
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transfer in superbase n-type doping of PCBM induced by deprotonation†
Chuan-Ding
Dong
 *a,
Fabian
Bauch
a,
Yuanyuan
Hu
b and
Stefan
Schumacher
ac
*a,
Fabian
Bauch
a,
Yuanyuan
Hu
b and
Stefan
Schumacher
ac
aDepartment of Physics and Center for Optoelectronics and Photonics Paderborn (CeOPP), Paderborn University, Warburger Strasse 100, Paderborn 33098, Germany. E-mail: cddong@mail.uni-paderborn.de
bInternational Science and Technology Innovation Cooperation Base for Advanced Display Technologies of Hunan Province, School of Physics and Electronics, Hunan University, Changsha 410082, China
cWyant College of Optical Sciences, University of Arizona, Tucson, AZ 85721, USA
First published on 11th January 2024
Abstract
N-type electronic doping of organic semiconductors (OSCs) by using superbase compounds shows high doping efficiency (H. Wei, Z. Cheng, T. Wu, Y. Liu, J. Guo, P.-A. Chen, J. Xia, H. Xie, X. Qiu, T. Liu, B. Zhang, J. Hui, Z. Zeng, Y. Bai and Y. Hu, Adv. Mater. 2023, 35, 2300084). While a deprotonation reaction is believed to trigger the doping process, the detailed mechanism therein is not yet fully understood. In the present work we theoretically study the electronic structure of the deprotonated Phenyl-C61-butyric acid methyl ester (PCBM) molecule, as well as the charge transfer (CT) between PCBM and its deprotonated species. We find that deprotonated PCBM without formation of a new bond between the deprotonated side chain and fullerene induces electronic structure with broken spin symmetry, where an in-gap state is singly occupied by an unpaired electron. A second scenario that we find to be possible is the formation of a new bond between the deprotonated side chain and a fullerene. This leads to a spin symmetric electronic structure with partially localized in-gap state, which is expected to contribute less to the effective doping. These results show that the deprotonated PCBM species without new bond formation predominantly accounts for the effective n-type doping of PCBM, an insight that will be useful for optimization of this recently discovered doping method.
1 Introduction
Electronic doping can enhance the conductivity of organic semiconductors (OSCs) by orders of magnitude,1,2 and is of fundamental importance for applications based on OSCs, such as in organic photovoltaics and organic electronics.3–8 Molecular doping, where p-type or n-type molecular dopants are mixed with OSCs to induce charge carriers, is widely used and receives tremendous research interest.9–12 Besides these more established approaches, efforts to explore alternative doping strategies with higher doping efficiency and improved electronic or structural properties are on the rise, leading to recent studies on Lewis acid doping,13–15 ion exchange doping,16 electrophilic p-type doping,17 and doping with the aid of noble metal clusters.18Different from the direct charge transfer (CT) between molecular dopants and OSC material, which in the simplest picture is driven by the energetic difference between HOMO and LUMO levels of the two entities, the mechanisms behind the more recently developed doping methods are more complex. In Lewis acid doping, it is proposed that an OSC polymer is first protonated by the Lewis acid and then attains an electron from a second polymer chain, leaving the latter in a p-doped state.13 Microscopic details including inter- and intrachain processes were ilucudated by use of atomistic quantum chemical approaches,19 and detailed studies of the chemical complexes involved in this process.20 A similar process is believed to account for electrophilic attack p-type doping by use of TrTPFB molecules. There the polymer is attacked by the trityl cation instead of a proton, followed by subsequent CT with a neighbouring polymer chain.17 Both Lewis-acid doping and electrophilic attack show remarkable efficiencies of p-type doping, and avoid typical issues caused by molecular dopants such as deterioration of film morphology.
Very recently, a new n-type doping method using superbase compounds was reported and shown to significantly enhance efficiency in doping the molecular OSCs PCBM and N2200 copolymer, compared to molecular n-type doping with N-DMBI.21 In analogy to the protonation in Lewis acid doping, a deprotonation of PCBM brought about by the superbase compound is believed to occur, with subsequent CT between deprotonated PCBM and ‘intact’ PCBM entities. However, as the species involved in the proposed process are difficult to experimentally identify, the exact mechanism remains to be verified. A more detailed understanding of the underlying microscopic mechanisms may allow further optimization of this new doping method and may also play a role in further optimization of electrophilic attack and Lewis-acid doping.
