
Excited state hydrogen or proton transfer pathways in microsolvated n-cyanoindole fluorescent probes†
Salsabil
Abou-Hatab
and
Spiridoula
Matsika
 *
*
Department of Chemistry, Temple University, Philadelphia, PA, USA. E-mail: smatsika@temple.edu; Fax: +1 215 204 1532; Tel: +1 215 204 7703
First published on 13th January 2024
Abstract
The sensitivity of the fluorescence properties of n-cyanoindole (n-CNI) fluorescent probes to the microenvironment makes them potential reporters of protein conformation and hydration. The fluorescence intensity of 5-CNI, 6-CNI, and 7-CNI is quenched when exposed to water solvent whereas substitution on position 4 of indoles dramatically increases it. A potential mechanism for this sensitivity to water may be similar to that found in indole. The fluorescence of indole is found to be quenched when interacting with water and ammonia solvent molecules via radiationless decay through an S1 (πσ*)/S0 conical intersection caused by excited state proton or hydrogen transfer to the solvent molecules. In this study we examine this fluorescence quenching mechanism along the N–H bond stretch of n-CNI probes using water cluster models and quantum mechanical calculations of the excited states. We find that n-CNI–(H2O)1−2 clusters form cyclic or non-cyclic structures via hydrogen bonds which lead to different photochemical reaction paths that can potentially quench the fluorescence by undergoing internal conversion from S1 to S0. However, the existence of a high energy barrier along the potential energy surface of the S1 state in most cases prevents this from occurring. We show that substitution on position 4 leads to the highest energy barrier that prevents the fluorophore from accessing these non-radiative channels, in agreement with its high intensity. We also find that the energy barrier in the S1 state of non-cyclic 5-CNI–(H2O)1−2 excited complexes decreases as the number of water molecules increases, which suggests great sensitivity of the fluorescence quenching on the aqueous environment.
1 Introduction
Aromatic amino acids are natural fluorophores in proteins that are commonly used in various technologies for imaging and monitoring of biological structures and dynamics.1–14 Analogues of tryptophan and its chromophore indole are extensively used in fluorescence imaging of biological systems due to the sensitivity of their fluorescence properties to changes in the local environment.2–5,15–35 The radiative and non-radiative decay pathways of the parent chromophore indole have been widely studied in gas phase and in solvents with varying polarities and acidities.15,36–49 It is accepted that the solvent polarity is responsible for the change in emission maximum wavelength of indole and its various functionalized analogues, primarily due to differences in the dipole moment of the two low-lying electronically excited ππ* transitions, known as the La and Lb states (Fig. 1).15,21,25,26,28,34,35,37,38,41–55 A bright intense fluorescence is observed when emission takes place from the polar La state, while fluorescence is weak when emission takes place from the less polar Lb state. The relative stability of these states depends on the polarity of the solvent.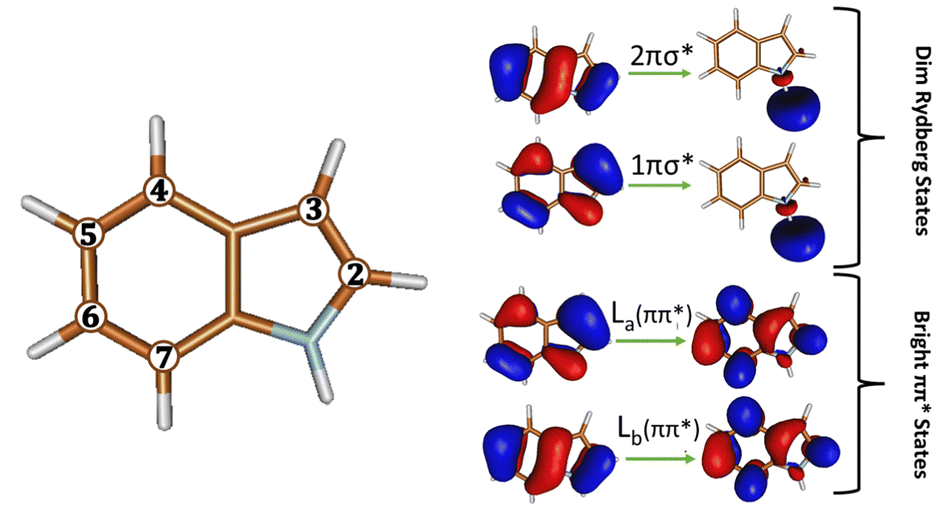 | ||
| Fig. 1 (Left): Illustration of the different positions on the bicyclic rings of indole, n, that can be functionalized. (Right): The orbitals of the ππ* and πσ* states involved in its radiative and non-radiative decay pathway. | ||
On the other hand, the dark Rydberg πσ* states (see Fig. 1) that lie above these ππ* states at absorption are found to play a vital role in quenching the fluorescence intensity.46,47,56 Gas phase computational studies have shown that excitation above the ππ* states promotes population to a Rydberg πσ* state. This Rydberg state dissociates along the N–H stretch coordinate and thus mediates N–H fission in indole and the ejection of a hydrogen atom or radiationless decay to the ground state.47,56 In solution, the polar solvent stabilizes the πσ* state such that it is now in close energetic proximity to the strongly emitting S1 (ππ*) state and forms a conical intersection (CI) with the electronic ground state S0.47,56 Radiationless decay via this S1 (πσ*)/S0 CI causes fluorescence quenching and a decrease in the fluorescence quantum yield as excited state hydrogen atom transfer (ESHT) from the NH group of indole to the hydrogen bonded (HB) solvent molecule takes place.46,47,56 Experimental and theoretical studies of indole and other aromatic chromophores with water clusters have found that ESHT and excited state proton transfer (ESPT) result in large emission Stokes's shift and are characterized by an ultrashort fluorescence lifetime.57 This acid–base photochemistry has also been observed in the parent probe tryptophan, other aromatic amino acids, and nucleic acids in polar protic solvents.39,58–63 Time resolved experiments have been performed to explore the dynamics in indole64,65 and microsolvated indole.66–68 The most recent ultrafast time resolved measurement on indole-(H2O)1 was able to capture the decay from the initially absorbed ππ* state to πσ* state and eventually to the ground state.68 The conversion from ππ* to πσ* happens in 445 fs while decay to the ground state was measured to be 13 ps.68
The fluorescence intensity, lifetime, and Stokes shift of n-cyanoindole (n-CNI) probes, were found to be largely and diversely dependent on the position of substitution of the cyano group on the indole rings (n) and on solvation (see Fig. 1 for position numbering).2 The fluorescence intensity is quenched when cyano substitution occurs on the six-membered ring (5-CNI, 6-CNI, 7-CNI) of the bicyclic assembly, with the exception of 4-CNI, which undergoes significant fluorescence enhancement.2–6 Static and time-resolved fluorescence measurements have shown that the fluorescence properties of 5-CNI and 5-cyanotryptophan are sensitive to changes in solvent polarity.4 The fluorescence quantum yield of 5-cyanotryptophan decreases from 0.11 to 0.01 when the solvent environment changes from 1,4-dioxane to H2O. Likewise, its lifetime rate decreases from 6.0 ns in 1,4-dioxane to 0.4 ns in H2O, respectively. In-turn, the fluorescence intensity becomes quenched upon hydration as the excited state non-radiative decay rate increases substantially. Similarly, for 5-CNI, an increase in Stokes' shift and drastic decrease in the fluorescence intensity is observed as the solvent changes from 1,4-dioxane to water solvent.4 This solvent-dependence of the fluorescence is also observed in 7-CNI. Studies using scattering methods have shown structural differences in 7-CNI dissolved in various ratios of organic-water binary mixtures that indicate the fluorescence intensity is sensitive to the formation of solvent clusters.5 In addition, fluorescence measurements from Hilaire et al.3 showed that the fluorescence lifetime of 7-CNI decreases from 14.5 ns to 2.0 ns when the probe is in dimethyl sulfoxide, a polar aprotic solvent to water, a protic and more polar solvent. Furthermore, the fluorescence intensity of 7-CNI fluorescent probe was found to decrease as the number of water molecules increased.4,5 The low quantum yield and fluorescence lifetime of these CNI fluorescent probes in aqueous solution is indicative of the existence of non-radiative pathways that quench the fluorescence intensity.
