
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First principles simulations of MgO(100) surface hydration at ambient conditions†
Michel
Sassi
 * and
Kevin M.
Rosso
* and
Kevin M.
Rosso
Physical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: michel.sassi@pnnl.gov
First published on 18th December 2023
Abstract
Developing a better understanding of water ordering and hydroxylation at oxide mineral surfaces is important across a breath of application spaces. Recent vibrational sum frequency generation (vSFG) measurements on MgO(100) surfaces at ambient conditions showed that water dissociates and hydroxylates the surface yielding a non-hydrogen bonded hydroxyl species. Starting from previously determined water hydroxylation patterns on MgO(100), we performed ab initio thermodynamic calculations and vibrational analysis to compare with the vSFG observations. At ambient conditions (i.e., T = 298.15 K and pH2O = 32 mbar), the most thermodynamically favorable surface hydroxylation is found to be p(3 × 2) – 8H2O, involving a dissociation of 25% of the adsorbed water. Analysis of the vibrational density of states for this hydroxylation configuration yielded three different hydrogen bonding environments with the frequency of the peaks in very good agreement with the vSFG measurements. However, the non-H-bonded spectral feature on this surface is predicted to be similar to that expected for Mg(OH)2, a thermodynamically downhill alteration of the surface that must be independently ruled out before one can be fully confident in the apparent theory/vSFG agreement. Our study provides more insights into the ordering and structure of water monolayer at MgO(100) surface at ambient conditions and completes previous theoretical and experimental analysis performed at low temperature and ultra-high vacuum conditions.
Introduction
Oxide–water interfaces play a key role in a number of important physical and chemical processes spanning a range of research fields such as materials science, biology, geochemistry, atmospheric science, and heterogeneous catalysis.1–9 Developing a molecular-level picture of water ordering and interactions at metal oxide interfaces is central to better understand processes such as adsorption, dissolution, heterogeneous nucleation and growth, and particle aggregation phenomena. In part due to its simple cubic rock-salt structure, the interaction of MgO surfaces with water under various conditions has been extensively studied both experimentally and computationally, particularly the (100) facet.10–31 At ambient conditions, in the presence of water vapor or liquid water, a challenging aspect of this interfacial system is that bulk MgO (periclase) is thermodynamically unstable with respect to its fully hydroxylated counterpart Mg(OH)2 (brucite).32 Therefore, studies of MgO(100)/water interaction intrinsically probe a quasi 2D free energy landscape for low energy configurations of adsorbed water and hydroxyls that is, in most cases, only metastable with respect to the 3D global minimum of Mg(OH)2.It is therefore unsurprising to find a lack of consensus in the literature regarding the hydration of MgO(100) when contacted by water. Using single crystal surfaces, early experiments using ultraviolet photoemission spectroscopy (UPS)33 and infra-red spectroscopy (IR)34 could not detect if water dissociates to form hydroxyl groups on the surface. In agreement with this experimental uncertainty, early density functional theory (DFT) simulations using the Car–Parrinello method concluded that water physisorbs on defect-free MgO(100) terraces, and that it only dissociates at surface defect sites such as steps.35 A few years later, ab initio investigations indicated that water strongly reacts with MgO(100) terraces and spontaneously dissociates at a ratio of two out of six water molecules adsorbed.13,15 This partial dissociation of water on well-ordered and low-defect MgO(100) surfaces was later experimentally confirmed.19–21
At low coverage, ultra-high vacuum (UHV) studies11 indicate formation of two ordered water monolayer phases, namely c(4 × 2), detected in the temperature range 100–180 K, and p(3 × 2), found at temperatures of 185–221 K. A more complete structural model of the p(3 × 2) phase was obtained from additional investigations using quantitative low energy electron diffraction (LEED) aided by semiempirical potential calculations performed at 0 K.36 For low temperature and UHV conditions, Włodarczyk et al.25 performed ab initio simulations and used a genetic algorithm to identify eight hydroxylation configurations for the p(3 × 2) and c(4 × 2) symmetries. Their predictions agreed well with the observed temperature dependence of surface phase changes and water desorption. The findings indicated that the low-temperature (T < 200 K) phase is composed of ten water molecules per unit cell with c(4 × 2) symmetry while at higher temperature (200 K ≤ T ≤ 202 K) the structure containing six water molecules per unit cell with p(3 × 2) symmetry is the most stable phase. At temperature higher than 202 K, water desorption from the surface was found to be more thermodynamically favorable. The most stable hydroxylated structure found for c(4 × 2) and p(3 × 2) respectively yielded 1/5 and 1/3 of the water molecules being dissociated. Extending this work to ambient conditions and for more than a monolayer of water coverage, Ončák et al.32 found that a fully hydroxylated surface is not energetically favorable while a reconstructed surface, involving hydrated/hydroxylated Mg2+ ions above the surface, are more stable.
Despite this comprehensive work, direct validation of the hydroxylation and hydration structure of MgO(100) at ambient conditions remains experimentally challenging. Recently, vibrational sum frequency generation (vSFG) spectroscopy in the OH stretching region showed a predominant non-hydrogen bonded peak at ∼3700 cm−1 in addition to a broad spectrum of states at lower frequencies consistent with an H-bonded network of surface hydroxyls and adsorbed water.31 Although the vSFG features alone do not unambiguously inform on the specific structure at the surface, they do provide a basis for comparison to theoretical vibrational spectra predicted for specific hydroxylation/hydration patterns.
