
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated acquisition of wideline solid-state NMR spectra of spin 3/2 nuclei by frequency-stepped indirect detection experiments†
Sujeewa N. S.
Lamahewage
ab,
Benjamin A.
Atterberry
ab,
Rick W.
Dorn
ab,
Eunbyeol
Gi
 ab,
Maxwell R.
Kimball
c,
Janet
Blümel
*c,
Javier
Vela
ab and
Aaron J.
Rossini
*ab
ab,
Maxwell R.
Kimball
c,
Janet
Blümel
*c,
Javier
Vela
ab and
Aaron J.
Rossini
*ab
aUS Department of Energy, Ames National Laboratory, Ames, Iowa 50011, USA. E-mail: arossini@iastate.edu; Tel: +1-515-294-8952
bIowa State University, Department of Chemistry, Ames, IA 50011, USA
cTexas A&M University, Department of Chemistry, College Station, Texas 77842, USA. E-mail: bluemel@tamu.edu; Tel: +1-979-845-7749
First published on 16th January 2024
Abstract
73% of all NMR-active nuclei are quadrupolar nuclei with a nuclear spin I > 1/2. The broadening of the solid-state NMR signals by the quadrupolar interaction often leads to poor sensitivity and low resolution. In this work we present experimental and theoretical investigations of magic angle spinning (MAS) 1H{X} double-echo resonance-echo saturation-pulse double-resonance (DE-RESPDOR) and Y{X} J-resolved solid-state NMR experiments for the indirect detection of spin 3/2 quadrupolar nuclei (X = spin 3/2 nuclei, Y = spin 1/2 nuclei). In these experiments, the spectrum of the quadrupolar nucleus is reconstructed by plotting the observed dephasing of the detected spin as a function of the transmitter offset of the indirectly detected spin. Numerical simulations were used to investigate the achievable levels of dephasing and to predict the lineshapes of indirectly detected NMR spectra of the quadrupolar nucleus. We demonstrate 1H, 31P and 207Pb detection of 35Cl, 81Br, and 63Cu (I = 3/2) nuclei in trans-Cl2Pt(NH3)2 (transplatin), (CH3NH3)PbCl3 (methylammonium lead chloride, MAPbCl3), (CH3NH3)PbBr3 (methylammonium lead bromide, MAPbBr3) and CH3C(CH2PPh2)3CuI (1,1,1-tris(diphenylphosphinomethyl)ethane copper(I) iodide, triphosCuI), respectively. In all of these experiments, we were able to detect megahertz wide central transition or satellite transition powder patterns. Significant time savings and gains in sensitivity were attained in several test cases. Additionally, the indirect detection experiments provide valuable structural information because they confirm the presence of dipolar or scalar couplings between the detected nucleus and the quadrupolar nucleus of interest. Finally, numerical simulations suggest these methods are also potentially applicable to abundant spin 5/2 and spin 7/2 quadrupolar nuclei.
Introduction
High-resolution magic angle spinning (MAS) solid-state nuclear magnetic resonance spectroscopy has played a pivotal role in determining the molecular structure and dynamics of inorganic materials, organic solids, and biomolecules within crystalline and amorphous systems.1–3 However, many NMR-active nuclei remain understudied due to technical difficulties in recording or interpreting their NMR spectra. For example, low natural abundances, low gyromagnetic ratios, large nuclear electric quadrupole moments (Q), long spin–lattice relaxation time constants (particularly for spin 1/2 nuclei), or some combination of these properties make acquisition of solid-state NMR spectra challenging.4 Approximately 73% of all NMR-active isotopes are quadrupolar nuclei, with the vast majority having half-integer spins (I = 3/2, 5/2, 7/2, or 9/2).5 Quadrupolar nuclei often give rise to extremely broad solid-state NMR spectra due to the first and second order quadrupolar interactions. When the second order quadrupolar interaction (SOQI) is sizable, only the central transition (CT) is typically observable, and the satellite transitions (ST) are usually broadened beyond detection. In such cases, the NMR spectrum of the central transition could be several megahertz broad, necessitating the use of wideline solid-state NMR techniques.6–9Slichter first reported the frequency-stepped acquisition of wideline solid-state NMR spectra for 195Pt (I = 1/2) by recording the intensity of Hahn echoes at variable frequency offsets.10 Following this work, additional wideline NMR studies on half-integer quadrupolar nuclei were performed for nuclei such as 91Zr,11,1269/71Ga,9 and 59Co.8 More recently, the quadrupolar Carr–Purcell–Meiboom–Gill (QCPMG) pulse sequence has found widespread application for acquisition of solid-state NMR spectra of nuclei such as 87Rb and 59Co,13,14 and low-γ quadrupolar metal nuclei such as 67Zn,15,1695Mo,1714N,1891Zr,18,1939K, 25Mg, and 87Sr.13,14 CPMG techniques have also been used to probe halogens like 35/37Cl (I = 3/2),20127I (I = 5/2),21 and 79Br (I = 3/2)22 in organic and inorganic materials.23 O’Dell and Schurko introduced the Wideband Uniform Rate Smooth Truncation (WURST)-QCPMG pulse sequence (Fig. 1A) and used it to obtain wideline solid-state NMR spectra of a variety of quadrupolar nuclei.6,19,24 WURST-CPMG offers improved excitation and refocusing bandwidths as compared to conventional QCPMG experiments with rectangular excitation and refocusing pulses.
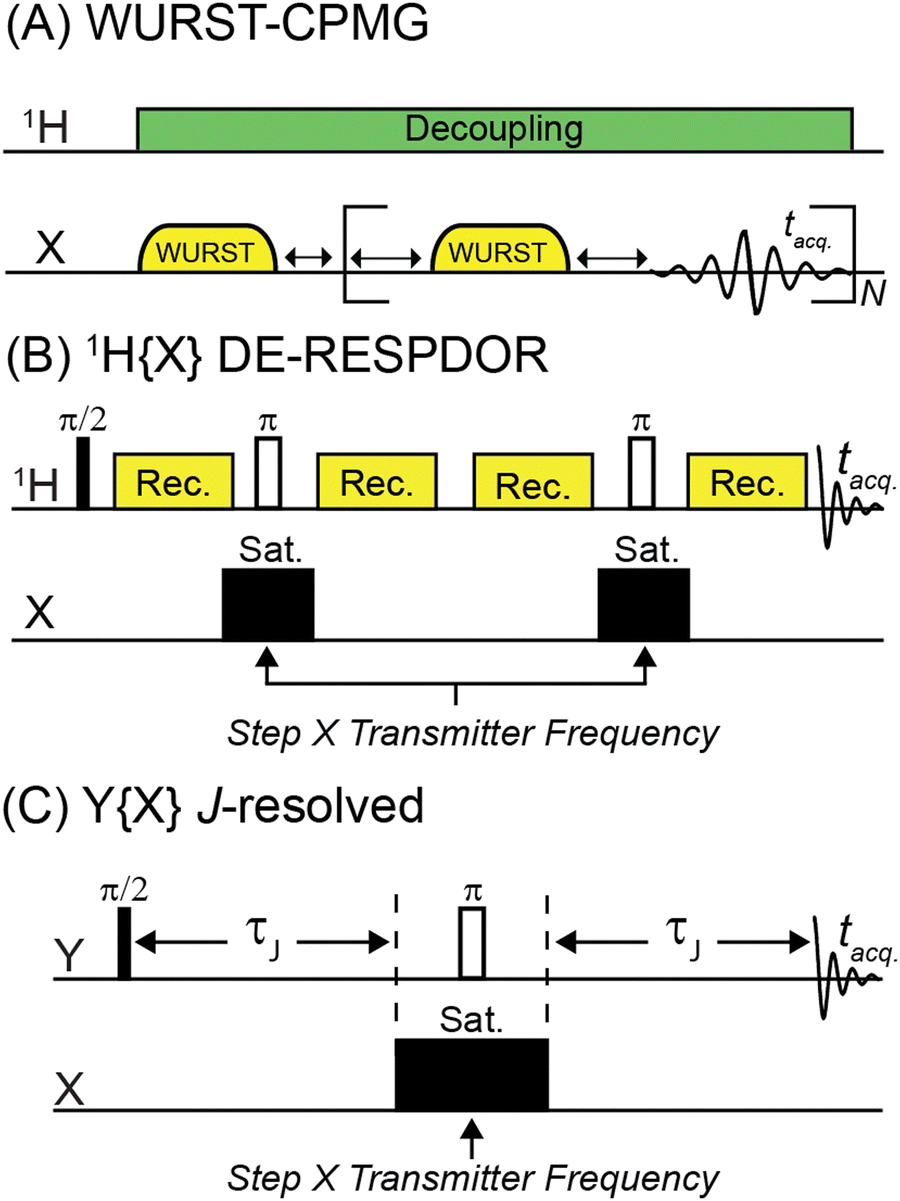 | ||
| Fig. 1 Pulse sequences used in this work. (A) WURST-CPMG pulse sequence used for direct detection of 35Cl and 81Br solid-state NMR spectra. (B) 1H{X} DE-RESPDOR pulse sequence used for indirect detection of 35Cl and 81Br. (C) Y{X} Heteronuclear spin echo (J-resolved) pulse sequence used for 207Pb{35Cl} and 31P{63Cu} NMR experiments. The frequency of the saturation pulse(s) in the DE-RESPDOR or J-resolved experiments can be varied in order to map the frequency dependence of the dephasing and reconstruct the solid-state NMR spectrum of the indirectly detected spin 3/2 nucleus. Phase cycles for pulse sequences are given in Table S1 (ESI†). | ||
Unfortunately, direct detection wideline solid-state NMR methods often suffer from inadequate sensitivity and long measurement times.8,11–15,17,22,25,26 An alternative to these direct detection methods is to use indirect detection experiments where the NMR signal from a sensitive “spy nucleus” that offers a narrow linewidth and/or high sensitivity is exploited. Indirect detection has been applied to a variety of quadrupolar nuclei. There are two ways of performing indirect detection. In the first approach, a standard two-dimensional (2D) NMR experiment is performed where a series of 1D NMR spectra are acquired with incrementation of the indirect dimension evolution time delay (t1). Fourier transformation of the indirect dimension time domain free induction decay yields the spectrum of the indirectly detected nucleus. For example, Gan and Bodenhausen et al. independently demonstrated the use of Heteronuclear Multiple Quantum Correlation (HMQC) pulse sequences27 for the indirect detection of 14N MAS quadrupolar powder patterns by 13C.28–30 With the advent of fast MAS probes, 1H{14N} HMQC solid-state NMR experiments have become routine.31–35 In the second approach, the transmitter offset of the pulses applied to the indirectly detected spin are incremented. The NMR spectrum of the indirectly detected spin is then reconstructed by plotting the response of the detected nucleus as a function of the pulse offset. This approach is especially useful for the quadrupolar nuclei because it can be applied even when the spectrum of the quadrupolar nucleus is megahertz broad, and it is not possible to coherently excite magnetization from the quadrupolar nucleus. These types of indirect detection experiments have been performed with pulse sequences such as Static Echo DOuble Resonance (SEDOR),36–38 TRAnsfer of Population in DOuble Resonance (TRAPDOR),39 Rotational Echo Adiabatic Passage DOuble Resonance (REAPDOR),40,41 Resonance Echo Saturation Pulse DOuble Resonance (RESPDOR),42–46 and PROgressive Saturation of the Proton Reservoir (PROSPR).47,48 These experiments have been used to indirectly detect NMR spectra of quadrupolar nuclei such as 14N, 27Al, 17O, 33S and 35Cl using 1H or 13C as the detected nucleus. We note that some of the examples cited above described acquisition of wideline solid-state NMR spectra of spin-1/2 nuclei broadened by chemical shift anisotropy (CSA).36–38,46 Recently, we showed that CT wideline 35Cl solid-state NMR spectra of partially Cl-terminated 2D silicon materials could be detected by plotting the dephasing observed in a 1H{35Cl} DE-RESPDOR experiments (Fig. 1B) as a function of the 35Cl pulse offset.42 Notably, for the 2D silicon materials, we were unable to observe a direct 35Cl WURST-CPMG signal, presumably due to an unfavorable 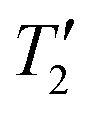 for the 35Cl nuclei.42
for the 35Cl nuclei.42
Here we provide a detailed investigation of frequency-stepped RESPDOR experiments for the detection of wideline solid-state NMR spectra of I = 3/2 nuclei, using 35Cl, 81Br and 63Cu as representative examples. We apply numerical SIMPSON simulations to investigate saturation pulse conditions that can be used to map out CT and ST wideline solid-state NMR spectra of I = 3/2 nuclei. We also simulate frequency-stepped dipolar dephasing experiments for higher spin quadrupolar nuclei.