Experiments on microsolvated CNI systems have also been performed where the focus was more on the character of the S1 state and its radiative behavior. Experimental measurements of the induced dipole moment using Stark effect for isolated indole and indole-(H2O)1 cluster have reported that fluorescence occurs from the non-polar excited Lb state.42 Similarly, combined Franck–Condon and rotational constants fitting of 2-CNI–(H2O)1 cluster69 and rotationally resolved electronic spectroscopy of 3-CNI–(H2O)1 cluster70 reported that they emit from the Lb state. Mass-selected resonant two-photon ionization (R2PI) and UV-UV hole-burning, of jet cooled 5-CNI and 3-CNI and their water clusters, were also measured.71–73 Similar to indole-water complex, electronic and vibrational jet-cooled spectroscopic studies complemented by theoretical calculations reported that the character of the emitting state of isolated 5-CNI–(H2O)1−2 clusters is La.71
Modeling the absorption and fluorescence of n-CNI using high-level coupled cluster, (EOM-CCSD), and multireference perturbation theory (CASPT2) quantum chemical calculations and polarizable continuum model in our previous study provided an explanation as to why the fluorescence intensity of 4-CNI is enhanced while others are quenched.41 However, while the energetics were consistent with experimental absorption and fluorescence spectra, the trend in the relative fluorescence intensities was not reproduced.41 In addition, the energy level switching between the La and Lb states at emission does not explain why these n-CNI probes exhibit short fluorescence lifetime and low quantum yield in aqueous solution. It is clear that other fluorescence quenching mechanisms are involved. However, due to the scaling problem that quantum mechanical methods face, it is difficult to model the entire solvent environment. Instead in this study we extend the previous study by using similar electronic structure approaches to study small water clusters (with one or two water molecules) to simulate the effects that position of cyano substitution on the rings of indole has on the non-radiative decay pathways of n-CNI probes in microsolvation. Specifically, we focus along the dissociative N–H bond stretch where hydrogen/proton transfer from the probes to the water solvents would take place.
2 Methods
The non-radiative decay pathways of n-CNI probes were examined by computing the electronic potential energy surface (PES) along the N–H bond stretch coordinate of n-CNI–(H2O)m clusters quantum mechanically, where the position of substitution on the indole ring, n, is 2-7, and the number of water molecules HB to the probe(s), m, is 1 or 2. In the n-CNI–(H2O)1 clusters, the water molecule is HB to the proton donating NH group of indole (depicted in Fig. 2(a)) and in the n-CNI–(H2O)2 cluster, a second water molecule is simultaneously HB to the proton accepting cyano group of the probe (depicted in Fig. 2(b)). | ||
| Fig. 2 Example of the two water cluster models illustrated using 4-CNI probe. (a) One water molecule (m = 1) is HB at the proton donating NH group and (b) two water molecules (m = 2) are simultaneously HB to the proton donating NH and accepting cyano site of the probe. | ||
2.1 S1 minima
In order to study how the fluorescence intensity of n-CNI probes is quenched, we start by modeling the emission. We obtain the S1 minimum geometry of each n-CNI–(H2O)m cluster where the calculated character of the emitting state corresponds to that which is measured by experiment as discussed in the introduction. The characters are indicated in Table 1. The S1 minimum geometry was found to be sensitive to the method and basis set used. For this reason, we used several approaches and benchmarked them against available experimental information. The results of benchmarking are shown in detail in ESI.† The time dependent density functional theory (TDDFT) with the CAM-B3LYP functional was used, in addition to CC2, and ADC(2) approaches. For most systems CC2/aug-cc-pVDZ and TD-CAM-B3LYP/6-311++G(d) gave the same results, and the TD-CAM-B3LYP/6-311++G(d) results are discussed in the main paper while the others are shown in ESI.† Indole and 3-CNI were more problematic. Indole has a small gap between the ππ* and Rydberg states, and adding diffuse functions led to overstabilization of the πσ* Rydberg state predicting this to be the minimum, which is contrary to experiment. So the optimized structure for Ind-(H2O)1 was obtained with CC2/cc-pVDZ. For 3-CNI–(H2O)1−2 we used CC2/aug-cc-pVDZ because TD-CAM-B3LYP/6-311++G(d) predicted the La state to be the S1 minimum in disagreement with experiment. Vibrational frequencies calculations were also performed at the same level of theory to confirm that these are true minima ensuring that all of the frequencies along all degrees of freedom of the system are positive.| Char | Oscillator strength | Energy barrier (eV) | |||
|---|---|---|---|---|---|
| 1H2O | 2H2O | 1H2O | 2H2O | ||
| a Indicates that the character has been confirmed experimentally. | |||||
| Ind | Lba | 0.047 | 0.11 | ||
| 2-CNI | Lba | 0.073 | 0.079 | 1.21 | 1.26 |
| 3-CNI | Lba | 0.140 | 0.173 | 0.55 | 0.65 |
| 4-CNI | La | 0.183 | 0.159 | 1.04 | 1.00 |
| 5-CNI | Laa | 0.039 | 0.035 | 0.92 | 0.60 |
| 6-CNI | La | 0.182 | 0.178 | 0.97 | 0.98 |
| 7-CNI | La | 0.109 | 0.110 | 0.76 | 1.30 |
The energies at the optimized S1 structures were recomputed using EOM-CCSD/6-311++G(d) and were used to calculate the adiabatic energies (energy of S1 at S1 minimum minus energy of S0 at S0 minimum). Ground state geometries were optimized at the B3LYP/6-311++G(d) level and the ground state CCSD/6-311++G(d) energies at these geometries were used in the calculation of adiabatic energies.
2.2 Potential energy surfaces
Starting from the S1 minimum geometry, constrained optimizations of the S1 excited state along the dissociative N–H bond coordinate were performed at the TD-CAM-B3LYP/6-311++G(d) level of theory, where N–H bond distances were displaced by increments of 0.1 Å and fixed, while all other nuclear coordinates were allowed to relax. Grimme's Dispersion (D3) correction was applied to these TDDFT calculations. It has been shown that these corrections are important when treating HB systems.74 The energies of the probe-water clusters were refined by performing single point excited state calculations at each converged geometry at the various N–H bond distances of the Ind-(H2O)1 and n-CNI–(H2O)1−2 clusters at the EOM-CCSD level of theory with the 6-311++G(d) basis set.Natural Transition Orbitals (NTO) were computed to characterize the excited states for each probe-water complex at this level of theory. Moreover, ChelpG charges were computed to assess if excited state charge transfer takes place. In order to test the accuracy of the geometries obtained at the CAM-B3LYP level, additional constrained optimizations were performed at the ADC(2)/aug-cc-pVDZ level followed by single point calculation of the excited states at the EOM-CCSD level of theory for comparison. The results based on ADC(2) optimizations are shown in the ESI.† Although it is well known that single reference methods poorly describe regions of configuration space where the S0 and S1 energies are close,75 we use the EOM-CCSD method cautiously to get a description of the barriers along S1 and a qualitative description of how the energy gap between the S1 and S0 changes as we stretch the N–H bond. The accuracy of the potential energy profiles predicted by EOM-CCSD is tested by reproducing the PES of these clusters at the CASSCF/aug-cc-pVDZ level of theory and at select cases using CASPT2/aug-cc-pVDZ. An active space of (12 e−, 11 orbitals) was used for Ind-(H2O)1 and (14 e−, 13 orbitals) for the n-CNI–(H2O)2 clusters respectively, except for 7-CNI. Efforts that were made to generate the PES of 7-CNI–(H2O)1−2 clusters at the CASSCF(14,13)/aug-cc-pVDZ level of theory were not successful because it was difficult to get the correct orbitals in the active space. The orbitals in the active space of each excited complex are depicted in Section S2 of the ESI.† A conical intersection search was performed for 5-CNI–(H2O)2 cluster starting from a geometry where the N–H distance is 1.52 Å using the CASSCF(14,13) and aug-pcseg-0, which is the segmented contracted version of the aug-pc-0 Jensen basis set.76–78 We used the aug-pcseg-0 basis set because the aug-cc-pVDZ basis set is not supported by Molpro for CI optimizations. Results of the CI optimization are shown in ESI,† Section S1.3. In addition, CASPT2/aug-cc-pVDZ calculations were performed on Ind-(H2O)1 and 4-CNI–(H2O)2 clusters using the same active space as CASSCF, to account for dynamic electronic correlation and obtain the correct excited state energetic ordering (results shown in ESI,† Section S1.1).