To make this comparison, here we report ab initio thermodynamics (AIT) calculations to examine the vibrational spectra for specific surface configurations of adsorbed water and hydroxyls on MgO(100), and to relate these characteristics to the relevant state variables that define the chemical potential of water, namely temperature and the partial pressures of O2 and H2. To do so we revisit the series of eight hydroxylated structures found by Włodarczyk et al.25 and expand the work performed by Ončák et al.32 for ambient conditions who limited their investigation to the two most stable hydroxylation structures for c(4 × 2) and p(3 × 2) identified by Włodarczyk et al.25 for low temperature and UHV conditions. By using the AIT approach, we investigate how the chemical potential of oxygen and hydrogen can affect the relative phase stability of different hydroxylated configurations. While four hydroxylation configurations have a favorable surface Gibbs free energy, we found that the most thermodynamically stable one under ambient conditions is different to that identified by Włodarczyk et al.25 for low temperature and UHV conditions. The predicted low energy hydroxylated configurations yield vibrational spectra that compare well to the vSFG results of Adhikari et al.31 However, we also show that the prominent non-H-bonded feature at ∼3700 cm−1 is overlapping with the spectrum predicted for a bulk-like Mg(OH)2 termination, a structure that is based entirely on non-H-bonded hydroxyls. Therefore, to unambiguously identify the hydroxylated surface structure in experiments requires careful elimination of the prospect of an Mg(OH)2-like termination.
Computational details
Density functional theory (DFT) simulations of bulk MgO, MgO(100) surface hydroxylation, and bulk Mg(OH)2 were performed using the VASP package.37,38 All the simulations used the exchange–correlation functional approximated by the generalized gradient approximation (GGA) as parametrized in the Revised PBE for solids (PBEsol).39 The cutoff energy for the plane-wave-basis set was fixed to 525 eV, and in each case, the total energy was converged to 10−8 eV per cell and the force components were relaxed to 10−5 eV Å−1. The sampling of the Brillouin zone for bulk MgO, and (1 × 1), p(3 × 2), and c(4 × 2) (100) surface slabs used a Γ-centered k-point mesh of 7 × 7 × 7, 7 × 7 × 1, 5 × 6 × 1, and 4 × 6 × 1 respectively. The calculated lattice parameter of bulk MgO is a = 4.213 Å, which is in very good agreement with the experimental value of 4.211 Å.40 The simulation of bulk Mg(OH)2 used a Γ-centered k-point mesh of 9 × 9 × 7 and the calculated lattice parameters were a = 3.141 Å and c = 4.552 Å, in good agreement with the experimental values of a = 3.142 Å and c = 4.766 Å.41In order to assess the accuracy of the PBEsol functional for the calculation of surface energies, some tests on surface energy of clean (i.e., defect-free) MgO(100) surface and slab size were performed as shown in Table 1. A comparison with the surface energy calculated using the Perdew, Burke, and Ernzerhof (PBE) functional42 with Grimme (D3) dispersion corrections43 and the Becke–Johnson (BJ) damping function44 (i.e., PBE + D3 (BJ)) is also provided. The calculated surface energy of MgO(100) with PBEsol is in very good agreement with the experimental value of 1.04 J m−2,45 while the PBE + D3 functional overestimates the surface energy by about 30%. Depending on the functional used, previous theoretical calculations estimated the MgO(100) surface energy in the range of 0.83 to 1.29 J m−2.46
| Clean (100) Surface | γ (J m−2) | ||
|---|---|---|---|
| (1 × 1) 8 Layers | p(3 × 2) 4 Layers | c(4 × 2) 4 Layers | |
| PBEsol | 0.99 | 1.00 | 1.00 |
| PBE + D3 (BJ) | 1.35 | 1.35 | 1.35 |
| Expt.45 | 1.04 | ||
The surface Gibbs free energy dependance to oxygen and hydrogen chemical potentials for various hydroxylation configurations of the MgO(100) surface has been evaluated within the ab initio thermodynamic framework. In these calculations, the surface Gibbs free energy is given by:
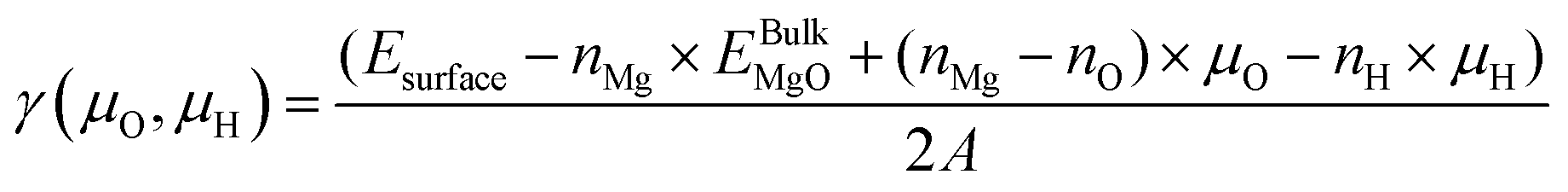 | (1) |
| Esurface = ETotalsurface + EZPEsurface + Δμ(T,P)surface | (2) |
| EBulkMgO = ETotalMgO + EZPEMgO + Δμ(T,P)MgO | (3) |
 | (4) |
 | (5a) |
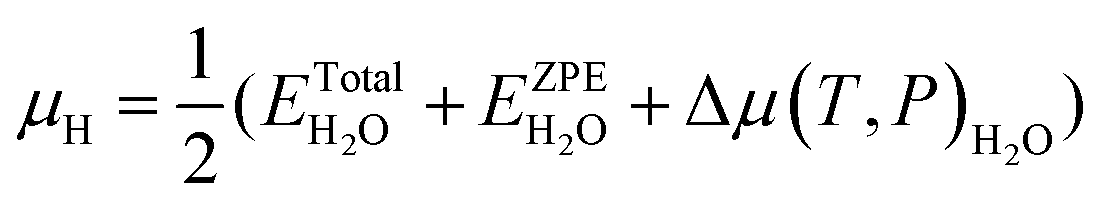 | (5b) |
A comparison of calculated and NIST-JANAF experimental48 Δμ(T,P) for bulk Mg and MgO crystals is shown in Fig. 1. The agreements between calculated and experimental values are within 2 kJ mol−1 across the temperature range.