Results and discussion
Pulse sequences used for direct detection and indirect detection solid-state NMR experiments
The most common pulse sequence used for direct detection of wideline solid-state NMR spectra of quadrupolar nuclei is WURST-CPMG (Fig. 1A).19 We used WURST-CPMG to acquire 35Cl and 81Br solid-state NMR spectra of transplatin (trans-Cl2Pt(NH3)2), MAPbCl3 ((CH3NH3)PbCl3), and MAPbBr3 ((CH3NH3)PbBr3). Fig. 1B illustrates the 1H{X} DE-RESPDOR pulse sequence.49 DE-RESPDOR is preferred over a conventional RESPDOR experiment because it is more robust to experimental MAS frequency fluctuations.49 We used DE-RESPDOR to perform 1H detected 35Cl and 81Br solid-state NMR experiments. The Y{X}-heteronuclear spin echo (J-resolved) pulse sequence shown in Fig. 1C was used to indirectly detect 35Cl and 63Cu NMR spectra, using 207Pb and 31P as the spy nucleus, respectively. In both the DE-RESPDOR experiments and the J-resolved experiments, solid-state NMR spectra are reconstructed by plotting the dephasing as a function of the pulse offset as illustrated below.Numerical simulations of dephasing in 1H{35Cl} DE-RESPDOR experiments
Prior to showing experimental NMR data, we have used SIMPSON50–52 to simulate 35Cl solid-state NMR spectra and illustrate how DE-RESPDOR can be used for indirect detection of 35Cl. Fig. 2A shows the SIMPSON simulated static 35Cl solid-state NMR spectra for transplatin with CQ = 37.0 MHz, ηQ = 0.12 and an applied magnetic field of 9.4 T. Fig. 2B illustrates a spectrum simulated with the same parameters but with a magic angle spinning frequency of 50 kHz. In this case, the width of the central transition (CT) MAS powder pattern (ca. 5 MHz) greatly exceeds the MAS frequency (50 kHz), consequently, overlap of the CT MAS powder pattern with its spinning sideband powder patterns yields a spectrum that is nearly identical in appearance to a conventional static spectrum. Comparison of Fig. 2A and B suggests that MAS NMR experiments can provide the same spectrum and line shape as static NMR experiments, provided the width of the quadrupolar powder pattern greatly exceeds the MAS frequency.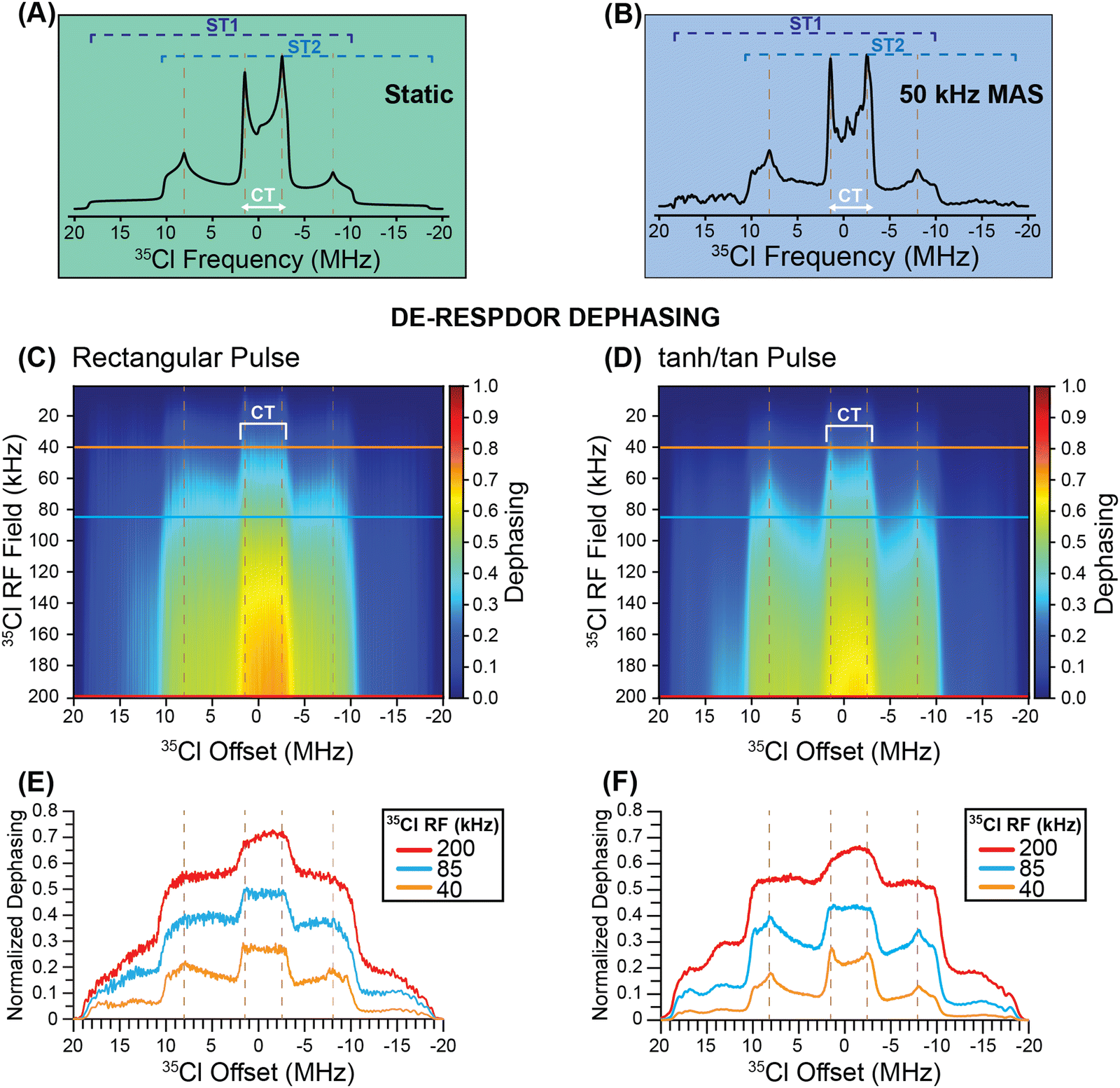 | ||
| Fig. 2 SIMPSON simulated 35Cl solid-state NMR spectra of transplatin (CQ = 37 MHz and ηQ = 0.12) shown for (A) a static (stationary) sample and (B) for a 50 kHz magic angle spinning (MAS) frequency with a magnetic field of 9.4 T. (C) and (D) Heat plots showing the simulated dephasing for 1H{35Cl} DE-RESPDOR experiments as a function of the 35Cl saturation pulse RF field and pulse offset. Simulations are shown for (C) conventional rectangular pulses 80 μs in duration and (D) frequency-swept tanh/tan pulses (ξ = 20, θ = 180°) using a frequency sweep of 5 MHz and 40 μs duration. All simulations used a 1H–35Cl spin system with a 2 kHz dipolar coupling constant. Dephasing profiles extracted from the heat plots for the 35Cl saturation pulse RF fields 200 kHz, 85 kHz, and 40 kHz with (E) rectangular saturation pulses and (F) tanh/tan pulses. | ||
Fig. 2C and D show SIMPSON simulated DE-RESPDOR dephasing heat maps. In the heat maps the offset of the saturation pulses is varied on the x-axis and the RF field of the saturation pulses is varied on the y-axis, while the color indicates the extent of dephasing. The dephasing is shown as a function of 35Cl pulse power and offset with 80 μs (4 rotor cycles) rectangular pulses (Fig. 2C) and 40 μs frequency-swept tanh/tan pulses53 with a 5 MHz sweep width (Fig. 2D). In Fig. 2E and F RESPDOR dephasing profiles are shown for 35Cl saturation pulses with radiofrequency fields (RF) of 200, 85, and 40 kHz. The heatmaps and dephasing profiles illustrate that the plot of the dephasing as a function of the offset can roughly trace the CT and ST powder patterns. Because the CT is narrower than the ST, it is more easily saturated, resulting in enhanced dephasing in the RESPDOR experiments and a contrast between the CT and ST powder patterns. However, increasing the saturation pulse RF field increases the dephasing for both CT and ST, but also reduces the contrast between the CT and ST. The use of higher RF field saturation pulses also results in a rounding of the powder pattern discontinuities. It is well known in NMR experiments such as CEST,54 that the saturation bandwidth will increase as the saturation pulse RF field is increased, resulting in broadening of the indirectly detected peaks. It is therefore unsurprising that the features in the dephasing profile become more rounded as the saturation pulses can create dephasing more effectively across the entire powder pattern when higher RF fields are used. Another interesting observation from Fig. 2 is that if shorter duration 35Cl saturation pulses are used, then the discontinuities or “horns” associated with the ST are sharper than when rectangular pulses are applied. The tanh/tan pulses employed in Fig. 2D had a pulse length of 40 μs (two rotor cycles). Additional simulations with 40 μs rectangular saturation show similarly sharp features in the RESPDOR dephasing profile as were seen for the tanh/tan pulses (Fig. S3, ESI†). The 40 μs tanh/tan pulses use less RF power due to their hyperbolic tangent amplitude profile, and thus could be preferred experimentally.
In summary, it is evident from Fig. 2 that the RESPDOR dephasing profiles roughly map out the MAS solid-state NMR spectrum, including both the CT and ST regions of the powder pattern. Therefore, the simulations suggest that RESPDOR experiments can be used to indirectly detect wideline solid-state NMR spectra of spin 3/2 nuclei.
Experimental comparison of 1H{35Cl} DE-RESPDOR and direct WURST-CPMG for acquisition of the 35Cl solid-state NMR spectrum of transplatin
Fig. 3A shows 1H{35Cl} DE-RESPDOR dephased spectrum (red, S) recorded with 35Cl saturation pulses and the control spectrum (black, S0) recorded without saturation pulses. All NMR spectra were acquired at B0 = 9.4 T, with a 35Cl saturation pulse that was four rotor cycles in duration (80 μs) with an 80 kHz RF field, and 3.84 ms of SR421 dipolar recoupling55 applied to the 1H spins. The NMR spectra shown in Fig. 3A were obtained with a 35Cl pulse offset of 1.2 MHz. Control (S0) and dephasing (S) 1H NMR spectra show signal-to-noise ratios of 60 and 18 with eight scans, respectively. The difference spectrum (S0−S, cyan) illustrated below has a signal-to-noise ratio of 30, which is adequate for indirect detection of 35Cl in transplatin. We did not optimize the total recoupling time to obtain optimal sensitivity for the 1H{35Cl} DE-RESPDOR experiments for transplatin. Fig. S2 (ESI†) illustrates how to choose the optimal recoupling duration using 1H{35Cl} DE-RESPDOR experiments, using MAPbCl3 as an example. In brief, the recoupling duration that maximizes the difference signal (ΔS = S0 − S) should be used. This optimal recoupling time can be found by making a plot of ΔS as a function of the recoupling duration (Fig. S2, ESI†).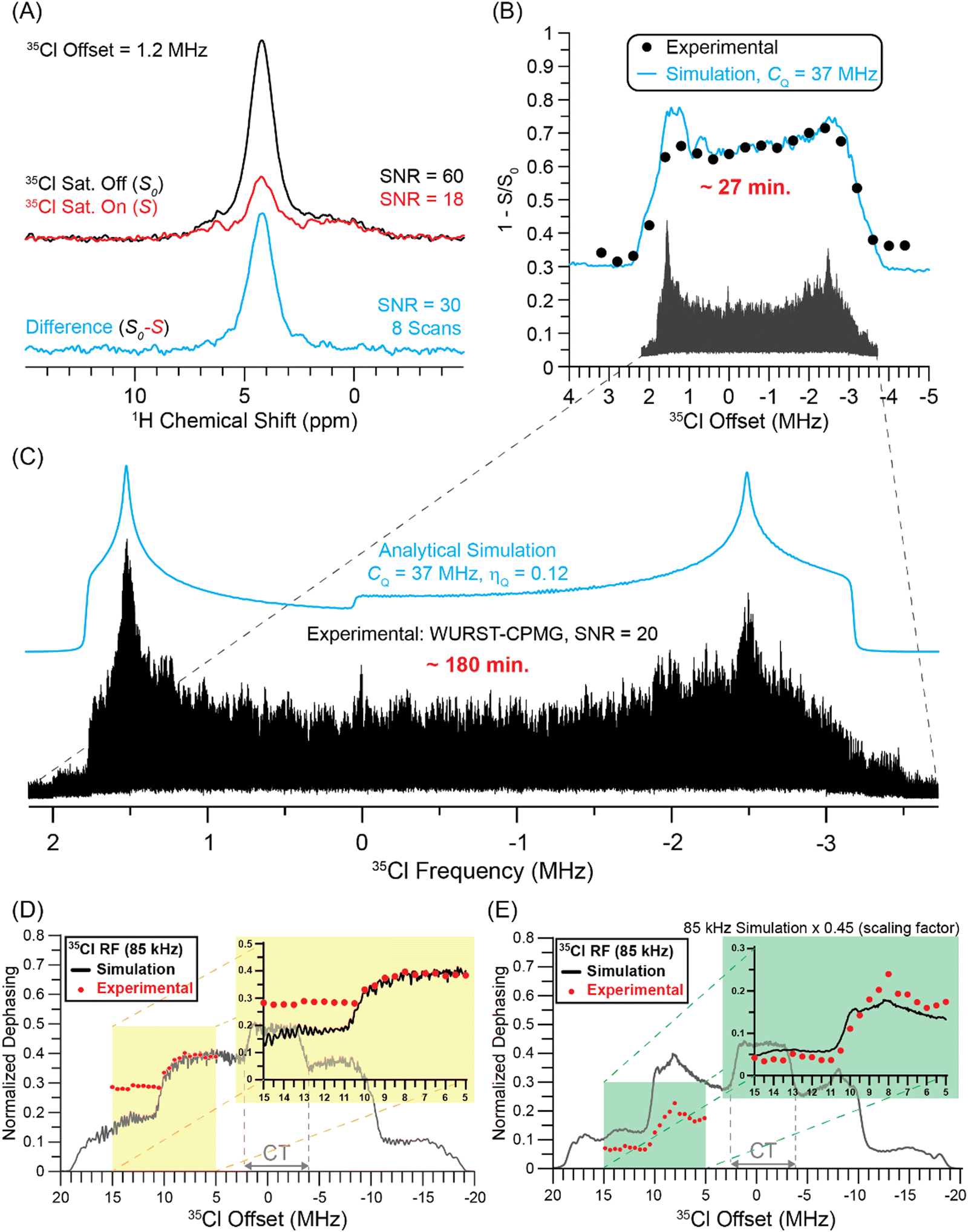 | ||
| Fig. 3 Summary of 35Cl solid-state NMR experiments on transplatin performed with a 9.4 T magnetic field. (A) 1D 1H{35Cl} DE-RESPDOR spectra recorded with (red) or without (black) 80 μs rectangular 35Cl saturation pulses. The difference spectrum (cyan) is shown below. 8 scans were acquired for each spectrum, the MAS frequency was 50 kHz, and the total duration of SR421 dipolar recoupling was 3.84 ms. (B) Plot of 1H{35Cl} DE-RESPDOR normalized signal dephasing (ΔS/S0) as a function of the 35Cl pulse offset over the CT region. The circles and lines correspond to experimental data points and numerical simulations, respectively. The same experimental parameters were used for the experiments shown in Fig. 3A and B. (C) VOCS WURST-CPMG static 35Cl NMR spectrum of transplatin and simulation (cyan). Comparison of experimental and SIMPSON simulated 1H{35Cl} DE-RESPDOR dephasing for (D) rectangular 35Cl saturation pulses 80 μs in duration and (E) tanh/tan pulses that are 40 μs in duration. The 35Cl pulse offset was varied in steps of 500 kHz over the range of +5 MHz to +15 MHz. | ||
1H{35Cl} DE-RESPDOR experiments on transplatin were performed where the offset of the 35Cl saturation pulse was varied in steps of 400 kHz over a range of +3.4 to −4.6 MHz (Fig. 3B). For each 35Cl pulse offset the probe needed to be manually retuned. Plotting the normalized signal dephasing (ΔS/S0) as a function of the 35Cl saturation pulse offset enables the reconstruction of the MAS 35Cl CT quadrupolar powder pattern for transplatin. Notably, the 1H detected central transition 35Cl solid-state NMR spectrum of transplatin was acquired in only ca. 27 minutes at B0 = 9.4 T. Numerical simulations of the 1H{35Cl} DE-RESPDOR dephasing profile suggest that the 35Cl CQ is ca. 37 MHz (with ηQ = 0.1), in good agreement with 35Cl CQ value of 36.6 MHz previously reported by Lucier et al.56 Note, that in order to fit the experimental RESPDOR saturation profile we must scale the simulated dephasing data. Scaling of the simulated dephasing is needed since we use two-spin simulations that assume 100% abundance of the quadrupolar nucleus and furthermore, the values of the 1H–35Cl dipolar coupling constants are unknown and challenging to measure. The variable offset cumulative spectrum (VOCS) 35Cl WURST-CPMG spectrum of transplatin (black) was constructed by co-adding 21 individual sub-spectra (Fig. 3C). The analytically simulated (ssNake)57 spectrum indicated the 35Cl CQ was 37 MHz with ηQ = 0.12. The co-added VOCS WURST-CPMG spectrum took 3 hours of spectrometer time to acquire (Fig. 3C).
