
Nonadiabatic quantum dynamics explores non-monotonic photodissociation branching of N2 into the N(4S) + N(2D) and N(4S) + N(2P) product channels†
Natalia
Gelfand
 *a,
Ksenia
Komarova
a,
Francoise
Remacle
ab and
R. D.
Levine
ac
*a,
Ksenia
Komarova
a,
Francoise
Remacle
ab and
R. D.
Levine
ac
aThe Fritz Haber Center for Molecular Dynamics, Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: natalia.gelfand@mail.huji.ac.il
bTheoretical Physical Chemistry, UR MolSys B6c, University of Liège, B4000 Liège, Belgium
cDepartment of Molecular and Medical Pharmacology, David Geffen School of Medicine and Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA
First published on 10th January 2024
Abstract
Vacuum ultraviolet (VUV) photodissociation of N2 molecules is a source of reactive N atoms in the interstellar medium. In the energy range of VUV optical excitation of N2, the N–N triple bond cleavage leads to three types of atoms: ground-state N(4S) and excited-state N(2P) and N(2D). The latter is the highest reactive and it is believed to be the primary participant in reactions with hydrocarbons in Titan's atmosphere. Experimental studies have observed a non-monotonic energy dependence and non-statistical character of the photodissociation of N2. This implies different dissociation pathways and final atomic products for different wavelength regions in the sunlight spectrum. We here apply ab initio quantum chemical and nonadiabatic quantum dynamical techniques to follow the path of an electronic state from the excitation of a particular singlet 1Σ+u and 1Πu vibronic level of N2 to its dissociation into different atomic products. We simulate dynamics for two isotopomers of the nitrogen molecule, 14N2 and 14N15N for which experimental data on the branching are available. Our computations capture the non-monotonic energy dependence of the photodissociation branching ratios in the energy range 108![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–116000 cm−1. Tracing the quantum dynamics in a bunch of electronic states enables us to identify the key components that determine the efficacy of singlet to triplet population transfer and therefore predissociation lifetimes and branching ratios for different energy regions.
000–116000 cm−1. Tracing the quantum dynamics in a bunch of electronic states enables us to identify the key components that determine the efficacy of singlet to triplet population transfer and therefore predissociation lifetimes and branching ratios for different energy regions.
Introduction
Space missions and radio astronomy can analyze the composition of upper levels of Earth's atmosphere, the atmospheres of other bodies in space and the interstellar medium. Up to nowadays, more than 200 individual molecular species from diatomic H2 and N2 to polyatomic Buckminsterfullerene and Rugbyballene were identified in space.1,2 These recent discoveries have formed the basis for astrochemistry, the study of molecular structure and processes in the interstellar medium.3–5The molecules that have been identified in space are composed of 19 different elements, but carbon, hydrogen, nitrogen, and oxygen predominate. Despite the diversity of space chemistry, the main components of interstellar medium are H2 and CO and only in the atmospheres of Earth, Pluto, Titan, and Triton have significant amounts of N2 been detected. Its high chemical stability makes N2 the most abundant nitrogen-containing molecule in the interstellar medium. However, the astrochemistry of nitrogen is diverse and rich: there are 92 different nitrogen-containing species1 from diatomic CN (cyano radical)6–8 and NO (nitric oxide)9,10 to triatomic CaNC (calcium isocyanide)11 and polyatomic CH2NH (methanimine)12,13 and (NH2)2CO (urea).14 This has resulted in a great interest in the chemistry of N2 in Earth's and Titan's atmospheres, where N2 molecules are the major constituent.15–19 A combination of lab experiments in plasma and computer simulations shows possible reactions of the activated forms of N2 with CH4 and C2H4 in Titan's atmosphere.20–22 To understand the mechanism of these reactions and their complexity, one should have a closer look at the electronic structure of N2.
The nitrogen molecule in its ground electronic state is a textbook example of high chemical stability: under standard conditions on Earth, its triple covalent bond is believed to be among the strongest known in nature. At the same time, nitrogen-containing molecules – purines, pyrimidines, pyrroles and indoles – play the essential role in life, and it is important to find out how the inert N2 molecule can be converted in nature into more chemically active forms. Early studies proposed a possible active nitrogen:23,24 N2 in its vibrationally excited ground state X1Σ+g and in the metastable electronically excited A3Σ+u state or excited-state N atoms. The optically excited states of N2 are high in energy: from 100000 to 120000 cm−1 (from 12.4 to 14.9 eV) relative to its ground state. Only in upper levels of Earth's atmosphere, which are not shielded by the ozone layer, the sunlight covers this energy range and nitrogen molecules can be excited and then dissociate into nitrogen atoms in their ground and excited states. The chemical reactivity of the resulting nitrogen atoms is not equal. The ground-state N(4S) is known to be rather inert while the excited atoms are chemically active and even further, among N(2P) and N(2D) the latter is known to be much more reactive.25–27 Today, excited N(2D) atoms are believed to play the principal role in reactions with H2 and hydrocarbons and forming HCN, C2N2 and other compounds in low-pressure space environments.28–32
The first step in generation of chemically active nitrogen atoms is the optical excitation of the molecule in the VUV spectral region. The absorption spectrum of N2 from 100000 to 120000 cm−1 was extensively studied both experimentally33,34 and theoretically.35 Following classification based on selection rules, two types of one-photon optically accessible singlet states are possible for excitation from the electronic ground state: 1Σ+u and 1Πu. These states are reached by a parallel or a perpendicular orientation of the molecule relative to the light field. They are in principle coupled by L-uncoupling interaction which is negligible for low rotational levels, so these states of different symmetry practically do not interact with one another. However, states of the same symmetry and multiplicity can be coupled by a rather strong nonadiabatic terms and so the electronic states of either 1Σ+u or 1Πu symmetry can be mixed among themselves. In the extensively studied lower energy region these states are bound.
In the energy range of interest here, the way to dissociation is through spin–orbit induced transfer from the singlets to the states of higher multiplicity which are dissociative. The photodissociation of N2 has been experimentally studied by, first, electron impact36 and further with fast-beams37–39 and wavelength selected VUV excitation.40,41 The latter was with special reference to isotopic selectivity. The most recent detailed work uses supersonic molecular beams coupled with velocity-mapped imaging method.42–45 In the energy range up to 120000 cm−1, there are three dissociation limits which lead to excited-state N atoms in the following states at the given thresholds:
N(4S) + N(2D), E > 12.143 eV (97938 cm−1)
N(4S) + N(2P), E > 13.335 eV (107553 cm−1)
N(2D) + N(2D), E > 14.526 eV (117162 cm−1)
The experiments consistently exhibit strong energy dependence in the photodissociation branching fractions: different predissociative N2 states with similar internal energies are found to follow different dissociation pathways, thereby suggesting that the unimolecular dissociation is not statistical.
Experimental studies provide the accurate information on the final dissociation products but do not explain the origin of their energy dependence. Here theoretical approaches can be essential. High-level ab initio quantum chemistry methods allow a close examination of a group of electronic states of comparable energies so that one can simulate a realistic dependence of the potential energy of different electronic states on changes in the molecular bond distance. Quantum chemical calculations have accompanied experiments in interpretation of the electronic structure of N2 in many directions: vibronic states energies and composition,46,47 nature of electronic transitions48,49 and band complexes.50,51 The potential energy curves were studied in detail52–59 as well as the spin–orbit56 and nonadiabatic couplings.60 Nowadays the quantum dynamical approaches61–64 have reached a level where they can offer a computational tool which allowed us to follow the time-evolution of an electronic wave packet travelling in the forest of electronic states, beyond the states directly accessed from the ground electronic state.65
The approach has been applied to explore the mechanism of dissociative ionization of N266–68 and ultrafast electronic excitation and dissociation.69–72 We recently computed a realistic energy dependence in the dissociation branching arising from excitation of different single vibrational levels of 1Σ+u electronic states of N2.65,73 These computations started from the molecule in the ground electronic state and excited it by a polarized laser pulse of a very long duration and of the required mean energy. In the present work we make a step further and present the results of the branching into the channels N(4S) + N(2D) (Channel 1) and N(4S) + N(2P) (Channel 2) accessed through the excited 1Πu states of N2. We highlight the differences in the results for these two distinct doorway excited states. First, following excitation of 1Σ+u states there is an early opening of Channel 2 at 110000 which is absent for the 1Πu singlets. Also, from 112000 to 115000 cm−1 there are oscillating changes in branching for 1Πu while for 1Σ+u the change is smooth. For both types of the singlets, a drop is present at 115000 cm−1, but then for excited 1Σ+u there is a preferable branching fraction into Channel 2 while for 1Πu the preference is for dissociation into Channel 1.