2.3 Bulk solvation effects
The PESs of the 7-CNI–(H2O)2 cluster were calculated with an implicit solvation model, i.e. the Polarizable Continuum Model (PCM),79 to evaluate how the electrostatic effects from the bulk properties of the solvent impact the geometries and the electronic potential energy surface. To assess the effect on the geometries, we performed constrained optimization of the excited S1 state along the N–H bond stretch of 7-CNI–(H2O)2 at the CAM-B3LYP/6-311++G(d) level of theory in gas phase and with equilibrium solvation using PCM. Optimizing on the excited state by default uses equilibrium state-specific approach of PCM which allows for solvent to undergo relaxation and the electronic density of the solvent to equilibrate with that of the excited state of the probe. Thus, the solute is polarized and influenced by both the fast electronic and slow nuclear components of the solvent.We also studied how the inclusion of the electrostatic effect of the solvent affects the potential energy of the electronic states by performing single point excited state calculations on the gas phase geometries using EOM-CCSD/6-311++G(d) and the linear-response non-equilibrium approach of PCM. In this approach, solvent relaxation is not considered and the probe experiences polarization from the fast electronic component of the solvent.
We have also tested the solvation effects using the Solvation Model Density (SMD) type of PCM,80 which in addition to electrostatics accounts for short-range interactions between the solute and solvent molecules in the first solvation shell. We used SMD for the S1 minimum of 7-CNI combined with CAM-B3LYP.
Geometry optimization and vibrational frequencies calculations using the CAM-B3LYP81 method were performed using Gaussian 1682 while the ADC2 and CC283,84 calculations were done using TURBOMOLE V6.5.85 The single point excited state calculations of these optimized geometries at the EOM-CCSD86 and CASSCF/CASPT287,88 levels were performed in Qchem 5.389 and Molpro 2021.1,90 respectively.
3 Results
We begin investigating the fluorescence behavior of n-CNI–(H2O)m complexes by starting at the S1 minimum. Our initial structures are motivated by the assumption that both NH and CN sites will be hydrogen bonded with water. This assumption is validated by MD simulations, which are detailed in ESI† (Section S6).We found that optimization of the electronically excited S1 state of these complexes resulted in the formation of cyclic and non-cyclic probe-water structures portrayed in Fig. 3(a) and (b) respectively. The cyclic structure is formed when the NH group of n-CNI is bridged by the HB network of waters to the cyano group. The excited state S1 minimum of Ind-(H2O)1 cluster results in a non-cyclic complex since there is only one site available for HB. Similarly, non-cyclic complexes are formed for indole analogues where positions n = 3–6 of indole's rings are functionalized with the electron-withdrawing cyano group, since the two HB sites are too far away from each other to form a cyclic structure (Fig. 3(a)). For 6-CNI the monohydrated complex does not form a cyclic structure but the dihydrated one, 6-CNI–(H2O)2, does. 2-CNI–(H2O)1−2 and 7-CNI–(H2O)1−2 clusters also form cyclic structures at the excited state S1 minimum.
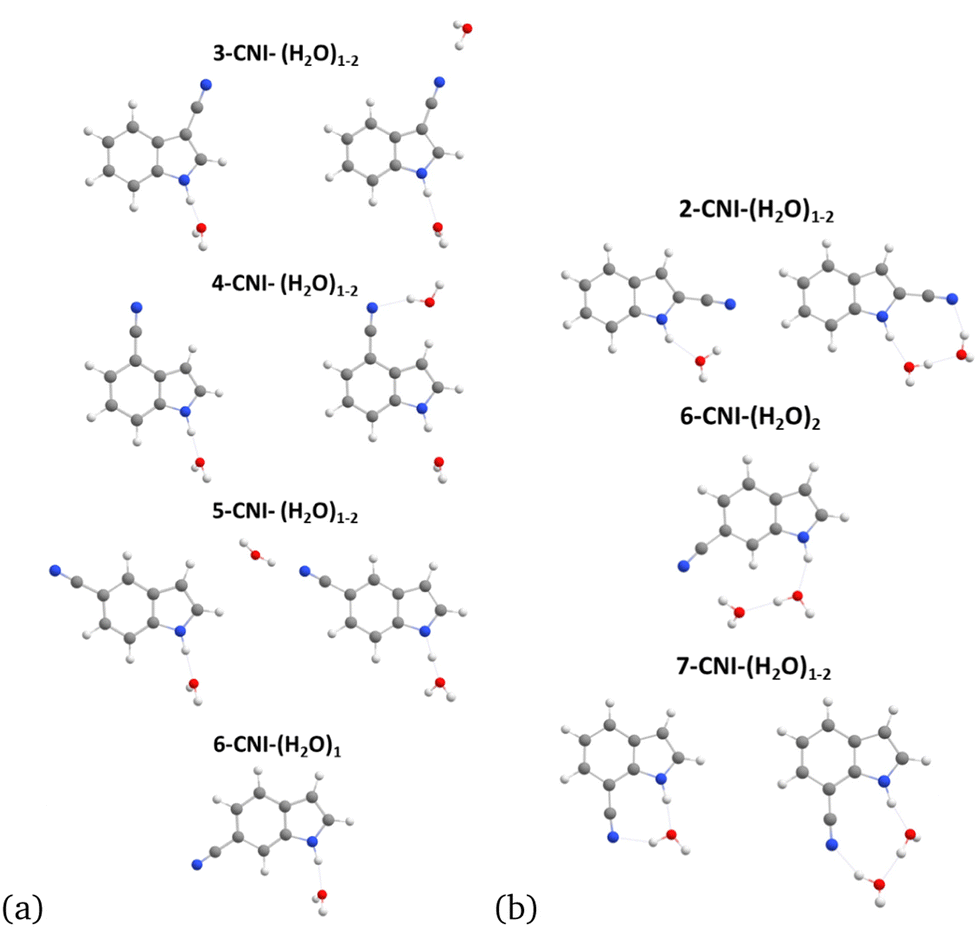 | ||
| Fig. 3 S1 minimum geometry of n-CNI–(H2O)m clusters for (a) non-cyclic and (b) cyclic structures. | ||
We investigated the non-radiative decay pathways that can potentially quench the fluorescence intensity of these probe-water clusters by computing the PES of the ground and excited states of the n-CNI–(H2O)m clusters as a function of N–H dissociation. We find that the formation of the cyclic n-CNI–(H2O)m complexes leads to an alternative solvent-assisted fluorescence quenching mechanism than that of the non-cyclic structures. For this reason, we will present the results of the non-radiative decay pathways along the N–H bond coordinate of these cyclic and non-cyclic structures separately in the next sections.
3.1 S1 minima
Before discussing the pathways, we will discuss comparisons with experiments so that we can benchmark our methodology. We will focus on the adiabatic excitation to the S1 minimum and the characters of the minimum, where there is available experimental information.We start with the character of the minima, where there is experimental information. Experimental information exists for monohydrated indole, 2-CNI, 3-CNI, and for mono and dihydrated 5-CNI.42,69–71 Our calculations predict that the character is the same in the mono and dihydrated systems, i.e. one additional water does not change the character. Both experiment and theory have demonstrated that this is the case for the addition of one water molecule to isolated n-CNI with respect to indole, 2-CNI, 3-CNI, and 5-CNI.69–71 In 5-CNI71 it is also found that addition of a second water molecule also retains the character of La. For 2-CNI and 3-CNI their fluorescence properties are not sensitive to the amount of water solvent.3,41 These systems have an Lb minimum isolated, monohydrated and in bulk solvent.3,41,69,70 In the case of the 4-, 6-, and 7-CNI–(H2O)m clusters, no experimental studies have been done that characterize the S1 minimum in solution or with water clusters. For these systems we rely on previous and current theoretical calculations to establish the character of the minimum.
Table 1 shows the character and oscillator strengths at the S1 minima for the monohydrated and dihydrated systems obtained at the EOM-CCSD/6-311++G(d) level of theory. The geometries were obtained at the CAM-B3LYP level of theory for all systems except indole and 3-CNI, where CC2/cc-pVDZ and CC2/aug-cc-pVDZ were used, respectively. In our calculations, Ind, 2-CNI and 3-CNI have Lb minima, while the 4,5,6,7-CNI have La character. Additional results using different electronic structure theory are given in ESI.† It is interesting to highlight that the oscillator strength is not always much smaller for the Lb state compared to the La. While this is true for indole, once substituents are introduced the electronic structure changes and this can be reflected in the oscillator strengths as well. For example, while the minimum for 3-CNI is Lb and for 5-CNI is La the oscillator strength is higher for 3-CNI.
