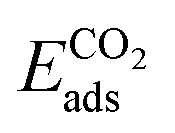A first-principles study of electro-catalytic reduction of CO2 on transition metal-doped stanene†
Received
6th October 2023
, Accepted 3rd January 2024
First published on 4th January 2024
Abstract
Employing first-principles calculations based on density functional theory, this work examines the activity of 3d transition metal-doped stanene for electro-catalytic CO2 reduction through the first two electron transfer steps to CO. Our results related to CO2 activation, the first and a crucial step of the reduction process revealed that, among the entire 3d transition metal row studied, only Ti- and Fe-doped stanene can bind and significantly activate the CO2 molecule, while the rest of the TM single atoms are inert in activating the molecule. The activation of the CO2 molecule on Ti- and Fe-doped stanene has been observed in the presence of water as well. In addition, the formation of OCHO has been observed to be energetically preferred over COOH formation as a reaction intermediate, indicating the preference for the formate path of the reduction reaction. Furthermore, despite the strong adsorption of H2O on the catalyst surface, the presence of water seems to enhance CO2 adsorption on the catalysts, contrary to what has been observed recently in graphene-based catalysts. Finally, our difference charge density and the Bader charge calculations reveal that the ability of Ti- and Fe-doped stanene in activating the CO2 molecule and their potential catalytic activity for CO2 reduction is to be attributed to the charge transfer between the catalyst and the molecule, providing new insights into the rational design of 2D catalysts beyond graphene.
1 Introduction
Electrochemical conversion of CO2 into value added products has emerged as a sustainable way to alleviate global warming, and as a promising solution to address the current energy crisis.1–6 The advantage of the electro-catalytic CO2 reduction reaction (CO2RR) lies in the fact that the reaction can proceed under ambient conditions on efficient electrocatalysts,7–20 and can be controlled by suitably changing the applied potential.21 Reduction of CO2 to CO is considered as the first step of this process22 and is highly important for the chemical industry.23 Nevertheless, the major bottleneck even for this two electron reduction reaction,22 is due to the poor selectivity, compromised stability, high cost and low transformation efficiency of the conventional catalysts.24–28 Design and development of non-precious and highly active catalysts with better selectivity and stability is hence highly necessary. Recently, two-dimensional (2D) nanomaterials have drawn immense attention due to their unique structural and electronic properties, high surface to volume ratio and higher maneuverability compared to their bulk counterparts,14,29–33 enabling their potential usage in an extensive range of applications. Lately, stanene, among the recently developed group iv monolayers, has exhibited efficiency for sensing triatomic gases such as CO2, NO2 and SO234–40 and its ability to attain higher room temperature conductivity compared to graphene and silicene through efficient doping.39 In spite of the unique structural and electronic properties of stanene and its already proven ability to capture and sense CO2 gas, the role of stanene as a catalyst for the CO2RR is not yet explored thoroughly. Employing first-principles calculations based on density functional theory (DFT), this work intends to investigate the activity of pure and 3d transition metal (TM: Sc to Zn)-doped stanene towards CO2 reduction.
This article is structured as follows: Section 2 provides the computational details. In Section 3 we discuss our results involving both pure and 3d TM-doped stanene as catalysts for the reduction of CO2 to CO, and Section 4 provides the conclusion.