The manuscript is organized as follows: first, we review the selection rules relevant to the one-photon excitation and dissociation of N2 and the computational scheme that we use for the simulations. Second, we compare the dissociation branching starting from excited 1Σ+u or 1Πu states and we discuss the predissociation lifetime and isotope effect on the dynamics and branching. We conclude with a prospective outlook on excited state dissociative dynamics in the N2 molecule in connection with reaction dynamics in the upper atmosphere and in the interstellar medium.
Computational details
Selection rules relevant for the one-photon excitation of N2
The 14N2 molecule belongs to D∞h point group. This means the presence of inversion through a center of symmetry and infinite number of horizontal mirror planes intersecting the principal symmetry Z axis along the N–N bond. Therefore, all the electronic states can be classified either “gerade” or “ungerade” relative to the inversion operation. In the present paper, we discus one photon VUV excitation of N2 from its ground state X1Σ+g. The dipole allowed transitions are those with the change of parity and so only 1Σ+u and 1Πu can be optically accessed. The symmetry of the final state depends on the orientation of the molecule relative to the laser: if the molecule aligns along X/Y axis, 1Πu states will be reached and if it aligns along Z axis – 1Σ+u states. These singlet states are coupled to other “ungerade” states of higher multiplicity via spin–orbit coupling.There are three Cartesian components of the spin–orbit coupling integrals that differ along X, Y and Z axes in the molecular frame: LSX, LSY, LSZ. According to their orbital momentum Λ, all the states can be labelled as Σ, Π, Δ etc. Triplet and quintet electronic states are degenerate states with different Sz projection (ms = −1, 0, 1 for triplets and ms = −2, −1, 0, 1, 2 for quintets). The selection rules of the spin–orbit coupling play a key role in the interconnections of manifolds of states of different multiplicities. Two states with ΔΛ = 0 are coupled by the Z component of the spin–orbit coupling, that we denote as LSZ and these two interacting states must have the same magnetic quantum number, Δms = 0. Two states with ΔΛ = ±1 are coupled by the LSX/LSY component of the coupling and must differ as Δms = ±1. The implication is that while both singlet 1Σ+u and 1Πu electronic states can interact with the triplets, the triplet states that they couple to are different. 1Σ+u singlet states are coupled by LSX/LSY component of the spin–orbit interaction to the 3Πu states and only mS = ±1 of the triplet states can be populated. 1Πu are coupled by LSZ spin–orbit interaction to the 3Πu states and for this type of coupling the selection rule is ΔmS = 0 and so mS = 0 of the triplet states can be populated. Similar considerations apply to coupling to the quintets: LSZ spin–orbit interaction between 3Πu and 5Πu and LSX/LSY spin–orbit coupling between 3Πu and 5Σ+u, 5Σ−u and 5Δu. Thus, considering different options for the magnetic quantum number, all the electronic states included into the Hamiltonian can be divided into two groups shown in Fig. 1 and therefore, a singlet state of 1Σ+u or 1Πu symmetry exits to the dissociative channels in a different manner. A more detailed scheme shown in Fig. S1 of ESI.†
 | ||
| Fig. 1 Schematic representation of spin–orbit coupling between the electronic states depending on their magnetic quantum number mS (indicated in parenthesis). There are two types of dipole-excited states shown in red: 1Σ+u and 1Πu singlet states. They are directly coupled to the specific triplet states shown in green (1st order spin–orbit coupling). The last ones are further linked to the electronic states depicted in blue (2nd order spin–orbit coupling). | ||
The states of the same symmetry and multiplicity are coupled by nonadiabatic interaction, and they can exchange population among themselves in the avoided crossings regions. We have included the nonadiabatic coupling among 1Σ+u, 1Πu, 3Πu states. More details on selection rules can be found in ref. 74.
Quantum chemical calculations
We consider the states in energy range 90000–120000 cm−1 where the dissociation channels 1 and 2 are open, see Fig. 2. Thus, the electronic basis we use consist of the ground X1Σ+g state and the following excited electronic states: three 1Σ+u, three 1Πu, one 1Σ−u, one 1Δu, one 3Σ+u, one 3Σ−u, four 3Πu, one 3Δu, one 5Σ+u, one 5Σ−u, two 5Πu and one 5Δu. 21 electronic states in all, and accounting for their magnetic degeneracy a total of 55 quantum electronic states. We represent the Hamiltonian in the adiabatic form and so the electronic states are labeled according to their energy. The reader should note that this classification is different from diabatic labeling commonly used in spectroscopy, however, diagonalizing the Hamiltonian in any given basis provides the same energy of the vibronic states. The following discussion will be given in the adiabatic representation which is the direct output of quantum chemical calculations.
 | ||
| Fig. 2 Potential energy curves of “ungerade” electronic states of the nitrogen molecule in energy range 90000–120000 cm−1. The colored singlets are the states that can be VUV optically accessible by one photon from the “gerade” ground electronic state. 1Σ+u singlet states are shown in red and 1Πu are in blue. Other singlet states are shown with solid black lines, the triplet states with dashed lines and quintets with dotted lines. The lowest dissociative channels are in blue and orange. | ||
The terms of the electronic Hamiltonian (electronic potential energy curves, nonadiabatic and spin–orbit couplings) of N2 in the VUV energy range are calculated for each value of internuclear distance using a state-averaged complete active space self-consistent field (CASSCF) approach followed by multi-reference configuration interaction (MRCI)75,76 calculations. This methodology was successfully applied to describe the electronic structure of N2 and its cation multiple times.49,50,56,57,68,77 An active space of 17 orbitals (4σu, 3σg, 4πu, 4πg, and 2δg) for 10 valence electrons is employed in all computations. The two lowest 1σu and 1σg orbitals are not included in the active space but are fully optimized in the CASSCF procedure. Restriction of only single occupancy for the higher Rydberg orbitals was used. We chose a doubly augmented cc-pVQZ basis set with additional bond-centered s and p diffuse functions for a proper description of Rydberg and valence singlet and triplet states according to the previously established methodology.35 All the quantum chemistry calculations are performed with the MOLPRO program package.78,79 The computed potential energy curves are shifted by 850 cm−1 to be in accordance with previously published data.35
The potential energy curves, nonadiabatic coupling and dipole moments of 1Πu states were taken in analytical form from ref. 35 and adiabatized as explained in ref. 65. The derived adiabatic data along with presently computed spin–orbit coupling elements are given in Fig. S1–S8 of the ESI.†
Nonadiabatic quantum dynamics
We use the time-dependent Schrödinger equation of motion to compute quantum dynamics on a grid of internuclear distance for the 54 excited and 1 ground electronic states of definite multiplicity and magnetic quantum number. The equation of motion is defined for the amplitudes Cng = Ψ(Rg) at a given electronic state n and grid point g for the internuclear distance R = Rg, see ref. 80. Diagonal and off-diagonal kinetic energy terms were evaluated using a five-point finite difference approximation.81 At large internuclear distances, R > 6.2 a.u., a complex absorbing potential, VCAP(R) = 0.01(R − 6.2)3, is applied for all the dissociative states. The nonadiabatic couplings τnk(R) between electronic states n and k are scaled by the finite difference momentum terms. The propagation of the equation of motion is solved via the Runge–Kutta method with the time step Δt = 10−4 fs and ΔR = 0.005 a.u. for the grid spacing. The propagation lasted for 5 ps in all the dynamical calculations. The lifetimes of the excited vibrational singlet states are estimated by a linear fit for the logarithm of their time-dependent population, |Cng(t)|2, assuming a unimolecular exponential decay.The interaction with the light field is governed by the transition dipole moment μnk(R) between the ground X1Σ+g and excited singlet electronic states. An explicit time-profile for the VUV light field is used with a very long duration
 | (1) |
Here εp is the polarization direction of the light field, set along the internuclear axis so as to access the 1Σ+u states and perpendicular to the internuclear axis to reach the 1Πu states. Emax is the maximum amplitude of the field; tp and σp are the time at which the pulse is centered and the width of the Gaussian envelope of the field.