The comparison of experimental time between indirectly detected 1H{35Cl} DE-RESPDOR experiments and directly detected 35Cl WURST-CPMG experiments shows six-fold time-saving in indirect detection using 1H as a spy nucleus. The signal-to-noise ratio was also 1.5 times higher in indirectly detected 35Cl NMR spectra compared with the direct detection experiment. The gains in sensitivity obtained are even more impressive if one considers the amounts of sample used in the two types of experiments. 1H{35Cl} DE-RESPDOR experiments were performed with a 1.3 mm rotor that holds approximately 5 μL of sample, while the static 35Cl WURST-CPMG experiments used ca. 100 μL of sample packed into a 4 mm rotor. Additionally, the 1H T1 determines the optimal recycle delay for the DE-RESPDOR experiments, while the 35Cl T1 determines the optimal recycle delay in the WURST-CPMG experiments. For transplatin we measured a 1H T1 of 17 s and used a recycle delay of 5 s for DE-RESPDOR experiments, while an experimentally optimized 35Cl recycle delay of 0.5 s was used for 35Cl WURST-CPMG experiments. Thus, samples with shorter and more favorable 1H T1 should show even higher gains in sensitivity for the indirect detection DE-RESPDOR NMR experiments.
One trade off with the wideline RESPDOR experiments is that the CT powder pattern discontinuities in the RESPDOR dephasing profiles are not as sharp as those seen in the direct acquisition WURST-CPMG NMR spectra, leading to reduced precision in the extraction of the CQ and ηQ values. Fitting of the WURST-CPMG spectrum was reported to gives uncertainties of 0.1 to 0.3 MHz and 0.02 on the measured CQ and ηQ values, respectively.56 Simulations of the RESPDOR dephasing profiles with variable CQ values suggest that the uncertainties on the measured values of CQ are on the order of 1 MHz (Fig. S4, ESI†). The simulations suggest the ηQ value is between 0.0 and 0.25. Thus, there is likely a loss of precision in measurement of CQ and ηQ as compared to direct detection methods. However, as is shown below, it is possible to improve the precision of the measured ηQ values by mapping out the ST powder patterns with RESPDOR experiments.
Additional 1H{35Cl} DE-RESPDOR experiments on transplatin were performed where the offset of the 35Cl saturation pulse varied in steps of 500 kHz over a range of +15 to +5 MHz in order to detect parts of the 35Cl ST powder patterns. Corresponding SIMPSON simulated normalized dephasing profiles with 85 kHz RF field saturation pulses (black) are shown in Fig. 3D. Plotting the signal dephasing as a function of the 35Cl saturation pulse offset enables the reconstruction of the partial MAS ST powder pattern for the transplatin. In Fig. 3D, there is a discrepancy between the experimental and simulated dephasing at different positions of the satellite transitions. The higher frequency part of the ST shows higher than expected dephasing. There are a number of reasons that explain this discrepancy. First, numerical simulations with a 1H–35Cl two-spin system show that the ratio of dephasing at the inner and outer part of the ST depends upon the recoupling duration (Fig. S5, ESI†). Second, the crystal structure of transplatin shows that each 1H spin is coupled to several 35Cl spins, meaning that a two-spin simulation is not an accurate representation of the experiments. Finally, the experimental 35Cl RF field likely changes as the probe is retuned. While all of these factors could change the dephasing at different offsets, the most important constraint on the ηQ value is the position of the “step” arising from the overlap of the two satellite transitions.
Fig. 3E shows the simulated and experimental RESPDOR dephasing profiles obtained with 35Cl tanh/tan saturation pulses with an 85 kHz RF field. The DE-RESPDOR experiments with the 40 μs tanh/tan pulses give well-defined ST discontinuities in both simulation and experiment, enabling ηQ to be more accurately determined. Fitting the ST region of the RESPDOR dephasing profile indicates that ηQ is between 0.0 and 0.1 (Fig. S3, ESI†). This is an important observation because the indirectly detected CT powder pattern is not very sensitive to ηQ. Therefore, if increased precision is required on measured values of ηQ then RESPDOR experiments can be performed to locate the ST discontinuities.
Comparison of direct detection and indirect detection for acquisition of the 35Cl solid-state NMR spectrum of MAPbCl3
Next, we investigated the use of indirect detection methods for acquisition of the 35Cl solid-state NMR spectrum of methylammonium lead chloride (MAPbCl3). Michaelis and co-workers have recently shown that 35Cl solid-state NMR spectroscopy can be used to probe the structure of lead chloride perovskites and related phases.58 The VOCS 35Cl WURST-CPMG spectrum of MAPbCl3 was obtained with 5 sub-spectra (Fig. 4A). Blue lines are numerically simulated spectra with a 35Cl CQ of 16.5 MHz (with ηQ = 0), which is consistent with the 16.34 MHz 35Cl CQ value reported by Sarkar et al.58 The total experimental time to acquire the WURST-CPMG spectrum was 51 minutes, and the signal-to-noise ratio is approximately 160 at the most intense part of the CT powder pattern.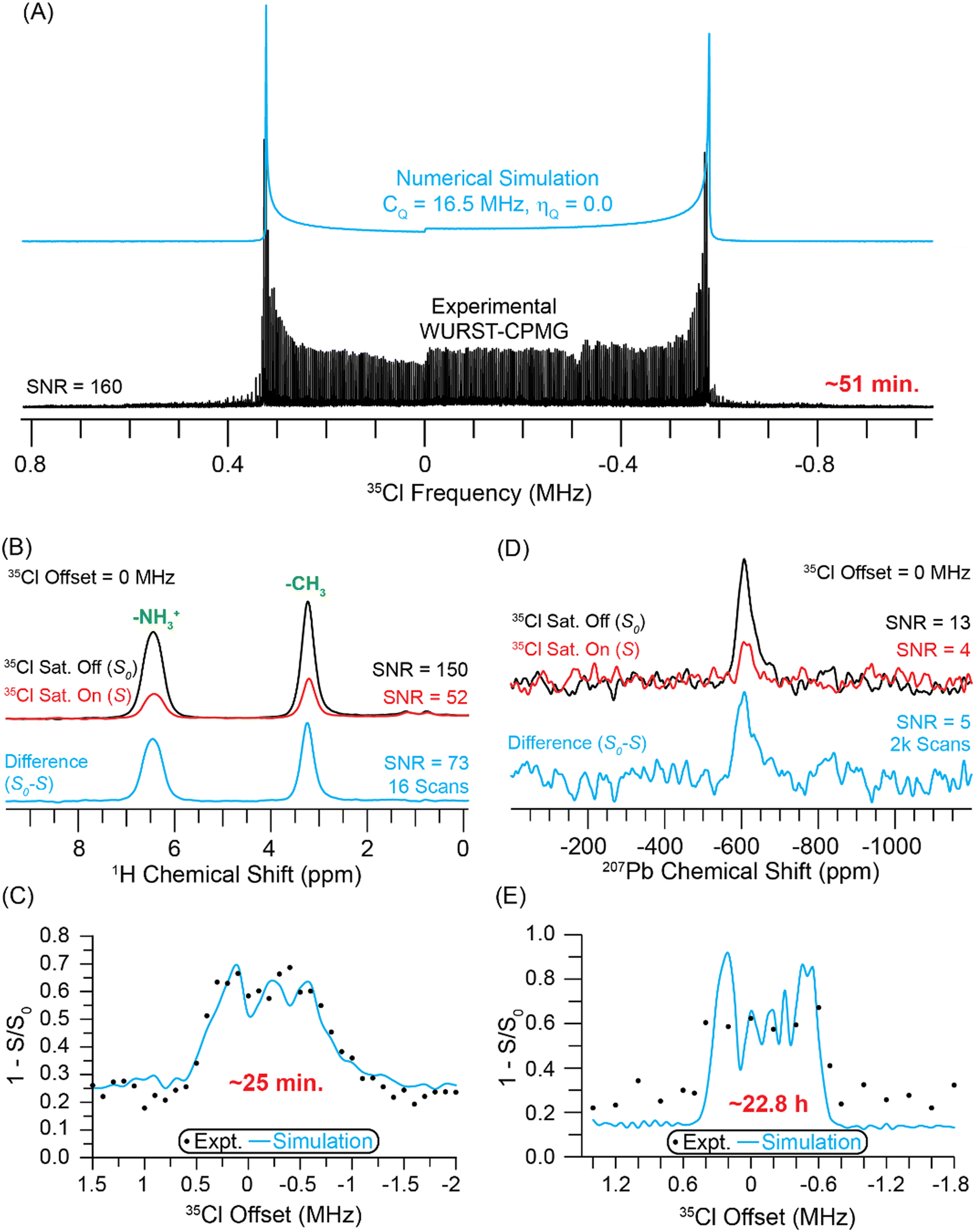 | ||
| Fig. 4 (A) Static VOCS WURST-CPMG 35Cl NMR spectrum of MAPbCl3. (B) 1D 1H{35Cl} DE-RESPDOR spectra recorded with (red) or without (black) a 35Cl saturation pulse and 3.84 ms of SR421 dipolar recoupling and a 50 kHz MAS frequency. A difference spectrum (cyan) is shown below. (C) Plot of 1H{35Cl} DE-RESPDOR signal dephasing as a function of the 35Cl transmitter. (D) 1D 207Pb{35Cl} J-resolved spectra recorded (red) with or (black) without a 35Cl saturation pulse and 0.64 ms of total J evolution time. (E) The plot of 207Pb{35Cl} J-resolved signal dephasing as a function of the 35Cl transmitter recorded with a 25 kHz MAS frequency. In (C) and (E), the circles and lines correspond to experimental data points and numerical simulations, respectively. The simulation used a 35Cl CQ of 16.5 MHz. All experiments were performed with a magnetic field of 9.4 T. | ||
Fig. 4B depicts 1H{35Cl} DE-RESPDOR spectra with saturation pulses (red) and spectra without saturation pulses (black). With 16 scans, the control and dephasing spectra have signal-to-noise ratios of 150 and 52, respectively. The difference spectrum (cyan) shown below has a signal-to-noise ratio of 73. The spectra shown in Fig. 4B were obtained with a 0 MHz 35Cl offset. All spectra were collected at B0 = 9.4 T using a 35Cl saturation pulse length of four rotor cycles (80 μs) with an 80 kHz RF field and 3.84 ms of SR421 dipolar recoupling.55 Fig. S2 (ESI†) illustrates how to choose the optimal recoupling duration. Fig. S6 (ESI†) shows how varying the recoupling duration affects the quality of the indirectly detected 35Cl NMR spectrum. The 35Cl spectrum was indirectly detected with 1H{35Cl} DE-RESPDOR by varying the offset of the 35Cl saturation pulse in 100 kHz steps over a range of +1.5 to −2 MHz (Fig. 4C). The 1H detected 35Cl NMR spectrum of MAPbCl3 was obtained in ca. 25 minutes at B0 = 9.4 T. However, as was seen above for transplatin, due to the RF field used for the 35Cl saturation pulses, the CT pattern seen in the RESPDOR dephasing profile does not perfectly match the directly acquired CT powder pattern. Numerical simulations of the 1H{35Cl} DE-RESPDOR dephasing profile indicate that the 35Cl CQ is ca. 16.5 MHz (with ηQ = 0), consistent with the static WURST-CPMG spectrum. We note that the offset of the 35Cl pulses was stepped in 100 kHz increments, resulting in lower resolution for the experimental RESPDOR dephasing profile as compared to the simulated one.