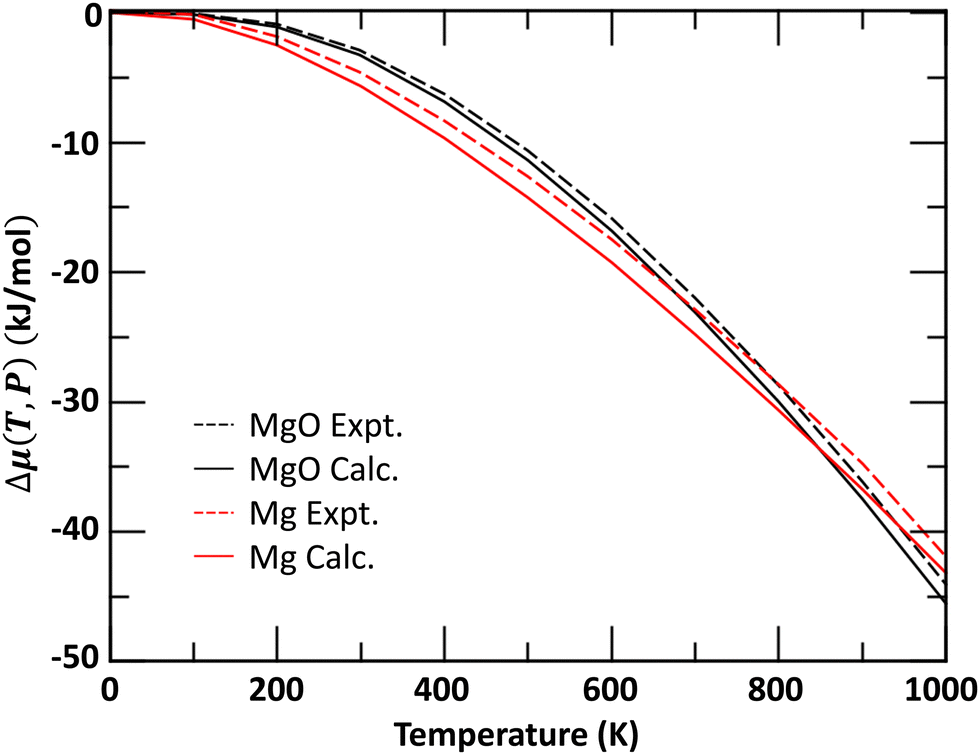 | ||
| Fig. 1 Comparison of experimental and calculated chemical potentials for Mg and MgO bulk materials as function of temperature at a pressure of P = 1 bar. | ||
In order to account for the atomization errors induced by GGA for O2, H2, and H2O, the DFT total energies of these species have been corrected by using their experimental, zero-point energy corrected, atomization energies.42,49 Therefore, the total energy of O2, H2, and H2O molecules were respectively corrected by 1.483 eV, −0.104 eV, and −0.515 eV.
Results and discussion
A total of nine hydroxylation configurations have been investigated for the hydrated MgO(100) surface. As shown in Fig. 2, those configurations have been classified into two groups depending on the translational symmetry of the water monolayer.36,50 The groups are labelled p(3 × 2) and c(4 × 2), with the numbers in brackets indicating the number of MgO primitive cell translations in the lateral plan. Eight hydroxylated surface structures, previously identified by Włodarczyk et al.25 using a genetic algorithm, have between 20% to 37% of dissociated water molecules at the surface. A ninth hydroxylation configuration, which has two water monolayers for a total of 12H2O (i.e., one H2O for each of the six surface Mg atom) at a (3 × 2) MgO(100) surface, has been additionally investigated because it does not involve water dissociation. Although not a monolayer, this additional hydroxylation configuration enables us to compare the relative energetics at ambient conditions for water coverage that does not involve dissociation.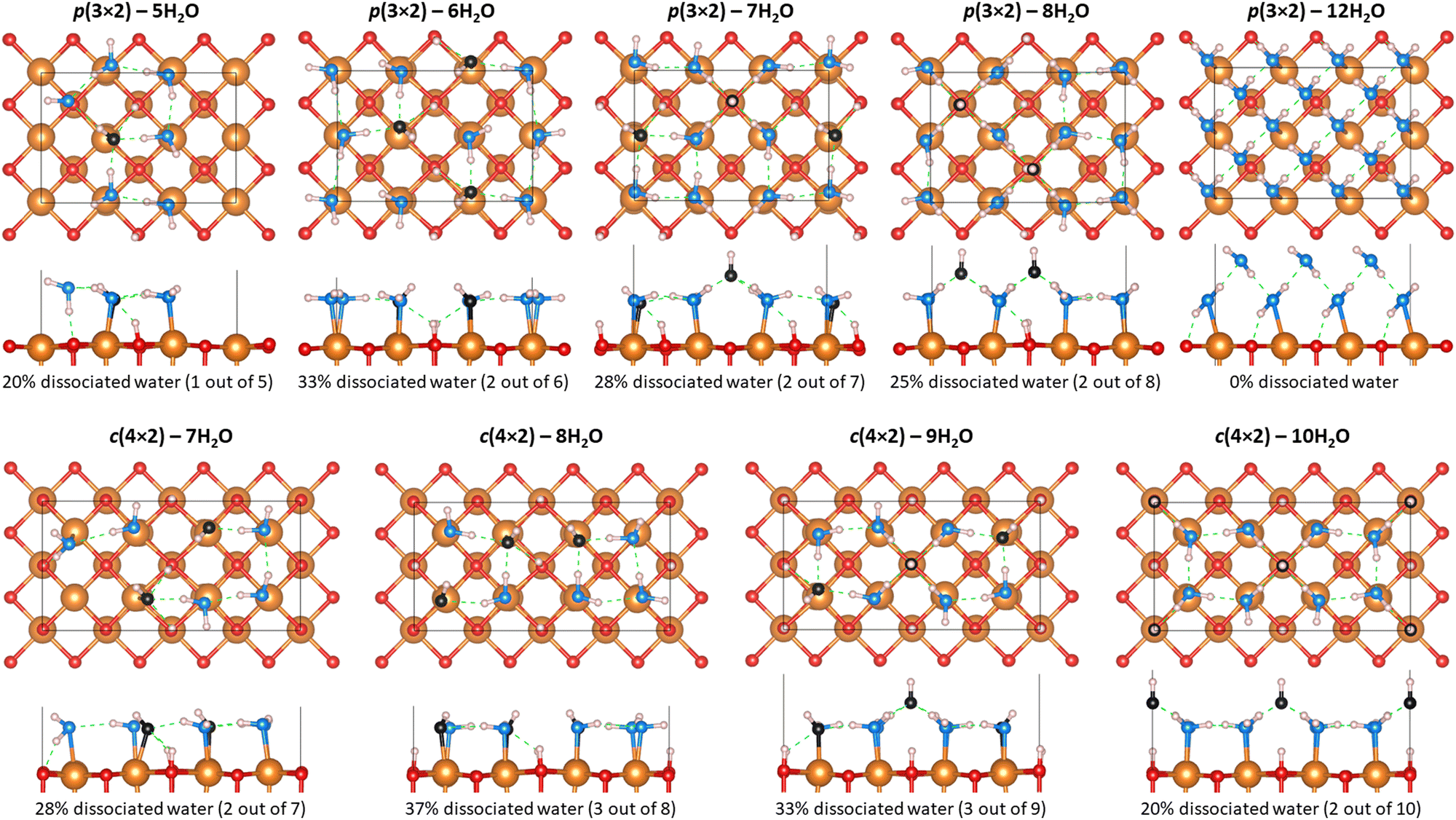 | ||
| Fig. 2 Top and side views of the nine surface hydration configurations investigated for the MgO(100) surface. The Mg and O atoms or MgO are represented by orange and red spheres. The water H and O atoms are represented by light pink and blue spheres respectively, while the O atoms from hydroxyl anions (OH−) are represented by black spheres. Hydrogen bonds are highlighted by green dashed lines. | ||
In the cases where water dissociates (into H+ and OH−) at the surface, Fig. 2 shows that OH groups are formed at the surface with a hydrogen atom forming a covalent bond with a surface O atom, while the remaining hydroxyl anion is only interacting with the water monolayer by forming hydrogen bonds with nearby water molecules. In the case where water does not dissociate, the water hydrogen of the first water monolayer forms a hydrogen bond with the oxygen atoms of the MgO(100) surface. The other water hydrogen of the first water monolayer is involved in hydrogen bonding with the water oxygen of the second water monolayer. The formation of such hydrogen bond network between first and second water monolayers helps prevent water dissociation.