Duration of the pulse is set to be long enough to selectively excite specific vibrational levels of the singlet states, σp = 160 fs, tp = 1200 fs and Emax = 0.0001 a.u. The carrier frequency ωp is varied from 108000 to 116000 cm−1 according to the energy of the vibrational levels obtained by diagonalization in the singlets manifold. The choice of this energy range is determined by the thresholds of two dissociation channels.
Given all the above information, the Hamiltonian can be represented as a sparse square matrix, as shown in Fig. 3. The potential energies of the electronic states form diagonal elements, and the various coupling terms are off-diagonal ones. These terms can link states of the same symmetry and multiplicity (nonadiabatic or spin–orbit couplings, shown in light-red, light-green, and light-blue) or states of different multiplicity (yellow and cyan off-diagonal blocks).
 | ||
| Fig. 3 A schematic representation of the Hamiltonian which includes the ground electronic state X1Σ+g, singlet 1Πu, triplet 3Πu and quintet 5Πu. The singlet 1Πu states are coupled by X/Y components of the transition dipole moments with the ground state X1Σ+g (the dark-grey block). In the calculations, such a block is comprised by more excited states, and it is formed for each internuclear N–N grid point from 1.0 to 7.5 a.u. with the grid spacing of 0.005 a.u., 1301 blocks in total for each time point along 5 ps propagation. The same structure of Hamiltonian is applied to the singlet 1Σ+u states which are coupled by Z components of the transition dipole moments with the ground state X1Σ+g. | ||
Results and discussion
VUV optically excited and dissociative states of N2
The N2 molecule dissociates indirectly and the excited 1Σ+u and 1Πu singlets are the (rather narrow) doorways in the population transfer from the ground X1Σ+g state to the dissociative electronic states.82 For both types of the singlets, there are two bound (Rydberg-type) and one delocalized (valence-type) states. However, the 1Πu states all are lower-lying and are stronger mixed by nonadiabatic interaction in the energy range of 100000–107000 cm−1 while the 1Σ+u states are more energetically separated from one another and from 23Πu and 33Πu states, see Fig. 4(a) and 5(b).
 | ||
| Fig. 4 Potential energy curves of singlet (a), triplet (b) and quintet (c) multiplicity color-coded according to the symmetry of the states: 1Σ+u, 3Σ+u and 5Σ+u are in orange-red; 1Σ−u, 3Σ−u, 5Σ−u are in light-blue; 1Πu, 3Πu, 5Πu are in mint green; 1Δu, 3Δu, 5Δu are in magenta. | ||
 | ||
| Fig. 5 (a) Potential energy curves of 1Σ+u (in black) and 3Πu (color-coded in the figure) and the absolute values of the spin–orbit coupling terms between them; (b) 1Πu and (in black) and 3Πu (color-coded in the figure) and the spin–orbit coupling terms between them. Sudden changes in the spin–orbit coupling curves determined by changes in the character of the adiabatic triplet 3Πu states at avoided crossings at 2.6, 3.5, 3.9 a.u. | ||
Triplet 3Πu states are believed to be the main doorway to the single photon VUV dissociation of the N2 molecule in the energy below 116000 cm−1. Three distinct dissociation channels can be seen in the energy range from 90000 to 120000 cm−1 and two lower ones are labeled as “Channel 1” and “Channel 2” in Fig. 2. The triplet states are composed of bound parts when the internuclear distance, R, is shorter than 2.7 au, the repulsive one from R > 3.5 a.u. and dissociative ones, R > 6 a.u. Among the other triplets, 23Σ+u also has a distinctive structure: in the short-range distances, it is very energetically close to the singlet 1Πu state and mirrors its shape, Fig. 4(b). The other triplets and quintet states are primarily repulsive: there are no avoided crossings or other distinctions in their manifold, Fig. 4(b) and (c).
The link between the excited states of different multiplicity is the spin–orbit coupling. The shape of the potentials predominantly affects the magnitude and sign of the spin–orbit induced transfer from the singlet to the triplet states. Fig. 5 shows the results of quantum chemical calculations for the spin–orbit coupling terms between 1Σ+u and 3Πu (panels a and c) and 1Πu and 3Πu states (panels b and d). All three lowest 1Πu adiabatic states are stronger connected to the 3Πu states: the coupling of 42 cm−1 against of 30 cm−1 in the case of 1Σ+u–3Πu. The delocalized 11Πu state exits to the higher N(2D) + N(2D) channel and so is coupled to 33Πu even in the region of its avoided crossing at 4 a.u.
The singlets are also coupled to other triplet states: 1Σ+u can interact with 3Σ−u while 1Πu interacts with 3Σ+u, 3Σ−u and 3Δu. However, our computations provide rather small couplings among the states mentioned above and the largest is of 12 cm−1 between 11Πu and 23Σ+u states, see Fig. S5–S8 of the ESI.†
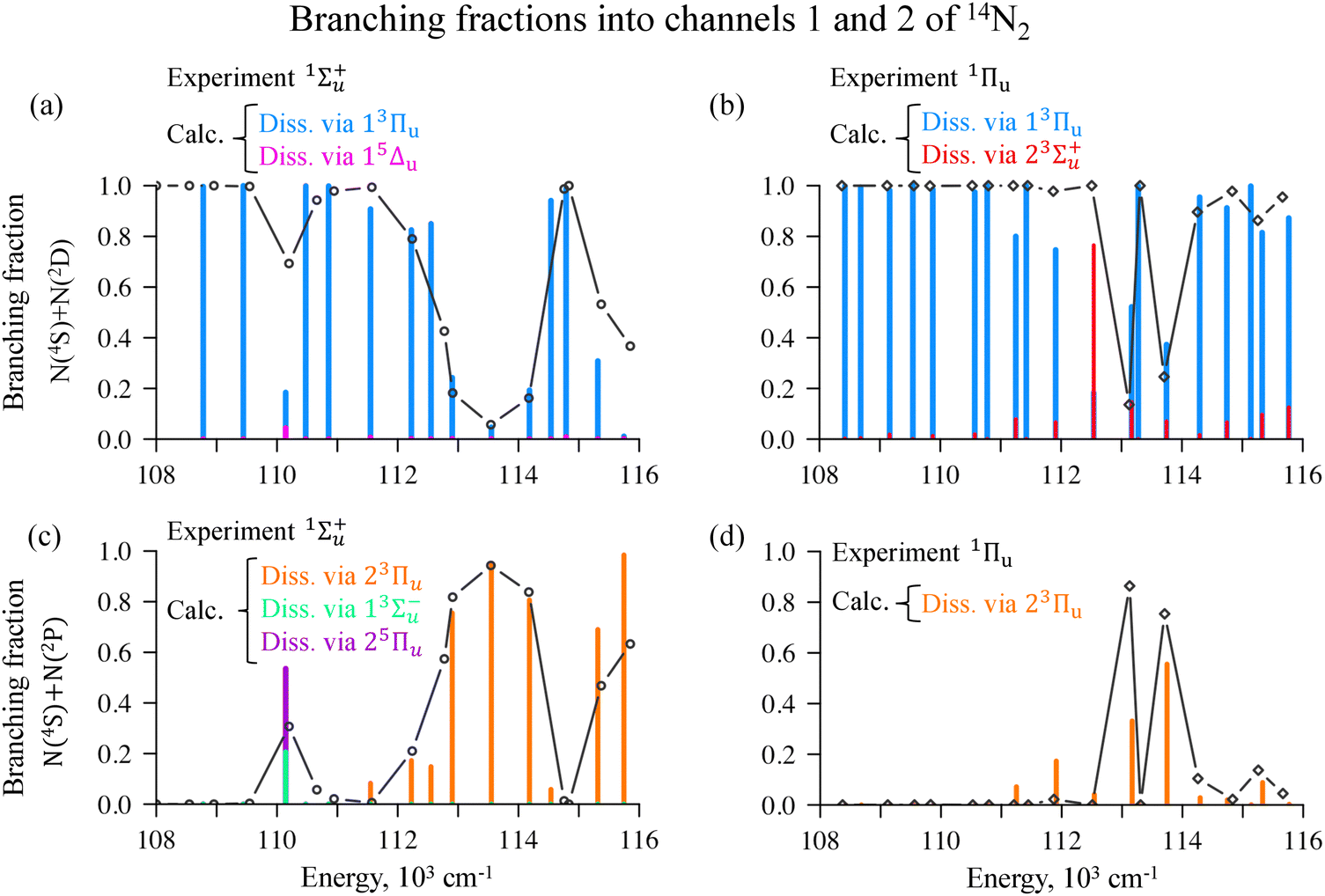 | ||
| Fig. 6 Experimental (black line)42 and calculated (histograms color-coded in the figure) branching fractions in Channel 1 (panels a and b) and in Channel 2 (panels c and d) obtained by exciting vibronic levels of 1Σ+u (panels a and c) or 1Πu (panels b and d) states of the 14N2 molecule. | ||
(1) In the case of 1Σ+u, there is an early opening of Channel 2 at 110000 cm−1 which is absent for 1Πu;
(2) Above the avoided crossing between 23Πu and 33Πu where the triplet repulsive potential is available (from 112000 to 115000 cm−1) there are oscillating changes in branching for 1Πu and a smooth change for 1Σ+u;
(3) In both cases, a drop is present at 115000 cm−1, but then for excited 1Σ+u there is a preferable branching into Channel 2 while for 1Πu the branching is favored into Channel 1.