In order to benchmark the energetics, Table 2 shows the calculated adiabatic energies to the S1 minima for isolated and monohydrated systems and compares them to the experimental 0–0 energies whenever available. Geometries of the ground state are optimized using B3LYP/6-311++G(d) while the S1 minima are optimized using either CC2/aug-cc-pvdz (CC2/cc-pvdz for Ind) (CC2 columns) or CAM-B3LYP/6-311++G(d) (DFT column) methods. The agreement between theory and experiment is very good, with an average error of 0.1 eV, while the maximum error is 0.24 eV for isolated indole. For the monohydrated system we can also compare the effect of using the CC2 or CAM-B3LYP methods for optimizing the S1 minimum. The energies differ by 0.1 eV or less, so the effect of the methodology for optimization is small. These results provide confidence to carry out the pathways along NH.
3.2 Non-cyclic clusters
 | ||
| Fig. 4 (a) Overlaid PES of S1 (dotted curve) and S0 (solid curve) of non-cyclic Ind-(H2O)1 and 3-, 4-, 5-CNI–(H2O)2 clusters along the dissociative N–H bond coordinate computed at EOM-CCSD/6-311++G(d) level of theory using geometries optimized at the CAM-B3LYP/6-311++G(d) level with the exception of the S1 minimum geometries of Ind and 3-CNI which were optimized using CC2/cc-pVDZ and CC2/aug-cc-pVDZ, respectively, to obtain the correct S1 minimum character. (b) The corresponding oscillator strengths of S1 overlaid for all of the non-cyclic clusters. (c) and (d) PES and oscillator strengths for clusters with one water molecule. | ||
The accuracy of these pathways was further tested by using multireference methods, CASSCF and CASPT2. The results are shown in ESI† Section S1, showing overall similar behavior qualitatively. Fig. S8 (ESI†) shows the overlaying PES for all non-cyclic n-CNI, similar to Fig. 4. The region where the methods deviate more is when the two surfaces approach each other at long N–H. The S0–S1 gap is computed to be smaller at the CASSCF level which can describe this region more accurately. The small gap, reaching 0.5 eV in some cases, indicates that conical intersections should be present nearby. We confirmed their existence for 5-CNI–(H2O)2 as an example, and the results are shown in ESI,† Section 1.4. The energy barriers at the CASSCF level for Ind, 3-, 4-, and 5-CNI–(H2O)2 clusters are 0.27, 0.00, 0.53, and 0.35 eV respectively, smaller in general than the EOM-CCSD values, but with similar trends, with the exception of 3-CNI–(H2O)2 which is predicted to be barrierless. Some caution should be given however in the accuracy of the CASSCF barriers, because the character of the S1 minimum may not always be the correct one, as discussed in ESI.† Additional information about the behavior and character of the S1 PES is given by examining the oscillator strengths. The oscillator strengths corresponding to S1 of these non-cyclic clusters depicted in Fig. 4(b) and (d), show that as the NH bond increases, the oscillator strength decreases and after the barrier it approaches zero for all of the systems. We characterized the S1 state at every point by computing NTOs at the EOM-CCSD level, and found that, at the starting point the S1 minimum is a ππ* transition (either La or Lb) and transforms to πσ* at the last point. This indicates that as the N–H is stretched the πσ* is stabilized and the ππ* becomes destabilized. The two states switch at the barrier and that is the reason for the very small oscillator strength which is characteristic of the πσ* state.
Furthermore, the net ChelpG atomic charges of the monomer probe and the one and two waters in the non-cyclic clusters were computed for the first and last geometry and reported in ESI,† Section S3. The results show that as the N–H bond is elongated, the net molecular charges of the probes and the water molecules do not change significantly. The lack of change in the net molecular charge of the fluorophore and water molecules indicates that this is a H atom transfer where the resulting products are radicals.
 | ||
| Fig. 5 (a) PES of S0 (solid curves) and S1 (dotted curves) of 5-CNI–(H2O)1 (brown) and 5-CNI–(H2O)2 (gray); (b) corresponding oscillator strength of S1 computed along the dissociative N–H coordinate at the EOM-CCSD/6-311++G(d) level of theory. The natural transition orbitals of the S1 minimum starting geometry of the 5-CNI–(H2O)2 cluster are depicted on the left-hand side bordered in gray and in brown is that of the 5-CNI–(H2O)1. Next to them on the right-hand side are the natural transition orbital for the last geometry of the clusters with 1 and 2H2O after ESHT has taken place. | ||
3.3 Cyclic clusters
 | ||
| Fig. 6 PES of the electronic ground state, S0 (solid curve), and first electronic excited state, S1 (dotted curve) computed at the EOM-CCSD/6-311++G(d) level for (a) 6CNI–(H2O)1 (blue) and 6-CNI–(H2O)2 (red) clusters; (c) 7CNI–(H2O)1 (blue) and 7-CNI–(H2O)2 clusters (red); (e) 2CNI–(H2O)1 (blue) and 2-CNI–(H2O)2 (red) clusters. Their corresponding oscillator strengths are plotted in (b), (d), and (f), respectively. The natural transition orbitals of the S1 minimum starting geometries of clusters are depicted on the left-hand side and the natural transition orbital for the last geometry of the clusters after proton transfer takes place are depicted on the right-hand side. Energies of S0 (green, square) and S1 (green, triangle) of the final geometry of 7-CNI–(H2O)1 with the addition of a second water molecule, denoted 7CNI–(H2O)2-G, are shown in (c). Natural transition orbitals corresponding to 7-CNI–(H2O)2 -G are shown in (b) bordered in green. | ||
In the case of 6-CNI, the proton is transferred to position n = 7 of indole's ring (Fig. 1), breaking the aromaticity of the benzene ring through the network of the two water molecules. An energy barrier of 0.98 eV is found prior to this proton transfer. The proton transfer is accompanied by a decrease in the oscillator strength of the S1 state (6(b)) from 0.18 to zero. The NTO of this last geometry shows a ππ* character. The electron density in the HOMO is found to be localized on the pyrrole ring of the 6-CNI monomer and no electron density is found on the water molecules portraying that the final products are neutral species consistent with the net molecular charge reported in ESI,† Section S3. Contrary to 6-CNI–(H2O)2, in 6-CNI–(H2O)1 the final point is a πσ* state, similar to what was discussed in the previous section.
7-CNI also shows two different behaviors along the NH stretching pathway. The first step involves a proton transfer to water. During the second step two possible mechanisms exist. In the monohydrated system during the second step of the mechanism, as the N–H bond distance increases to 2.04 Å, a proton is transferred from the H3O+ ion to the CN group of the 7-CNI− ion changing the triple bond to a double bond and increasing the length of the C–C bond connecting to the ring. The geometry is shown in Fig. 7. The S1 state at the final point is characterized as  state where the
state where the  is a π-orbital that belongs to the CN group and is in-plane with the molecular structure (a′ symmetry) unlike the other π molecular orbitals which are out-of-plane (a′′ symmetry). Following the proton transfer to the CN of 7-CNI, the S0–S1 gap decreases substantially, and the oscillator strength becomes zero. This same mechanism is observed for both mono and dihydrated 2-CNI–water clusters. PES are shown in Fig. 6(e) and structures in Fig. 8. In Fig. 7 and 8 we show the S1 minimum starting geometry where emission takes place (left-hand) and the final geometry after proton transfer has occurred (right-hand) for 7-CNI and 2-CNI with (a) 1H2O and (b) 2H2O clusters. It is clear in the figure that 7-CNI–(H2O)1, 2-CNI–(H2O)1, and 2-CNI–(H2O)2 behave similarly.
is a π-orbital that belongs to the CN group and is in-plane with the molecular structure (a′ symmetry) unlike the other π molecular orbitals which are out-of-plane (a′′ symmetry). Following the proton transfer to the CN of 7-CNI, the S0–S1 gap decreases substantially, and the oscillator strength becomes zero. This same mechanism is observed for both mono and dihydrated 2-CNI–water clusters. PES are shown in Fig. 6(e) and structures in Fig. 8. In Fig. 7 and 8 we show the S1 minimum starting geometry where emission takes place (left-hand) and the final geometry after proton transfer has occurred (right-hand) for 7-CNI and 2-CNI with (a) 1H2O and (b) 2H2O clusters. It is clear in the figure that 7-CNI–(H2O)1, 2-CNI–(H2O)1, and 2-CNI–(H2O)2 behave similarly.