The relative stability of each hydroxylation configuration has been evaluated with ab initio thermodynamics, in which the surface Gibbs free energy was calculated as function of the oxygen and hydrogen chemical potentials at T = 298.15 K. A negative surface Gibbs free energy indicates that the surface is energetically and thermodynamically favorable to form while a surface with positive Gibbs free energy suggests that the formation of the surface is not favorable. As shown in Fig. 3, the most thermodynamically favorable surface configuration for μO ≤ −3.52 eV is the clean MgO(100) surface. For μO values between −3.52 eV and −2.72 eV, the surface hydroxylation configuration c(4 × 2) with 10H2O is found to be more thermodynamically favorable than the clean surface. For μO ≥ −2.72 eV, the surface hydroxylation configuration p(3 × 2) with 8H2O is found to be more thermodynamically favorable than c(4 × 2) with 10H2O.
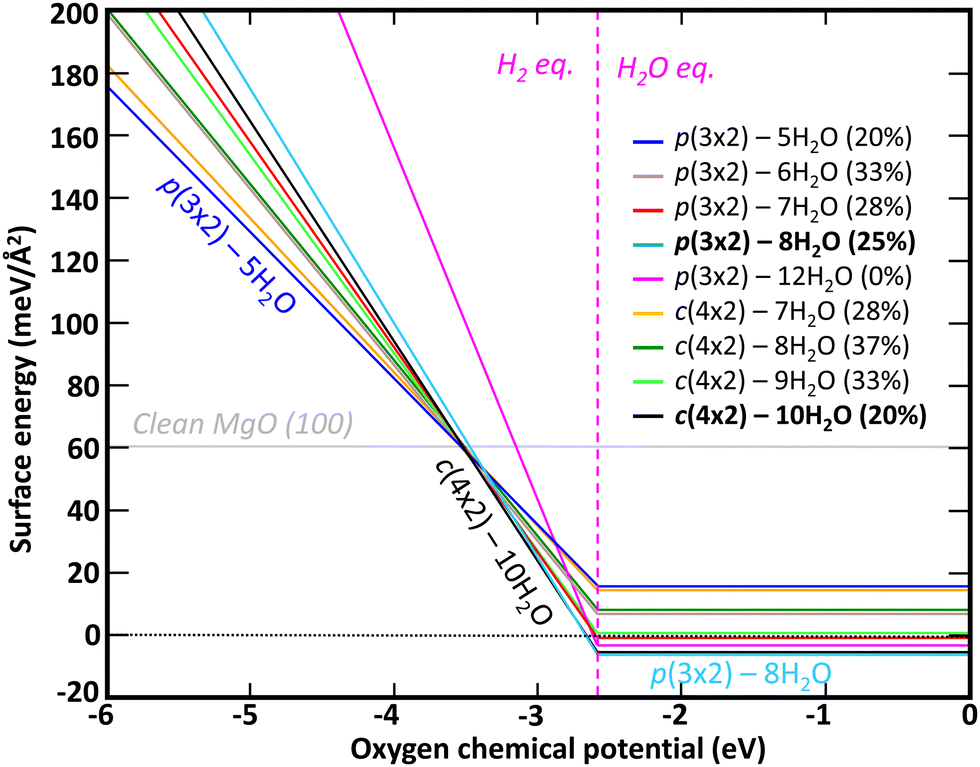 | ||
| Fig. 3 Thermodynamic stability at T = 298.15 K of different surface hydration configurations for MgO(100) as function of oxygen chemical potential. The transition of hydrogen chemical potential from H2 equilibrium to H2O equilibrium is symbolized by a magenta vertical dashed line. | ||
Beside the influence of oxygen chemical potential, the chemical potential of H also impacts the relative stabilities. Interestingly, the clean MgO(100) and the c(4 × 2) – 10H2O surfaces are energetically competitive only when hydrogen is in equilibrium with H2. For hydrogen in equilibrium with H2O, the surface configuration p(3 × 2) with 8H2O is the most energetically favorable surface. More detailed surface Gibbs free energy values for H in equilibrium with H2O (i.e., μO ≥ −2.72 eV) at T = 298.15 K are provided in Table 2. Of the nine hydroxylation configurations investigated, four have a negative surface Gibbs free energy, p(3 × 2) – 8H2O, c(4 × 2) – 10H2O, p(3 × 2) – 12H2O, and p(3 × 2) – 7H2O with a surface energy of −6.191 meV Å−2, −5.546 meV Å−2, −3.328 meV Å−2, and −0.888 meV Å−2 respectively. While the bilayer p(3 × 2) – 12H2O configuration, for which no water dissociation occurs, is energetically favorable (γ(μO, μH) < 0) at room temperature, two monolayer surface configurations for which water dissociation is predicted to occur are lower in energy, indicating that water dissociation should be preferrable at ambient conditions (i.e., T = 298.15 K and P = pH2O = 32 mbar). Interestingly, Fig. 3 and Table 2 show that all nine hydroxylated surface configurations investigated are more energetically favorable than clean MgO(100) surface when H is in equilibrium with H2O. This is in good agreement with MgO being a hydrophilic mineral and thus strongly interacting with water.