To investigate an intricate pathway of electronic subsystem on its way from excitation of a particular vibronic level of singlets to dissociation, we performed quantum dynamical calculations on a grid of the N–N internuclear coordinate. These calculations are based on the results of ab initio quantum chemical computations at CASSCF/MRCI level. The states of triplet and quintet multiplicity of u parity from 90000 to 120000 cm−1 were included in the Hamiltonian, more details are given in section “Computational details”.
Fig. 6 shows the computed branching fractions into Channel 1 and Channel 2 versus the experimental data. The calculations have captured key features of the experiment and therefore allow us to deliver a detailed explanation on the questions pointed above.
Both Channel 1 and Channel 2 are accessible in the energy range from 108000 to 116000 cm−1 and, statistically, up to electronic degeneracies, one would expect to see the dissociation primarily into the lowest in energy channel. However, at some energies the molecule dissociates into the higher exit. Fig. 6 shows the branching into Channel 1 in panels (a) and (b) for 1Σ+u and 1Πu, respectively. Various electronic states of triplet and quintet multiplicity feed the channels, and all the significantly contributing states (more than 5% of the total dissociation) are depicted in Fig. 6. The major contributor to the dissociation is the 3Πu triplet states: 13Πu leads to Channel 1 (the blue columns in Fig. 6) and 23Πu leads to Channel 2 (the orange columns in Fig. 6). When 1Σ+u states are excited, 13Σ−u, 13Δu and 25Πu contribute at 110000 cm−1 only, Fig. 6(c). When 1Πu states are excited, 23Σ+u plays a major role due to a higher spin–orbit coupling comparing with 13Σ−u and 13Δu (Fig. S7 of ESI†).
Channel 1 is preferable at lower energies almost up to 113000 cm−1 with an exception at 110000 cm−1 exclusively for the excited 1Σ+u. At the higher energies, the picture becomes more complicated due to a competition between the channels. To understand the origin of these oscillations, one should look closely at the dynamics of the wave packet. Employing ultraslow laser pulses, which are narrow in energy, one can reach a particular vibronic level and so the population will not redistribute after the pulse is over but will slightly decrease with time due to a slow dissociation. There is a discussion on this in ref. 65, 83 and 84. This allows us to examine the wave packet redistribution when the pulse is already over, but the molecule is far from being fully dissociated, as we show in the snapshots of the dynamics at 5 ps in Fig. 7. We have selected four energy levels for both 1Σ+u and 1Πu where drastic changes can be seen in the branching ratio. Let us first note the similarity between the branching starting from the vibronic states of the two singlets, 1Σ+u are presented in the top row and 1Πu in the bottom row in the Fig. 7.
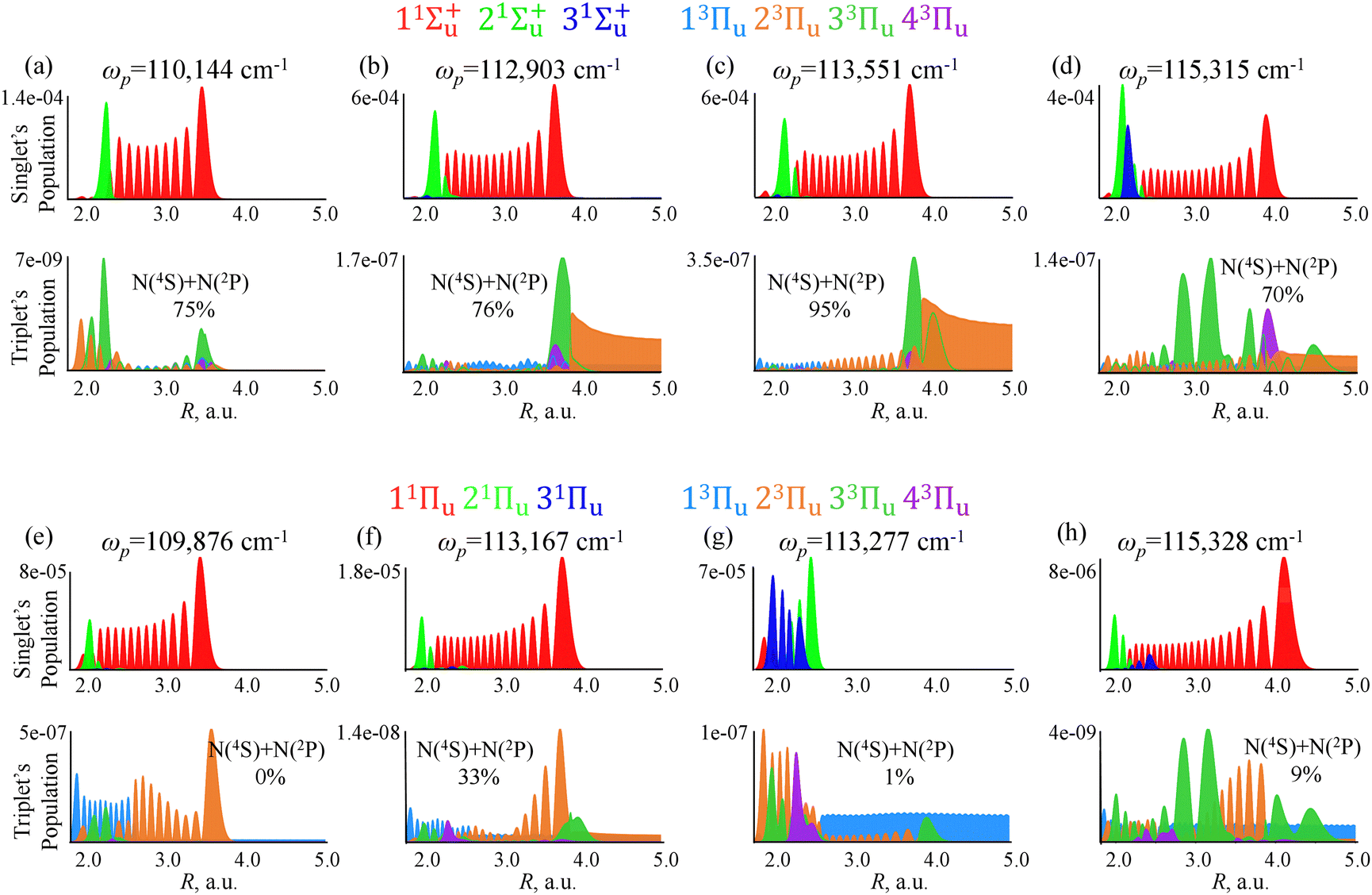 | ||
| Fig. 7 Population redistribution in the singlet and triplet manifolds at 5 ps obtained by exciting vibronic levels of 1Σ+u (panels a–d) or 1Πu (panels e–h) states of the 14N2 molecule. ωp is the carrier frequency of the exciting pulse (eqn (1)). Top two rows show population of four selected 1Σ+u vibronic singlet and correspondingly populated triplet 3Πu states. Each column compares the dynamics for the two singlets at similar energy range. The branching fraction into N(4S) + N(2P) (Channel 2) is indicated in the figure for each energy. | ||
There are clear differences between the two singlet sates. Under excitation of 1Σ+u at 110144 cm−1, Channel 2 opens. As one can see in Fig. 7(a), the population of the triplets 3Πu is very low and so even other low populated states become important. In a previous study,73 we proposed that the reason of this low population is the notably long lifetime of the excited singlet state.
