functionals, formation of H2 is strongly exothermic, by 59–107 kJ mol−1 for the best structures (i.e. leading to H2 and E1 with S2B protonated; least for TPSS and most for TPSSh, ΔEH2 in Table 2). The only exception is B3LYP, for which the C3 structure is 49 kJ mol−1 more stable than E1 + H2, i.e. an energy that is larger than the entropy gain from H2 dissociation.
| Struct. | H1 | H2 | H3 | BS | TPSS | r2SCAN | TPSSh | B3LYP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H–H | ΔE | ΔEH2 | ΔE‡ | ΔE | ΔEH2 | H–H | ΔE | ΔEH2 | ΔE‡ | ΔE | ΔEH2 | |||||
| a BS-136. b Barrier >250 kJ mol−1. c S2B dissociated from Fe2. d Protonated S2B dissociated from Fe2. | ||||||||||||||||
| 352_ | S2B(3) | Fe2/6(5) | Fe3/7(2) | 147 | 2.02 | 57 | −116 | 39 | 77 | −169 | 2.03 | 62 | −170 | 34 | 265 | −216 |
| 35_2 | S2B(3) | Fe2/6(5) | S5A(2) | 147 | 4.29 | 55 | −115 | 136 | 56 | −148 | 4.26 | 32 | −139 | 124 | 178 | −129 |
| 35_3 | S2B(3) | Fe2/6(5) | S5A(3) | 147 | 3.47 | 46 | −105 | 45 | −137 | 3.50 | 21 | −128 | 187 | −138 | ||
| 55_2 | S2B(5) | Fe2/6(5) | S5A(2) | 147 | 2.26 | 74 | −133 | 75 | 77 | −168 | 2.27 | 51 | −158 | 115 | 212 | −163 |
| 55_3 | S2B(5) | Fe2/6(5) | S5A(3) | 147 | 2.26 | 63 | −122 | 65 | −156 | 2.26 | 40 | −147 | 206 | −157 | ||
| 345 | S2B(3) | Fe4 | Fe5 | 147 | 5.86 | 68 | −127 | 52 | 58 | −150 | 5.91 | 58 | −165 | 29 | 248 | −199 |
| 335 HP | S2B(3) | Fe2/6(3) | Fe5 | 147 | 2.43 | 61 | −120 | 101 | 69 | −161 | 2.47 | 56 | −163 | 110 | 258 | −209 |
| HH | Fe2/6(3) | Fe5 | 3.99 | 55 | 3.91 | 46 | ||||||||||
| 355 | S2B(3) | Fe2/6(5) | Fe5 | 147 | 5.61 | 55 | −115 | 58 | −150 | 5.65 | 44 | −152 | 242 | −193 | ||
| S2 | S2B(H2) | Fe2/6(3) | Fe2/6(5) | 147 | 1.94 | 59 | −118 | 67 | −158 | 1.93 | 52 | −159 | 263 | −214 | ||
| S6 HP | S2B(H6) | Fe2/6(3) | Fe2/6(5) | 147 | 1.90 | 0 | −72 | 118 | 0 | −91 | 1.90 | 0 | −107 | 86 | 195 | −146 |
| HH | S2B(H6) | Fe2/6(3) | 2.94 | −59 | 57 | 2.92 | 29 | |||||||||
| C3 | C2367 | C2456 | C3457 | 147 | 1.84 | 189 | −248 | 141 | −233 | 1.80 | 4a | −111 | 0 | 49b | ||
| Product states = E1 + H2 | ||||||||||||||||
| 352_ | S2B(3) | H2 on Fe7 | 147 | 0.85 | 77 | −136 | 139 | 104 | −195 | 0.84 | 74 | −181 | 187 | 233 | −184 | |
| 55_2 | S5A(2) | H2 dissociated | 147 | 0.76 | 31 | −90 | 92 | 12 | −103 | 0.76 | −1 | −107 | 113 | 151 | −102 | |
| 345 | S2B(3) | H2 dissociated | 147 | 0.77 | −61 | 2 | 0 | −109 | 18 | 0.76 | −114 | 6 | 0 | 42 | 7 | |
| 335 HP | Fe5 | H2 dissociated | 147 | 0.76 | −25 | −34 | 37 | −55 | −36 | 0.76 | −46 | −61 | 68 | 134 | −85 | |
| 335 HH | S2B(3) | H2 dissociated | 147 | 0.76 | −49 | −11 | 13 | −97 | 5 | 0.76 | −101 | −6 | 13 | 56 | −7 | |
| S6 HP | Fe2,6(5) | H2 on Fe2c | 147 | 0.79 | 49 | −108 | 110 | 28 | −119 | 0.78 | 34 | −142 | 148 | 222 | −173 | |
| S6 HH | S2B(H6) | H2 on Fe2d | 147 | 0.76 | 9 | −68 | 70 | −38 | −54 | 0.77 | −42 | −65 | 71 | 115 | −67 | |
We studied H2 formation for seven different structures. Interestingly, the activation barrier for the reactions depends mainly on what types of H atoms are involved. Connecting two Fe-bound hydride ions give small barriers. The two hydride ions in the S6 structure (at a H–H distance of 1.90 Å) can be connected with an activation energy of only 57 (TPSS) or 29 (TPSSh) kJ mol−1. The reason for the large difference between the two functionals is that the reaction is much more exothermic with TPSSh than with TPSS (42 and 5 kJ mol−1), because the product state contains S2B dissociated from Fe2 (and H2 bound to Fe2). The transition state is later for TPSS (the H–H distances are 1.1 and 1.3 Å, respectively). Likewise, the barrier to connect the two Fe2/6 and Fe3/7 bridging hydride ions in the 352_ structure, at an initial distance of 2.02 Å, is only 34–39 kJ mol−1.