2 Computational details
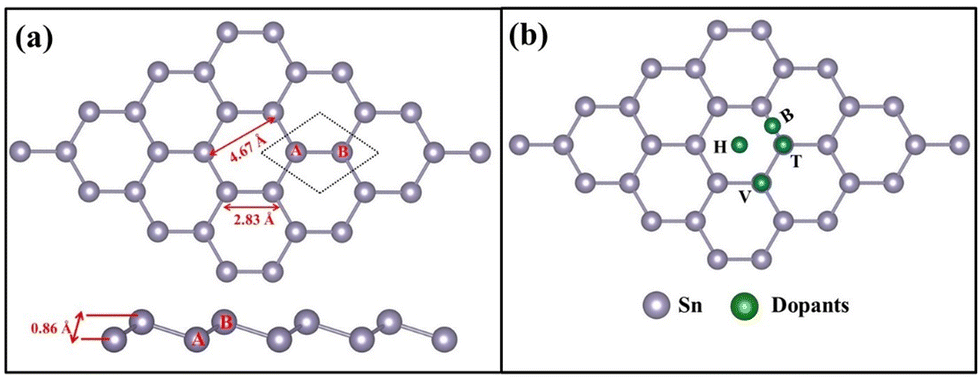
Spin polarized density functional theory calculations have been performed using the Vienna ab initio simulation package (VASP)41 employing the projected augmented wave (PAW) method42 and a plane wave basis43 set. The generalized gradient approximation (GGA) as implemented in the Perdew–Burke–Ernzerhof (PBE)44 functional has been employed and D3 Grimme-dispersion correction45 to address the van der Waals interaction is incorporated in all our calculations. The total energy of the stanene layer per atom is calculated as −3.48 eV per atom, 0.09 eV less than the energy reported using the HSE06 functional.46 A fully optimized unit cell of stanene has a = b = 4.67 Å, a Sn–Sn bond length of 2.83 Å, and a buckling height of 0.86 Å. The calculated values are all within a reasonable agreement with previous reports.40,47–50 A 4 × 4 supercell constructed from the unit cell with 20 Å vacuum introduced along the z direction was further used to construct doped stanene structures. TM single atom (SA)-doped stanene with a doping concentration of 3.12% is depicted in Fig. 1a. All the structures are optimized using the conjugate gradient scheme with a 5 × 5 × 1 Monkhorst–Pack51k-mesh and a plane wave energy cut-off of 450 eV. The calculations are considered converged when the energy difference between successive iterations becomes less than 10−5 eV and the force exerted on each atom is less than 0.001 eV Å−1. The stability of a TM single atom in stanene is calculated from its binding energy (B.E.) calculated as,
| B.E. = ETM@Stanene − EStanene − ETM |
where ETM@Stanene, ETM and EStanene are the energies of TM doped stanene, of a single TM atom, and of the energy of the pristine stanene monolayer, respectively. The adsorption energy (Eads) of a molecule adsorbed on the catalyst has been computed using the following equation,52
| Eads = Ecat+mol − Ecat − Emol |
Ecat+mol, Ecat and Emol are the energies of the catalyst with the adsorbed molecule, of the catalyst and of the isolated molecule, respectively.
 |
| | Fig. 1 (a) Top and side view of an optimized 4 × 4 stanene supercell. The dotted area indicates a single unit cell. The buckling height of 0.86 Å is the distance observed between the A and B sublattices. (b) Possible sites to add the TM dopants. | |
The reaction free energy change ΔG is calculated as ΔG = ∑G(products) − ∑G(reactants) where the free energy for each adsorbed species is calculated using the following equation,53
Here,
EDFT is the DFT calculated electronic energy,
54S and ZPE are the entropy and zero-point energy correction, respectively, and the corresponding values for the reaction intermediates are taken from the literature.
52,55 For all the proton coupled electron transfer (PCET) steps of the CO
2RR, the chemical potential of a proton and electron pair is considered as ½ of the chemical potential of gaseous hydrogen, as per the CHE model.
54 The limiting potential (
UL) is determined as,
56where
e is electronic charge and Δ
Gmax is the highest change in free energy observed in the potential limiting step (PLS) of the reaction.
3 Results and discussion
3.1 The binding sites for TM in stanene
Four possible binding sites, namely (i) hollow (H): center of hexagon, (ii) top (T): top of an Sn atom in the upper sublattice, (iii) valley (V): top of an Sn atom in the lower sublattice, and (iv) bridge site (B): in between two adjacent Sn atoms,48 were investigated to find the most preferred binding position for the TM atoms (Sc to Zn) doped in stanene (Fig. 1b). The binding energy (B.E.) value for each TM dopant in stanene is listed in Table 1.