The next difference in the branching is a rapid change at 113277 cm−1 for 1Πu. The upper panel in Fig. 7(g) shows the singlet vibronic state 1Πu which is of Rydberg type. Unlike Rydberg 21Σ+u and 31Σ+u states, localized 21Πu and 31Πu are strongly coupled to 3Πu triplets at short distances (see the bottom panel of Fig. 5(d)), so 1Πu Rydberg singlet state is effectively populating the bound triplet, see the bottom panel in Fig. 7(g).
The third difference is at 115300 cm−1 where 1Σ+u occurs to be in resonance with the repulsive 3Πu potential while 1Πu is primarily coupled to the bound 3Πu. However, both singlets contribute to Channel 2 at 113000 cm−1 which is just above the avoided crossing between 23Πu and 33Πu states.
The principal similarity of the two singlet states is the dissociation into Channel 2 at 113000 cm−1: the repulsive triplet 3Πu is accessible here for both singlets and the prompt dissociation occurs (Fig. 7(b) and 7(f)). At other lower energies, Channel 1 is almost always preferable and also at 115000 cm−1 when the triplet is trapped (a rapid decrease at 115000 cm−1 in Fig. 6(c) and (d)).
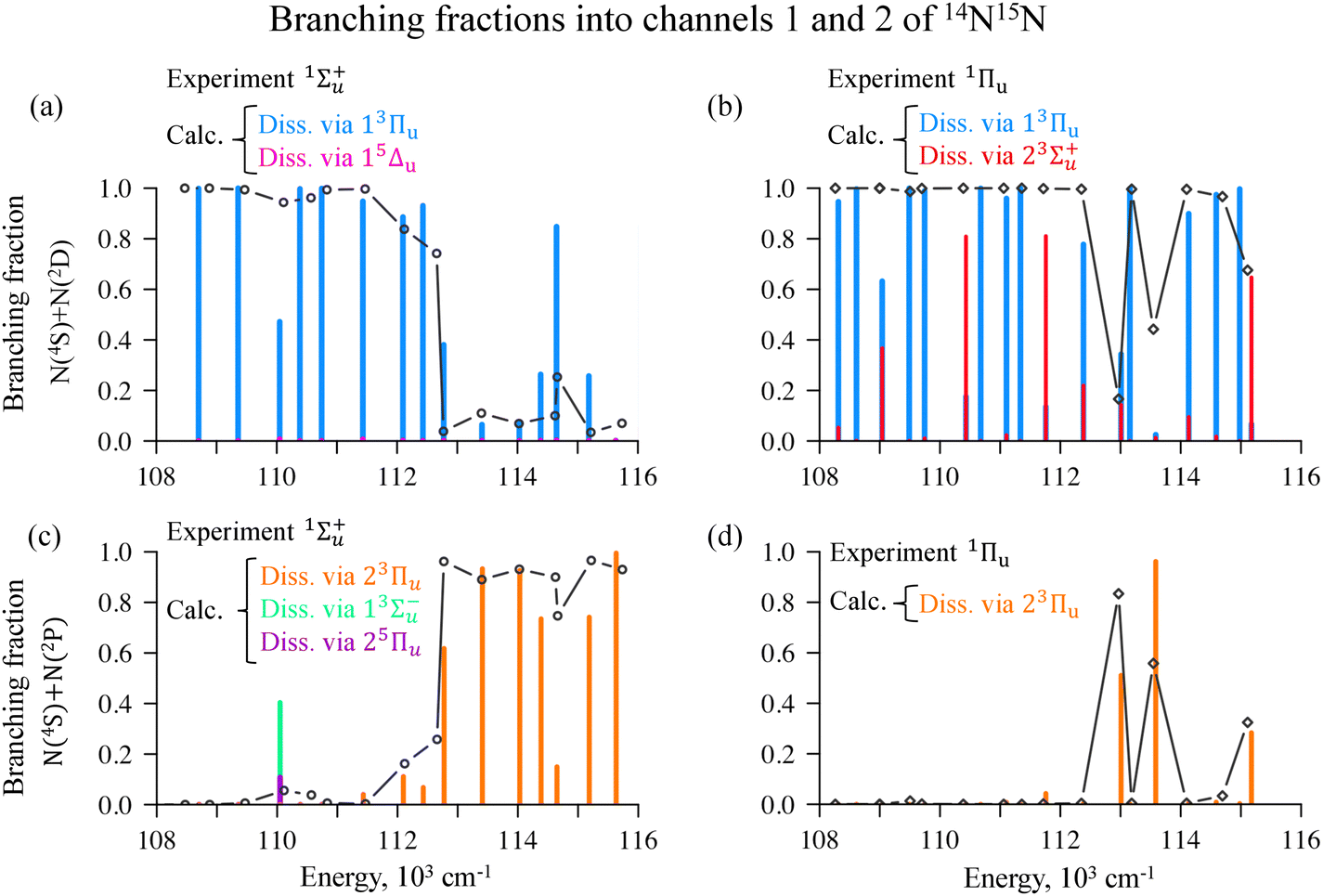 | ||
| Fig. 8 Experimental (black line with circles)43 and calculated (histograms are color-coded in the figure) branching fractions in Channel 1 (panels a and b) and in Channel 2 (panels c and d) obtained by exciting vibronic levels of 1Σ+u (panels a and c) or 1Πu (panels b and d) states of the 14N15N molecule. | ||
Another characteristic of the photodissociation is the predissociation singlet lifetime which indicates how fast or slow the initial wave packet will be transferring from the excited singlet state to the dissociation product channel via the triplets.
Fig. 9 presents the calculated lifetimes for both 1Σ+u (Fig. 9a) and 1Πu (Fig. 9b) of the two isotopomers. A key result is that the Y-axes is ten-times larger for 1Σ+u: because of a weaker coupling, these states dissociate much more slowly. In particular, at the specific energy of 110144 cm−1 the singlets 1Σ+u will take much longer to decay.