More complicated reactions involving two hydride ions also give relatively low barriers. The Fe2/6 and Fe5 hydride ions in the 335 structure are initially at a distance of 3.91–3.99 Å. However, they can form H2 by first moving the Fe2/6 hydride ion also to Fe5 (in the endo position) (i.e. almost bridging to Fe4) and then connect the two hydride ions. The first step has a barrier of 46–55 kJ mol−1, whereas the second step is facile with a barrier of only 30–31 kJ mol−1. Likewise, the two terminal hydride ions on Fe4 and Fe5 in the 345 structure, initially 5.9 Å apart, can be connected via an intermediate with the Fe5 hydride ion moved to S5A and a net barrier of 52 kJ mol−1 with TPSS and only 29 kJ mol−1 for TPSSh (in both cases for the first step).
On the other hand, reactions involving a proton and a hydride ion have appreciably higher activation energies. The proton on S2B and the hydride ion bridging Fe2/6 in the 55_2 structure are initially at a distance of 2.26–2.27 Å. They can form H2via an activation barrier of 75 (TPSS) to 115 kJ mol−1 (TPSSh). Likewise, the same two H atoms in the 335 structure (on the other side of the cluster), with an initial distance of 2.43–2.47 Å, can be connected with a barrier of 101–110 kJ mol−1. The same applies to the proton on S2B and the closest Fe2/6 hydride ion in the S6 structure (initially 2.92–2.94 Å apart). They can be connected passing a barrier of 118 (TPSS) or 86 kJ mol−1 (TPSSh).
However, we failed to find any reaction of the two closes protons on the central carbide in the C3 structure with both B3LYP and TPSSh. Although the two protons are only 1.76 Å apart at the start, the reaction was monotonously uphill to over 200 kJ mol−1.
We also studied some proton-transfer reactions within the cluster. For example, the Fe hydride ion bridging Fe3/7 could be moved to S5A in the 352_ structure (forming the 35_2 structure) with a barrier of 53 (TPSS) or 26 kJ mol−1 (TPSSh). Again, the reason for the lower barrier with TPSSh is that the reaction is more exothermic with that functional.
Among the product structures, the one started from the 345 structure was most stable, because it resulted in a E1 structure with a proton on S2B(3) and H2 in the second coordination sphere. It was 61–114 kJ mol−1 more stable than the best E3 state with the TPSS, r2SCAN and TPSSh functionals, but 42 kJ mol−1 less stable than the C3 structure with B3LYP. Three structures had H2 coordinated to the cluster (either to Fe2 or Fe7), but these structures were at least 70–73 kJ mol−1 less stable than the best structure with H2 dissociated. It should be noted, however, that we have not made any systematic investigation of H2-bound (or H2-dissociated) structures.
H2 dissociation from the E4 state
Finally, we studied H2 formation for the E4 state. We studied 24 reactions, starting from 14 different structures, described in Table 3 and in Fig. 5. Most of the structures are of two different types. The first have two protons on S2B and S5A and two hydride ions bridging Fe2/6 and Fe3/7, as suggested by Hoffman and coworkers.38 As for the E3 structures, they are named according to the direction of the four H atoms (towards S3A, S2B or S5A), given by a number in the order S2B–Fe2/6–Fe3/7–S5A, e.g. 5522 (cf.Fig. 5). We also studied two structures with S2B dissociated from Fe2 (3323H6 and 3523H6) The second group is related to the S2 and S6 structures for E3. They also have protons on S2B and S5A, and two hydride ions, but both hydride ions bridge Fe2 and Fe6, and S2B has dissociated from either Fe2 (S6) or Fe6 (S2). Both structures have two local minima depending on the direction of the proton on S2B (cf.Table 3). In addition, we studied two differing structures: 3H has a proton on S2B and three hydride ions on Fe5, Fe6 and bridging Fe2/6. C3 has three protons on the central carbide and one on S2B.| Structure | H1 | H2 | H3 | H4 | TPSS | r2SCAN | TPSSh | B3LYP | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BS | H–H | ΔE | ΔEH2 | ΔE‡ | BS | ΔE | ΔEH2 | BS | H–H | ΔE | ΔEH2 | ΔE‡ | BS | ΔE | ΔEH2 | |||||