Table 1 Binding energies of 3d TM atoms on all four possible doping sites
| TM |
B.E. (eV) |
| Top (T) |
Hollow (H) |
Valley (V) |
Bridge (B) |
| Sc |
−3.28 |
−3.99 |
−3.74 |
Unstable |
| Ti |
−2.10 |
−4.33 |
−3.90 |
Unstable |
| V |
−2.72 |
−3.58 |
−3.11 |
Unstable |
| Cr |
−1.11 |
−2.33 |
−1.94 |
Unstable |
| Mn |
−1.11 |
−2.38 |
−2.19 |
Unstable |
| Fe |
−1.37 |
−3.14 |
−3.05 |
Unstable |
| Co |
−3.25 |
−3.52 |
−3.56 |
Unstable |
| Ni |
−3.32 |
−3.78 |
−3.64 |
Unstable |
| Cu |
−1.57 |
−2.57 |
−2.39 |
Unstable |
| Zn |
−0.41 |
0.10 |
−0.44 |
−0.42 |
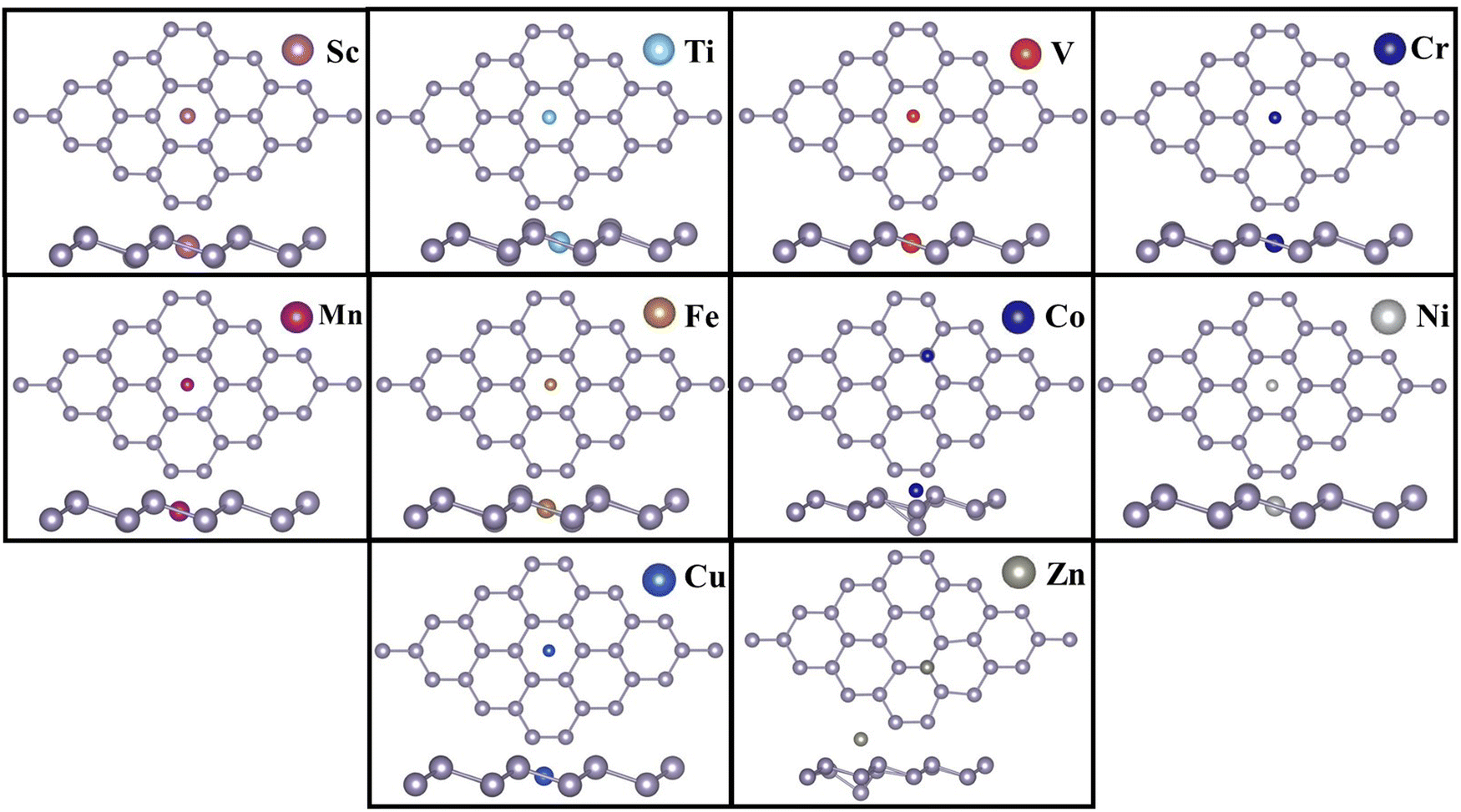
Table 1 shows that the hollow site (H) is the most preferred binding site for all TM atoms studied, except Co and Zn. For Co and Zn, the valley site (V) is the preferred site for TM binding. It has also been observed that the bridge site is unstable for all TM atoms, except Zn. For Zn, however, the difference in binding energies between the valley and bridge sites is very small (0.02 eV). For other TM atoms, our simulations revealed that if doped at the bridge site, the TM atom will eventually migrate to the valley site. It has also been noticed that, TM atoms, when added at the valley site, move closer to the plane of the stanene monolayer and push the Sn atom slightly away from the plane. The negative binding energies indicate the strong binding of TM-SACs on stanene.57,58Fig. 2 depicts the TM single atoms at the most preferred doping sites in stanene.