Here we present an in-depth investigation of the electronic structure of the deprotonated PCBM and its formation of CT complexes with PCBM molecules. We find that the electronic behavior of deprotonated PCBM crucially depends on the possible formation of a new C–C bond between the deprotonated side chain and the fullerene as illustrated in Fig. 1. The structures with new bond formation exhibit an electronic structure and an in-gap state, which is partially localized around this new bond and is therefore expected to contribute less to electronic transport. On the other hand, in the structures without formation of a new bond, the unpaired electron on the deprotonated PCBM migrates from the local C–H bond on the side chain to the conjugated electronic system on the fullerene. In that case, we further show that CT to a neighbouring PCBM, important for effective n-type doping, is generally possible.
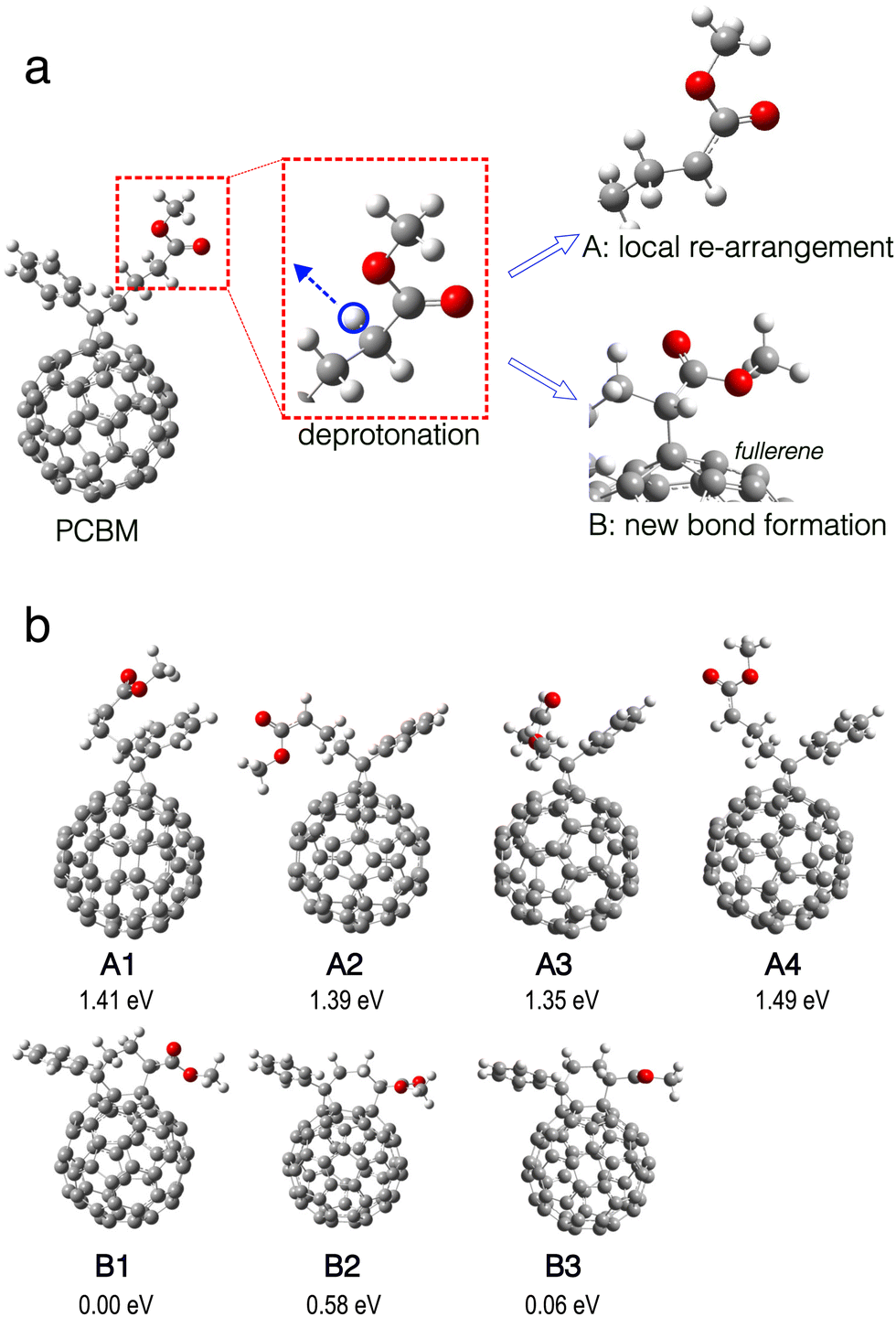 | ||
| Fig. 1 (a) Illustration of deprotonation on the side chain of PCBM. After deprivation of the proton marked by the blue circle (left), either the local structure of the side chain re-arranges (upper right) or a new bond forms between the side chain and the fullerene part (lower right), leading to different structures, examples of which are shown in panel (b). (b) Different local minima structures resulting from deprotonation as shown in (a) including total energies relative to structure B1. A1–A4 have local structural re-arrangement in the side chain. B1–B3 result from new bond formation. Color code: grey for carbon, white for hydrogen, and red for oxygen. | ||
2 Computational details