| 3H | S2B(3) | Fe2/6(3) | Fe5 | Fe6 | 14 | 4.02 | 40 | −112 | 75 | 14 | 125 | −228 | 147 | 3.90 | 112 | −207 | 72 | 147 | 333 | −330 |
| Fe2/6(3) | Fe6 | 2.40 | 25 | 2.30 | 15 | |||||||||||||||
| S2B(3) | Fe2/6(3) | 2.39 | 59 | 2.39 | 57 | |||||||||||||||
| 3322 | S2B(3) | Fe2/6(3) | Fe3/7(2) | S5A(2) | 14 | 2.36 | 15 | −87 | 70 | 147 | 52 | −156 | 147 | 2.57 | 39 | −134 | 80 | 147 | 292 | −289 |
| Fe3/7(2) | S5A(2) | 2.55 | 101 | 2.48 | 105 | |||||||||||||||
| 3323 | S2B(3) | Fe2/6(3) | Fe3/7(2) | S5A(3) | 14 | 2.37 | 3 | −75 | 72 | 135 | 27 | −131 | 135 | 2.45 | 11 | −107 | 81 | 135 | 233 | −230 |
| 3323H6 | S2B(H6S) | Fe2/6(3) | Fe3/7(2) | S5A(3) | 147 | 2.37 | 39 | −111 | 76 | 147 | 44 | −147 | 147 | 2.36 | 49 | −145 | 88 | 147 | 313 | −311 |
| Fe2/6(3) | Fe3/7(2) | 3.24 | 92 | 3.09 | 56 | |||||||||||||||
| 3522 | S2B(3) | Fe2/6(5) | Fe3/7(2) | S5A(2) | 14 | 2.04 | 24 | −96 | 75 | 14 | 76 | −179 | 14 | 2.09 | 48 | −143 | 53 | 147 | 266 | −264 |
| Fe3/7(2) | S5A(2) | 2.48 | 99 | 2.51 | 74 | |||||||||||||||
| 3523 | S2B(3) | Fe2/6(5) | Fe3/7(2) | S5A(3) | 14 | 2.08 | 14 | −86 | 80 | 147 | 25 | −129 | 135 | 2.26 | 18 | −113 | 43 | 346 | 192 | −189 |
| 3523H6 | S2B(H6S) | Fe2/6(5) | Fe3/7(2) | S5A(3) | 147 | 2.36 | 39 | −111 | 77 | 147 | 44 | −147 | 147 | 2.35 | 49 | −145 | 88 | 147 | 293 | −291 |
| Fe2/6(5) | Fe3/7(2) | 3.16 | 91 | 3.09 | 56 | |||||||||||||||
| 5322 | S2B(5) | Fe2/6(3) | Fe3/7(2) | S5A(2) | 14 | 2.53 | 22 | −93 | 98 | 147 | 59 | −162 | 147 | 2.56 | 48 | −143 | 102 | 147 | 305 | −303 |
| S2B(5) | Fe3/7(2) | 2.96 | 81 | 2.97 | 72 | |||||||||||||||
| 5323 | S2B(5) | Fe2/6(3) | Fe3/7(2) | S5A(3) | 14 | 2.77 | 11 | −83 | 78 | 147 | 45 | −148 | 147 | 3.09 | 37 | −132 | 67 | 147 | 296 | −293 |
| 5522 | S2B(5) | Fe2/6(5) | Fe3/7(2) | S5A(2) | 14 | 2.24 | 41 | −112 | 61 | 147 | 59 | −162 | 147 | 2.32 | 51 | −147 | 49 | 147 | 305 | −303 |
| Fe2/6(5) | Fe3/7(2) | 2.09 | 85 | 2.17 | 60 | |||||||||||||||
| Fe3/7(2) | S5A(2) | 2.51 | 102 | 2.51 | 77 | |||||||||||||||
| 5523 | S2B(5) | Fe2/6(5) | Fe3/7(2) | S5A(3) | 14 | 2.25 | 30 | −101 | 63 | 147 | 43 | −146 | 147 | 2.32 | 40 | −136 | 48 | 147 | 296 | −294 |
| Fe2/6(5) | Fe3/7(2) | 2.13 | 84 | 2.19 | 46 | |||||||||||||||
| S2 | S2B(H2) | Fe2/6(3) | Fe2/6(5) | S5A(3) | 147 | 1.88 | 54 | −126 | 147 | 79 | −182 | 147 | 1.86 | 56 | −152 | 147 | 293 | −290 | ||
| S64 | S2B(H64) | Fe2/6(3) | Fe2/6(5) | S5A(3) | 147 | 1.85 | 1 | −72 | 47 | 147 | 29 | −132 | 147 | 1.87 | 18 | −113 | 38 | 147 | 257 | −254 |
| S61 | S2B(H61) | Fe2/6(3) | Fe2/6(5) | S5A(3) | 147 | 1.82 | 0 | −72 | 42 | 346 | 0 | −103 | 157 | 1.74 | 0 | −95 | 34 | 347 | 229 | −226 |
| C3 | S2B(3) | C2367 | C2456 | C3457 | 234 | 1.83 | 192 | −263 | 346 | 146 | −249 | 234 | 1.83 | 1 | −97 | 234 | 0 | 3 | ||
| C1 | S2B(H6) | Fe2/6 | C2367 | S5A(3) | 346 | 158 | −156 | |||||||||||||
| Product states | ||||||||||||||||||||
| 3H Fe2/6–5 | S2B(3) | Fe5 | 146 | 0.76 | −34 | −38 | 147 | −56 | −48 | 147 | 0.76 | −56 | −40 | 147 | 113 | −111 | ||||