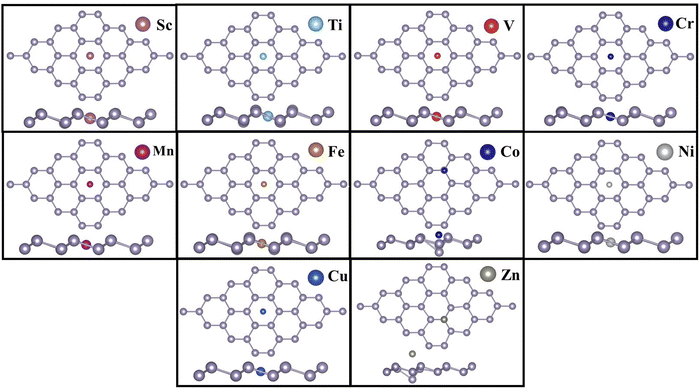 |
| | Fig. 2 TM SAs at their preferred doping sites in stanene. | |
From our calculations it is evident that, the TM binding energy is stronger for the early transition metals (Sc to V), with Ti, which shares the same number of valence electrons as Sn, exhibiting the highest binding energy amongst all the TMs studied. The binding energy, however, decreases in Cr and Mn where both the TMs possess a half-filled 3d shell. The B.E. increases again for Fe, Co and Ni, and decreases in late TMs Cu and Zn. The structural geometries of the TM embedded stanene layer and the corresponding TM magnetizations are listed in Table 2. It is apparent that incorporation of the TM SA in stanene results in local distortion with the maximum distortion observed in Co-doped stanene, where the Co atom pushes the Sn atom away from the plane towards the valley site and the local Sn–Sn bond length stretches to 3.29 Å. For the other TMs, the Sn–Sn bond length varies from 2.85 Å in Ni-, to 3.03 Å in Sc-doped stanene. While most of the TM SAs exhibit strong chemisorption with the stanene sheet, with the TM–Sn distance varying from 2.43 Å to 2.88 Å, for Zn, physisorption is observed with a Zn–Sn distance of 4.01 Å and a binding energy of −0.44 eV. The weak binding of Zn in stanene compared to other TMs can be attributed to its closed shell structure.