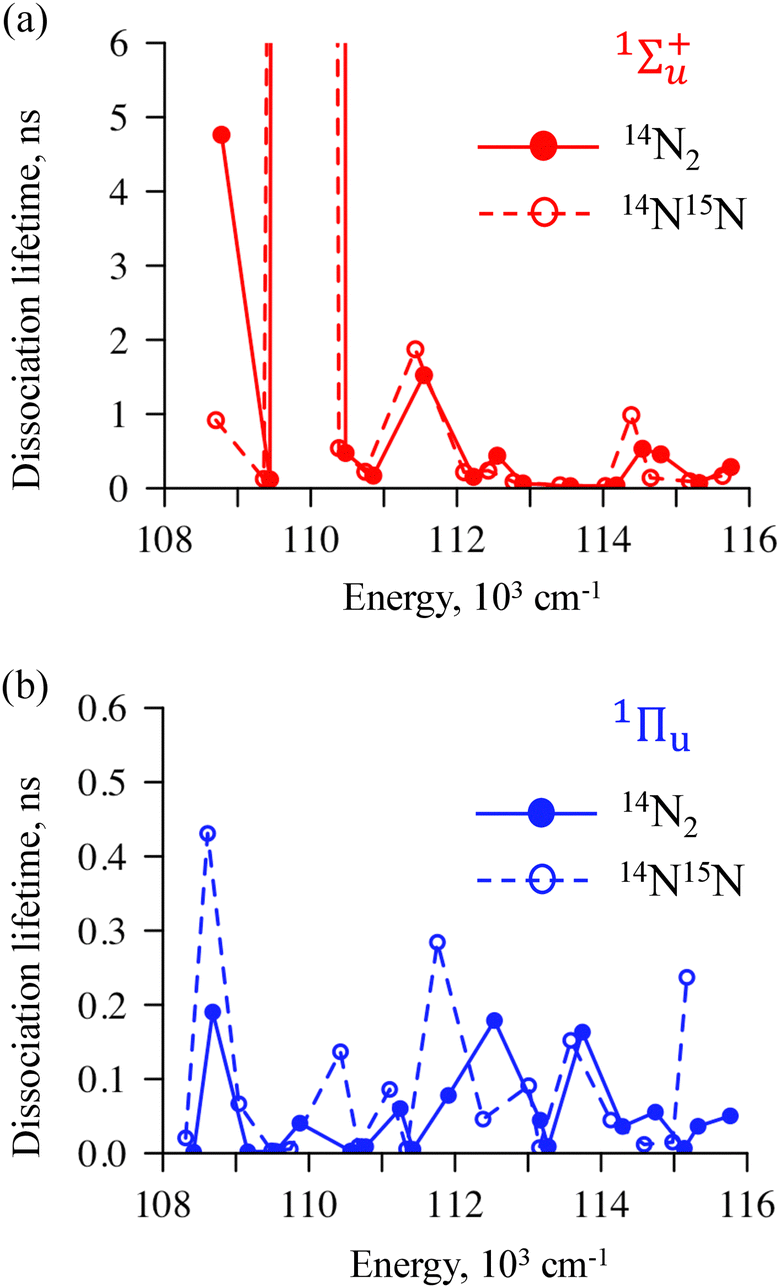 | ||
| Fig. 9 Calculated predissociation lifetimes obtained by exciting vibronic levels of 1Σ+u (panel a) or 1Πu (panel b) states of the 14N2 (full circles) or 14N15N (empty circles) molecule. | ||
Conclusions
Photodissociation is the most direct way to obtain a chemically reactive nitrogen, however, the atomic composition of dissociation products cannot be easily predicted if more than one product channel is accessible. Combining the results of accurate experiments and state-of-art computer modelling, a better understanding of energy-dependence in the dissociation of N2 and its fractionation mechanism has been achieved. Our simulations show that the character of singlets, valence or Rydberg, and the strength of their spin–orbit coupling to the triplets determine the efficiency of population transfer and therefore predissociation lifetime. As a result of stronger coupling, excited 1Πu states dissociate much faster than 1Σ+u. However, we also found the repulsive shape of the triplet 3Πu potentials is in control of the prompt dissociation from 112000 to 115000 cm−1. For some energies, contribution of other states – 23Σ+u, 3Σ−u, 25Πu – have been found significant.
Therefore, the route to producing reactive N(2D) is not straightforward and the fraction will be especially low when it is the vibronic levels of 1Σ+u above 115000 cm−1 that are excited. The somewhat lower in energy vibronic states of 1Πu symmetry are on the contrary better coupled to the bound 3Πu triplets and dissociative 23Σ+u which allows N(2D) to be the more favourable product. Shielding of the VUV light in the upper atmosphere can therefore play a major role in the preferential formation of the reactive N(2D) atoms.
Author contributions
NG (conceptualization; formal analysis; investigation; visualization; writing – original draft; writing – review & editing), KK (formal analysis; investigation; writing – review & editing), FR (formal analysis; investigation; writing – review & editing), RDL (conceptualization; formal analysis; funding acquisition; investigation; supervision; writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support of the US–Israel NSF–BSF grant no. 2019722. F. R. acknowledges the support of the Fonds National de la Recherche (F. R. S.-FNRS, Belgium), grant no. T0205.20 and COST action ATTOCHEM (CA18222).References
- B. A. McGuire, Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules, Astrophys. J., Suppl. Ser., 2021, 2022(259), 30 Search PubMed.
- J. Cami, J. Bernard-Salas, E. Peeters and S. E. Malek, Detection of C60 and C70 in a Young Planetary Planetary Nebula, Science, 2010, 329, 1180–1182 CrossRef CAS PubMed.
- D. A. Williams and C. Cecchi-Pestellini, Astrochemistry: Chemistry in Interstellar and Circumstellar Space, Royal Society of Chemistry, 1st edn, 2023, p. 248 Search PubMed.
- E. A. Bergin, W. D. Langer and P. F. Goldsmith, Gas-phase chemistry in dense interstellar clouds including grain surface molecular depletion and desorption, Astrophys. J., 1995, 441, 222–243 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, Atmospheric Chemistry of Titan: Ab Initio Study of the Reaction between Nitrogen Atoms and Methyl Radicals, J. Am. Chem. Soc., 1992, 114, 9118–9122 CrossRef CAS.
- A. McKellar, Evidence for the molecular origin of some hitherto unidentified interstellar lines, Astron. Soc. Pac., 1940, 52, 187–192 CrossRef CAS.
- W. S. Adams, Some Results with the COUDÉ Spectrograph of the Mount Wilson Observatory, Astrophys. J., 1941, 93, 11 CrossRef CAS.
- K. B. Jefferts, A. A. Penzias and R. W. Wilson, Observation of the CN Radical in the Orion Nebula and W51, Astrophys. J., 1970, 161, 87–89 CrossRef.
- J. J. Gallagher and C. M. Johnson, Uncoupling Effects in the Microwave Spectrum of Nitric Oxide, Phys. Rev., 1956, 103, 1727–1737 CrossRef CAS.
- H. S. Liszt and B. E. Turner, Microwave Detection of Interstellar NO, Astrophys. J., 1978, 224, 73–76 CrossRef.
- J. Cernicharo, L. Velilla-Prieto, M. Agúndez, J. R. Pardo, J. P. Fonfría, G. Quintana-Lacaci, C. Cabezas, C. Bermúdez and M. Guélin, Discovery of the first Ca-bearing molecule in space: CaNC, Astron. Astrophys., 2019, 627, L4 CrossRef CAS PubMed.
- D. R. Johnson and F. J. Lovas, Microwave Detection of the Molecular Transient Methyleneimine (CH2 = NH), Chem. Phys. Lett., 1972, 15, 65–68 CrossRef CAS.
- P. D. Godfrey, R. D. Brown, B. J. Robinson and S. M. W. Discovery, of Interstellar Methanimine (Formaldimine), Astrophys. Lett., 1973, 13, 119–121 CAS.
- A. Belloche, R. T. Garrod, H. S. P. Müller, K. M. Menten, I. Medvedev, J. Thomas and Z. Kisiel, Re-exploring Molecular Complexity with ALMA (ReMoCA): Interstellar detection of urea, Astron. Astrophys., 2019, 628, A10 CrossRef CAS.
- V. Vuitton, R. V. Yelle and V. G. Anicich, The Nitrogen Chemistry of Titan's Upper Atmosphere Revealed, Astrophys. J., 2006, 647, 175–178 CrossRef.
- C. He and M. A. Smith, Identification of nitrogenous organic species in Titan aerosols analogs: Nitrogen fixation routes in early atmospheres, Icarus, 2013, 226, 33–40 CrossRef CAS.
- C. He and M. A. Smith, Identification of nitrogenous organic species in Titan aerosols analogs: Implication for prebiotic chemistry on Titan and early Earth, Icarus, 2014, 238, 86–92 CrossRef CAS.
- V. Vuitton, R. V. Yelle, S. J. Klippenstein, S. M. Hörst and P. Lavvas, Simulating the density of organic species in the atmosphere of Titan with a coupled ion-neutral photochemical model, Icarus, 2019, 324, 120–197 CrossRef CAS.
- J. Cui, R. V. Yelle, V. Vuitton, J. H. Waite, W. T. Kasprzak, D. A. Gell, H. B. Niemann, I. C. F. Müller-Wodarg, N. Borggren, G. G. Fletcher, E. L. Patrick, E. Raaen and B. A. Magee, Analysis of Titan's neutral upper atmosphere from Cassini Ion Neutral Mass Spectrometer measurements, Icarus, 2009, 200, 581–615 CrossRef CAS.