| Surface configuration | γ(μO, μH) (meV Å−2) |
|---|---|
| Clean MgO(100) | 60.426 |
| p(3 × 2) – 5H2O | 15.736 |
| p(3 × 2) – 6H2O | 6.844 |
| p(3 × 2) – 7H2O | −0.888 |
| p(3 × 2) – 8H2O | −6.191 |
| p(3 × 2) – 12H2O | −3.328 |
| c(4 × 2) – 7H2O | 14.452 |
| c(4 × 2) – 8H2O | 8.119 |
| c(4 × 2) – 9H2O | 0.670 |
| c(4 × 2) – 10H2O | −5.546 |
Previous thermodynamic calculations by Włodarczyk et al.25 carried out for similar hydroxylated surface configurations as function of temperature and at P = 10−10 mbar, indicated that c(4 × 2) – 10H2O and p(3 × 2) – 6H2O were the most stable surfaces configurations for temperatures below 202 K. For temperatures above 202 K, a complete desorption of water from the MgO(100) surface was predicted. For the thermodynamic conditions selected (i.e., low temperature and pressure), these theoretical results were in good agreement with ultra-high vacuum (UHV) experiments performed at 85 K during water adsorption. The work performed in our study completes the theoretical analysis of Włodarczyk et al.25 by focusing on the hydroxylation of MgO(100) surface at ambient conditions (i.e., T = 298.15 K and P = pH2O =32 mbar). At those conditions, we found that p(3 × 2) – 8H2O is the most thermodynamically stable surface configuration.
In connection with the recent vSFG study performed by Adhikari et al.31 at ambient laboratory conditions, we calculated the phonon dispersion relationship using the Phonopy code47 for the two most thermodynamically favorable surface hydroxylation configurations. The vibrational density of states (VDOS) for p(3 × 2) – 8H2O and c(4 × 2) – 10H2O are shown in Fig. 4 along with the experimental frequency range of different hydrogen bonding environments, as detected by vSFG. While a direct comparison of the calculated VDOS and experimental vSFG spectral intensity is not possible because the VDOS does not have any selection rule and it would require the computation of more complex quantities, such as the second order susceptibility which can be calculated from molecular dynamic simulations,51 one can still compare the frequencies of the peaks. From the calculated VDOS for p(3 × 2) – 8H2O, three types of hydrogen bonds can be distinguished with frequencies higher than 3300 cm−1. Those correspond to hydrogen bonded surface hydrogen (labelled H-bonded Hs), non-hydrogen bonded surface hydrogen (labelled non-H-bonded Hs), and non-hydrogen bonded hydrogen of the hydroxyl anion (labelled non-H-bonded HOH) with frequency peaks located at 3390 cm−1, 3642 cm−1, and 3778 cm−1 respectively.
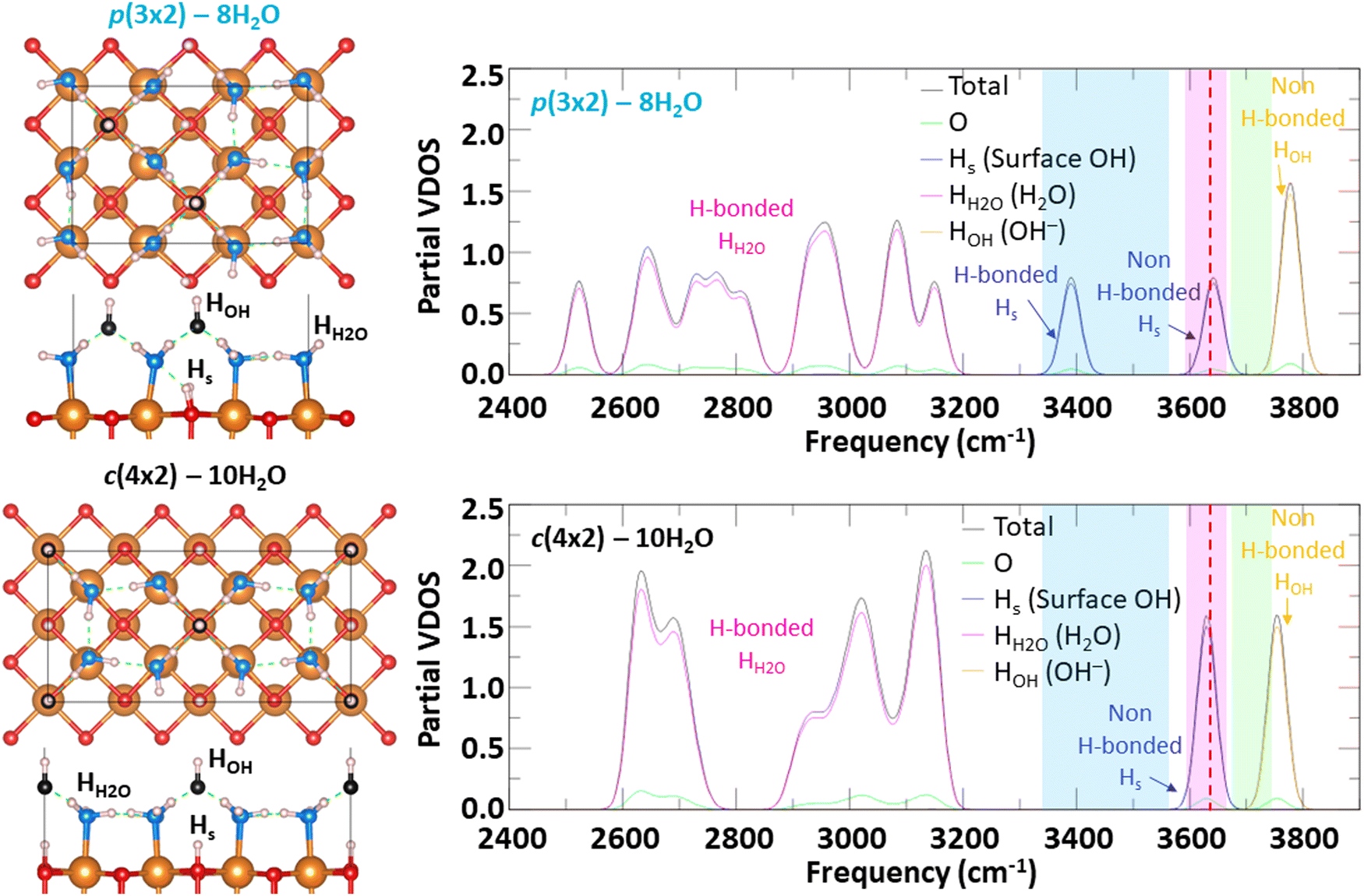 | ||
| Fig. 4 Vibrational density of state of the two most thermodynamically stable hydroxylated configurations of MgO(100). Vertical blue, pink, and green bands delimit the experimental frequency range of different H bonding environments, as detected by vSFG.31 The red dashed line indicates the calculated frequency of OH stretch in bulk Mg(OH)2. | ||