For MAPbCl3 the sensitivity with 1H detection is worse than the directly detected 35Cl NMR spectrum. The gains in signal-to-noise ratio and time savings provided by 1H detection is likely limited because the 1H T1 of MAPbCl3 (T1 = 30 s) is much longer than the 35Cl T1 (optimal recycle delays of 0.15 s). Furthermore, the 35Cl homogeneous transverse relaxation time constant 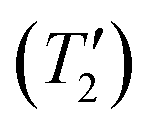 is long enough to allow CPMG echoes to be acquired for ca. 50 ms, making CPMG detection efficient for MAPbCl3. The relatively narrow breadth of the CT selective spectrum also means that only 5 WURST-CPMG sub-spectra need to be acquired. Thus, the results for MAPbCl3 suggest that if the CT is less than 1 MHz in breadth and the 35Cl
is long enough to allow CPMG echoes to be acquired for ca. 50 ms, making CPMG detection efficient for MAPbCl3. The relatively narrow breadth of the CT selective spectrum also means that only 5 WURST-CPMG sub-spectra need to be acquired. Thus, the results for MAPbCl3 suggest that if the CT is less than 1 MHz in breadth and the 35Cl 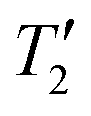 is sufficiently long to allow efficient CPMG detection then direct detection is likely to be the preferred acquisition method. But we note that the 1H{35Cl} RESPDOR experiments may still be worthwhile since they provide valuable structural information by confirming that the 1H spins of the MA cations are dipole coupled (spatially proximate) to the Cl anions.
is sufficiently long to allow efficient CPMG detection then direct detection is likely to be the preferred acquisition method. But we note that the 1H{35Cl} RESPDOR experiments may still be worthwhile since they provide valuable structural information by confirming that the 1H spins of the MA cations are dipole coupled (spatially proximate) to the Cl anions.
We also attempted 207Pb{35Cl} J-resolved experiments to establish if 207Pb could also be used as a spy nucleus for 35Cl detection. Fig. 4D shows 207Pb{35Cl} J-resolved spectra recorded with (red) saturation pulses and without (black) saturation pulses. The pulse sequence for the J-resolved experiments is shown in Fig. 1C. The J-resolved experiments were performed at a field of 9.4 T with an MAS frequency of 25 kHz. The control and dephasing spectra have signal-to-noise ratios of 13 and 4, respectively. Each spectrum was obtained with 2048 scans and a 2 s recycle delay. The difference spectrum (cyan) shown below has an adequate signal-to-noise ratio of 5. The 35Cl saturation pulse was two rotor periods in duration (80 μs) with a 41.7 kHz RF field, and the spin echo had a total duration of 0.64 ms. In this sample, the 207Pb 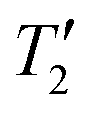 limits the echo duration and the achievable dephasing. 207Pb{35Cl} J-resolved experiments on MAPbCl3 were performed with 35Cl saturation pulse offsets varying in 200 kHz steps over a range of +1.4 to −1.8 MHz. Plotting the signal dephasing as a function of the 35Cl saturation pulse offset enables reconstruction of the MAS 35Cl CT quadrupolar powder pattern for MAPbCl3. The 207Pb{35Cl} J-resolved experiments required ca. 22.8 hours at B0 = 9.4 T. In this case, 207Pb detection would not be used to enhance the sensitivity of the 35Cl NMR experiments, rather it would be used to obtain structural information because the 207Pb{35Cl} J-resolved experiments confirm that the lead atoms are covalently bound to chlorine atoms and that the CQ of the chlorine atoms matches that observed in the directly acquired 35Cl solid-state NMR spectrum.
limits the echo duration and the achievable dephasing. 207Pb{35Cl} J-resolved experiments on MAPbCl3 were performed with 35Cl saturation pulse offsets varying in 200 kHz steps over a range of +1.4 to −1.8 MHz. Plotting the signal dephasing as a function of the 35Cl saturation pulse offset enables reconstruction of the MAS 35Cl CT quadrupolar powder pattern for MAPbCl3. The 207Pb{35Cl} J-resolved experiments required ca. 22.8 hours at B0 = 9.4 T. In this case, 207Pb detection would not be used to enhance the sensitivity of the 35Cl NMR experiments, rather it would be used to obtain structural information because the 207Pb{35Cl} J-resolved experiments confirm that the lead atoms are covalently bound to chlorine atoms and that the CQ of the chlorine atoms matches that observed in the directly acquired 35Cl solid-state NMR spectrum.
1H{81Br} DE-RESPDOR and direct WURST-CPMG experiments on MAPbBr3
We investigated the use of indirect detection methods for acquisition of the 81Br solid-state NMR spectrum of methylammonium lead bromide (MAPbBr3). Previously, it was reported that the total 79Br CT solid-state NMR spectrum of MAPbBr3 required days of spectrometer time to acquire.22 Due to the breadth of the 81Br CT solid-state NMR spectrum (ca. 16 MHz), we only acquired a few VOCS sub-spectra close to the discontinuities of the 81Br CT powder pattern (Fig. 5A). Blue lines are numerically simulated 81Br solid-state NMR spectra with a 81Br CQ of 118 MHz (with ηQ = 0). The experimental time to acquire the two WURST-CPMG spectra was 41 minutes and the signal-to-noise ratio is approximately 117 at the most intense horn.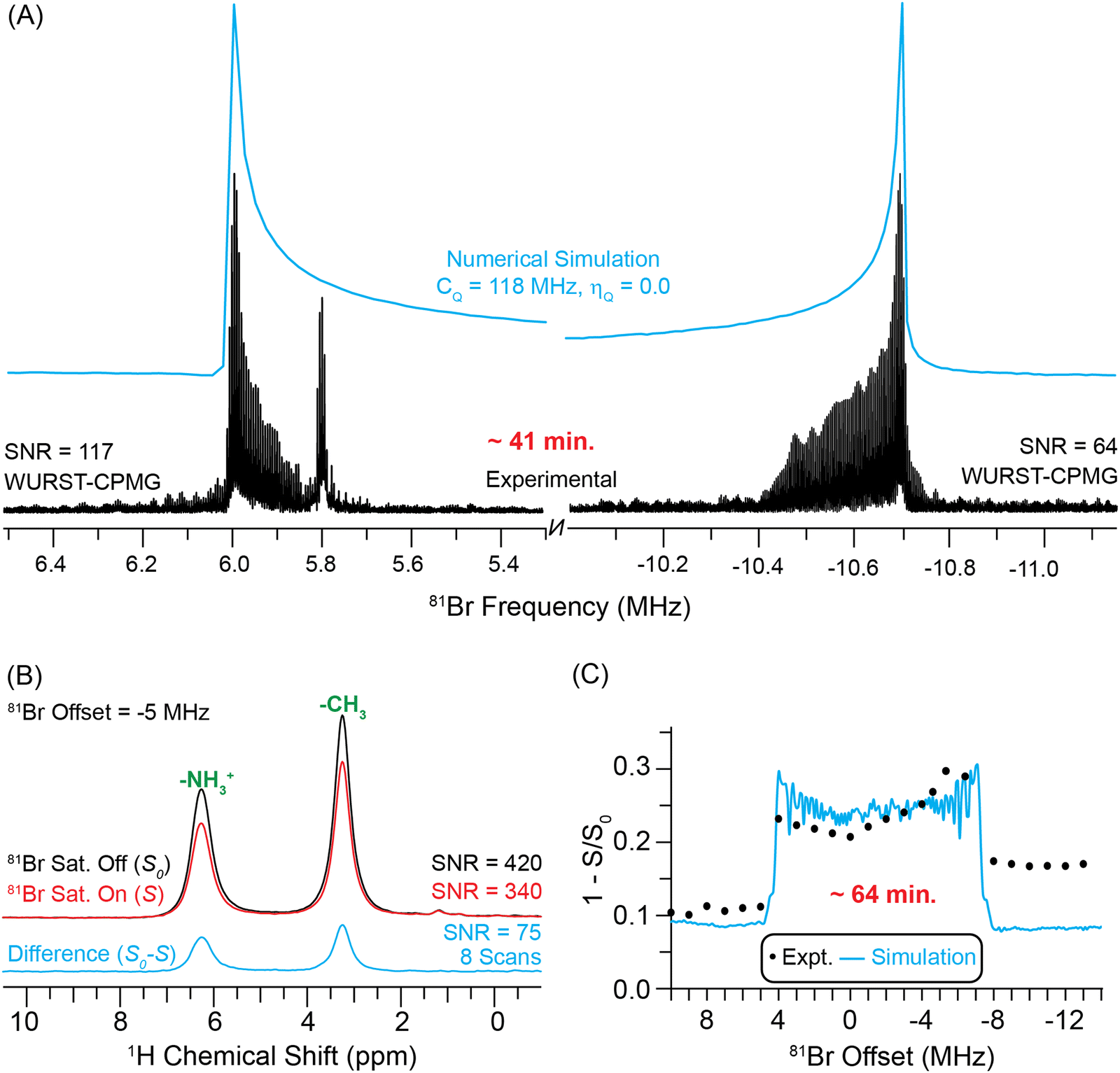 | ||
| Fig. 5 (A) Static WURST-CPMG 81Br NMR spectra of MAPbBr3 obtained at a magnetic field of 9.4 T. (B) 1D 1H{81Br} DE-RESPDOR spectra recorded with (red) or without (black) a 81Br saturation pulse, 1.92 ms of SR421 dipolar recoupling, a 25 kHz MAS frequency and a 14.1 T magnetic field. A difference spectrum (cyan) is shown below. (C) Plot of 1H{81Br} DE-RESPDOR dephasing as a function of the 81Br transmitter offset. The circles and a line correspond to experimental data points and numerical simulations, respectively. The simulation used a 81Br CQ of 118 MHz. | ||
Fig. 5B depicts 1H{81Br} DE-RESPDOR spectra with (red) and without (black) 81Br saturation pulses. All spectra were collected at B0 = 14.1 T using a 81Br saturation pulse length of four rotor cycles (80 μs) with a 50 kHz RF field and 1.92 ms of SR42155 dipolar recoupling. With 8 scans, the control and dephasing spectra have signal-to-noise ratios of 420 and 340, respectively. The difference spectrum (cyan) shown below has a signal-to-noise ratio of 75. The spectra shown in Fig. 5B were obtained with a 81Br pulse offset of −5 MHz. The 81Br spectrum was then indirectly detected with 1H{81Br} DE-RESPDOR by varying the offset of the 81Br saturation pulse in 1 MHz steps over a range of +10 to −14 MHz (Fig. 5C). The complete 1H detected 81Br solid-state NMR spectrum of MAPbBr3 was obtained in ca. 64 minutes. Numerical simulations of the 1H{81Br} DE-RESPDOR dephasing profile indicate that the 81Br CQ is ca. 118 MHz (with ηQ = 0), consistent with the static VOCS WURST-CPMG spectrum. Note, a simulation of the 1H{81Br} DE-RESPDOR dephasing profile that also accounts for additional dephasing from 79Br explains why the dephasing profile in Fig. 5C is asymmetric; at lower 81Br transmitter offsets there is dephasing from the 79Br ST (Fig. S7, ESI†).
Comparing the sensitivity of the indirectly detected 1H{81Br} DE-RESPDOR and the directly detected 81Br WURST-CPMG reveals a slight sensitivity gain with 1H detection. While the 81Br WURST-CPMG spectrum of the high frequency horn has a signal-to-noise ratio of 117, this sub-spectrum required 20.5 minutes to acquire, corresponding to a sensitivity of 26 min−1/2. In comparison, the 1H{81Br} DE-RESPDOR difference spectrum had a signal-to-noise ratio of 75 in 3.2 minutes, corresponding to a sensitivity of 42 min−1/2. Therefore, in this case the RESPDOR experiments provide a 1.6-fold improvement in sensitivity and 2.6-fold reduction in experiment time. For MAPbBr3 the gain in signal-to-noise provided by 1H detection is likely limited because the 1H T1 of MAPbBr3 (T1 = 24 s) is much longer than the 81Br T1 (optimal recycle delays of 0.12 s). However, we stress again that indirect detection also gives additional structural information by confirming that the 1H spins of the MA+ cations are dipole-coupled to the Br− anions. We also note that the 1H detected experiments required only a few μL of material, while we used 100 μL of material for the direct 81Br WURST-CPMG experiments.