Following our and other researchers’ previous works,11,22–25 we performed the broken-symmetry DFT calculations. The B3LYP hybrid functional with dispersion correction in Grimme's D3 form26 was used for single molecules (Fig. 1–3). The range separated hybrid functional ωB97XD with default ω was used for bi-molecular complexes (Fig. 4 and 5) for more reliable coverage of CT behavior.23 In the DFT calculations, the initialization of charge density was realized by mixing spin-up (α) and spin-down (β) orbitals, to allow the separation of α and β states energetically and spatially, but without enforcing it. We note that this methodology generally gives good result for charge density distributions but is less accurate for the spin density due to possible spin contamination.27 We note that ω B97XD generally gives a lower HOMO level and a higher LUMO level with respect to B3LYP, thus also overestimating the HOMO–LUMO gap. However, for the molecules and complexes studied in the present work we find that electronic structure and in particular ordering and character of relevant electronic states turns out to be very similar comparing results obtained with the two functionals (not shown). The 6-31g(d,p) basis set was used for all calculations except for the bond length scanning calculations in 3, where a smaller basis set 6-31g(d) was used. The geometries of single molecules and bi-molecular complexes were all optimized. | ||
| Fig. 2 Electronic structures of two different local minima structures of deprotonated PCBM as introduced in Fig. 1. (a) Upper left: A1 structure without formation of new intramolecular bond. Lower left: spin-up α SOMO molecular orbital. Right: Calculated density of states (DOS) in spin-up α (left, indigo) and spin-down β (right, green) channels, showing broken spin symmetry. DOS of occupied states is filled with color. (b) Same as panel (a) but for B1 structure with new bond formation. Lower left: HOMO molecular orbital. Right: DOS, showing spin symmetry. | ||
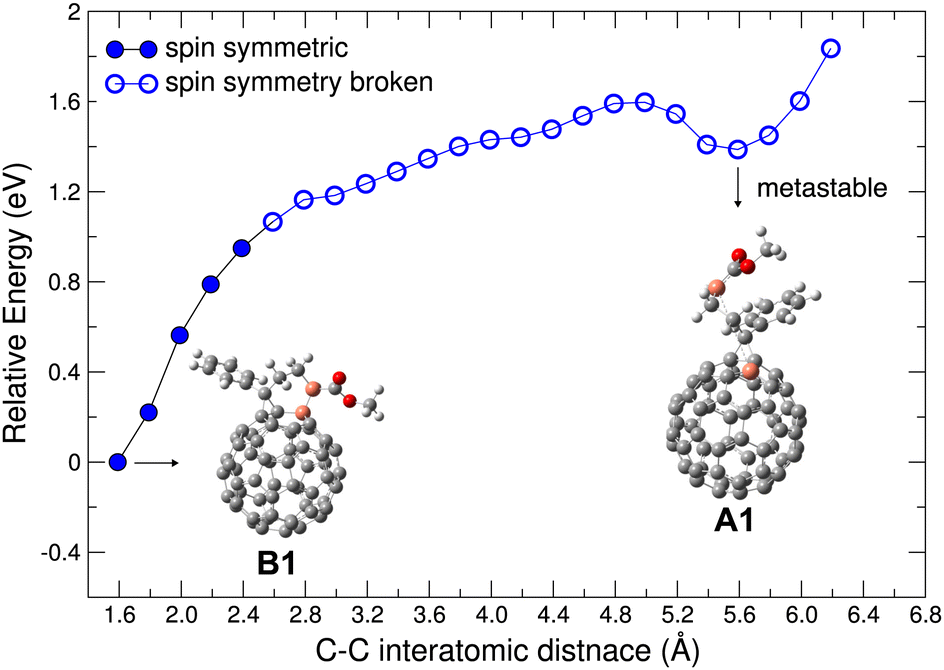 | ||