| 3H Fe2/6–6 | Fe5 | Fe6 | H2 on Fe6 | 14 | 0.87 | 80 | −152 | 147 | 131 | −234 | 147 | 0.85 | 132 | −227 | 147 | 423 | −420 | |||
| 3322 | Fe3/7 | S5A(2) | 147 | 0.76 | −46 | −26 | −31 | −73 | 0.76 | −32 | −64 | 228 | −225 | |||||||
| 3322 | S2B(3) | Fe2/6(3) | 147 | 0.76 | −28 | −43 | −44 | −59 | 0.76 | −54 | −41 | 183 | −181 | |||||||
| 3323 | Fe3/7 | S5A(3) | 147 | 0.76 | −60 | −11 | −47 | −57 | 0.76 | −44 | −51 | 217 | −214 | |||||||
| 3522 | S2B(3) | S5A(2) | 147 | 0.76 | 21 | −93 | 11 | −114 | 0.76 | −11 | −84 | 151 | −148 | |||||||
| 3522 | S2B(3) | Fe2/6(5) | 147 | 0.75 | −6 | −66 | −28 | −75 | 0.75 | −34 | −61 | 179 | −177 | |||||||
| 3523 | S2B(3) | S5A(3) | 146 | 0.75 | 5 | −77 | −9 | −94 | 0.76 | −24 | −71 | 142 | −139 | |||||||
| 5322 | S5A(2) | Fe2/6(3) | 235 | 0.76 | 41 | −112 | 58 | −162 | 0.75 | 48 | −143 | 267 | −265 | |||||||
| 5322 | S2B(5) | Fe2/6(3) | 147 | 0.76 | −16 | −56 | −30 | −73 | 0.76 | −41 | −54 | 198 | −196 | |||||||
| 5323 | S5A(3) | Fe2/6(3) | 146 | 0.76 | 47 | −119 | 147 | 56 | −159 | 147 | 0.76 | 36 | −132 | 147 | 246 | −243 | ||||
| 5522 | Fe3/7 | S5A(2) | 147 | 0.76 | 1 | −73 | 35 | −138 | 0.75 | 30 | −125 | 298 | −295 | |||||||
| 5522 | S2B(5) | S5A(2) | 147 | 0.76 | 41 | −112 | 29 | −132 | 0.77 | 8 | −103 | 187 | −185 | |||||||
| 5522 | S2B(5) | Fe2/6(5) | 147 | 0.75 | 7 | −79 | −12 | −91 | 0.75 | −29 | −66 | 195 | −192 | |||||||
| 5523 | Fe3/7 | S5A(3) | 147 | 0.76 | −10 | −61 | 20 | −123 | 0.75 | 18 | −113 | 288 | −285 | |||||||
| 5523 | S2B(5) | S5A(3) | 146 | 0.76 | 21 | −93 | 8 | −111 | 0.76 | −6 | −89 | 147 | 131 | −129 | ||||||
| 3323H6 | Fe3/7(2) | S5A(3) | 147 | 0.75 | −34 | −38 | −44 | −59 | 0.76 | −41 | −55 | 220 | −217 | |||||||
| 3323H6 | S2B(5) | S5A(3) | 147 | 0.76 | 30 | −101 | 17 | −121 | 0.77 | −4 | −91 | 178 | −176 | |||||||
| 3523 | Fe3/7(2) | S5A(3) | 147 | 0.75 | −67 | −5 | −44 | −59 | 0.76 | −41 | −55 | 255 | −252 | |||||||
| 3523H6 | S2B(5) | S5A(3) | 147 | 0.76 | 39 | −110 | 17 | −121 | 0.76 | −2 | −94 | 172 | −170 | |||||||
| S64 | S2B(H6M) | S5A(3) | H2 on Fe2 | 147 | 0.94 | 40 | −112 | 12 | −116 | 0.80 | 12 | −107 | 346 | 109 | −106 | |||||
| S61 | S2B(H6S) | S5A(3) | H2 on Fe2 | 147 | 0.83 | 26 | −98 | 9 | −113 | 0.79 | −5 | −91 | 84 | −82 | ||||||
 | ||
| Fig. 5 The 15 considered E4 structures, showing the structures obtained with TPSS (B3LYP for C3). The labels are explained in the text. | ||
The relative stabilities of the various E4 states were discussed in a previous study,32 showing that S61 is most stable with TPSS, TPSSh and r2SCAN, but C3 with B3LYP (which is 233 kJ mol−1 more stable than S61 with this functional, whereas C3 is 192, 146 and 1 kJ mol−1 less stable than S61 with TPSS, TPSSh and r2SCAN, respectively). However, with TPSS, several Hoffman-type structures are competitive, in particular 3323, which is only 3 kJ mol−1 less stable than S61 (27 and 11 kJ mol−1 less stable with r2SCAN and TPSSh).
With the TPSS, TPSSh and r2SCAN functionals, all studied E4 structures are less stable than the best E2 structure and H2 in a COSMO solvent (i.e. ΔEH2 in Table 3) by at least 72, 103 and 95 kJ mol−1, respectively. On the other hand, the C3 structure is 3 kJ mol−1 more stable than the dissociated state with B3LYP. Thus, we can again conclude that formation and dissociation of H2 from the FeMo cluster is highly thermodynamically favourable and should be kinetically controlled. Therefore, we performed a thorough investigation of the formation of H2 from all pairs of H atoms on the same face of the FeMo cluster in 14 different starting structures.