Indirect detection of the 63Cu solid-state NMR spectrum of triphosCuI complex by 1H → 31P{63Cu} J-resolved experiment
We studied the use of indirect detection methods for acquisition of the 63Cu solid-state NMR spectrum of the copper(I) phosphine complex, CH3C(CH2PPh2)3CuI (triphosCuI, Fig. S8, ESI†). Phosphines are common ligands coordinated to copper centers and phospine copper complexes are important catalysts, for example, for Sonogashira C–C cross-coupling reactions.59 Therefore, it is interesting to determine whether 31P-detection can be used as a method to obtain 63Cu NMR spectra of copper complexes with phosphine ligands. The 63Cu solid-state NMR signals of triphosCuI were narrow enough to obtain a CT MAS NMR spectrum that was free of spinning sidebands (Fig. 6A). Note, to acquire this spectrum we used the DEPTH pulse sequence60 (with CT selective pulses) because the conventional spin echo NMR spectrum showed an intense 63Cu NMR signal from copper metal components of the probe. A fit of the 63Cu MAS NMR spectrum gave 63Cu CQ = 3.2 MHz, ηQ = 0.57 and δiso = 322 ppm. The total experimental time to acquire the DEPTH NMR spectrum was 205 minutes, and the signal-to-noise ratio was approximately 50. In this case, the 63Cu NMR spectrum is quite narrow, and therefore, 31P detection is not technically needed to study this compound. However, it is difficult to accurately determine the value of ηQ by only fitting the CT lineshape. Below, we show that indirect detections allow both the 63Cu CT and ST powder patterns to be obtained and confirms the accuracy of the fitted value of ηQ.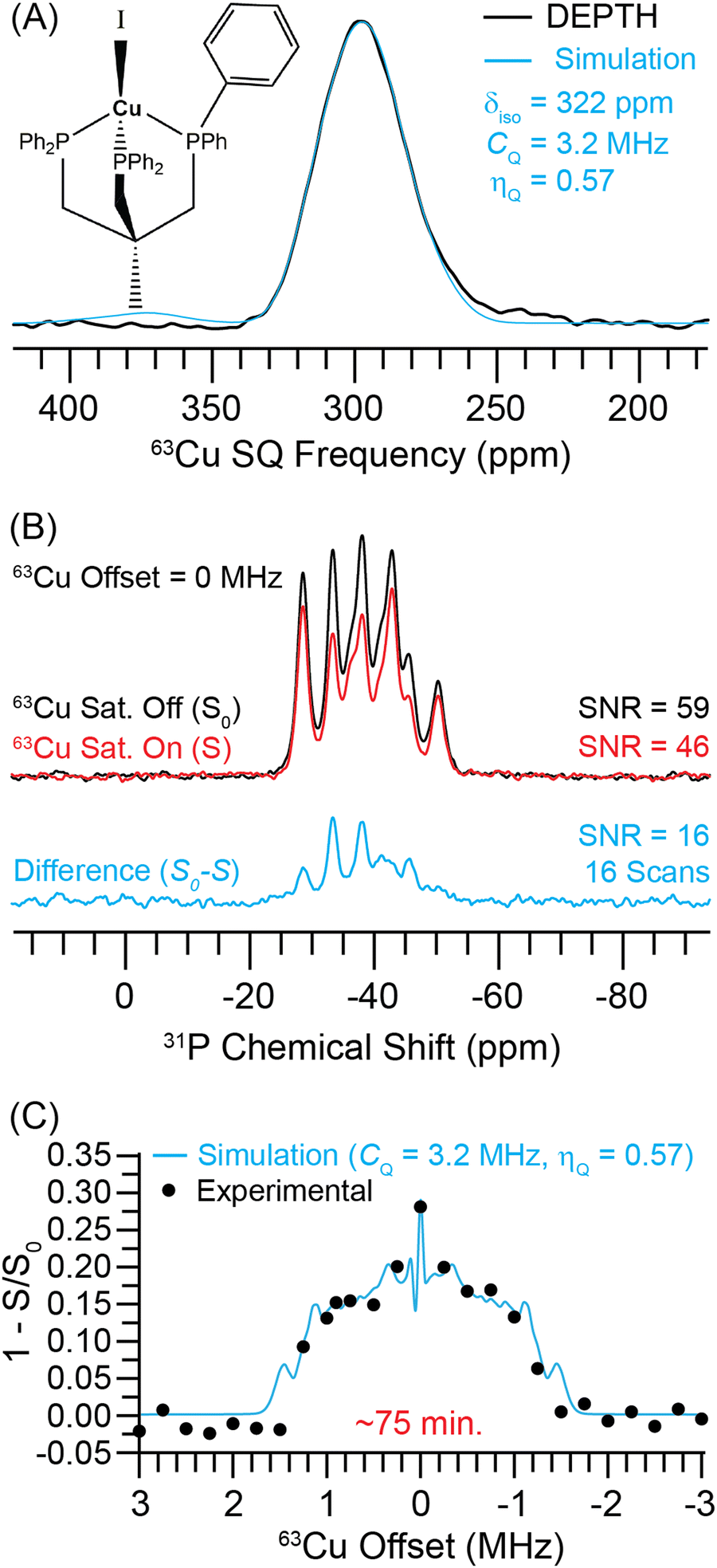 | ||
| Fig. 6 (A) Experimental 63Cu DEPTH NMR spectrum (black) and a simulation with a 63Cu CQ = 3.2 MHz and ηQ = 0.57 (cyan). (B) 1H → 31P{63Cu} J-resolved NMR spectra recorded with (red) or without (black) a 63Cu saturation pulse and 1.44 ms of total J evolution time. (C) 1H → 31P{63Cu} J-resolved signal dephasing as a function of the 63Cu transmitter recorded at B0 = 9.4 T with 25 kHz MAS frequency. The circles and a line correspond to experimental data points and numerical simulation for a 63Cu CQ = 3.2 MHz and ηQ = 0.57 (cyan). | ||
Fig. 6B shows the MAS 1H → 31P{63Cu} J-resolved NMR spectra obtained with (red) and without (black) a 63Cu saturation pulse. The 31P NMR spectrum shows two sets of NMR signals centered at −35 ppm and −43 ppm. Each set of signals in the 31P NMR spectrum is composed of a 4-component multiplet that arises due to the combined effects of J-coupling and residual dipolar coupling to 63Cu and 65Cu nuclei that experience quadrupolar broadening (see a simulated spectrum in Fig. S10, ESI†). 31P solid-state NMR spectra similar in appearance to the ones shown in Fig. 6B have been reported for other Cu(I) phosphine compounds.61–63 The single crystal X-ray structure of triphosCuI shows that all three P atoms are crystallographically distinct (CCDC: 2250183†). The Cu–P bonds are similar in length for all P atoms (2.274 Å to 2.288 Å). However, two of the phosphorus atoms have similar I–Cu–P bond angles (126.1° and 123°), while the other I–Cu–P bond angle is 115.8°. Gauge-including projector-augmented wave (GIPAW)64 calculations implemented in the CASTEP65 program was used to predict the 31P magnetic shielding and differences in chemical shifts. The calculation predicts that two of the 31P nuclei have similar magnetic shielding (σiso = 318.6 ppm and 320.5 ppm), while the third 31P nucleus which has the I–Cu–P bond angle of 115.8° is approximately 15 ppm more shielded (σiso = 335.0 ppm). The predicted shieldings are in reasonable agreement with the experimental spectrum which shows higher intensity for the higher frequency 31P NMR signals.
With 16 scans, the control and dephased 31P NMR spectra have signal-to-noise ratios of 59 and 46, respectively. The difference spectrum (cyan) shown below has a signal-to-noise ratio of 16. The spectra shown in Fig. 6B were obtained with a 0 MHz 63Cu offset. All spectra were collected at B0 = 9.4 T using a 63Cu saturation pulse length of one rotor cycle (40 s) and a total spin echo duration of 1.44 ms. The 63Cu NMR spectrum was then indirectly detected with 1H → 31P{63Cu} J-resolved experiments by varying the offset of the 63Cu saturation pulse in 250 kHz steps over a range of +3 to −3 MHz (Fig. 6C). In Fig. 6C, we have integrated over all 31P peaks. The 31P detected 63Cu NMR spectrum of triphosCuI was obtained in ca. 75 minutes at B0 = 9.4 T. Numerical simulations of the 1H → 31P{63Cu} J-resolved dephasing profile indicate that the 63Cu CQ is ca. 3.2 MHz (with ηQ = 0.57), in good agreement with the 63Cu DEPTH NMR spectrum. Numerical simulations with different 63Cu saturation pulse RF fields and different values of ηQ are shown in the ESI† (Fig. S9). With the current optimized saturation pulse condition of 30 kHz RF field, the dephasing profile remains sensitive to the value of ηQ.
Given the narrow width of the central transition 63Cu NMR spectrum of triphosCuI, 31P detection is not useful for sensitivity enhancement. However, the J-resolved experiments are still helpful in this case because they confirm the presence of coordinative bonds between the Cu and P atoms and confirms that the copper observed in the directly detected 63Cu NMR spectrum gives rise to the observed dephasing.
Numerical simulations of dephasing in 1H{35Cl} DE-RESPDOR experiments as a function of 35Cl CQ
The dependence of the dephasing in a 1H{35Cl} DE-RESPDOR experiment on 35Cl CQ was investigated with numerical SIMPSON simulations. Fig. 7 shows normalized dephasing as a function of 35Cl CQ for a 50 kHz MAS frequency with a magnetic field of 9.4 T for 200 kHz, 80 kHz, and 40 kHz saturation pulse RF fields. The 35Cl saturation pulse was 80 μs in duration. Each curve was constructed by varying CQ from 1 to 80 MHz with 1 MHz increments while maintaining ηQ constant at 0.5 and considering the dephasing at 35Cl transmitter offset = 0 MHz for each data point. The dephasing is significantly higher at 200 kHz RF field for the 35Cl CQ range considered in this simulation. However, such a high 35Cl RF field may not be possible to achieve experimentally. For the other two saturation pulse RF fields of 80 kHz and 40 kHz, the dephasing gradually decreases as the 35Cl CQ increases. But, even if the 35Cl pulse RF field was limited to 40 kHz appreciable dephasing is still predicted. Notably, Cl covalently bonded to Cl has a CQ on the order of 75 MHz.66 Therefore, the 1H{35Cl} DE-RESPDOR experiments could even be applicable to detection of covalently bonded chlorine atoms in organic systems.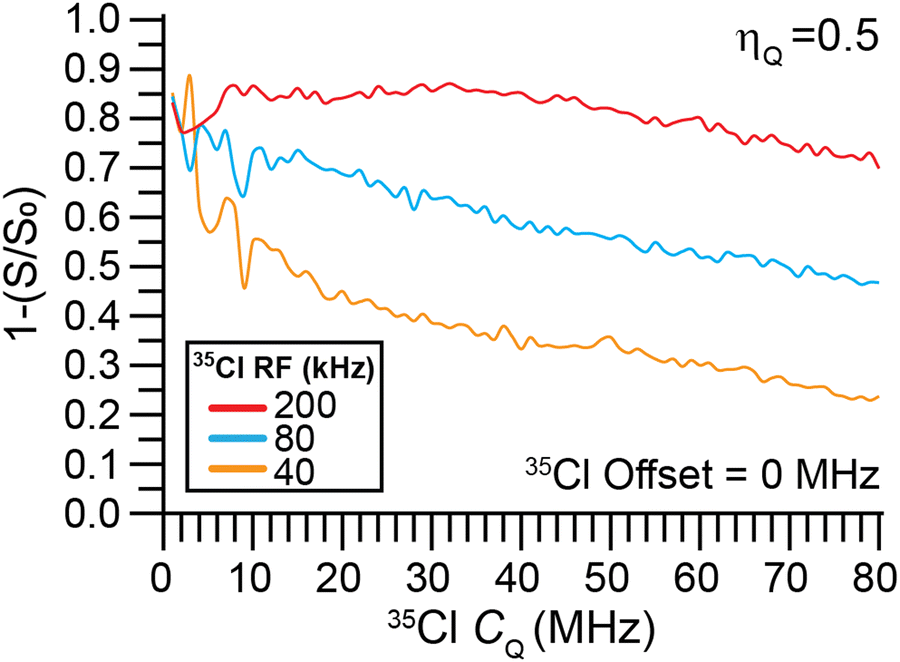 | ||
| Fig. 7 SIMPSON simulated dependence of the normalized 1H{35Cl} DE-RESPDOR dephasing (1 − S/S0) on the 35Cl CQ. Simulations were performed with a 50 kHz magic angle spinning (MAS) frequency with a magnetic field of 9.4 T. Simulations used 80 μs 35Cl saturation pulses with RF fields of 200 kHz, 80 kHz, or 40 kHz. All simulations used a 1H–35Cl spin system with a 2 kHz dipolar coupling constant and ηQ = 0.5. | ||
Numerical simulations of RESPDOR experiments with I = 5/2 and 7/2 nuclei
One obvious question that arises from the previous results is whether or not the indirect detection methods can potentially be applied to quadrupolar nuclei with spins greater than 3/2. To address this question, we have performed SIMPSON simulations of DE RESPDOR experiments for representative 5/2 (127I, Fig. 8) and 7/2 nuclei (51V, Fig. 8). Fig. 8 shows SIMPSON simulated 127I and 51V NMR spectra and DE RESPDOR dephasing plots for the two test cases of BaI2·2H2O26 and V2O2(OH)3,67 respectively. The 127I CQ and ηQ of BaI2·2H2O has been previously measured, while the CQ and ηQ of V2O2(OH)3 were obtained from a plane-wave DFT calculation. The SIMPSON simulations clearly suggest that the frequency stepped RESPDOR experiments should be applicable to higher spin nuclei. However, we note that it is possibly more challenging to resolve the CT from the ST since there are more ST for higher spin quadrupolar nuclei. Additionally, there may be significant broadening of the powder patterns, especially if higher RF fields are used for the saturation pulses. For example, in the simulated 1H{51V} RESPDOR dephasing profiles the powder pattern is significantly broadened due to off-resonance saturation effects. We plan to experimentally test these approaches for I = 5/2 and I = 7/2 nuclei in future work.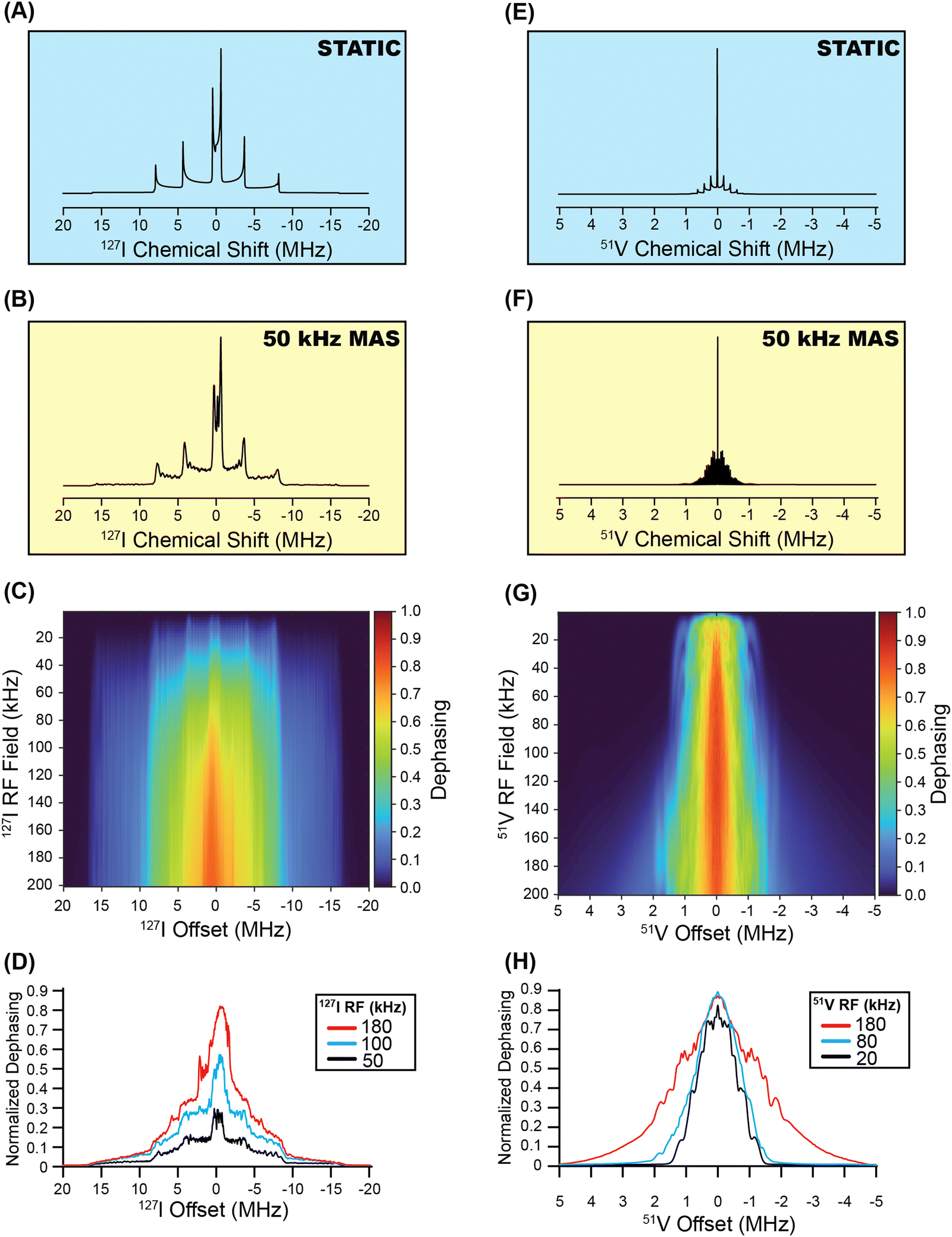 | ||
| Fig. 8 SIMPSON simulated 127I solid-state NMR spectra of BaI2·2H2O (CQ = 53.8 MHz and ηQ = 0) shown for (A) a stationary sample and (B) for a 50 kHz MAS frequency. (C) Heat plot showing the simulated dephasing for 1H{127I} DE-RESPDOR experiments as a function of the 127I saturation pulse RF field with conventional rectangular pulses that are 80 μs in duration. (D) Dephasing profiles extracted from the heat plot for 127I saturation pulse RF fields of 180 kHz, 100 kHz, and 50 kHz. SIMPSON simulated 51V solid-state NMR spectra of V2O2(OH)3 (CQ = 5.8 MHz and ηQ = 0.33) shown for (E) a stationary sample and (F) for a 50 kHz MAS frequency. (G) Heat plot showing the simulated dephasing for 1H{51V} DE-RESPDOR experiments as a function of the 51V saturation pulse RF field with conventional rectangular pulses that are 80 μs in duration. (H) Dephasing profiles extracted from the heat plot for 51V saturation pulse RF fields 180 kHz, 80 kHz, and 20 kHz. | ||
3. Conclusions