In the case of surface hydroxyl groups (OHs) originating from water dissociation, only the Hs located right under a hydroxyl anion are not bonded to any surrounding water molecules as those water molecules preferentially form hydrogen bonds with the hydroxyl anions. This feature gives rise to two types of hydrogen bonding environments for surface hydroxyl (OHs) groups, with the hydrogen bonded ones having a lower frequency than the non-hydrogen bonded ones. The frequencies associated with the three types of hydrogen bonding environments are in good agreement with the vSFG frequency peaks detected experimentally,31 as shown by the colored zones in Fig. 4. While c(4 × 2) – 10H2O is the second most thermodynamically stable hydroxylation surface configuration, only two peaks can be distinguished for frequencies higher than 3300 cm−1. These peaks correspond to non-hydrogen bonded OHs (3630 cm−1) and to hydrogen bonded hydroxyl anions (3754 cm−1). In this hydroxylated surface configuration, there are no hydrogen bonded OHs, and thus, lower frequencies in the range from about 3350 cm−1 to 3550 cm−1 are not present. This suggests that under ambient conditions, the MgO(100) surface is hydroxylated similarly to p(3 × 2) – 8H2O, which is the most thermodynamically favorable surface configuration and shows a vibrational spectra with frequency positions above 3300 cm−1 in good correlation with vSFG experiments.
For the calculated VDOS with frequencies lower than 3300 cm−1, which would mainly correspond to the hydrogen bond network between water molecules, we note that the experimental vSFG has intensity in this frequency region. However, the vSFG peaks are at least 100 times less intense than the probed OH stretches, which have a strong vSFG signal. The small vSFG intensity of the hydrogen bonded water frequencies can be observed in Fig. S4 and S7 (ESI†) of the study by Adhikari et al.31
However, conceptually there is another way to produce non-H-bonded hydroxyl spectral signal at the hydrated MgO surface – by converting it wholly or in part to more thermodynamically favorable Mg(OH)2.32 For example, using the same DFT methods we calculated that the thermodynamic driving force to convert bulk MgO to Mg(OH)2 at ambient conditions (i.e., T = 298.15 K and pH2O = 32 mbar) is −0.71 eV (or −68.42 kJ mol−1). In the bulk structure of Mg(OH)2, every OH group can be considered non-H-bonded, as shown in Fig. S1 (ESI†), as they each point toward the center of a triangle made of three octahedral Mg atoms. The frequency associated to the OH stretch is calculated to be 3636 cm−1 and has been reported in Fig. 4 by a red dashed line. As this frequency overlaps with the non-H-bonded Hs range of frequencies, chemical transformation of the MgO surface to an Mg(OH)2-like termination would likely yield a prominent non-H-bonded feature (pink band) in the vSFG spectrum. The remaining hydroxylation/hydration spectral weight at lower frequencies would in turn depend on the structural details of the Mg(OH)2-like termination onto which water adsorbs and reacts at a given temperature and pressure, which is beyond the scope of the current study. Nonetheless, these results suggest that although our findings predict a good correspondence between the p(3 × 2) – 8H2O hydroxylated MgO(100) configuration, and that this configuration is low in energy, a contribution from an Mg(OH)2-like termination cannot yet be entirely ruled out.