| Fig. 3 Energy variation in scanning the C–C interatomic distance between the C atoms marked in orange. Local minima structures, B1 (with new bond formation) and A1 (without new bond formation) as discussed in Fig. 1 and 2 and in the text are shown. Spin symmetric electronic structure is denoted by filled circles, broken spin symmetry by open circles. | ||
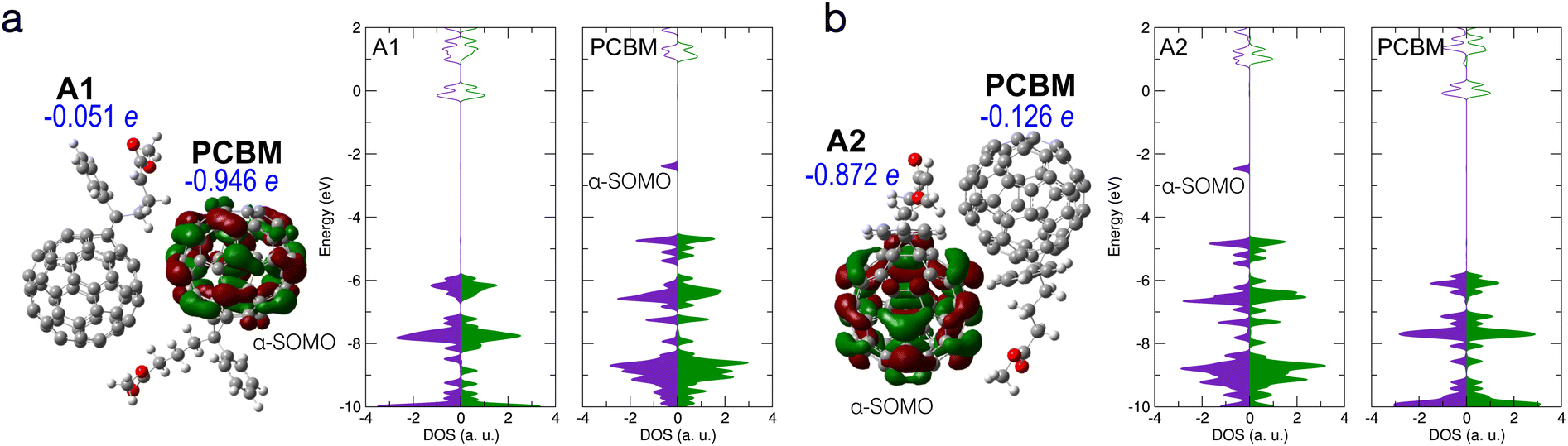 | ||
| Fig. 4 Electronic structure of bi-molecular complexes consisting of one PCBM and one deprotonated PCBM without new bond formation. (a) Left: A1/PCBM complex, with the calculated α-SOMO and Hirshfeld net charge on each molecule given. Right: Partial DOS of each molecule. (b) A2/PCBM complex with α-SOMO and Hirshfeld net charges and partial DOS as in panel (a). Notation and colors same as in Fig. 2 above. | ||
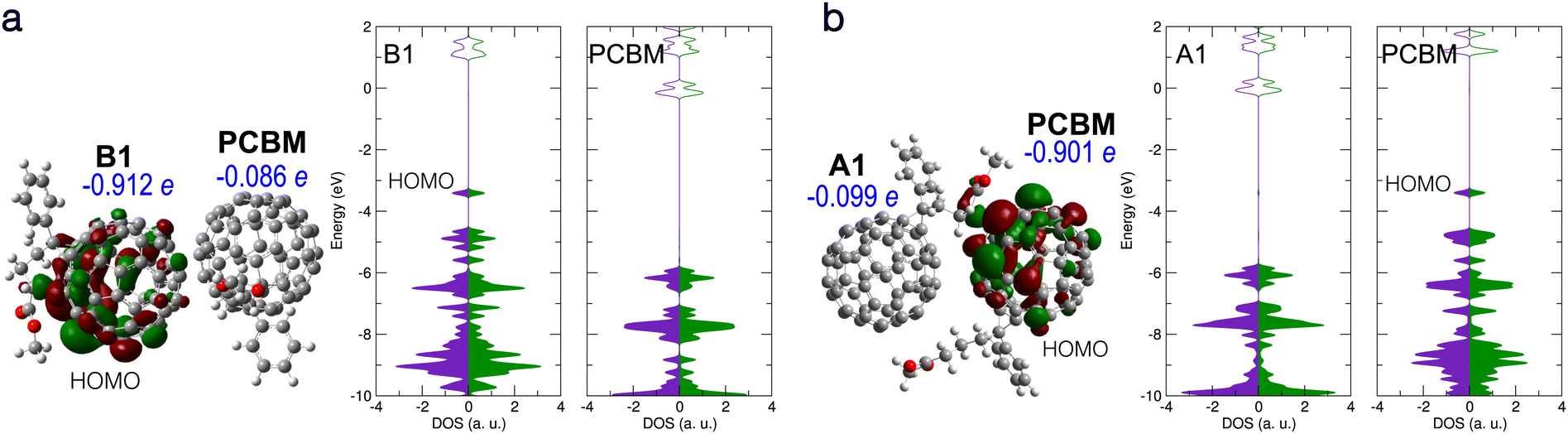 | ||
| Fig. 5 (a) Electronic structure of B1/PCBM complex. Left: B1/PCBM structure, with HOMO, Hirshfeld net charges, and partial DOS given. (b) Electronic structure of A1/PCBM complex with an intermolecular bond forming between the molecules. HOMO, Hirshfeld net charges, and partial DOS are given. Notation and colors same as in Fig. 2 above. | ||
The DFT calculations were performed using the Gaussian1628 package. The Hirshfeld charge decomposition presented in Fig. 4 and 5, which was previuosly shown to agree well with other charge decomposition schemes,23,29 as well as the calculation of density of states (DOS) were performed by using the Multiwfn software.30
3 Results and discussion
The deprotonation reaction caused by the superbase is well known and considered crucial for the highly effective n-type doping observed for PCBM.21 It is generally accepted that the deprotonation takes place at the methyl ester side chain, as shown in Fig. 1. The deprotonated PCBM molecule still possesses the same number of