In conclusion, we have demonstrated that frequency-stepped dephasing experiments can be used for the indirect detection of wideline solid-state NMR spectra of abundant spin-3/2 nuclei. For the test cases considered in this paper, significant time savings and gains in sensitivity were realized in some cases. The exact gains in sensitivity that are achievable with indirect detection depend upon the ratio of the 1H T1 and quadrupolar nucleus T1 and the spin quantum number and γ of the quadrupolar nucleus. Additional factors that influence the gains in sensitivity include the magnitude of the dipolar or scalar couplings and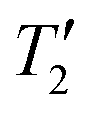 of the quadrupolar nucleus, with the latter parameter determining how efficient CPMG detection can be. The breadth of the quadrupolar powder pattern is also an important consideration, with indirect detection likely preferred for wider spectra and direct detection preferred for narrower quadrupolar powder patterns. Even in cases where the indirect detection dephasing experiments do not provide absolute gains in sensitivity, they may still be helpful as they provide valuable structural information by confirming the presence of dipolar or scalar couplings between the detected nucleus and the quadrupolar nucleus of interest. We also note that much smaller sample volumes are required for the indirect detection experiments, so they should be useful for limited quantity samples. Numerical simulations suggest that the RESPDOR methods are applicable to samples with very large CQ.
of the quadrupolar nucleus, with the latter parameter determining how efficient CPMG detection can be. The breadth of the quadrupolar powder pattern is also an important consideration, with indirect detection likely preferred for wider spectra and direct detection preferred for narrower quadrupolar powder patterns. Even in cases where the indirect detection dephasing experiments do not provide absolute gains in sensitivity, they may still be helpful as they provide valuable structural information by confirming the presence of dipolar or scalar couplings between the detected nucleus and the quadrupolar nucleus of interest. We also note that much smaller sample volumes are required for the indirect detection experiments, so they should be useful for limited quantity samples. Numerical simulations suggest that the RESPDOR methods are applicable to samples with very large CQ.
We anticipate that the indirect detection experiments demonstrated here can be readily combined with dynamic nuclear polarization (DNP) to further enhance sensitivity.68 Under MAS conditions DNP-enhanced 1H spin polarization can be efficiently transferred to spin-1/2 nuclei such as 1H, 13C and 31P, whereas direct DNP or indirect DNP of quadrupolar nuclei is often more challenging. Hence, the indirect detection approach is potentially easier to implement in DNP experiments. We are pursuing further research along these lines. Another interesting direction to consider would be to incorporate phase-modulated (PM) saturation pulses69,70 into the DE-RESPDOR experiments. The PM saturation pulses have been shown to provide enhanced dephasing in REDOR experiments with quadrupolar nuclei that exhibit large quadrupolar coupling constants.69,70 For samples with very large CQ, the PM pulses may be beneficial. We are actively using these indirect detection methods to study materials that contain quadrupolar nuclei that give rise to wideline solid-state NMR spectra.
4. Experimental
Transplatin was purchased from Alfa Aesar and used without further purification. Methylammonium bromide (CH3NH3Br) was purchased from DyeSol. Methylammonium chloride (CH3NH3Cl, 99.0%) and lead chloride (PbCl2, 99.999%) were purchased from Sigma-Aldrich. Lead bromide (PbBr2, 98 + %) was purchased from Acros. N,N-Dimethylformamide (DMF) and toluene were purchased from Fisher Scientific. Copper(I) iodide (CuI, 95 + %) was purchased from Alfa Aesar and used after purification. 1,1,1-Tris(diphenylphosphinomethyl)ethane (triphos, 97 + %) was purchased from Strem. Chloroform was purchased from BeanTown Chemical and used after vacuum distillation over sieves. MAPbCl3, MAPbBr3 and triphosCuI were prepared according to the following procedures:Synthesis of methylammonium lead halides
CH3NH3PbBr3 and CH3NH3PbCl3 were synthesized by a slightly modified literature procedure.71,72 Briefly, CH3NH3Br (1.5 mmol) and PbBr2 (1.5 mmol) were dissolved in DMF (30 mL). After stirring until the mixture became homogeneous (<30 min), excess toluene (100 mL) was added. Crystalline CH3NH3PbBr3 was collected by centrifugation at 4000 rpm for 10 min. After washing 3 times with toluene (30 mL), the material was dried under vacuum. CH3NH3PbCl3 was synthesized from CH3NH3Cl and PbCl2 following a similar procedure. Synthesis of CH3NH3PbBr3 and CH3NH3PbCl3 was confirmed by powder X-ray diffraction (PXRD, Fig. S11, ESI†). PXRD patterns were recorded using Cu Kα radiation on a Rigaku Ultima IV diffractometer (40 kV, 44 mA).Synthesis of the complex triphosCuI
A flask was charged with 1,1,1-tris(diphenylphosphinomethyl)ethane (111.3 mg, 0.1782 mmol), CuI (32.5 mg, 0.171 mmol), and 25 mL of chloroform under nitrogen. The reaction mixture was stirred overnight, forming a crystalline precipitate. The mixture was filtered, and the solids were washed with 3 mL of chloroform, then dried in vacuo. A colorless powder (103.5 mg, 0.1270 mmol, 71.3%) was collected. Solution 1H NMR spectra and single crystal X-ray diffraction data are included in the ESI.† The single-crystal X-ray diffraction structure of triphosCuI can be accessed from the Cambridge Crystallographic Data Centre (CCDC code: 2250183).Solid-state NMR spectroscopy
Solid-state NMR spectroscopy experiments were performed on a 9.4 T Bruker wide-bore magnet equipped with a Bruker AVANCE III HD console (1H{35Cl} DE-RESPDOR,4935Cl and 81Br WURST-CPMG,19207Pb{35Cl} J-resolved, 63Cu DEPTH60 and 1H → 31P{63Cu} J-resolved) and equipped with a Bruker 1.3 mm HX (1H{35Cl} experiments of transplatin and MAPbCl3), Bruker 2.5 mm HXY probe (207Pb{35Cl} of MAPbCl3 and 1H → 31P{63Cu} of triphosCuI complex) or a Bruker 4.0 mm HXY magic angle spinning (MAS) NMR probe (all WURST-CPMG experiments). 1H{81Br} experiments were performed with a 14.1 T Bruker wide-bore magnet equipped with a Bruker AVANCE NEO console and a Bruker 2.5 mm HXY MAS NMR probe. All experiments utilized N2 gas for spinning. 1H chemical shifts were referenced to neat tetramethylsilane by using adamantane (δiso(1H) = 1.72 ppm) as a secondary chemical shift reference. 207Pb and 31P chemical shifts were indirectly referenced to neat TMS using the IUPAC recommended relative NMR frequency.73 NMR spectra were processed and analyzed with Bruker Topspin version 3.6.4 (AVANCE III HD data) or 4.1.4 (AVANCE NEO data) software.The following experimental details are with respect to data acquired at B0 = 9.4 T with the 1.3 mm HX NMR probe (1H{35Cl} experiments), 2.5 mm HXY NMR probe (207Pb{35Cl} and 1H → 31P{63Cu} experiments) and 4 mm HXY NMR probe (WURST-QCPMG experiments). 1H{35Cl} DE-RESPDOR spectra of transplatin were recorded with the 1.3 mm HX NMR probe and a 50 kHz MAS frequency. The 1H longitudinal relaxation time constant (T1) of transplatin was ca.17 s; all experiments utilized a 5 s recycle delay. 1H{35Cl} DE-RESPDOR experiments were performed with 35Cl saturation pulses that were 80 μs (4 × τrot) in duration with an 80 kHz RF field.49 Additional experiments on transplatin were performed with tanh/tan 35Cl saturation pulses 40 μs in duration. The sweep width of the tanh/tan pulses was set to 5 MHz. The SR421 heteronuclear dipolar recoupling sequence was applied to the 1H spins to reintroduce the 1H–35Cl dipolar interaction under MAS.55 A control (without a 35Cl saturation pulse) and dephased (with a 35Cl saturation pulse) point were recorded at each 35Cl offset. Similarly, 1H{35Cl} DE-RESPDOR spectra of MAPbCl3 were recorded with the 1.3 mm HX NMR probe and a 50 kHz MAS frequency. The 1H T1 of MAPbCl3 was ca. 24 s and 30 s for high-frequency (NH+3) and low-frequency 1H NMR signal (–CH3), respectively; all experiments utilized a 5 s recycle delay and the high-frequency 1H NMR signal was used to map out the 35Cl spectrum. 1H{35Cl} DE-RESPDOR experiments were performed with 35Cl saturation pulses that were 80 μs (4 × τrot) in duration with an 80 kHz radio frequency (RF) field.49 The SR421 heteronuclear dipolar recoupling sequence was applied to the 1H spins to reintroduce the 1H–35Cl dipolar interaction under MAS.55 A control (without a 35Cl saturation pulse) and dephased (with a 35Cl saturation pulse) point was recorded at each 35Cl offset. 207Pb{35Cl} J-resolved spectra of MAPbCl3 were recorded with the 2.5 mm HXY NMR probe and a 25 kHz MAS frequency. All experiments utilized a 2 s recycle delay. 207Pb{35Cl} J-resolved experiments were performed with 35Cl saturation pulses that were 80 μs (2 × τrot) in duration with a 14 kHz RF field. A control (without a 35Cl saturation pulse) and dephased (with a 35Cl saturation pulse) point were recorded at each 35Cl offset. 1H → 31P{63Cu} J-resolved spectra of triphosCuI were recorded with the 2.5 mm HXY NMR probe and a 25 kHz MAS frequency. The 1H T1 of triphosCuI complex was ca. 6.4 s; all experiments utilized an 8 s recycle delay. 1H → 31P{63Cu} J-resolved experiments were performed with 63Cu saturation pulses that were 40 μs (1 × τrot) in duration with a 30 kHz RF field. A control (without a 63Cu saturation pulse) and dephased (with a 63Cu saturation pulse) point were recorded at each 63Cu offset. The 63Cu DEPTH60 spectrum was recorded with 3 s recycle delay and 4096 scans. Static WURST-QCPMG spectra of MAPbCl3 were recorded with the 4 mm HXY NMR probe with WURST pulses that were 25 μs in duration and with a sweep width of 800 kHz.19,24,74 Each echo in the CPMG train was 50 μs in duration. A total of 30 echoes were acquired. 50 kHz 1H RF field SPINAL-64 decoupling was performed throughout the entire static CPMG experiments.75 Similarly, static WURST-QCPMG spectra of transplatin were recorded with the 4 mm HXY NMR probe with WURST pulses that were 50 μs in duration and with a sweep width of 800 kHz.19,24,74 Each echo in the CPMG train was 90 μs in duration. A total of 100 echoes were acquired. High-frequency and low-frequency ends were recorded for static WURST-QCPMG spectra of MAPbBr3 with the 4 mm HXY NMR probe with WURST pulses that were 25 μs in duration and with a sweep width of 800 kHz.19,24,74 Each echo in the CPMG train was 50 μs in duration. A total of 10 echoes were acquired.