The barriers of all 24 studied reactions are collected in Table 3. The reactions can be divided into five groups. The smallest barrier, 15–25 kJ mol−1, is found for the reaction of two hydride ions bound to the same Fe6 ion, one terminal and the other bridging to Fe2 in the 3H structure. This gives an initial H–H distance of 2.30–2.40 Å. Likewise, reactions from the S61 and S64 structures, where the two hydride ions both bridge Fe2 and Fe6 also give quite small barriers of 34–47 kJ mol−1. In the starting structure, the two hydride ions are only 1.74–1.87 Å apart.
Five reactions have barriers of 59–72 kJ mol−1 with TPSS and 48–81 kJ mol−1 with TPSSh. They all involve reactions between a proton on S2B and hydride ion bridging Fe2 and Fe6. They have initial H–H distances of 2.24–2.37 Å, irrespectively of whether the two H atoms are on the same side as S5A or S3A. The analogous reaction involving the Fe2/6 hydride and a proton on a half-dissociated S2B give a slightly higher barrier of 76–77 kJ mol−1 and 88 kJ mol−1, respectively. Two reactions also involve the proton on S2B but the hydride ion bridging Fe3 and Fe7. They have slightly larger initial H–H distances, 2.77–3.09 Å and they give slightly larger barriers of 78–81 kJ mol−1 with TPSS, but lower barriers with TPSSh, 67–72 kJ mol−1.
There are four reactions that involve the proton on S5A and the hydride ion bridging Fe3/7. They have initial H–H distances of 2.48–2.55 Å, but the barriers are appreciably larger, 98–105 kJ mol−1 (except two barriers with TPSSh, 74–77 kJ mol−1). Thus, the barriers depend more on the involved H atoms than on the initial H–H distances.
Finally, seven reactions involve two hydride ions on different Fe ions. Four of them involve hydrides on the Fe2/6 and Fe3/7 pairs, having initial H–H distances of 2.04–2.13 Å. They give barriers of 75–85 kJ mol−1 with TPSS and 43–60 kJ mol−1 with TPSSh. Two involve hydrides in the same positions, but with S2B dissociated from Fe2. This gives longer initial distances (3.16–3.24 Å) and higher barriers with TPSS (91–92 kJ mol−1), but not with TPSSh (56 kJ mol−1). The seventh reaction involves hydride ions on Fe2/6 and Fe5 in the 3H complex. In this case, the initial H–H distance is much larger, 4.02 Å. This gives a more complicated reaction, in which the Fe2/6 hydride ion first moves to Fe5, via a Fe5/6 bridging position (with a barrier of 72–75 kJ mol−1) and then the two hydride ions on the Fe5 ion (with a H–H distance of 2.20 Å) connect with a barrier of 49–58 kJ mol−1 relative to the starting structure.
We also tried to connect the closest protons on the central carbide in the C3 structure. However, as for the corresponding E2 and E3 states, this was not be possible – the energy rose monotonically to >170 kJ mol−1. On the other hand, it was possible to move the Fe3/7 hydride ion in 3523H6 structure to the central carbide ion. The barrier was only 47 kJ mol−1 when studied with B3LYP and the product (with protons also on S2B(H6), Fe2/6 and S5A(5)) was 33 kJ mol−1 more stable than the reactant (and therefore the second-best B3LYP structure), although it is still 158 kJ mol−1 less stable than the C3 structure. The corresponding reaction with TPSSh was not possible, mainly owing to the low stability of the product, but also to the fact that it was started from the 3523 structure with S2B still bridging and therefore the hydride on Fe2/6 was partly in the way for the movement of the Fe3/7 hydride ion.
In addition, we tried to convert the S61 structure to the 3323 structure. This requires two steps. First, one of the Fe2/6 hydrides needs to move to Fe3/7. This can be done with a barrier of 54–69 kJ mol−1. Then, S2B should bind back to Fe2, which can be done with a minimal barrier of 5–7 kJ mol−1.
As a result of the study of the H2-formation reactions, we obtained 24 different product structures. Most of them have H2 in the second coordination sphere of the FeMo cluster. The best structure with TPSS has a proton on S5A and a hydride bridging Fe3 and Fe7. With B3LYP, the best structure has two protons on S2B and S5A. With r2SCAN and TPSSh, a structure with a proton on S2B and a terminal hydride ion on Fe5 is best. This is quite different from what was observed for the E2 state without H2, indicating that the second-sphere H2 molecule has a significant influence on the relative energies and reflecting that we have not performed any systematic investigation of this type of complexes. This is also confirmed by the fact that none of the structures is more stable than the fully dissociated E2 + H2 state (ΔEH2 is −5 to −111 kJ mol−1 for the best structures with the four DFT methods). In four of the product structures, H2 coordinates side-on to Fe2 or Fe6. These structures have appreciably larger H–H bond lengths than the other structures (0.80–0.94 Å, compared to 0.75–0.76 Å). The best structure has H2 bound to Fe2, S2B protonated and bound only to Fe6, and S5A protonated. However, this structure is 24–93 kJ mol−1 less stable than be best structure with H2 in the second sphere.