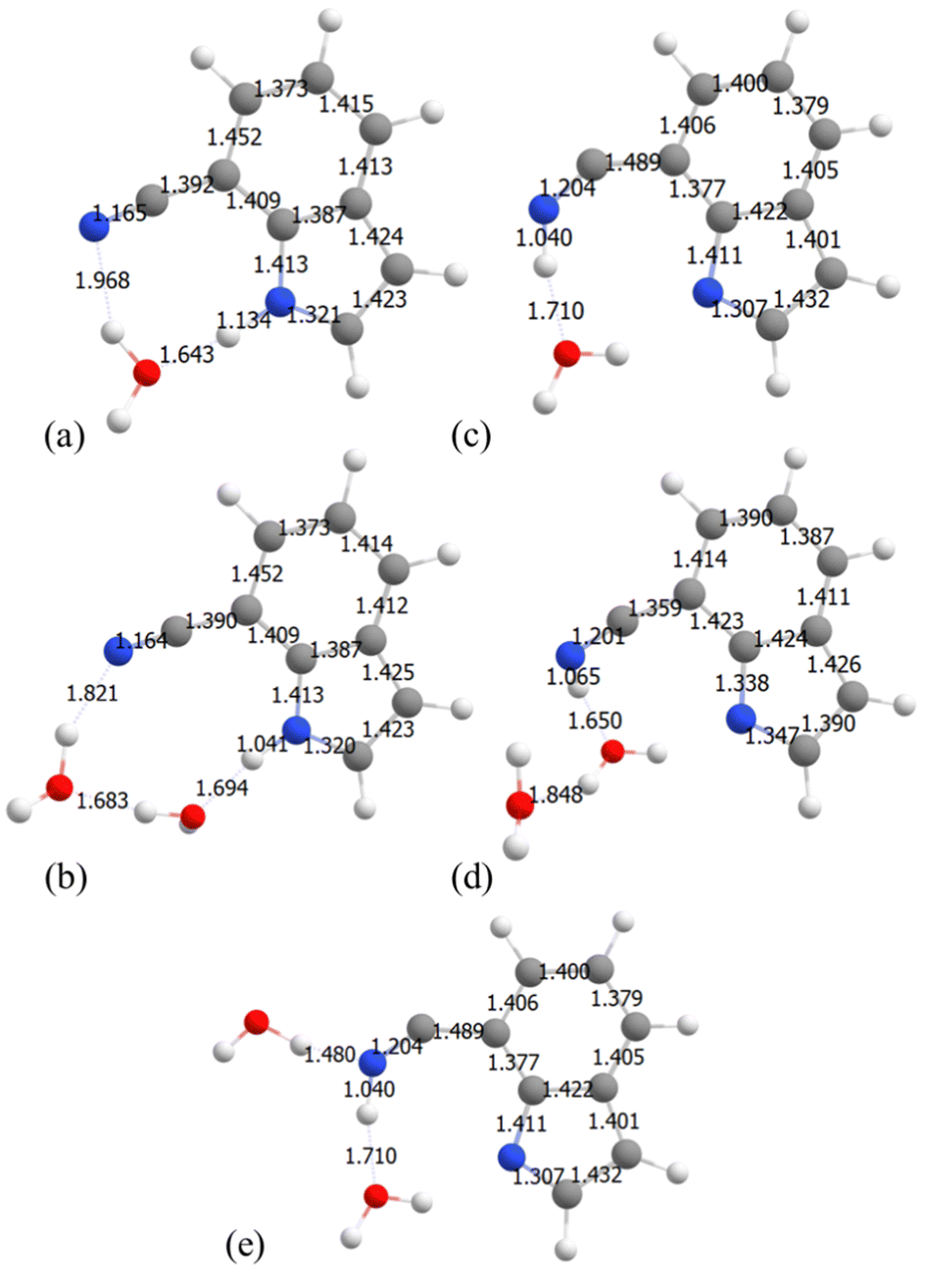 | ||
| Fig. 7 7-CNI geometries at S1 PES before ESPT (a) and (b) and after ESPT (c) and (d) obtained from constrained optimizations at the CAM-B3LYP/6-311++G(d) level of theory. (a) and (c) 2-CNI with 1H2O and (b) and (d) 2CNI with 2H2O clusters. (e) 7-CNI–(H2O)2-G: Addition of 1H2O to the last geometry in the 7-CNI–(H2O)1 cluster. | ||
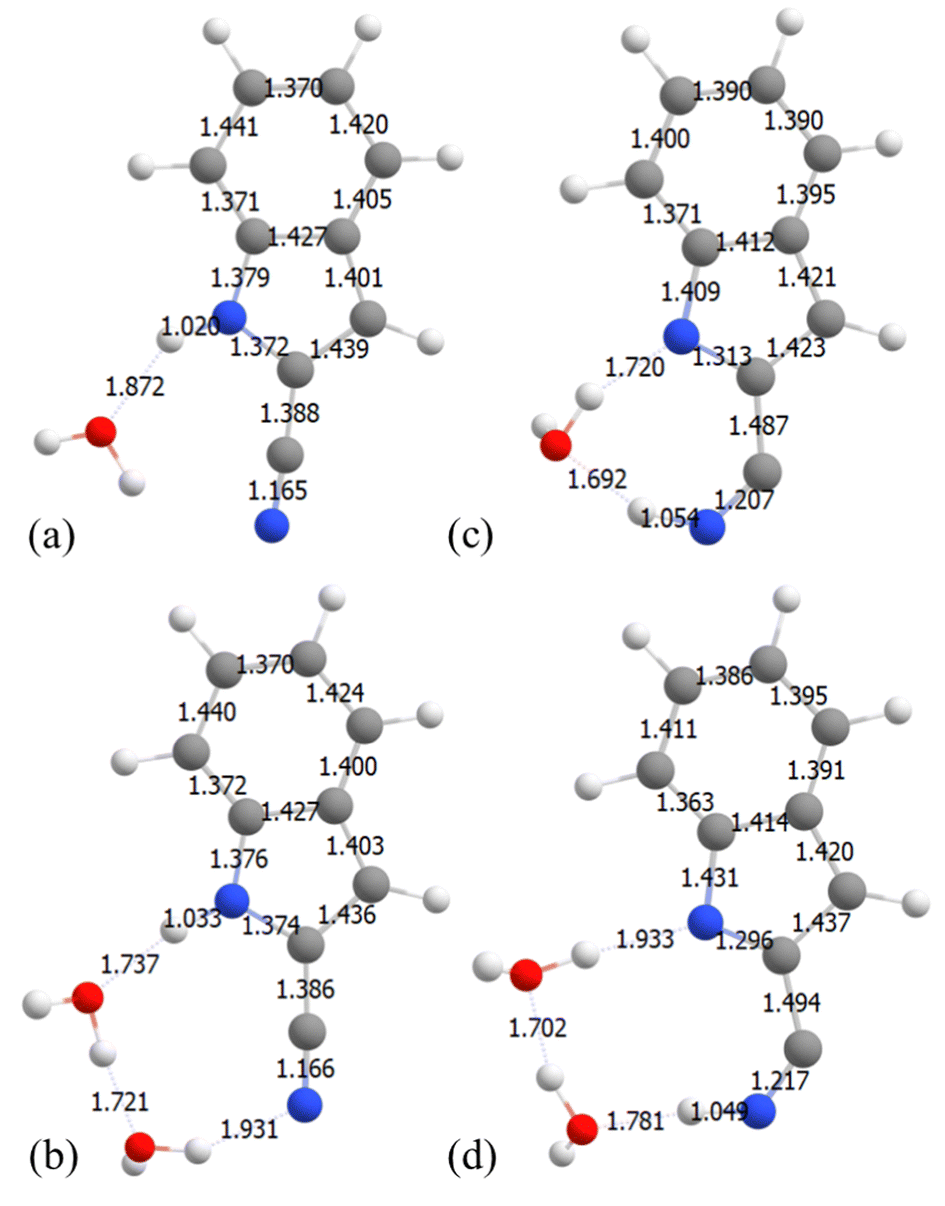 | ||
| Fig. 8 2-CNI geometries at S1 PES before ESPT (a) and (b) and after ESPT (c) and (d) obtained from constrained optimizations at the CAM-B3LYP/6-311++G(d) level of theory. (a) and (c) 2-CNI with 1H2O and (b) and (d) 2-CNI with 2 H2O clusters. | ||
In dihydrated 7-CNI however a different pathway is observed. Although the second step still involves the proton transferring back to 7-CNI, the energies and final structure look different. The energy of the S1 state does not become stabilized but it continues to increase, and the oscillator strength does not go to zero. As can be seen in Fig. 6(c), as we stretch the N–H bond from 1.04 to 2.54 Å, the S1 energy continues to increase parallel to that of the S0 PES and the energy gap between them remains large. The difference in energies can be explained by the differences in the final structures. We do this by examining the bond length differences in the S1 minimum and final geometry of the 7-CNI–(H2O)1−2 complexes after proton transfer has occurred. In Fig. 7 at the S1 minimum both clusters form a cyclic clusters. Although proton transfer takes place for 7-CNI modeled with both the 1H2O and 2H2O clusters we find that the final geometries after the proton has transferred to the CN are different. In the 1H2O cluster, the C8-CN bond is elongated from 1.392 to 1.489 Å which allows the C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– angle to become bent at a 141° angle, and the CNH substituent is loosely connected to the ring. On the other hand, in the 2H2O cluster, the C8-CN bond length of the 7-CNI probe decreases from 1.390 Å (Fig. 7(c)) to 1.359 Å (Fig. 7(d)) after H is transferred to the CN group which results in a near linear CCN– structure at an angle of 168°.