Conclusion
A thermodynamic and vibrational analysis of several hydroxylation configurations of the MgO(100) surface have been performed with DFT. It was found that a water monolayer on adsorbed MgO can transform from a c(4 × 2) to a p(3 × 2) symmetry depending on the oxygen chemical potential. For surface hydration of one monolayer at ambient conditions, the most thermodynamically favorable surface hydroxylation is found to be p(3 × 2) – 8H2O, which involves 25% water dissociation. The calculation of the vibrational density of states for this hydroxylation configuration provided insights about the local environment of hydrogen. Three different bonding environments could be identified, such as hydrogen bonded and non-hydrogen bonded surface OH groups, and hydrogen bonded hydroxyl anions, all hydroxyl groups originating from water dissociation at MgO(100) surface. While the intensity cannot be directly compared between calculated VDOS and experimental vSFG spectra, the calculated frequency position for each hydrogen bonding environment shows very good agreement with recent vibrational SFG measurements performed at ambient conditions, with the caveat that a contribution from Mg(OH)2-like domains cannot be ruled out. Our study provides more insights into the structure of the water monolayer on MgO(100) surface at ambient conditions and complete previous theoretical and experimental analysis performed at low temperature and ultra-high vacuum conditions.Associated content
Visual representation of the bulk structure of Mg(OH)2 is provided in Fig. S1 (ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division through its Geosciences program at Pacific Northwest National Laboratory (FWP 56674). PNNL is a multi-program national laboratory operated for the DOE by Battelle Memorial Institute under Contract No. DE-AC05-76RL01830. Computational resources were provided by PNNL Institutional Computing (PIC).References
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel and M. I. McCarthy, et al., Metal Oxide Surfaces and Their Interactions with Aqueous Solutions and Microbial Organisms, Chem. Rev., 1999, 99, 77–174 CrossRef CAS PubMed.
- A. Verdaguer, G. M. Sacha, H. Bluhm and M. Salmeron, Molecular Structure of Water at Interfaces: Wetting at the Nanometer Scale, Chem. Rev., 2006, 106, 1478–1510 CrossRef CAS PubMed.
- G. E. Ewing, Ambient Thin Film Water on Insulator Surfaces, Chem. Rev., 2006, 106, 1511–1526 CrossRef CAS PubMed.
- J. Atkinson, B. Murray, M. Woodhouse, T. F. Whale, K. J. Baustian, K. S. Carslaw, S. Dobbie, D. O’Sullivan and T. L. Malkin, The importance of feldspar for ice nucleation by mineral dust in mixed-phase clouds, Nature, 2013, 498, 355–358 CrossRef CAS PubMed.
- A. Kiselev, F. Bachmann, P. Pedevilla, S. J. Cox, A. Michaelides, D. Gerthsen and T. Leisner, Active sites in heterogeneous ice nucleation - the example of K-rich feldspars, Science, 2017, 355, 367–371 CrossRef CAS PubMed.
- Z.-J. Zhao, Z. Li, Y. Cui, H. Zhu, W. F. Schneider, W. N. Delgass, F. Ribeiro and J. Greeley, Importance of metal-oxide interfaces in heterogeneous catalysis: A combined DFT, microkinetic, and experimental study of water-gas shift on Au/MgO, J. Catal., 2017, 345, 157–169 CrossRef CAS.
- S. Pletincx, L. L. I. Fockaert, J. M. C. Mol, T. Hauffman and H. Terryn, Probing the formation and degradation of chemical interactions from model molecule/metal oxide to buried polymer/metal oxide interfaces, npj Mater. Degrad., 2019, 3, 23 CrossRef.
- S. N. Kerisit and J. J. De Yoreo, Effect of Hydrophilicity and Interfacial Water Structure on Particle Attachment, J. Phys. Chem. C, 2020, 124, 5480–5488 CrossRef CAS.
- J. L. Bañuelos, E. Borguet, G. E. Brown, R. T. Cygan, J. J. DeYoreo, P. M. Dove, M.-P. Gaigeot, F. M. Geiger, J. M. Gibbs and V. H. Grassian, et al., Oxide– and Silicate–Water Interfaces and Their Roles in Technology and the Environment, Chem. Rev., 2023, 123, 6413–6544 CrossRef PubMed.
- P. J. Anderson, R. F. Horlock and J. F. Oliver, Interaction of Water with Magnesium Oxide Surface, Trans. Faraday Soc., 1965, 61, 2754–2762 RSC.
- D. Ferry, A. Glebov, V. Senz, J. Suzanne, J. P. Toennies and H. Weiss, Observation of the Second Ordered Phase of Water on the MgO(100) Surface: Low Energy Electron Diffraction and Helium Atom Scattering Studies, J. Chem. Phys., 1996, 105, 1697–1701 CrossRef CAS.
- P. Liu, T. Kendelewicz, G. E. Gordon and G. A. Parks, Reaction of Water with MgO(100) Surfaces. Part I: Synchrotron X-ray Photoemission Studies of Low-Defect Surfaces, Surf. Sci., 1998, 412–13, 287–314 CrossRef.
- L. Giordano, J. Goniakowski and J. Suzanne, Partial Dissociation of Water Molecules in the (3 × 2) Water Monolayer Deposited on the MgO (100) Surface, Phys. Rev. Lett., 1998, 81, 1271–1273 CrossRef CAS.
- D. Abriou and J. Jupille, Self-Inhibition of Water Dissociation on Magnesium Oxide Surfaces, Surf. Sci., 1999, 430, L527–L532 CrossRef CAS.
- M. Odelius, Mixed Molecular and Dissociative Water Adsorption on MgO(100), Phys. Rev. Lett., 1999, 82, 3919–3922 CrossRef CAS.
- L. Delle Site, A. Alavi and R. M. Lynden-Bell, The Structure and Spectroscopy of Monolayers of Water on MgO: An Ab Initio Study, J. Chem. Phys., 2000, 113, 3344–3350 CrossRef CAS.
- J. H. Cho, J. M. Park and K. S. Kim, Influence of Intermolecular Hydrogen Bonding on Water Dissociation at the MgO(001) Surface, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 9981–9984 CrossRef CAS.
- R. M. Lynden-Bell, L. Delle Site and A. Alavi, Structures of Adsorbed Water Layers on MgO: an ab Initio Study, Surf. Sci., 2002, 496, L1–L6 CrossRef CAS.
- Y. D. Kim, J. Stultz and D. W. Goodman, Dissociation of Water on MgO(100), J. Phys. Chem. B, 2002, 106, 1515–1517 CrossRef CAS.
- Y. D. Kim, R. M. Lynden-Bell, A. Alavi, J. Stulz and D. W. Goodman, Evidence for Partial Dissociation of Water on Flat MgO(100) Surfaces, Chem. Phys. Lett., 2002, 352, 318–322 CrossRef CAS.
- Y. H. Yu, Q. L. Guo, S. Liu, E. G. Wang and P. J. Møller, Partial Dissociation of Water on a MgO(100) Film, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115414 CrossRef.