Table 2 TM–Sn distance (dTM–Sn), Sn–Sn distance (dSn–Sn), and magnetization for TM, doped at the most preferred binding site in stanene
| TM |
d
TM–Sn (Å) |
d
Sn–Sn (Å) |
μ
d
|
| Sc |
2.88 |
3.03 |
0.00 |
| Ti |
2.80 |
3.03 |
0.84 |
| V |
2.80 |
2.95 |
2.40 |
| Cr |
2.82 |
2.92 |
3.64 |
| Mn |
2.80 |
2.94 |
3.71 |
| Fe |
2.73 |
2.92 |
2.60 |
| Co |
2.43 |
3.29 |
1.46 |
| Ni |
2.72 |
2.85 |
0.00 |
| Cu |
2.77 |
2.89 |
0.00 |
| Zn |
4.01 |
2.91 |
0.00 |
3.2 CO2 reduction to CO on bare catalysts
3.2.1 CO2 activation.
CO2 activation is the first and a crucial step of CO2RR, although largely ignored in most of the studies pertaining to CO2RR.59–63 Thus, as a first step to understand the CO2RR on TM@Sn, we analysed the efficiency of the TM SAs to bend and therefore to activate the CO2 molecule. The structural geometry of the complex, the adsorption energies and the reaction free energies of the CO2 molecule, and the TM magnetization after CO2 adsorption are listed in Table 3.
Table 3 TM–C distance, O–C–O angle, adsorption energies (Eads) and free energies (ΔG) for CO2 adsorption. TM magnetization is also listed
| TM |
d
TM–C (Å) |
∠O–C–O |
E
ads (eV) |
ΔG (eV) |
μ
d
|
| Pure stanene |
3.64 |
179.7 |
−0.09 |
0.59 |
0.00 |
| TM@Sn |
| Sc |
4.61 |
179.7 |
−0.29 |
0.39 |
0.00 |
| Ti |
2.23 |
149.4 |
0.09 |
0.77 |
0.60 |
| V |
4.16 |
179.6 |
−0.16 |
0.52 |
2.38 |
| Cr |
4.49 |
179.9 |
−0.21 |
0.47 |
3.76 |
| Mn |
4.41 |
179.9 |
−0.19 |
0.49 |
3.67 |
| Fe |
2.06 |
148.4 |
−0.12 |
0.56 |
0.84 |
| Co |
3.44 |
179.6 |
−0.11 |
0.57 |
1.40 |
| Ni |
3.53 |
179.1 |
−0.27 |
0.41 |
0.00 |
| Cu |
4.03 |
179.7 |
−0.14 |
0.54 |
−0.02 |
| Zn |
3.41 |
179.6 |
−0.15 |
0.53 |
0.00 |
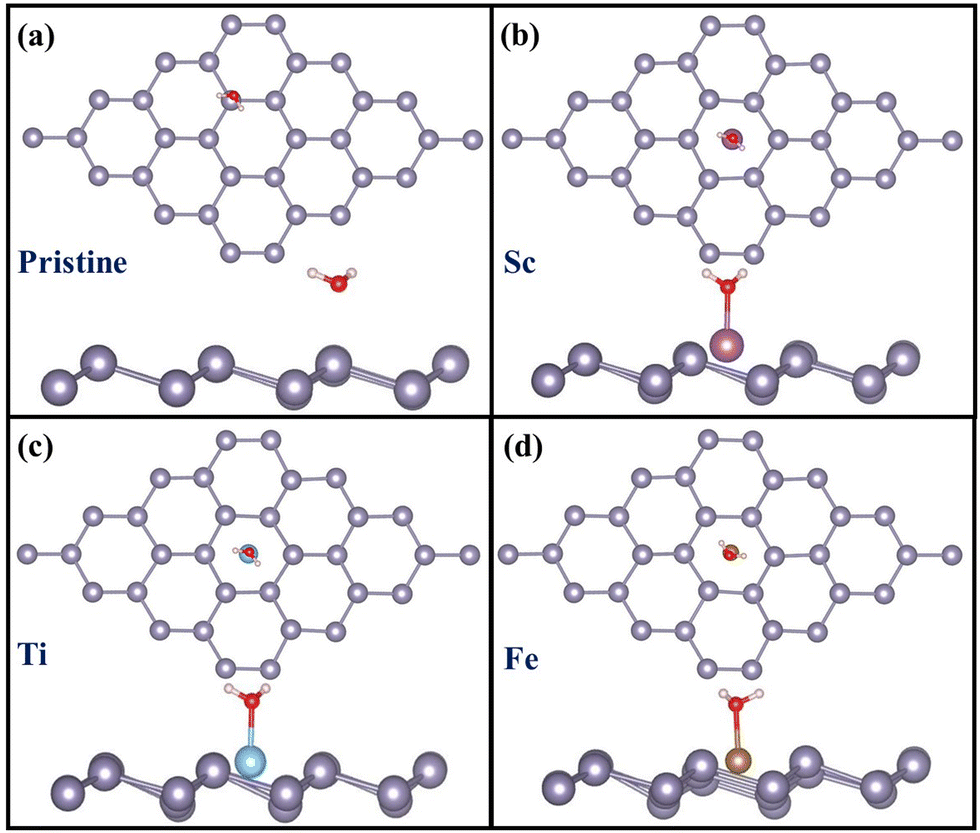
For CO2 adsorption on pristine stanene (Fig. 3a), a weak physisorption (Eads −0.09 eV) is observed with no activation of the molecule. Our simulations for the entire 3d TM row studied revealed that, only Ti and Fe can activate the CO2 molecule. For Ti, the CO2 molecule is activated with ∠O–C–O of 149°, and the C–O bond is elongated to 1.22 Å. For Fe, the activation of the CO2 molecule is observed with ∠O–C–O reducing to 148° and the bond length is increased to 1.22 Å. For the rest of the TM SAs, no activation of the CO2 molecule is observed. For Sc@Sn, the Sn atom is pushed slightly out of the plane of the stanene monolayer and the CO2 molecule moves further away from the plane. The same trend has been observed for other TMs (V, Cr, Mn, Co and Cu) with no activation of the CO2 molecule. For Ni@Sn as well, no activation in CO2 is observed, and our calculated CO2 adsorption energy of −0.27 eV is consistent with previous reports involving Ni SAC in similar materials.64