N– angle to become bent at a 141° angle, and the CNH substituent is loosely connected to the ring. On the other hand, in the 2H2O cluster, the C8-CN bond length of the 7-CNI probe decreases from 1.390 Å (Fig. 7(c)) to 1.359 Å (Fig. 7(d)) after H is transferred to the CN group which results in a near linear CCN– structure at an angle of 168°.
To assess whether having this bent C–CN- angle is essential for the small S1–S0 gap observed in 7-CNI–(H2O)1, 2-CNI–(H2O)1, 2-CNI–(H2O)2 but not in 7-CNI–(H2O)2, we added a water molecule near the cyano group of the final geometry of the 7-CNI–(H2O)1 cluster (shown in the right-hand panel of Fig. 6(c)), where C–C is elongated and S1–S0 gap is small (geometry shown in Fig. 6(d) in green and in Fig. 7(e)) and computed the energy of the S0 and S1. We found that indeed the bent C–CN– angle results in a small S1–S0 energy gap and an oscillator strength of zero.
In summary, these results suggest that the photochemical pathways that the 2-CNI, 6-CNI and 7-CNI can take are very sensitive to explicit hydration, providing different results depending on the exact patterns of hydration.
On the other hand implicit solvation seems to be insignificant. We included electrostatic effects using the implicit solvation model, PCM, and examined whether this can result in a different photochemical reaction path or if inclusion of long-range polarization from the solvent can stabilize the S1 energy and decrease the S1–S0 energy gap. We tested this on 7-CNI–(H2O)2 as a representative system. We examined the effect of solvation on the geometries and on the excitation energies separately. In the first approach, the constrained optimization of the S1 PES along the N–H bond stretch was computed with CAM-B3LYP/6-311++G(d) and PCM, which uses equilibrium solvation allowing the solvent to equilibrate and relax with the solute. In the second approach, we used the excited state geometries of the isolated 7-CNI–(H2O)2 clusters and recomputed the energies with EOM-CCSD/6-311++G(d) and linear response (LR)-PCM where the fast electronic component of the solvent polarizes the solute and thus only the energy is affected and not the geometry. The PES of the S1 and S0 and corresponding oscillator strengths which were computed using these two approaches are shown in ESI,† in Section S5. Both the geometries and energies were not impacted by the inclusion of PCM. Finally, we examined whether non-electrostatic effects were important by calculating the energies at the S1 minimum of 7-CNI–(H2O)2 by comparing to the SMD variation of IEFPCM solvation. The energy changed by 0.004 eV and the oscillator by 0.01, so the effect is negligible.
4 Discussion
It was observed experimentally that substitution of a cyano group on different carbons of the bicyclic ring of indole leads to diverse fluorescence properties in aqueous solution. Substitution on position 4 enhances the fluorescence intensity and increases the fluorescence lifetime and quantum yield.2,3 On the other hand, substitution on positions 5, 6, and 7 was found to quench the relative fluorescence intensity and decrease the fluorescence lifetime and quantum yield.2,4,5 Furthermore, substitution of the cyano group on the pyrrole ring of indole resulted in weakly fluorescent probes in aqueous solution.2 In our previous study we investigated how the emission of these n-CNI positional isomers depends on the solvent by modeling the absorption and fluorescence using EOM-CCSD and CASPT2 and implicit solvation.41 Our calculations in that study did not reproduce the trend of the relative fluorescence intensity or Stokes's shift, which suggested that non-radiative pathways are also involved. In the current study we explored the non-radiative pathways of n-CNI–water clusters quantum mechanically to uncover the fluorescence quenching mechanism of the probes in microsolvation. We found that the fluorescence properties of n-CNI–(H2O)1−2 clusters depend on three factors, which include (1) the formation of a cyclic probe-water structure, (2) the energy barrier in the S1 PES, and (3) the oscillator strength of the S1 excited state minimum at emission.4.1 Formation of cyclic structures
Optimization of the S1 minimum of the n-CNI–(H2O)1−2 clusters showed that in some positional isomers the fluorophores formed a cyclic complex with the water molecules in the excited state by bridging the NH group to the cyano group of the n-CNI probes via hydrogen bond. The non-cyclic structures include Ind-(H2O)1, 3-, 4-, and 5-CNI–(H2O)1−2, as well as 6-CNI–(H2O)1. The S1 and S0 PES of these non-cyclic structures were found to behave and share the same solvent-mediated excited state pathways as microsolvated indole. It was found that as the N–H bond is stretched, ESHT takes place where a H atom is transferred from the NH group of the CNI moiety to the HB water molecule resulting in the formation of radicals. Along this reaction path the Rydberg πσ* state becomes stabilized at long bond distances decreasing the energy gap between the S1 and S0 PESs and most likely leading to a conical intersection where the fluorophore could relax non-radiatively.In contrast, when a cyclic structure is formed such as in the case of 2-, 7-CNI–(H2O)1−2, and 6-CNI–(H2O)2, a different photochemical reaction along the N–H bond coordinate exists. In these cyclic structures we find a two step ESPT mechanism where in the first step the acidic proton of the CNI is transferred to the H2O molecules forming CNI− and H3O+ ion pairs and then a proton is transferred back to the CNI− in the second step. In 2- and 7-CNI isomers, the proton transfers to the CN substituent, reducing the triple bond to a double bond. Unlike the non-cyclic structures, a  state which lies higher in energy than the πσ* at emission becomes stabilized along the pathway. We find that elongation of the C7-CN bond, which allows the –CN–H angle to become bent, is significant for the S1–S0 gap to decrease. Similarly, the 6-CNI–(H2O)2 is also found to have this double excited state proton transfer mechanism, however the second proton does not get transferred to the CN group because it is too far away. Instead, the second proton is transferred to the C7 of indole, breaking the aromaticity of the benzene ring. In all cases, along the ESPT reaction path, the oscillator strength approaches zero after the first proton transfer. The S1–S0 energy gap then decreases when the S1 becomes stabilized in response to the second proton being transferred back to the CNI− neutralizing the ions. If the energy gap becomes small enough it could lead to internal conversion under favorable conditions.
state which lies higher in energy than the πσ* at emission becomes stabilized along the pathway. We find that elongation of the C7-CN bond, which allows the –CN–H angle to become bent, is significant for the S1–S0 gap to decrease. Similarly, the 6-CNI–(H2O)2 is also found to have this double excited state proton transfer mechanism, however the second proton does not get transferred to the CN group because it is too far away. Instead, the second proton is transferred to the C7 of indole, breaking the aromaticity of the benzene ring. In all cases, along the ESPT reaction path, the oscillator strength approaches zero after the first proton transfer. The S1–S0 energy gap then decreases when the S1 becomes stabilized in response to the second proton being transferred back to the CNI− neutralizing the ions. If the energy gap becomes small enough it could lead to internal conversion under favorable conditions.