electrons as an ‘intact’ PCBM and therefore carries a negative net charge. We find that the molecular structure of this anion either stays similar to PCBM with just modest local structural re-arrangement, or the structure changes significantly with a new C–C bond forming between the deprotonated side chain and the fullerene. In the structural optimization, we identify several local minima structures, examples of which are shown in Fig. 1(b). Representative structures from each class found, namely A1 without and B1 with new bond formation, are shown in Fig. 2 together with the calculated HOMO orbitals and electronic DOS.Interestingly, the two structures show dramatically different electronic structures: the DOS of A1 shows broken spin symmetry with a singly occupied in-gap state around −1.05 eV, namely the α-SOMO (singly occupied molecular orbital). As shown in Fig. 2(a), this in-gap state is an electronically well conjugated orbital on the fullerene. The Hirshfeld net charge on the fullerene part of the deprotonated PCBM is −0.84 e, in comparison with −0.05 e in the case of neutral PCBM. This indicates that the residual electron migrates to the fullerene after deprotonation of the side chain. In contrast, the structure B1 shows a spin-symmetric electronic structure. The formation of the new bond induces re-organization of the conjugated electronic system on the fullerene and gives rise to a doubly occupied in-gap state. This in-gap state is less delocalized and partially localized around the new bond compared to the singly occupied in-gap state in A1, indicating a significant effect of the new bond formation on the electronic system of the deprotonated PCBM. In addition, the energy separation between the in-gap state and the LUMO is significantly smaller in A1 than in B1, implying a more active electronic behavior for the former.