- N. Balucani, D. Skouteris, F. Leonori, R. Petrucci, M. Hamberg, W. D. Geppert, P. Casavecchia and M. Rosi, Combined crossed beam and theoretical studies of the N(2D) + C2H4 reaction and implications for atmospheric models of titan, J. Phys. Chem. A, 2012, 116, 10467–10479 CrossRef CAS PubMed.
- N. Balucani and D. Skouteris, in Prebiotic Photochemistry: From Urey–Miller-like Experiments to Recent Findings, ed. F. Saija and G. Cassone, The Royal Society of Chemistry, 1st edn, 2021, pp. 37–59 Search PubMed.
- C. He, J. Serigano, S. M. Hörst, M. Radke and J. A. Sebree, Titan Atmospheric Chemistry Revealed by Low-Temperature N2-CH4Plasma Discharge Experiments, ACS Earth Sp. Chem., 2022, 6, 2295–2304 CrossRef CAS.
- R. Brown and C. A. Winkler, The Chemical Behavior of Active Nitrogen, Angew. Chem., Int. Ed. Engl., 1970, 9, 181–196 CrossRef CAS.
- J. T. Herron, Evaluated Chemical Kinetics Data for Reactions of N(2D), N(2P), and N2(A3Σu +) in the Gas Phase, J. Phys. Chem. Ref. Data, 1999, 28, 1453–1483 CrossRef CAS.
- T. Suzuki, Y. Shihira, T. Sato, H. Umemoto and S. Tsunashima, Reactions of N(22D) and N(22P) with H2 and D2, J. Chem. Soc., Faraday Trans., 1994, 89, 995–999 RSC.
- R. J. Donovan and D. Husain, Recent advances in the chemistry of electronically excited atoms, Chem. Rev., 1970, 70, 489–516 CrossRef CAS.
- K. Schofield, Critically evaluated rate constants for gaseous reactions of several electronically excited species, J. Phys. Chem. Ref. Data, 1979, 8, 723–798 CrossRef CAS.
- G. Vanuzzo, D. Marchione, L. Mancini, P. Liang, G. Pannacci, P. Recio, Y. Tan, M. Rosi, D. Skouteris, P. Casavecchia and N. Balucani, The N(2D) + CH2CHCN (Vinyl Cyanide) Reaction: A Combined Crossed Molecular Beam and Theoretical Study and Implications for the Atmosphere of Titan, J. Phys. Chem. A, 2022, 126, 6110–6123 CrossRef CAS PubMed.
- L. Mancini, G. Vanuzzo, D. Marchione, G. Pannacci, P. Liang, P. Recio, M. Rosi, D. Skouteris, P. Casavecchia and N. Balucani, The Reaction N(2D) + CH3CCH (Methylacetylene): A Combined Crossed Molecular Beams and Theoretical Investigation and Implications for the Atmosphere of Titan, J. Phys. Chem. A, 2021, 125, 8846–8859 CrossRef CAS PubMed.
- P. Recio, D. Marchione, A. Caracciolo, V. J. Murray, L. Mancini, M. Rosi, P. Casavecchia and N. Balucani, A crossed molecular beam investigation of the N(2D) + pyridine reaction and implications for prebiotic chemistry, Chem. Phys. Lett., 2021, 779, 138852 CrossRef CAS.
- K. M. Hickson, C. Bray, J. C. Loison and M. Dobrijevic, A kinetic study of the N(2D) + C2H4reaction at low temperature, Phys. Chem. Chem. Phys., 2020, 22, 14026–14035 RSC.
- L. Mancini, M. Rosi, D. Skouteris, G. Vanuzzo, G. Pannacci, P. Casavecchia and N. Balucani, A computational characterization of the reaction mechanisms for the reactions N(2D) + CH3CN and HC3N and implications for the nitrogen-rich organic chemistry of Titan, Comput. Theor. Chem., 2023, 1229, 114341 CrossRef CAS.
- J. Geiger and B. Schröder, Intensity perturbations due to configuration interaction observed in the electron energy-loss spectrum of N2, J. Chem. Phys., 1969, 50, 107–119 CrossRef.
- M. Liu, P. Jiang, M. Cheng and H. Gao, Vacuum ultraviolet photoexcitation and photofragment spectroscopic studies of 14N15N between 109000 and 117500 cm−1, J. Chem. Phys., 2021, 155, 234305 CrossRef CAS PubMed.
- D. Spelsberg and W. Meyer, Dipole-allowed excited states of N2: Potential energy curves, vibrational analysis, and absorption intensities, J. Chem. Phys., 2001, 115, 6438–6449 CrossRef CAS.
- E. C. Zipf and R. W. McLaughlin, On the dissociation of nitrogen by electron impact and by EUV photo-absorption, Planet. Sp. Sci., 1977, 26, 449–462 CrossRef.
- H. Helm and P. C. Cosby, Product branching in predissociation of the e 1Πu, e′1Σu +, and b′ 1Σu+ states of N2, J. Chem. Phys., 1989, 90, 4208–4215 CrossRef CAS.
- C. W. Walter, P. C. Cosby and H. Helm, N(4S0), N(2D0), and N(2P0) yields in predissociation of excited singlet states of N2, J. Chem. Phys., 1993, 99, 3553–3561 CrossRef CAS.
- C. W. Walter, P. C. Cosby and H. Helm, Predissociation quantum yields of singlet nitrogen, Phys. Rev. A, 1994, 50, 2930–2936 CrossRef CAS PubMed.
- S. Chakraborty, B. H. Muskatel, T. L. Jackson, M. Ahmed, R. D. Levine, M. H. Thiemens and T. E. Cerling, Massive isotopic effect in vacuum UV photodissociation of N2 and implications for meteorite data, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14704–14709 CrossRef CAS PubMed.
- S. Chakraborty, T. L. Jackson, B. Rude, M. Ahmed and M. H. Thiemens, Nitrogen isotopic fractionations in the low temperature (80 K) vacuum ultraviolet photodissociation of N2, J. Chem. Phys., 2016, 145, 114302 CrossRef.
- Y. Song, H. Gao, Y. C. Chang, D. Hammouténe, H. Ndome, M. Hochlaf, W. M. Jackson and C. Y. Ng, Quantum-State Dependence of Product Branching Ratios in Vacuum Ultraviolet Photodissociation of N2, Astrophys. J., 2016, 819, 23 CrossRef.
- M. Liu, P. Jiang, L. Lu, T. Yin, L. Ma, M. Cheng, Q. Yin and H. Gao, Strong Isotope-dependent Photodissociation Branching Ratios of N2 and Their Potential Implications for the 14N/15N Isotope Fractionation in Titan's Atmosphere, Astrophys. J., 2021, 923, 196 CrossRef CAS.
- P. Jiang, L. Lu, M. Liu and H. Gao, Multi-channel photodissociation dynamics of 14N2 in its b′ 1Σ + u(ν = 20) state, Phys. Chem. Chem. Phys., 2022, 24, 11544–11551 RSC.
- L. Lu, P. Jiang and H. Gao, Accurate measurements of the bond dissociation energies of 14N2, 14N15N and 15N2, Fundam. Res., 2022 DOI:10.1016/j.fmre.2022.08.003.
- S. W. Sharpe and P. M. Johnson, Triplet Rydberg states in molecular nitrogen, J. Chem. Phys., 1986, 85, 4943–4948 CrossRef CAS.
- B. R. Lewis, A. N. Heays, S. T. Gibson, H. Lefebvre-Brion and R. Lefebvre, A coupled-channel model of the 3Πu states of N2: Structure and interactions of the 3sσgF 3 3Πu and 3pπuG3 3Πu Rydberg states, J. Chem. Phys., 2008, 129, 164306 CrossRef CAS PubMed.
- B. Minaev, P. Norman, D. Jonsson and H. Ågren, Response theory calculations of singlet-triplet transitions in molecular nitrogen, Chem. Phys., 1995, 190, 11–29 CrossRef CAS.
- Z. Qin, J. Zhao and L. Liu, Radiative transition probabilities between low-lying electronic states of N2, Mol. Phys., 2019, 117, 2418–2433 CrossRef CAS.