4.2 Energy barrier in the S1 state
Although several pathways were revealed, a major condition for their accessibility is the presence and height of the energy barrier in the PES of the S1 excited state along the pathways. The energy barriers predicted at the EOM-CCSD level of theory for clusters with 1 and 2 water molecules are reported in Table 1.Among the non-cyclic structures, 4-CNI–(H2O)1−2 was found to have the highest energy barrier along the S1 PES. When the energy barrier is large, the excited molecule becomes trapped in the S1 minimum. This barrier would limit access to the CI and in-turn increase the excited state lifetime. This further contributes to our explanation for why the relative fluorescence lifetime and quantum yield is higher for 4-CNI in solution in addition to the reasons discussed in our previous work. Ind-(H2O) has the lowest barrier, in agreement with the time-resolved experiments that measure fast nonradiative decay.68 Moreover, the energy barrier along the S1 PES of the 5-CNI is found to decrease significantly as the number of water molecules in the first solvation shell increases from 1 to 2. This suggests that the energy barrier could further decrease in solution, thus increasing accessibility to the CI. This prediction is in good agreement with experimental observations of the fluorescence response of 5-CNI in aqueous solution.
Furthermore, probe-water complexes that form cyclic structures were also found to have significantly large energy barriers in the S1 PES, which would make radiationless decay unlikely, or nearly impossible. In the cases of 6-CNI and 7-CNI, we do see that the type of pathway found depends on the number of water molecules hydrogen bonded to the chromophore. This sensitivity agrees with experimental results, however the height of barriers we found would suggest that all of these pathways are inaccessible. More calculations are thus needed to explain how the fluorescence intensity of 7-CNI and 6-CNI is affected by solvation.
These results suggest that like 4-CNI, 6-, 7-, and 2-CNI fluorescent probes would have an increased fluorescence lifetime and quantum yield in aqueous solution, which is not the case. Thus we must explore other factors that can distinguish the unique fluorescence behavior of these probes from one another. It is likely that microsolvation is not sufficient to describe the observed behavior in solution.
4.3 Radiative decay at emission
Since the energy barrier would trap the fluorophores in the S1 minimum from which emission takes place, the character of the S1 state and the radiative decay become significant in explaining the experimental trends.The oscillator strength of the complexes are reported in Table 1. An oscillator strength less than 0.1 in general corresponds to an Lb state, whereas values greater than 0.1 correspond to La. Exceptions exist however. 3-CNI–(H2O)2 is found to have a relatively high oscillator strength although the NTOs show that this state is Lb(ππ*). 5-CNI is also an exception and it is an La state despite small oscillator strength. This is because after substitution the oscillator strengths can be very different from indole. Nonetheless, 2-CNI was found to emit from the dim Lb excited state whereas 6- and 7-CNI emit from a bright La excited state. Although the character of the S1 minimum helps distinguish these probes apart, it still does not explain the ultrafast fluorescence decay observed for 6- and 7-CNI isomers.
5 Conclusion
Overall, the fluorescence quenching mechanisms of n-CNI fluorescent probes in microsolvation were studied by calculating the PES of the electronic ground and excited states of n-CNI–(H2O)m clusters along the N–H bond coordinate using high level theories of quantum mechanics. Understanding how the modulation of the molecular structure of the fluorescent probes and their microsolvation can tune the energy barrier on the S1 surfaces can be used to design probes with the desired fluorescence properties in solvated biological systems.We show that substitution of the cyano group on the rings of indole gives rise to different photochemical processes involving non-cyclic and cyclic probe-water clusters. We find that the n-CNI fluorescent probes can undergo ESHT and ESPT upon formation of non-cyclic and cyclic structures, respectively, via a HB network with H2O molecules. We also find that the height of the energy barrier in the S1 PES dictates whether these non-radiative decay channels can be accessed and thus influences the fluorescence lifetime and quantum yield. Lastly, we show that substitution on position 4, and formation of cyclic clusters leads to large energy barriers that prevent the fluorophores from accessing radiationless decay pathways where the fluorescence would become quenched via internal conversion from the S1 to the S0. Low barriers exist in Ind and to a lesser extend in 3-CNI making radiationless decay through ESHT possible for these molecules. The barrier in 5-CNI decreases with the addition of an additional water molecule indicating that ESHT in this case depends on the number of water molecules, in agreement with experimental observations. On the other hand, the behavior we see in 6-CNI and 7-CNI is not sufficient to explain the experimental dependence of their fluorescence on aqueous solvation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the National Science Foundation (NSF, grant number: CHE-2303111) for funding. We also thank Prof. Feng Gai and his group for developing these probes which motivated our research study and Prof. Tolga Karsili for great discussion on this topic. This research includes calculations carried out on HPC resources at Temple University supported in part by the National Science Foundation through major research instrumentation grant number 1625061 and by the US Army Research Laboratory under contract number W911NF-16-2-0189. This work used Expanse at the San Diego Supercomputer Center through allocation CHE140114 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296.Notes and references
- B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965–970 CrossRef CAS PubMed.
- M. R. Hilaire, I. A. Ahmed, C.-W. Lin, H. Jo, W. F. DeGrado and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6005–6009 CrossRef CAS PubMed.
- M. R. Hilaire, D. Mukherjee, T. Troxler and F. Gai, Chem. Phys. Lett., 2017, 685, 133–138 CrossRef CAS PubMed.
- B. N. Markiewicz, D. Mukherjee, T. Troxler and F. Gai, J. Phys. Chem. B, 2016, 120, 936–944 CrossRef CAS PubMed.
- D. Mukherjee, L. I. Ortiz Rodriguez, M. R. Hilaire, T. Troxler and F. Gai, Phys. Chem. Chem. Phys., 2018, 20, 2527–2535 RSC.
- P. Talukder, S. Chen, B. Roy, P. Yakovchuk, M. M. Spiering, M. P. Alam, M. M. Madathil, C. Bhattacharya, S. J. Benkovic and S. M. Hecht, Biochemistry, 2015, 54, 7457–7469 CrossRef CAS PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- J. Zhang, R. Campbell, A. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 906–918 CrossRef CAS PubMed.
- D. Coling and B. Kachar, Curr. Protoc. Mol. Biol., 1998, 44, 14.10.1–14.10.11 Search PubMed.
- M. Sauer, J. Hofkens and J. Enderlein, Handbook of Fluorescence Spectroscopy and Imaging, 2011, pp. 1–30 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- T. Terai and T. Nagano, Pfluegers Arch., 2013, 465, 347–359 CrossRef CAS PubMed.
- E. C. Jensen, Anat. Rec., 2012, 295, 2031–2036 CrossRef CAS PubMed.
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- P. R. Callis, Methods Enzymol., 1997, 278, 113–150 CAS.
- S. M. Twine and A. G. Szabo, Biophotonics, Part A, Academic Press, Methods Enzymol, 2003, vol. 360, pp. 104–127 Search PubMed.
- A. B. T. Ghisaidoobe and S. J. Chung, Int. J. Mol. Sci., 2014, 15, 22518–22538 CrossRef CAS PubMed.
- H.-T. Yu, M. A. Vela, F. R. Fronczek, M. L. McLaughlin and M. D. Barkley, J. Am. Chem. Soc., 1995, 117, 348–357 CrossRef CAS.
- J. Alexander Ross, A. G. Szabo and C. W. Hogue, Flourescence Spectroscopy, Academic Press, Methods in Enzymology, 1997, vol. 278, pp. 151–190 Search PubMed.
- D. Creed, Photochem. Photobiol., 1984, 39, 537–562 CrossRef CAS.
- J. Catalán, Phys. Chem. Chem. Phys., 2016, 18, 15170–15176 RSC.
- D. W. Pierce and S. G. Boxer, Biophys. J., 1995, 68, 1583–1591 CrossRef CAS PubMed.
- J.-J. Aaron, THEOCHEM, 1986, 135, 105–116 CrossRef.
- J.-j J. Aaron, A. Tine, C. Villiers, C. Parkanyi and D. Bouin, Croat. Chem. Acta, 1983, 56, 157–168 CAS.
- J. Catalán, Phys. Chem. Chem. Phys., 2015, 17, 12515–12520 RSC.
- J. Catalán and J. P. Catalán, Phys. Chem. Chem. Phys., 2011, 13, 15022–15030 RSC.
- M. S. Walker, T. W. Bednar and R. Lumry, J. Chem. Phys., 1966, 45, 3455 CrossRef CAS PubMed.
- M. Sun and P. Song, Photochem. Photobiol., 1977, 25, 3–9 CrossRef CAS.
- R. M. Levy, J. D. Westbrook, D. B. Kitchen and K. Krogh-jespersen, J. Phys. Chem., 1991, 95, 6756–6758 CrossRef CAS.
- E. H. Strickland, J. Horwitz and C. Billups, Biochem., 1970, 9, 4914–4921 CrossRef CAS PubMed.