 |
| | Fig. 3 CO2 adsorption on (a) pristine, (b) Sc-, (c) Ti- and (d) Fe-doped stanene monolayers: top view and side view. | |
Henceforth, we focus our attention on Ti and Fe-doped stanene as they activate the CO2 molecule and hence are likely to be active in the CO2RR. In addition, Sc-doped stanene is also studied for comparison, Sc being the representative of the TMs that does not activate the CO2 molecule. The optimized geometries of CO2 adsorption on TM@Sn (Sc, Ti and Fe) are shown in Fig. 3.
Our ab initio molecular dynamics (AIMD) simulation at 300 K for 5 ps verifies the kinetic stability of the catalysts that activate CO2. As depicted in Fig. S1 (ESI†), the energy and temperature profiles of Ti- and Fe-doped stanene oscillate within a very small range, indicating the stability of these catalytic systems. Additionally, the thermodynamic and electrochemical stability of the catalysts have also been investigated (ESI†). Our calculated formation energy per unit area for pure stanene within the GGA-PBE framework is −0.26 eV Å−2 with a deviation of +0.09 eV Å−2 from the HSE06 value of −0.35 eV Å−2 reported earlier.46 The stability further increases in TM-doped stanene with the order ΔETi@Sn < ΔEFe@Sn < ΔESn (Table S2, ESI†). Furthermore, our dissolution potential calculations for the catalysts (Table S2, ESI†) indicate the electrochemical stability of TM@Sn as well. Our results are in range with the existing reports on similar 2D materials65 and the absolute deviations from the reported values for a specific TM are attributed to the different levels of theory involved in the respective work.
3.2.2 Reaction intermediates.
CO2RR is an intricate process involving multiple proton-electron transfer steps, and various reaction intermediates. The pathway involving a two-electron transfer mechanism is explored in our study (reduction of CO2 to CO) and the subsequent equations describing the possible intermediates for the same are given below.66
| *CO2 + H+ + e− → *COOH(*OCHO) |
| *COOH + H+ + e− → *CO + H2O |
where the catalyst is indicated by an asterisk and the asterisk placed prior to an adsorbate signifies its binding on the catalyst.