- B. F. Minaev, O. O. Panchenko, V. A. Minaeva and H. Ågren, Triplet state harvesting and search for forbidden transition intensity in the nitrogen molecule, Front. Chem., 2022, 10, 1–10 Search PubMed.
- B. Minaev, R. S. Da Silva, O. Panchenko and H. Ågren, Prediction of new spin-forbidden transitions in the N2 molecule - The electric dipole A′5Σg + → A3Σu+ and magnetic dipole a′1Σu-← A3Σu+ transitions, J. Chem. Phys., 2023, 158, 084304 CrossRef CAS PubMed.
- F. R. Gilmore, Potential energy curves for N2, NO, O2 and corresponding ions, J. Quant. Spectrosc. Radiat. Transf., 1965, 5, 369–389 CrossRef CAS.
- H. Lefebvre-Brion and C. M. Moser, Calculation of Rydberg levels in the nitrogen molecule, J. Chem. Phys., 1965, 43, 1394–1399 CrossRef CAS.
- D. A. Little and J. Tennyson, An ab initio study of singlet and triplet Rydberg states of N2, J. Phys. B: At., Mol. Opt. Phys., 2013, 46, 145102 CrossRef.
- W. C. Ermler, J. P. Clark and R. S. Mulliken, Ab initio calculations of potential energy curves and transition moments of 1Σg+ and 1Σ u+ states of N2, J. Chem. Phys., 1987, 86, 370–375 CrossRef CAS.
- M. Hochlaf, H. Ndome and D. Hammoutne, Quintet electronic states of N2, J. Chem. Phys., 2010, 132, 104310 CrossRef CAS PubMed.
- M. Hochlaf, H. Ndome, D. Hammoutène and M. Vervloet, Valence-Rydberg electronic states of N2: Spectroscopy and spin-orbit couplings, J. Phys. B: At., Mol. Opt. Phys., 2010, 43, 245101 CrossRef.
- S. O. Adamson, V. V. Kuverova, G. K. Ozerov, G. V. Golubkov, S. S. Nabiev and M. G. Golubkov, Ab Initio Calculation of the Lowest Singlet and Triplet Excited States of the N2 Molecule, Russ. J. Phys. Chem. B, 2018, 12, 620–631 CrossRef CAS.
- D. Bhattacharya, K. R. Shamasundar and A. Emmanouilidou, Potential Energy Curves of Molecular Nitrogen for Singly and Doubly Ionized States with Core and Valence Holes, J. Phys. Chem. A, 2021, 125, 7778–7787 CrossRef CAS PubMed.
- D. Stahel, M. Leoni and K. Dressler, Nonadiabatic representations of the 1Σu+ and 1Πu states of the N2 molecule, J. Chem. Phys., 1983, 79, 2541–2558 CrossRef CAS.
- B. F. E. Curchod and T. J. Martínez, Chem. Rev., 2018, 118, 3305–3336 CrossRef CAS PubMed.
- H. B. Schlegel, Ab Initio Direct Dynamics, Acc. Chem. Res., 2021, 54, 3749–3759 CrossRef CAS PubMed.
- M. Nisoli, P. Decleva, F. Calegari, A. Palacios and F. Martín, Attosecond Electron Dynamics in Molecules, Chem. Rev., 2017, 117, 10760–10825 CrossRef CAS PubMed.
- Y. Lassmann, D. Hollas and B. F. E. Curchod, Extending the Applicability of the Multiple-Spawning Framework for Nonadiabatic Molecular Dynamics, J. Phys. Chem. Lett., 2022, 13, 12011–12018 CrossRef CAS PubMed.
- N. Gelfand, K. Komarova, F. Remacle and R. D. Levine, Nonadiabatic dynamics in a forest of coupled states: Electronic state branching in the VUV photodissociation of N2, J. Chem. Phys., 2023, 158, 164302 CrossRef CAS PubMed.
- A. Trabattoni, M. Klinker, J. González-Vázquez, C. Liu, G. Sansone, R. Linguerri, M. Hochlaf, J. Klei, M. J. J. Vrakking, F. Martín, M. Nisoli and F. Calegari, Mapping the dissociative ionization dynamics of molecular nitrogen with attosecond time resolution, Phys. Rev. X, 2015, 5, 041053 Search PubMed.
- M. Klinker, C. Marante, L. Argenti, J. González-Vázquez and F. Martín, Electron Correlation in the Ionization Continuum of Molecules: Photoionization of N2 in the Vicinity of the Hopfield Series of Autoionizing States, J. Phys. Chem. Lett., 2018, 9, 756–762 CrossRef CAS PubMed.
- T. Ayari, M. Desouter-Lecomte, R. Linguerri, G. A. Garcia, L. Nahon, A. Ben Houria, H. Ghalila, R. Ben Said and M. Hochlaf, State-to-state dissociative photoionization of molecular nitrogen: the full story, Adv. Phys. X, 2020, 5, 1831955, DOI:10.1080/23746149.2020.1831955.
- B. H. Muskatel, F. Remacle and R. D. Levine, AttoPhotoChemistry. Probing ultrafast electron dynamics by the induced nuclear motion: The prompt and delayed predissociation of N2, Chem. Phys. Lett., 2014, 601, 45–48 CrossRef CAS.
- J. Ajay, J. Šmydke, F. Remacle and R. D. Levine, Probing in Space and Time the Nuclear Motion Driven by Nonequilibrium Electronic Dynamics in Ultrafast Pumped N2, J. Phys. Chem. A, 2016, 120, 3335–3342 CrossRef CAS PubMed.
- J. S. Ajay, K. G. Komarova, F. Remacle and R. D. Levine, Time-dependent view of an isotope effect in electron-nuclear nonequilibrium dynamics with applications to N2, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5890–5895 CrossRef CAS PubMed.
- K. G. Komarova, F. Remacle and R. D. Levine, Time resolved mechanism of the isotope selectivity in the ultrafast light induced dissociation in N2, J. Chem. Phys., 2019, 151, 114308 CrossRef PubMed.
- N. Gelfand, K. Komarova, F. Remacle and R. D. Levine, On the energy-specific photodissociation pathways of 14N2 and 14N15N isotopomers to N atoms of different reactivity: a quantum dynamical perspective, Astrophys. J., 2023, 948, 58 CrossRef.
- H. Lefebvre-Brion and R. W. Field, The Spectra and Dynamics of Diatomic Molecules, Elsevier, Amsterdam, 2004 Search PubMed.
- H. J. Werner and P. J. Knowles, A second order multiconfiguration SCF procedure with optimum convergence, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS.
- P. J. Knowles and H. J. Werner, An efficient second-order MC SCF method for long configuration expansions, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS.
- S. L. Guberman, Spectroscopy above the ionization threshold: Dissociative recombination of the ground vibrational level of N2, J. Chem. Phys., 2012, 137, 074309 CrossRef PubMed.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: A general-purpose quantum chemistry program package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- H. J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- J. S. Ajay, K. G. Komarova, S. Van Den Wildenberg, F. Remacle and R. D. Levine, in Theoretical and Computational Chemistry Series, ed. M. J. J. Vrakking and F. Lepine, The Royal Society of Chemistry, Cambridge, 2018, pp. 308–347 Search PubMed.
- B. Fornberg, Generation of Finite Difference Formulas on Arbitrarily Spaced Grids, Math. Comput., 1988, 51, 699 CrossRef.
- X. Li, A. N. Heays, R. Visser, W. Ubachs, B. R. Lewis, S. T. Gibson and E. F. Van Dishoeck, Photodissociation of interstellar N2, Astron. Astrophys., 2013, 555, A14 CrossRef.
- N. Gelfand, F. Remacle and R. D. Levine, Recombination of N Atoms in a Manifold of Electronic States Simulated by Time-Reversed Nonadiabatic Photodissociation Dynamics of N2, J. Phys. Chem. Lett., 2023, 14, 4625–4630 CrossRef CAS PubMed.
- N. Gelfand, K. Komarova, F. Remacle and R. D. Levine, Photodissociation via weak intersystem crossing: Incoherent versus coherent excited-state nonadiabatic dynamics in N2, Phys. Rev. A, 2023, 108, 053116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04854c |
| This journal is © the Owner Societies 2024 |