- K. Jug, B. Heidberg and T. Bredow, Cyclic Cluster Study of Water Adsorption Structures on the MgO(100) Surface, Surf. Sci., 2007, 601, 1529–1535 CrossRef CAS.
- K. Jug, B. Heidberg and T. Bredow, Molecular dynamics study of water adsorption structures on the MgO(100) surface, J. Phys. Chem. C, 2007, 111, 6846–6851 CrossRef CAS.
- E. Carrasco, M. A. Brown, M. Sterrer, H.-J. Freund, K. Kwapien, M. Sierka and J. Sauer, Thickness-Dependent Hydroxylation of MgO(001) Thin Films, J. Phys. Chem. C, 2010, 114, 18207–18214 CrossRef CAS.
- R. Włodarczyk, M. Sierka, K. Kwapień, J. Sauer, E. Carrasco, A. Aumer, J. F. Gomes, M. Sterrer and H.-J. Freund, Structures of the Ordered Water Monolayer on MgO(001), J. Phys. Chem. C, 2011, 115(14), 6764–6774 CrossRef.
- J. T. Newberg, D. E. Starr, S. Yamamoto, S. Kaya, T. Kendelewicz, E. R. Mysak, S. Porsgaard, M. B. Salmeron, G. E. Brown and A. Nisson, et al., Autocatalytic Surface Hydroxylation of MgO(100) Terrace Sites Observed under Ambient Conditions, J. Phys. Chem. C, 2011, 115, 12864–12872 CrossRef CAS.
- J. T. Newberg, D. E. Starr, S. Yamamoto, S. Kaya, T. Kendelewicz, E. R. Mysak, S. Porsgaard, M. B. Salmeron, G. E. Brown and A. Nilsson, et al., Formation of Hydroxyl and Water Layers on MgO Films Studied with Ambient Pressure XPS, Surf. Sci., 2011, 605, 89–94 CrossRef CAS.
- D. P. Woodruff, Quantitative Structural Studies Of Corundum and Rocksalt Oxide Surfaces, Chem. Rev., 2013, 113, 3863–3886 CrossRef CAS PubMed.
- E. Carrasco, A. Aumer, J. F. Gomes, Y. Fujimori and M. Sterrer, Vibrational Spectroscopic Observation of Ice Dewetting on MgO(001), Chem. Commun., 2013, 49, 4355–4357 RSC.
- J. P. Coulomb, B. Demirdjian, D. Ferry and M. Trabelsi, Thermodynamic and Structural Properties of Water Adsorbed Film on MgO(100) Ionic Surface, Adsorption, 2013, 19, 861–867 CrossRef CAS.
- N. M. Adhikari, A. Tuladhar, Z. Wang, J. J. De Yoreo and K. M. Rosso, No Hydrogen Bonding between Water and Hydrophilic Single Crystal MgO Surfaces?, J. Phys. Chem. C, 2021, 125, 26132–26138 CrossRef CAS.
- M. Ončák, R. Włodarczyk and J. Sauer, Water on the MgO(001) Surface: Surface Reconstruction and Ion Solvation, J. Phys. Chem. Lett., 2015, 6, 2310–2314 CrossRef PubMed.
- H. Onishi, C. Egawa, T. Aruga and Y. Iwasawa, Adsorption of Na atoms and oxygen-containing molecules on MgO(100) and (111) surfaces, Surf. Sci., 1987, 191, 479–491 CrossRef CAS.
- C. F. Jones, R. A. Reeve, R. Rigg, R. L. Segall, R. S. C. Smart and P. S. Turner, Surface area and the mechanism of hydroxylation of ionic oxide surfaces, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2609–2617 RSC.
- W. Langel and M. Parrinello, Hydrolysis at stepped MgO surfaces, Phys. Rev. Lett., 1994, 73, 504–507 CrossRef CAS PubMed.
- D. Ferry, S. Picaud, P. N. M. Hoang, C. Girardet, L. Giordano, B. Demirdjian and J. Suzanne, Water monolayers on MgO(100): structural investigations by LEED experiments, tensor LEED dynamical analysis and potential calculations, Surf. Sci., 1998, 409, 101–116 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- R. M. Hazen, Effects of temperature and pressure on the cell dimension and X-ray temperature factors of periclase, Am. Mineral., 1976, 61, 266–271 CAS.
- F. Zigan and R. Rothbauer, Neutronenbeugungsmessungen am Brucit, Neues Jahrb. Mineral., Monatsh., 1967, 137–143 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Jura and C. W. Garland, The Experimental Determination of the Surface Tension of Magnesium Oxide, J. Am. Chem. Soc., 1952, 74, 6033–6034 CrossRef CAS.
- A. J. Logsdail, D. Mora-Fonz, D. O. Scanlon, C. R. A. Catlow and A. A. Sokol, Structural, energetic and electronic properties of (100) surfaces for alkaline earth metal oxides as calculated with hybrid density functional theory, Surf. Sci., 2015, 642, 58–65 CrossRef CAS.
- A. Togo and I. Tanaka, First Principles Phonon Calculations in Materials Science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- NIST-JANAF Thermochemical Tables, ed. J. M. W. Chase, American Chemical Society, Washington, DC, 4th edn, 1998, pp. 1529–1536 Search PubMed.
- P. Janthon, S. Luo, S. M. Kozlov, F. Viñes, J. Limtrakul, D. G. Truhlar and F. Illas, Bulk Properties of transition metals: A challenge for the design of universal density functionals, J. Chem. Theory Comput., 2014, 10, 3832–3839 CrossRef CAS PubMed.
- J. Heidberg, B. Redlich and D. Wetter, Adsorption of Water Vapor on the MgO(100) Single Crystal Surface, Bunsen-Ges. Phys. Chem., Ber., 1995, 99, 1333–1337 CrossRef CAS.
- A. Moriata and T. Ishiyama, Recent progress in theoretical analysis of vibrational sum frequency generation spectroscopy, Phys. Chem. Chem. Phys., 2008, 10, 5801–5816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04848a |
| This journal is © the Owner Societies 2024 |