The following experimental details are for data collected with the Bruker 2.5 mm HXY NMR probe and B0 = 14.1 T (1H{81Br} experiments). 1H{81Br} DE-RESPDOR spectra of MAPbBr3 were recorded with a 25 kHz MAS frequency. The 1H T1 of MAPbBr3 was ca. 24 s and 23 s for high-frequency (–NH+3) and low-frequency signal (–CH3), respectively. All experiments applied a 10 s recycle delay and considered high-frequency signal to map out the 81Br spectrum. 1H{81Br} DE-RESPDOR experiments were performed with 81Br saturation pulses that were 80 μs (2 × τrot) in duration with a 50 kHz RF field.49 The SR421 heteronuclear dipolar recoupling sequence was applied to the 1H spins to reintroduce the 1H–81Br dipolar interaction under MAS.55 A control (without a 81Br saturation pulse) and dephased (with a 81Br saturation pulse) point was recorded at each 81Br offset.
Numerical solid-state NMR spectroscopy simulations
SIMPSON v4.1.1 was used to perform numerical solid-state NMR simulations.50–52 The archived data includes the SIMPSON input codes. Except for the 1H π/2 pulses, all the pulses in the files were finite in duration. All 2D heat maps were created using the rep678 crystal file. Static and MAS ideal 35Cl NMR spectra were simulated using the zcw28656 crystal file. The number of gamma angles was 15 for the MAS spectrum simulations. In Fig. 2C and D, the 35Cl offset was incremented in steps of 50 kHz over a frequency range of +20 MHz to −20 MHz while the 35Cl saturation RF field was increased in steps of 2 kHz from 0 to 200 kHz. The simulations in Fig. 2E were extracted from the simulation corresponding to Fig. 2C. The simulation in Fig. 2F was extracted from corresponding simulations shown in Fig. 2D. MATLAB R2021B was used to create all heat maps. The simulated spectra were processed in ssNake v1.3.57 The DE-RESPDOR heat maps were normalized by dividing each dephasing intensity by the maximum dephasing exhibited in each heat map. All the other numerical simulations were run with identical parameters (RF fields, transmitter offsets, and B0) to those used experimentally. The other numerical simulations for 1H{35Cl} DE-RESPDOR in Fig. 2B and 4C were performed with a two-spin 1H–35Cl spin system, the zcw4180 crystal file, 13 γ-angles, and a 0.5 μs time step where the Hamiltonian was considered time-independent. 1H{81Br} DE-RESPDOR numerical simulations were done with a two-spin 1H–81Br spin system, the zcw4180 crystal file, 13 γ-angles, and a 0.1 μs time step where the Hamiltonian was considered time-independent. Numerical simulations of static 35Cl/81Br solid-state NMR spectra were performed with the zcw28656 crystal file and assumed ideal excitation of the powder pattern (start operator I1x, detect operator I1c). In Fig. 8, all 2D heat maps were created using the rep678 crystal file. Static and MAS ideal 35Cl NMR spectra were simulated using the zcw28656 crystal file. The number of gamma angles was 15 for the MAS spectrum simulations. In Fig. 8C, the 127I offset was incremented in steps of 50 kHz over a frequency range of +20 MHz to −20 MHz while the 127I saturation RF field was increased in steps of 2 kHz from 0 to 200 kHz. The simulation in Fig. 8D was extracted from the simulation corresponding to Fig. 8C.In Fig. 8G, the 51V offset was incremented in steps of 50 kHz over a frequency range of +5 MHz to −5 MHz while the 51V saturation RF field was increased in steps of 1 kHz from 0 to 200 kHz. The simulation in Fig. 8H was extracted from the simulation corresponding to Fig. 8G. MATLAB R2021B was used to create all heat maps. The simulated spectra were processed in ssNake v1.3.57
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Material synthesis, solid-state NMR experiments and simulations were supported by the US Department of Energy (DOE), Office of Science, Basic Energy Sciences, Materials Science and Engineering Division (A. J. R., J. V., S. N. S. L, B. A. A., R. W. D., and E. G.). The Ames Laboratory is operated for the US DOE by Iowa State University under Contract DE-AC02-07CH11358. Synthesis, characterization and NMR experiments on triphosCuI were supported by grant CHE-1900100 from the US NSF (J. B. and M. R. K). A. J. R. acknowledges additional support from the Alfred P. Sloan Foundation through a Sloan research fellowship.References
- S. P. Brown and H. W. Spiess, Advanced solid-state NMR methods for the elucidation of structure and dynamics of molecular, macromolecular, and supramolecular systems, Chem. Rev., 2001, 101, 4125–4155 CrossRef CAS PubMed.
- P.-H. Chien, K. J. Griffith, H. Liu, Z. Gan and Y.-Y. Hu, Recent Advances in Solid-State Nuclear Magnetic Resonance Techniques for Materials Research, Annu. Rev. Mater. Res., 2020, 50, 493–520 CrossRef CAS.
- T. K. Karamanos, A. P. Kalverda and S. E. Radford, Generating Ensembles of Dynamic Misfolding Proteins, Front. Neurosci., 2022, 16, 1–14 Search PubMed.
- D. L. Bryce, New frontiers for solid-state NMR across the periodic table: A snapshot of modern techniques and instrumentation, Dalton Trans., 2019, 48, 8014–8020 RSC.
- S. E. Ashbrook, Solid-state NMR spectroscopy, Phys. Chem. Chem. Phys., 2009, 11, 6875 RSC.
- R. W. Schurko, Ultra-wideline solid-state NMR spectroscopy, Acc. Chem. Res., 2013, 46, 1985–1995 CrossRef CAS PubMed.
- S. E. Ashbrook and S. Sneddon, New methods and applications in solid-state NMR spectroscopy of quadrupolar nuclei, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef CAS PubMed.
- A. Medek, V. Frydman and L. Frydman, Central Transition Nuclear Magnetic Resonance in the Presence of Large Quadrupole Couplings: Cobalt-59 Nuclear Magnetic Resonance of Cobaltophthalocyanines, J. Phys. Chem. A, 1999, 103, 4830–4835 CrossRef CAS.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. P. Coutures, 71Ga and 69Ga nuclear magnetic resonance study of beta-Ga2O3: resolution of four- and six-fold coordinated Ga sites in static conditions, Solid State Nucl. Magn. Reson., 1995, 4, 241–248 CrossRef CAS PubMed.
- C. P. Slichter, NMR Study of Platinum Catalysis, Surf. Sci., 1981, 106, 382–396 CrossRef CAS.
- T. J. Bastow, M. E. Smith and S. N. Stuart, Observation of 91Zr NMR in zirconium-based metals and oxides, Chem. Phys. Lett., 1992, 19, 125–129 CrossRef.
- T. J. Bastow and M. E. Smith, 91Zr NMR characterisation of phases in transformation toughened zirconia, Solid State Nucl. Magn. Reson., 1992, 1, 165–174 CrossRef CAS PubMed.
- F. H. Larsen, J. Skibsted, H. J. Jakobsen and N. C. Nielsen, Solid-state QCPMG NMR of low-γ quadrupolar metal nuclei in natural abundance, J. Am. Chem. Soc., 2000, 122, 7080–7086 CrossRef CAS.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, High-field QCPMG-MAS NMR of half-integer quadrupolar nuclei with large quadrupole couplings, Mol. Phys., 1998, 95, 1185–1195 CrossRef CAS.
- A. S. Lipton, M. D. Smith, R. D. Adams and P. D. Ellis, Zn solid-state and single-crystal NMR spectroscopy and X-ray crystal structure of zinc formate dihydrate, J. Am. Chem. Soc., 2002, 124, 410–414 CrossRef CAS PubMed.
- A. S. Lipton, R. W. Heck, M. Hernick, C. A. Fierke and P. D. Ellis, Residue ionization in LpxC directly observed by 67Zn NMR spectroscopy, J. Am. Chem. Soc., 2008, 130, 12671–12679 CrossRef CAS PubMed.
- H. Nagashima, J. Trébosc, Y. Kon, K. Sato, O. Lafon and J. P. Amoureux, Observation of Low-γ Quadrupolar Nuclei by Surface-Enhanced NMR Spectroscopy, J. Am. Chem. Soc., 2020, 142, 10659–10672 CrossRef CAS PubMed.
- B. E. G. Lucier, K. E. Johnston, W. Xu, J. C. Hanson, S. D. Senanayake, S. Yao, M. W. Bourassa, M. Srebro, J. Autschbach and R. W. Schurko, Unravelling the structure of Magnus’ pink salt, J. Am. Chem. Soc., 2014, 136, 1333–1351 CrossRef CAS PubMed.
- L. A. O’Dell and R. W. Schurko, QCPMG using adiabatic pulses for faster acquisition of ultra-wideline NMR spectra, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- A. J. Rossini, R. W. Mills, G. A. Briscoe, E. L. Norton, S. J. Geier, I. Hung, S. Zheng, J. Autschbach and R. W. Schurko, Solid-state chlorine NMR of group IV transition metal organometallic complexes, J. Am. Chem. Soc., 2009, 131, 3317–3330 CrossRef CAS PubMed.
- D. Laurencin, F. Ribot, C. Gervais, A. J. Wright, A. R. Baker, L. Campayo, J. V. Hanna, D. Iuga, M. E. Smith, J. M. Nedelec, G. Renaudin and C. Bonhomme, 87Sr, 119Sn, 127I Single and {1H/19F}-Double Resonance Solid-State NMR Experiments: Application to Inorganic Materials and Nanobuilding Blocks, ChemistrySelect, 2016, 1, 4509–4519 CrossRef CAS.
- L. Piveteau, M. Aebli, N. Yazdani, M. Millen, L. Korosec, F. Krieg, B. M. Benin, V. Morad, C. Piveteau, T. Shiroka, A. Comas-Vives, C. Copéret, A. M. Lindenberg, V. Wood, R. Verel and M. V. Kovalenko, Bulk and Nanocrystalline Cesium Lead-Halide Perovskites as Seen by Halide Magnetic Resonance, ACS Cent. Sci., 2020, 6, 1138–1149 CrossRef CAS PubMed.
- D. L. Bryce and G. D. Sward, Solid-state NMR spectroscopy of the quadrupolar halogens: Chlorine-35/37, bromine-79/81, and iodine-127, Magn. Reson. Chem., 2006, 44, 409–450 CrossRef CAS PubMed.