Given the distinctly different electronic behavior without and with new bond formation (cf. results for A1 and B1), it is interesting to examine in more detail the evolution of the molecular structure associated with the new bond. To this end, starting from B1, we scanned the relevant C–C interatomic distance from 1.59 Å up to 6.2 Å and investigated the electronic states and energies of each optimized structures (with the scanned C–C distance fixed). As shown in Fig. 3, the total energy rises with increasing C–C distance (as expected). The energy minimum structure at C–C distance of 5.6 Å is the A1 structure. Along the scanning path this energy minimum has a depth of about 0.2 eV. We note that a rigorous investigation of energy minimum depth require a full potential surface sampling, which is computationally challenging and out of the interest of the present work. For the purposes of the present work, we believe it is sufficient to note that this meta-stable state we identify is already associated with other important structural parameters of PCBM such as the rotation of the methyl ester around the side chain. Compared with B1, the structure A1 is energetically less favorable, which may indicate lower stability and in turn a reduced doping efficiency. Nevertheless, we believe that this aspect might be mitigated by several factors: (i) Besides A1 the deprotonated PCBM appears in multiple other configurations with similar electronic behavior, such as structures A2–A4 shown in Fig. 1b, which can also contribute to the doping. Our calculations showed that structure A2–A4 can all induce CT to neighboring PCBM as well as A1. (ii) The scanning results shown in Fig. 3 did not take into account the effect of the molecular surroundings, where the solvent molecules and their dynamics will also have an effect on potential energy landscapes and influence occurrence and lifetimes of A1–A4 and other related structure.
Interestingly, when the C–C distance is stretched to about 2.6 Å, we find that the electronic structure turns from spin symmetric to spin symmetry-broken and keeps this property in each structure for the remainder of the scanning path, including the energy minimum around 5.6 Å. These results strongly suggest that the broken spin symmetry will be an important feature of the deprotonated PCBM as long as the new C–C bond is not formed and the system remains in a meta-stable state, which is likely to occur and play a role in a realistic setting with partially hindered relaxation pathways and environmental influences.
The CT between PCBM and deprotonated PCBM is believed to contribute directly to the superbase-induced n-type doping of PCBM.21 To examine the possibility of the CT, we analyze the electronic structure of bi-molecular complexes containing one PCBM and one deprotonated PCBM. To this end, different local minima structures of the latter were investigated. Fig. 4 shows two molecular complexes containing deprotonated PCBM without new bond formation. The A1/PCBM complex contains the local minimum structure A1, while in Fig. 4(b) the complex consists of PCBM and another local minimum of deprotonated PCBM, A2. (see Fig. 1).
The DOS and Hirshfeld charge decomposition show that A1/PCBM does undergo integer CT, while A2/PCBM does not. These results show that CT is possible without the formation of a new bond, but depends on the structure of the deprotonated PCBM as well as the relative position of the two molecules. Both complexes in Fig. 4(a) and (b) show electronic structures of broken spin symmetry with a singly occupied in-gap state. In complex A2/PCBM (Fig. 4(b)) the in-gap state (α-SOMO) is a conjugated state based on the deprotonated PCBM. The Hirshfeld net charge on the deprotonated PCBM reads −0.87 e, showing a behavior similar to the individual deprotonated PCBM (A1) without significant CT. On the other hand, in complex A1/PCBM the in-gap state, also α-SOMO, is based on the PCBM and the net negative charge is mostly on PCBM (−0.94 e), indicating a clear integer CT. In the DOS in Fig. 4(a), it is interesting to notice that the state β-SOMO is lying energetically close to α-(SOMO-1) and forms the energy gap bottom, so that it may contribute less to the effective doping.