Conclusions
We have studied the formation of H2 in the E2–E4 states of Mo-nitrogenase. Even if there are quite some variations between the individual structures and the DFT functionals employed, there are some general trends. The lowest barrier for H2 formation is obtained when combining two hydride ions, one terminal on Fe6 and one bridging Fe2/6, 15–25 kJ mol−1. Combining two hydride ions both bridging Fe2/6 also give low barriers, 29–57 kJ mol−1. Combining hydride ions bridging Fe2/6 and Fe3/7, give higher barriers with TPSS, 75–85 kJ mol−1, except in the E3 state (39 kJ mol−1). With TPSSh, those barriers are lower, 34–60 kJ mol−1. The reaction between a proton and a hydride ion gives similar barriers. With TPSS, combining a proton on S2B and a hydride ion bridging Fe2/6 typically gives lower barriers than combining two hydride ions on Fe2/6 and Fe3/7 or a proton on S5A and a hydride ion bridging Fe3/7. With TPSSh, barriers for two hydride ions are lower and there are smaller differences between the two proton–hydride reactions. Instead, there is some difference between whether the S2B proton and the Fe2/6 hydride ion are on the same side as S5A (lower barrier) or as S3A (higher barrier). For both functionals, it seems that the proton–hydride reactions give a lower barrier as the number of added electrons and protons increase (i.e. E2 > E3 > E4). In general, TPSSh gives lower barriers than TPSS (by 9 kJ mol−1 on average), but for proton–hydride reactions, the opposite is often true.We have also studied some other reactions, moving around the H atoms within the FeMo cluster. In agreement with previous studies of proton transfers in the FeMo cluster during the later (E4–E8) steps of the reaction mechanism,60 we find that individual barriers are rather small, 5–69 kJ mol−1. The calculations show that a proton on S2B can rotate rather freely, either moving from the S2B(3) to the S2B(5) side (55–62 kJ mol−1) when S2B is bridging Fe2/6 or rotating around the Fe6–S2B (12–17 kJ mol−1) axis when it is dissociated from Fe6. The dissociation of S2B from Fe2 also has a low barrier (35–39 kJ mol−1). In fact, it is also possible to move a Fe3/7 hydride ion to S5A (26–53 kJ mol−1) or to move a bridging hydride ion from Fe2/6 to Fe3/7 (54–69 kJ mol−1). A hydride ion can move to the central carbide with B3LYP (47 kJ mol−1), but only in the E4 state and if the starting structure is appropriate. This shows that the various isomers of the En states can interconvert rather freely. Therefore, it is unlikely that the FeMo cluster may avoid unwanted side reactions by placing the H atoms on different faces of the FeMo cluster. Likewise, it seems impossible to avoid the most stable structures for each En state, i.e. to stabilise metastable states by kinetic pathways and barriers.
Finally, we discuss the implications of the current calculations on the reaction mechanism of nitrogenase. From a functional point of view, it is clear that the enzyme should avoid the formation of H2 before N2 binds, i.e. at least for the E2 and E3 states. For the E2 state, B33 is the most stable structure with TPSS. Formation of H2 from this state has an activation barrier of 86–91 kJ mol−1. This corresponds to a rate of 0.0008–0.0072 s−1 (3–26 h−1; using classical transition-state theory with a pre-exponential factor of 6.2 × 1012 s−1), which is smaller than the turnover rate of the enzyme 1–5 s−1.2,85 Thus, the protein should be able to avoid this side reaction if the electron flow is proper, but the rate constant is somewhat smaller than what has been estimated from kinetic measurements, 0.2 s−1,85 corresponding to 77 kJ mol−1. For the B55 structure, the activation energy for H2 formation is lower, 51–55 kJ mol−1. However, this structure is 33–42 kJ mol−1 less stable than the best state with TPSS or TPSSh, giving net barriers of 84–97 kJ mol−1, corresponding to rates of 0.2–65 h−1, still much lower than the turnover rate. For the H6 structure, which is the most stable structure with TPSSh and r2SCAN, the activation barrier for H2 formation is quite high, 79–104 kJ mol−1. This corresponds to rates of 0.13–5 × 10−6 s−1 (0.02–460 h−1), which are less than the turnover rate of the enzyme. For the best structure with B3LYP, C2, no H2 formation could be obtained. Thus, we can conclude that H2 formation does not seem to be any serious problem at the E2 level of the protein.
Our calculated activation barrier for the H6 structure of E2 is similar to that reported by Thorhallsson and Bjornsson, 86 kJ mol−1, with TPSSh.33 On the other hand, our barrier for the B55 structure (51 kJ mol−1) is somewhat larger than that reported by Khadka and coworkers, 29 kJ mol−1.52 This might be caused by the difference in DFT methods (TPSS and BP86) but pure GGA functionals often give similar results for nitrogenase models.18 It is more likely that the reason for the difference is that Khadka and coworkers used QM-cluster calculations, whereas we have performed QM/MM calculations. In fact, Thorhallsson and Bjornsson reported much lower activation energies with cluster models than QM/MM,33 indicating that the surrounding enzyme disfavours the H2 formation.