- L. A. O’Dell, A. J. Rossini and R. W. Schurko, Acquisition of ultra-wideline NMR spectra from quadrupolar nuclei by frequency stepped WURST-QCPMG, Chem. Phys. Lett., 2009, 468, 330–335 CrossRef.
- A. S. Lipton, J. A. Sears and P. D. Ellis, A general strategy for the NMR observation of half-integer quadrupolar nuclei in dilute environments, J. Magn. Reson., 2001, 151, 48–59 CrossRef CAS PubMed.
- C. M. Widdifield and D. L. Bryce, Solid-state 127I NMR and GIPAW DFT study of metal iodides and their hydrates: Structure, symmetry, and higher-order quadrupole-induced effects, J. Phys. Chem. A, 2010, 114, 10810–10823 CrossRef CAS PubMed.
- L. Müller, Sensitivity Enhanced Detection of Weak Nuclei Sensitivity Enhanced Detection of Weak Nuclei Using Heteronuclear Multiple Quantum Coherence, J. Am. Chem. Soc., 1979, 101, 4481–4484 CrossRef.
- S. Cavadini, A. Abraham and G. Bodenhausen, Proton-detected nitrogen-14 NMR by recoupling of heteronuclear dipolar interactions using symmetry-based sequences, Chem. Phys. Lett., 2007, 445, 1–5 CrossRef CAS.
- S. Cavadini, A. Lupulescu, S. Antonijevic and G. Bodenhausen, Nitrogen-14 NMR spectroscopy using residual dipolar splittings in solids, J. Am. Chem. Soc., 2006, 128, 7706–7707 CrossRef CAS PubMed.
- Z. Gan, Measuring amide nitrogen quadrupolar coupling by high-resolution 14N/13C NMR correlation under magic-angle spinning, J. Am. Chem. Soc., 2006, 128, 6040–6041 CrossRef CAS PubMed.
- Y. Nishiyama, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi, K. Hioka and T. Asakura, Very fast magic angle spinning 1H-14N 2D solid-state NMR: Sub-micro-liter sample data collection in a few minutes, J. Magn. Reson., 2011, 208, 44–48 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, Indirect detection of nitrogen-14 in solids via protons by nuclear magnetic resonance spectroscopy, J. Magn. Reson., 2006, 182, 168–172 CrossRef CAS PubMed.
- S. Cavadini, A. Abraham, S. Ulzega and G. Bodenhausen, Evidence for dynamics on a 100 ns time scale from single- and double-quantum nitrogen-14 NMR in solid peptides, J. Am. Chem. Soc., 2008, 130, 10850–10851 CrossRef CAS PubMed.
- M. Grüne, R. Luxenhofer, D. Iuga, S. P. Brown and A. C. Pöppler, 14N-1H HMQC solid-state NMR as a powerful tool to study amorphous formulations-an exemplary study of paclitaxel loaded polymer micelles, J. Mater. Chem. B, 2020, 8, 6827–6836 RSC.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, Probing hydrogen bonding in cocrystals and amorphous dispersions using 14N-1H HMQC solid-state NMR, Mol. Pharm., 2013, 10, 999–1007 CrossRef CAS PubMed.
- C. D. Makowka, C. P. Slichter and J. H. Sinfelt, NMR of platinum catalysts: Double NMR of chemisorbed carbon monoxide and a model for the platinum NMR line shape, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 5663–5679 CrossRef CAS PubMed.
- C. D. Makowka, C. P. Slichter and J. H. Sinfelt, Probe of the Surface of a Heterogeneous Catalyst: Double NMR of Carbon Monoxide Chemisorbed on Highly Dispersed Platinum, Phys. Rev. Lett., 1982, 49, 379–382 CrossRef CAS.
- P. K. Wang, J. P. Ansermet, S. L. Rudaz, Z. Wang, S. Shore, C. P. Slichter and J. H. Sinfelt, NMR Studies of Simple Molecules on Metal Surfaces, Science, 1986, 234, 35–41 CrossRef CAS PubMed.
- C. P. Grey and A. J. Vega, Determination of the Quadrupole Coupling Constant of the Invisible Aluminum Spins in Zeolite HY with 1H/27A1 TRAPDOR NMR, J. Am. Chem. Soc., 1995, 117, 8232–8242 CrossRef CAS.
- M. Kalwei and H. Koller, Quantitative comparison of REAPDOR and TRAPDOR experiments by numerical simulations and determination of H-Al distances in zeolites, Solid State Nucl. Magn. Reson., 2002, 21, 145–157 CrossRef CAS.
- C. Schroeder, V. Siozios, C. Mück-Lichtenfeld, M. Hunger, M. R. Hansen and H. Koller, Hydrogen Bond Formation of Brønsted Acid Sites in Zeolites, Chem. Mater., 2020, 32, 1564–1574 CrossRef CAS.
- R. W. Dorn, B. J. Ryan, S. N. S. Lamahewage, M. V. Dodson, J. B. Essner, R. Biswas, M. G. Panthani and A. J. Rossini, Chlorination of Hydrogenated Silicon Nanosheets Revealed by Solid-State Nuclear Magnetic Resonance Spectroscopy, Chem. Mater., 2023, 35, 539–548 CrossRef CAS.
- N. T. Duong and Y. Nishiyama, Detection of remote proton-nitrogen correlations by 1H-detected 14N overtone solid-state NMR at fast MAS, Phys. Chem. Chem. Phys., 2022, 24, 10717–10726 RSC.
- Z. Gan, Rotary resonance echo double resonance for measuring heteronuclear dipolar coupling under MAS, J. Magn. Reson., 2006, 183, 235–241 CrossRef CAS PubMed.
- C. P. Grey, W. S. Veeman and A. J. Vega, Rotational echo 14N/13C/1H triple resonance solid-state nuclear magnetic resonance: A probe of 13C-14N internuclear distances, J. Chem. Phys., 1993, 98, 7711–7724 CrossRef CAS.
- A. Venkatesh, D. Gioffrè, B. A. Atterberry, L. Rochlitz, S. L. Carnahan, Z. Wang, G. Menzildjian, A. Lesage, C. Copéret and A. J. Rossini, Molecular and Electronic Structure of Isolated Platinum Sites Enabled by the Expedient Measurement of 195Pt Chemical Shift Anisotropy, J. Am. Chem. Soc., 2022, 144, 13511–13525 CrossRef CAS PubMed.
- T. Wolf, A. Eden-Kossoy and L. Frydman, Indirectly detected satellite-transition quadrupolar NMR via progressive saturation of the proton reservoir, Solid State Nucl. Magn. Reson., 2023, 125, 101862 CrossRef CAS PubMed.
- M. J. Jaroszewicz, A. R. Altenhof, R. W. Schurko and L. Frydman, Sensitivity Enhancement by Progressive Saturation of the Proton Reservoir: A Solid-State NMR Analogue of Chemical Exchange Saturation Transfer, J. Am. Chem. Soc., 2021, 143, 19778–19784 CrossRef CAS PubMed.
- B. A. Atterberry, S. L. Carnahan, Y. Chen, A. Venkatesh and A. J. Rossini, Double echo symmetry-based REDOR and RESPDOR pulse sequences for proton detected measurements of heteronuclear dipolar coupling constants, J. Magn. Reson., 2022, 336, 107147 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- Z. Tošner, T. Vosegaard, C. Kehlet, N. Khaneja, S. J. Glaser and N. C. Nielsen, Optimal control in NMR spectroscopy: Numerical implementation in SIMPSON, J. Magn. Reson., 2009, 197, 120–134 CrossRef PubMed.
- Z. Tošner, R. Andersen, B. Stevensson, M. Edén, N. C. Nielsen and T. Vosegaard, Computer-intensive simulation of solid-state NMR experiments using SIMPSON, J. Magn. Reson., 2014, 246, 79–93 CrossRef PubMed.
- T. L. Hwang, P. C. M. Van Zijl and M. Garwood, Fast Broadband Inversion by Adiabatic Pulses, J. Magn. Reson., 1998, 133, 200–203 CrossRef CAS PubMed.
- A. Pankowska, K. Kochalska, A. Łazorczyk, K. Dyndor, P. Kozioł, B. Zieńczuk, M. Toborek and R. Pietura, Chemical exchange saturation transfer (CEST) as a new method of signal obtainment in magnetic resonance molecular imaging in clinical and research practice, Pol. J. Radiol., 2019, 84, e147–e152 CrossRef PubMed.
- A. Brinkmann and A. P. M. Kentgens, Proton-selective 17O-1H distance measurements in fast magic-angle-spinning solid-state NMR spectroscopy for the determination of hydrogen bond lengths, J. Am. Chem. Soc., 2006, 128, 14758–14759 CrossRef CAS PubMed.
- B. E. G. Lucier, A. R. Reidel and R. W. Schurko, Multinuclear solid-state NMR of square-planar platinum complexes Cisplatin and related systems, Can. J. Chem., 2011, 89, 919–937 CrossRef CAS.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, ssNake: A cross-platform open-source NMR data processing and fitting application, J. Magn. Reson., 2019, 301, 56–66 CrossRef CAS PubMed.
- D. Sarkar, R. W. Hooper, A. Karmakar, A. Bhattacharya, A. Pominov, V. V. Terskikh and V. K. Michaelis, Metal Halide Perovskite and Perovskite-like Materials through the Lens of Ultra-wideline 35/37Cl NMR Spectroscopy, ACS Mater. Lett., 2022, 4, 1255–1263 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 50, 4467–4470 CrossRef.
- D. G. Cory and W. M. Ritchey, Suppression of Signals from the Probe in Bloch Decay Spectra, J. Magn. Reson., 1988, 80, 128–160 Search PubMed.
- B. E. G. Lucier, J. A. Tang, R. W. Schurko, G. A. Bowmaker, P. C. Healy and J. V. Hanna, Solid-state 65Cu and 31P NMR spectroscopy of bis(triphenylphosphine) copper species, J. Phys. Chem. C, 2010, 114, 7949–7962 CrossRef CAS.
- J. A. Tang, B. D. Ellis, T. H. Warren, J. V. Hanna, C. L. B. Macdonald and R. W. Schurko, Solid-state 63Cu and 65Cu NMR spectroscopy of inorganic and organometallic copper(I) complexes, J. Am. Chem. Soc., 2007, 129, 13049–13065 CrossRef CAS PubMed.
- H. Yu, X. Tan, G. M. Bernard, V. V. Terskikh, J. Chen and R. E. Wasylishen, Solid-State 63Cu, 65Cu, and 31P NMR Spectroscopy of Photoluminescent Copper(I) Triazole Phosphine Complexes, J. Phys. Chem. A, 2015, 119, 8279–8293 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, All-electron magnetic response with pseudopotentials: NMR chemical shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 2451011 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard Ii, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 CAS.
- F. A. Perras and D. L. Bryce, Direct investigation of covalently bound chlorine in organic compounds by solid-state 35Cl NMR spectroscopy and exact spectral line-shape simulations, Angew. Chem., Int. Ed., 2012, 51, 4227–4230 CrossRef CAS PubMed.
- A. M. Callegari and M. Boiocchi, Häggite from the Gambatesa mine, Liguria, Italy: a refinement of the crystal structure, Neues Jahrb. Mineral., Abh., 2015, 192, 33–38 CAS.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K. N. Hu, C. G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A. Van Der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, Dynamic nuclear polarization at high magnetic fields, J. Chem. Phys., 2008, 128, 052211 CrossRef PubMed.
- E. Nimerovsky and A. Goldbourt, Efficient rotational echo double resonance recoupling of a spin-1/2 and a quadrupolar spin at high spinning rates and weak irradiation fields, J. Magn. Reson., 2010, 206, 52–58 CrossRef CAS PubMed.
- E. Nimerovsky, R. Gupta, J. Yehl, M. Li, T. Polenova and A. Goldbourt, Phase-modulated LA-REDOR: A robust, accurate and efficient solid-state NMR technique for distance measurements between a spin-1/2 and a quadrupole spin, J. Magn. Reson., 2014, 244, 107–113 CrossRef CAS PubMed.
- B. A. Rosales, L. Men, S. D. Cady, M. P. Hanrahan, A. J. Rossini and J. Vela, Persistent Dopants and Phase Segregation in Organolead Mixed-Halide Perovskites, Chem. Mater., 2016, 28, 6848–6859 CrossRef CAS.
- F. Zhu, L. Men, Y. Guo, Q. Zhu, U. Bhattacharjee, P. M. Goodwin, J. W. Petrich, E. A. Smith and J. Vela, Shape evolution and single particle luminescence of organometal halide perovskite nanocrystals, ACS Nano, 2015, 9, 2948–2959 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001), Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- A. J. Rossini, R. W. Mills, G. A. Briscoe, E. L. Norton, S. J. Geier, I. Hung, S. Zheng, J. Autschbach and R. W. Schurko, Solid-state chlorine NMR of group IV transition metal organometallic complexes, J. Am. Chem. Soc., 2009, 131, 3317–3330 CrossRef CAS PubMed.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Shows additional solid-state NMR spectroscopy spectra, PXRD patterns and numerical simulations. Raw NMR data associated and SIMPSON simulation files associated with the main text figures are available for download at DOI: https://doi.org/10.5281/zenodo.10014989. The CCDC 2250183 contains the supplementary crystallographic data for the copper complex CH3C(CH2PPh2)3CuI (triphosCuI) for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp05055f |
| This journal is © the Owner Societies 2024 |