- X. Meng, T. Harricharran and L. J. Juszczak, Photochem. Photobiol., 2013, 89, 40–50 CrossRef CAS PubMed.
- S. R. Meech, D. Phillips and A. G. Lee, Chem. Phys., 1983, 80, 317–328 CrossRef CAS.
- G. Kumar, A. Roy, R. S. Mcmullen, S. Kutagulla and S. E. Bradforth, Faraday Discuss., 2018, 212, 359–381 RSC.
- H. Lami, Chem. Phys. Lett., 1977, 48, 447–450 CrossRef CAS.
- H. Lami and N. Glasser, J. Chem. Phys., 1986, 84, 597–604 CrossRef CAS.
- K. S. Yu Harabuchi and S. Maeda, Photochem. Photobiol. Sci., 2018, 17, 315 CrossRef PubMed.
- D. Brisker-klaiman and A. Dreuw, Chem. Phys. Chem., 2015, 16, 1695–1702 CrossRef CAS PubMed.
- L. Serrano-Andrés and B. O. Roos, J. Am. Chem. Soc., 1996, 118, 185–195 CrossRef.
- D. Shemesh, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2010, 12, 4899–4905 RSC.
- A. Giussani, M. Merchán, D. Roca-Sanjuán and R. Lindh, J. Chem. Theory Comput., 2011, 7, 4088–4096 CrossRef CAS PubMed.
- S. Abou-Hatab and S. Matsika, J. Phys. Chem. B, 2019, 123, 7424–7435 CrossRef CAS PubMed.
- C. Kang, T. M. Korter and D. W. Pratt, J. Chem. Phys., 2005, 122, 174301 CrossRef PubMed.
- P. R. Callis, J. Chem. Phys., 1991, 95, 4230–4240 CrossRef CAS.
- L. S. Slater and P. R. Callis, J. Phys. Chem., 1995, 99, 8572–8581 CrossRef CAS.
- A. L. Sobolewski, Phys. Chem. Chem. Phys., 2008, 10, 1243–1247 RSC.
- A. L. Sobolewski and W. Domcke, Chem. Phys. Lett., 2000, 329, 130–137 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2007, 111, 11725–11735 CrossRef CAS PubMed.
- W. Caminati and S. Di Bernardo, J. Mol. Struct., 1990, 240, 253 CrossRef CAS.
- C. Chang, C. Wu, A. R. Muirhead and J. R. Lombardi, Photochem. Photobiol., 1974, 19, 347–351 CrossRef CAS PubMed.
- J. R. Lombardi, J. Phys. Chem. A, 1998, 102, 2817–2823 CrossRef CAS.
- J. R. Lombardi, J. Phys. Chem. A, 1999, 103, 6335–6338 CrossRef CAS.
- S. Arulmozhiraja and M. L. Coote, J. Chem. Theory Comput., 2012, 8, 575–584 CrossRef CAS PubMed.
- N. Sharma, S. K. Jain and R. C. Rastogi, Spectrochim. Acta, Part A, 2007, 66, 171–176 CrossRef PubMed.
- C. Song and Y. M. Rhee, Int. J. Quantum Chem., 2011, 111, 4091–4105 CrossRef CAS.
- M. R. Eftink, L. R. A. Selvidge, P. R. Callis and A. A. Rehms, J. Phys. Chem., 1990, 94, 3469–3479 CrossRef CAS.
- C. D.-L. A. L. Sobolewski, W. Domcke and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- J. Jankowska and A. L. Sobolewski, Molecules, 2021, 26, 5140 CrossRef CAS PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122–127 CrossRef CAS PubMed.
- Y. Zhong, Y. Chen, X. Feng, Y. Sun, S. Cui, X. Li, X. Jin and G. Zhao, J. Mol. Liq., 2020, 302, 112562 CrossRef.
- B. Heggen, Z. Lan and W. Thiel, Phys. Chem. Chem. Phys., 2012, 14, 8137–8146 RSC.
- V. Ludwig, M. S. do Amaral, Z. M. da Costa, A. C. Borin, S. Canuto and L. Serrano-Andrés, Chem. Phys. Lett., 2008, 463, 201–205 CrossRef CAS.
- S. Chai, G.-J. Zhao, P. Song, S.-Q. Yang, J.-Y. Liu and K.-L. Han, Phys. Chem. Chem. Phys., 2009, 11, 4385–4390 RSC.
- C. Manca, C. Tanner and S. Leutwyler, Int. Rev. Phys. Chem., 2005, 24, 457–488 Search PubMed.
- S. T. Park, A. Gahlmann, Y. He, J. S. Feenstra and A. H. Zewail, Angew. Chem. Int. Ed., 2008, 47, 9496–9499 CrossRef CAS PubMed.
- T. J. Godfrey, H. Yu, M. S. Biddle and S. Ullrich, Phys. Chem. Chem. Phys., 2015, 17, 25197–25209 RSC.
- H. Lippert, V. Stert, L. Hesse, C. Schulz, I. Hertel and W. Radloff, Chem. Phys. Lett., 2003, 376, 40–48 CrossRef CAS.
- A. Peralta Conde, V. Ovejas, R. Montero, F. Castaño and A. Longarte, Chem. Phys. Lett., 2012, 530, 25 CrossRef CAS.
- J. Onvlee, S. Trippel and J. Kuepper, Nat. Commun., 2022, 13, 7462 CrossRef CAS PubMed.
- C. Henrichs, S. Zimmermann, M.-L. Hebestreit and M. Schmitt, J. Mol. Struct., 2021, 1233, 130055 CrossRef CAS.
- M. Schneider, M.-L. Hebestreit, M. M. Lindic, H. Parsian, A. Y. Torres-Boy, L. Alvarez-Valtierra, W. L. Meerts, R. Kühnemuth and M. Schmitt, Phys. Chem. Chem. Phys., 2018, 20, 23441 RSC.
- A. Min, C. J. Moon, A. Ahn, J. H. Lee, S. K. Kim and M. Y. Choi, Chem. Phys. Lett., 2016, 658, 63–70 CrossRef CAS.
- A. Min, A. Ahn, C. J. Moon, J. H. Lee, M. Y. Choi and S. K. Kim, Chem. Phys. Lett., 2014, 614, 263–268 CrossRef CAS.
- A. Ahn, A. Min, C. J. Moon, J. H. Lee and M. Y. Choi, Chem. Phys. Lett., 2014, 616–617, 55–60 CrossRef CAS.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- S. Matsika, Chem. Rev., 2021, 121, 9407–9449 CrossRef CAS PubMed.
- F. Jensen, J. Chem. Theory Comput., 2014, 10, 1074–1085 CrossRef CAS PubMed.
- F. Jensen, WIREs Comput. Mol. Sci., 2013, 3, 273–295 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS PubMed.
- B. Mennucci, WIREs Comput. Mol. Sci., 2012, 2, 386–404 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian–16 Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- K. Sneskov and O. Christiansen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 566–584 CAS.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 4 Search PubMed.
- TURBOMOLE V6.5 2013, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com.
- J. F. Stanton and R. J. Bartlett, J. Chern. Phys., 1993, 98, 9 Search PubMed.
- B. O. Roos, Adv. Chem. Phys., 1987, 399–445 CrossRef CAS.
- P. Pulay, Int. J. Quantum Chem., 2011, 111, 3273–3279 CrossRef CAS.
- Y. Shao, L. Fusti-Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. V. Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Daschel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C. Hsu, G. Kedziora, R. Z. Khaliullin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer III, J. Kong, A. I. Krylov, P. M. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172 RSC.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2015.1, a package of ab initio programs, 2015 Search PubMed.
- M.-L. Hebestreit, M. Schneider, H. Lartian, V. Betz, M. Heinrich, M. Lindic, M. Y. Choi and M. Schmitt, Phys. Chem. Chem. Phys., 2019, 21, 14766–14774 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: In Section 1 potential energy surfaces of all isomers studied are shown using alternative methods to the ones in the main paper. Section 2 shows the orbitals used in the CASSCF calculations. Section 3 discusses the charge transfer analysis used to determine whether hydrogen or proton transfer occurs. Section 4 discusses additional benchmarking studies used to determine the validity of our approach. Section 5 discusses implicit solvent results, and Section 6 shows the MD simulations used to determine the frequency of hydrogen bonding in aqueous solution. See DOI: https://doi.org/10.1039/d3cp04844f |
| This journal is © the Owner Societies 2024 |