For the E3 state, the results are more problematic. The S6 structure, which is preferred with the TPSS, r2SCAN and TPSSh functionals, has two hydride ions both bridging Fe2/6, and the barrier of H2 formation from these two hydrides is small 29–57 kJ mol−1, corresponding to 7 × 102 to 5 × 107 s−1, much faster than the turnover of the enzyme. We see no way for the enzyme to avoid this problematic side reaction with the current results. Either the TPSS, r2SCAN and TPSSh methods give gravely unreliable results or we have not yet found the proper lowest-energy structure or BS state for E3. Alternatively, the B3LYP results are trusted, which indicates that C3 structure is most stable. In our calculations, no H2 formation is observed from this structure. On the other hand, B3LYP gives very weak N2 binding in the E4,32 which has led Siegbahn to suggest that four additional reductions are needed before N2 may bind,46,83 something that does not find any experimental support.13
For the E4 state, the situation is similar: The best structure with TPSS, r2SCAN and TPSSh is S61, with two hydride ions both bridging Fe2/6. These can be connected with a barrier of only 34–47 kJ mol−1, corresponding to rates of 5 × 104–7 × 106 s−1, i.e. much faster than the turnover rate of the enzyme. However, with TPSS, the Hoffman-type structures are competitive in stability, in particular 3323, which is only 3 kJ mol−1 less stable than S61. From this structure, H2 formation has a higher barrier of 72–81 kJ mol−1, i.e. similar to the turnover rate of the enzyme. There are other Hoffman-type of complexes with lower barriers (down to 61 kJ mol−1 with TPSS and 46 kJ mol−1 with TPSSh), but they are too unstable compared to the best structure so that the net barrier (counted from the best structure) becomes prohibitively high. Finally, the C3 structure is best with B3LYP and competitive with TPSSh, but no H2 formation could be obtained from this structure. In conclusion, H2 formation from the best E4 structures is possible. In this case, it is unclear whether this reaction should take place before or after binding of N2, i.e. if it should be avoided or not. The results are in accordance with a recent steady-state kinetic model of nitrogenase, indicating that the rate of H2 formation is much higher from the E4 state than from E2.85 In future studies, we will study the binding of N2 to E4 structures with H2 and whether the binding of N2 can be enhanced by the concerted formation or dissociation of H2.
Author contributions
HJ performed all calculations and contributed to the analysis and the writing of the manuscript. UR directed the research, analysed the results and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This investigation has been supported by grants from the Swedish research council (projects 2018-05003 and 2020-06176) and from China Scholarship Council. The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) and by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at Lunarc at Lund University, NSC at Linköping University and HPC2N at Umeå University, partially funded by the Swedish Research Council (grants 2018-05973 and 2022-06725).References
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983 Search PubMed.
- B. Schmid et al. , Handbook of Metalloproteins, John Wiley & Sons, Ltd, 2006, p. 1025 Search PubMed.
- B. M. Hoffman, et al. , Chem. Rev., 2014, 114, 4041 Search PubMed.
- L. C. Seefeldt, et al. , Chem. Rev., 2020, 120, 5082 Search PubMed.
- T. Spatzal, et al. , Science, 2011, 334, 940 Search PubMed.
- J. Kim and D. C. Rees, Science, 1992, 257, 1677 Search PubMed.
- O. Einsle, et al. , Science, 2002, 297, 1696 Search PubMed.
- T. Spatzal, et al. , Science, 2014, 345, 1620 Search PubMed.
- O. Einsle, J. Biol. Inorg. Chem., 2014, 19, 737 Search PubMed.
- R. N. F. Thorneley, and D. J. Lowe, in Molybdenum Enzymes, ed. T. G. Spiro, Wiley, New York, 1985, p. 221 Search PubMed.
- Z. Y. Yang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16327 Search PubMed.
- D. Lukoyanov, et al. , J. Am. Chem. Soc., 2016, 138, 10674 Search PubMed.
- V. Hoeke, et al. , J. Am. Chem. Soc., 2019, 141, 11984 Search PubMed.
- H. Yang, et al. , Inorg. Chem., 2018, 57, 12323 Search PubMed.
- R. Y. Igarashi, et al. , J. Am. Chem. Soc., 2005, 127, 6231 Search PubMed.
- C. Van Stappen, et al. , Chem. Rev., 2020, 120, 5005 Search PubMed.
- I. Dance, ChemBioChem, 2020, 21, 1671 Search PubMed.
- L. Cao and U. Ryde, Phys. Chem. Chem. Phys., 2019, 21, 2480 Search PubMed.
- H. Jiang, O. K. G. Svensson and U. Ryde, Inorg. Chem., 2022, 61, 18067 Search PubMed.
- L. Cao, O. Caldararu and U. Ryde, J. Chem. Theory Comput., 2018, 14, 6653 Search PubMed.
- T. Spatzal, et al. , Nat. Commun., 2016, 7, 10902 Search PubMed.
- B. Benediktsson and R. Bjornsson, Inorg. Chem., 2017, 56, 13417 Search PubMed.
- R. Bjornsson, F. Neese and S. DeBeer, Inorg. Chem., 2017, 56, 1470 Search PubMed.
- S. J. Yoo, et al. , J. Am. Chem. Soc., 2000, 122, 4926 Search PubMed.
- C. Van Stappen, et al. , Inorg. Chem., 2019, 58, 12365 Search PubMed.
- C. Van Stappen, et al. , Chem. Sci., 2019, 10, 9807 Search PubMed.
- D. A. Lukoyanov, et al. , Inorg. Chem., 2022, 61, 5459 Search PubMed.
- H. Jiang, K. J. M. Lundgren and U. Ryde, Inorg. Chem., 2023, 62(48), 19433–19445 Search PubMed.
- J. Tao, et al. , Phys. Rev. Lett., 2003, 91, 146401 Search PubMed.