It is intriguing that the frontier orbitals in these two cases without and with CT, namely the energy levels and the nature of the in-gap states and the overall DOS profile around the HOMO and the LUMO, are actually very similar to each other and barely distinguishable from an energetic point of view. Indeed, in both cases the in-gap state is located on the fullerene and therefore less impacted by the proton missing from the side chain, while the other molecule, either the PCBM in A2/PCBM or the deprotonated PCBM in A1/PCBM, stays virtually neutral in charge and has less electrostatic effect. Furthermore, in both A1/PCBM and A2/PCBM, the same orbital character of the in-gap states implies similar behavior of the induced electron as a charge carrier. For completeness, the possible effect of chlorobenzene solvent, which was used in ref. 21 was also addressed by using the PCM model with ε = 5.69. The CT results read −0.075 e (A1)/−0.925 e (PCBM) for A1/PCBM and −0.930 e (A2)/−0.068 e (PCBM) for A2/PCBM, which deviate only slightly from the results without dielectric environment.
The CT between the PCBM and the deprotonated PCBM with new bond formation was also studied by using different local minimum structures of the latter. However, no CT with the neighbouring PCBM was found in our calculations. Fig. 5(a) shows a typical case of a bi-molecular complex containing PCBM and the B1 structure. As in the case of the individual B1 structure, a doubly occupied in-gap state forms, which turns out to be a partially localized orbital located on B1, while the PCBM stays nearly neutral. We also considered the possibility of the structure A1 forming a bond with the neighbouring PCBM, as presented in Fig. 5(b). In this case the new bond induces an in-gap state, as well as a net charge of −0.90 e, on the side of PCBM. Similar to the complex B1/PCBM, the in-gap state of A1/PCBM (with new intermolecular bond) shows also partial localization around the new bond. Therefore we propose that in both complexes the in-gap state contributes only little to the effective doping.
4 Conclusion
In the present work we theoretically studied the molecular structure and electronic behavior of deprotonated PCBM, as well as the possibility of CT between deprotonated PCBM and an ‘intact’ PCBM, by using broken-symmetry DFT. We find that deprotonated PCBM can form different local minima structures, largely depending on the arrangement of the deprotonated side chain. Structures with the side chain bending towards the fullerene and forming a new C–C bond are favorable in energy, while the structures without formation of a new intramolecular bond are found to be meta stable. The structures with new intramolecular bond have spin symmetric electronic structure with a doubly occupied in-gap state. In that case, the in-gap states are partially localized around the new bond and CT to a neighbouring PCBM turns out to be difficult, effectively hindering electronic doping. On the other hand, the deprotonated PCBMs without new intramolecular bond formation show broken spin symmetry with a singly occupied in-gap state that is electronically well conjugated and delocalized on the fullerene. In that case, CT to a neighbouring PCBM is shown to be possible, with details depending on geometrical arrangement. Also with occurrence of CT, the conjugated character and energy of the in-gap state are preserved, such that efficient contribution to doping is expected. With these results, our work reveals that electronic behavior of deprotonated PCBM strongly depends on intramolecular bonding. Those structures without intramolecular bonding appear to play the most important role in the investigated doping with superbases. In turn, inhibition of intramolecular and intermolecular bonding of PCBM after superbase deprotonation may be an effective approach to further optimize efficiency of superbase n-type doping.