- J. W. Furness, et al. , J. Phys. Chem. Lett., 2020, 11, 8208 Search PubMed.
- V. N. Staroverov, et al. , J. Chem. Phys., 2003, 119, 12129 Search PubMed.
- H. Jiang and U. Ryde, Dalton Trans., 2023, 52, 9104 Search PubMed.
- A. T. Thorhallsson and R. Bjornsson, Chem. – Eur. J., 2021, 27, 16788 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 Search PubMed.
- Y. Pang and R. Bjornsson, Phys. Chem. Chem. Phys., 2023, 25, 21020 Search PubMed.
- S. Raugei, L. C. Seefeldt and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10521 Search PubMed.
- L. Cao and U. Ryde, J. Chem. Theory Comput., 2020, 16, 1936 Search PubMed.
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, Chem. Sci., 2019, 10, 11110 Search PubMed.
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2016, 138, 10485 Search PubMed.
- P. E. M. Siegbahn, J. Comput. Chem., 2018, 39, 743 Search PubMed.
- J. B. Varley, et al. , Phys. Chem. Chem. Phys., 2015, 17, 29541 Search PubMed.
- M. Rohde, et al. , Biochemistry, 2018, 57, 5497 Search PubMed.
- W.-L. Li, et al. , Chem. Catal., 2023, 3(7), 100662 Search PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2023, 25, 23602 Search PubMed.
- D. Sippel, et al. , Science, 2018, 359, 1484 Search PubMed.
- W. Kang, et al. , Science, 2020, 368, 1381 Search PubMed.
- I. Dance, J. Am. Chem. Soc., 2007, 129, 1076 Search PubMed.
- I. Dance, Z. Anorg. Allg. Chem., 2015, 641, 91 Search PubMed.
- I. Dance, Dalton Trans., 2021, 50, 18212 Search PubMed.
- N. Khadka, et al. , J. Am. Chem. Soc., 2017, 139, 13518 Search PubMed.
- L. Cao, O. Caldararu and U. Ryde, J. Phys. Chem. B, 2017, 121, 8242 Search PubMed.
- B. M. Barney, et al. , Biochemistry, 2007, 46, 6784 Search PubMed.
- J. A. Maier, et al. , J. Chem. Theory Comput., 2015, 11, 3696 Search PubMed.
- W. L. Jorgensen, et al. , J. Chem. Phys., 1983, 79, 926 Search PubMed.
- L. Hu and U. Ryde, J. Chem. Theory Comput., 2011, 7, 2452 Search PubMed.
- C. I. Bayly, et al. , J. Phys. Chem., 1993, 97, 10269 Search PubMed.
- F. Furche, et al. , Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91 Search PubMed.
- H. Jiang, et al. , Angew. Chem., Int. Ed., 2022, 61, e202208544 Search PubMed.
- B. Benediktsson and R. Bjornsson, J. Chem. Theory Comput., 2022, 18, 1437 Search PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 Search PubMed.
- K. Eichkorn, et al. , Chem. Phys. Lett., 1995, 240, 283 Search PubMed.
- K. Eichkorn, et al. , Theor. Chem. Acc., 1997, 97, 119 Search PubMed.
- E. Caldeweyher, et al. , J. Chem. Phys., 2019, 150, 154122 Search PubMed.
- R. Bjornsson, et al. , Chem. Sci., 2014, 5, 3096 Search PubMed.
- D. J. Lowe, R. R. Eady and R. N. F. Thorneley, Biochem. J., 1978, 173, 277 Search PubMed.
- D. Lukoyanov, et al. , Inorg. Chem., 2014, 53, 3688 Search PubMed.
- D. A. Lukoyanov, et al. , Inorg. Chem., 2018, 57, 6847 Search PubMed.
- T. Lovell, et al. , J. Am. Chem. Soc., 2001, 123, 12392 Search PubMed.
- L. Cao and U. Ryde, Int. J. Quantum Chem., 2018, 118, e25627 Search PubMed (16 pages).
- C. Greco, et al. , Int. J. Quantum Chem., 2011, 111, 3949 Search PubMed.
- R. K. Szilagyi and M. A. Winslow, J. Comput. Chem., 2006, 27, 1385 Search PubMed.
- L. Cao and U. Ryde, J. Biol. Inorg. Chem., 2020, 25, 521 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 Search PubMed.
- A. Schäfer, et al. , Phys. Chem. Chem. Phys., 2000, 2, 2187 Search PubMed.
- A. Klamt, et al. , J. Phys. Chem. A, 1998, 102, 5074 Search PubMed.
- U. Ryde, J. Comput.-Aided Mol. Des., 1996, 10, 153 Search PubMed.
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335 Search PubMed.
- N. Reuter, et al. , J. Phys. Chem. A, 2000, 104, 1720 Search PubMed.
- L. Hu, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2011, 7, 761 Search PubMed.
- L. Cao and U. Ryde, Front. Chem., 2018, 6, 89 Search PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2019, 21, 15747 Search PubMed.
- W.-J. Wei and P. E. M. Siegbahn, Chem. – Eur. J., 2022, 28, e202103745 Search PubMed.
- D. F. Harris, A. Badalyan and L. C. Seefeldt, Biochemistry, 2022, 61, 2131 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05181a |
| This journal is © the Owner Societies 2024 |