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A grant for computing time at the Paderborn Center for Parallel Computing (PC2) is gratefully acknowledged. C. D. thanks Dr Yugang Bai for enlightening discussions.Notes and references
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed
.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed
- L. Sun, K. Fukuda and T. Someya, npj Flexible Electron., 2022, 6, 89 CrossRef
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed
- T.-H. Han, Y. Lee, M.-R. Choi, S.-H. Woo, S.-H. Bae, B. H. Hong, J.-H. Ahn and T.-W. Lee, Nat. Photonics, 2012, 6, 105–110 CrossRef CAS
- H. Kleemann, K. Krechan, A. Fischer and K. Leo, Adv. Funct. Mater., 2020, 30, 1907113 CrossRef CAS
- G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed
- D. Di Nuzzo, C. Fontanesi, R. Jones, S. Allard, I. Dumsch, U. Scherf, E. von Hauff, S. Schumacher and E. Da Como, Nat. Commun., 2015, 6, 6460 CrossRef PubMed
- M. Schwarze, C. Gaul, R. Scholz, F. Bussolotti, A. Hofacker, K. S. Schellhammer, B. Nell, B. D. Naab, Z. Bao, D. Spoltore, K. Vandewal, J. Widmer, S. Kera, N. Ueno, F. Ortmann and K. Leo, Nat. Mater., 2019, 18, 242–248 CrossRef CAS PubMed
- B. Yurash, D. X. Cao, V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, N. Graham, K. R. abd Koch, G. C. Bazan and T.-Q. Nguyen, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed
- P. Pingel, M. Arvind, L. Kölln, R. Steyrleuthner, F. Kraffert, J. Behrends, S. Janietz and D. Neher, Adv. Electron. Mater., 2016, 2, 1600204 CrossRef
- E. H. Suh, J. G. Oh, J. Jung, S. H. Noh, T. S. Lee and J. Jang, Adv. Energy Mater., 2020, 10, 2002521 CrossRef CAS
- Y. Yamashita, J. Tsurumi, M. Ohno, R. Fujimoto, S. Kumagai, T. Kurosawa, T. Okamoto, J. Takeya and S. Watanabe, Nature, 2019, 572, 634–638 CrossRef CAS PubMed
- J. Guo, Y. Liu, P.-A. Chen, X. Wang, Y. Wang, J. Guo, X. Qiu, Z. Zeng, L. Jiang, Y. Yi, S. Watanabe, L. Liao, Y. Bai, T.-Q. Nguyen and Y. Hu, Adv. Sci., 2022, 9, 2203111 CrossRef CAS PubMed
- H. Guo, C.-Y. Yang, X. Zhang, A. Motta, K. Feng, Y. Xia, Y. Shi, Z. Wu, K. Yang, J. Chen, Q. Liao, Y. Tang, H. Sun, H. Y. Woo, S. Fabiano, A. Facchetti and X. Guo, Nature, 2021, 599, 67–73 CrossRef CAS PubMed
- F. Bauch, C.-D. Dong and S. Schumacher, RSC Adv., 2022, 12, 13999–14006 RSC
- P. S. Marqués, G. Londi, B. Yurash, T.-Q. Nguyen, S. Barlow, S. R. Marder and D. Beljonne, Chem. Sci., 2021, 12, 7012–7022 RSC
- H. Wei, Z. Cheng, T. Wu, Y. Liu, J. Guo, P.-A. Chen, J. Xia, H. Xie, X. Qiu, T. Liu, B. Zhang, J. Hui, Z. Zeng, Y. Bai and Y. Hu, Adv. Mater., 2023, 35, 2300084 CrossRef CAS PubMed
- T. Bathe, C.-D. Dong and S. Schumacher, J. Phys. Chem. A, 2022, 126, 2075–2081 CrossRef CAS PubMed
- C.-D. Dong and S. Schumacher, J. Phys. Chem. C, 2019, 123, 30863–30870 CrossRef CAS
- C.-D. Dong and S. Schumacher, J. Mater. Chem. C, 2020, 8, 11929–11935 RSC
- D. Fazzi, S. Fabiano, T.-P. Ruoko, K. Meerholza and F. Negri, J. Mater. Chem. C, 2019, 7, 12876–12885 RSC
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed
- C.-D. Dong and S. Schumacher, J. Phys. Chem. C, 2021, 125, 21824–21830 CrossRef CAS
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05105f |
| This journal is © the Owner Societies 2024 |