The subsequent step after CO2 activation entails the first hydrogenation process, leading to the formation of COOH or OCHO. This step holds substantial significance as it plays a crucial role in determining both the end products and the desired reaction pathway, as the formation of COOH (OCHO) indicates the preference for the carboxylic (formate) pathway.52,67 Hence in our study following the CO2 activation over Ti and Fe doped stanene, we compared the probabilities of formation of both COOH and OCHO as reaction intermediates. For Ti@Sn, our calculation revealed that OCHO formation has higher energy preference (ΔG = −1.51 eV) over COOH (ΔG = −0.70 eV). Subsequently, the next step of the reaction process, the formation of the intermediate HCOOH, has also been studied and the additional energy input required for the conversion of OCHO to HCOOH is calculated to be 1.37 eV. Interestingly, had COOH been the favored intermediate in the 1st hydrogenation step, the further conversion of COOH to CO would be an exothermic process with a free energy change of −0.38 eV. The energy preference of OCHO over COOH has also been observed in previous reports.52,68
For Fe@Sn, we found a similar trend, where OCHO emerges as the energetically preferred intermediate in the 1st hydrogenation step over COOH (ΔG = −0.88 eV and 0.16 eV respectively). For the next step of the reaction, an energy input of 1.32 eV is required for HCOOH formation from OCHO. Similar to Ti@Sn, had COOH been the preferred intermediate in the initial step, the subsequent CO formation would have a free energy change of −1.17 eV.
Hence, for both Ti- and Fe-doped Sn, OCHO to HCOOH formation is the potential limiting step (PLS), with the limiting potentials (UL) being −1.37 V and −1.32 V respectively. Fig. 4(a) and (b) display the optimal geometries and the corresponding free energy diagrams along the carboxylic and formate pathways for both the catalysts. For each reaction step the change in ΔG as shown in Fig. 4(b) is considered with respect to the previous reaction step.69–71 All the energy parameters are summarized in Table 4.
 |
| | Fig. 4 (a) Optimized structures and (b) free energy profiles along the carboxylic and formate pathways for Ti- and Fe-doped stanene. | |
Table 4 Change in free energy along the reaction pathway and limiting potentials for Ti- and Fe-doped stanene
| TM |
COOH |
CO |
OCHO |
HCOOH |
PLS |
U
L (V) |
|
E
ads (eV) |
ΔG (eV) |
E
ads (eV) |
ΔG (eV) |
E
ads (eV) |
ΔG (eV) |
E
ads (eV) |
ΔG (eV) |
| Ti |
−3.09 |
−0.70 |
−1.85 |
−0.38 |
−4.04 |
−1.51 |
−0.73 |
1.37 |
*OCHO + H+ + e− → *HCOOH |
−1.37 |
| Fe |
−2.45 |
0.16 |
−2.00 |
−1.17 |
−3.62 |
−0.88 |
−0.36 |
1.32 |
*OCHO + H+ + e− → *HCOOH |
−1.32 |
However, apart from the reaction intermediates mentioned above, H* may also emerge as a part of the hydrogen evolution reaction (HER) that can occur simultaneously with CO2RR.72 Hence, free energy changes related to H* adsorption on the catalysts have been calculated and compared with that of COOH and OCHO, reaction intermediates from the 1st hydrogenation step of CO2RR.72 The free energy change (ΔGH) related to H-adsorption along the reaction pathway of HER came out to be −0.09 eV and 0.16 eV for Ti- and Fe doped stanene, respectively. Hence, we found that ΔGOCHO < ΔGCOOH < ΔGH for Ti-doped stanene, and ΔGOCHO < ΔGCOOH = ΔGH for Fe@Sn. The lowest Gibbs free energy change has been observed for OCHO formation in both the cases, indicating that the formation of OCHO is most preferred among all the reactants considered, and the catalysts are selective towards CO2RR over HER.72
3.3 CO2 reduction in water
As CO2RR occurs in aqueous medium, CO2 activation in the presence of water has also been studied for these systems. However, in order to understand CO2 adsorption on wet catalysts, first the catalysts’ ability to bind water molecules has been investigated.
3.3.1 H2O adsorption on TM@Sn.
The optimized catalyst structures with adsorbed H2O are shown in Fig. 5 and the adsorption energies and the structural geometries are listed in Table 5.
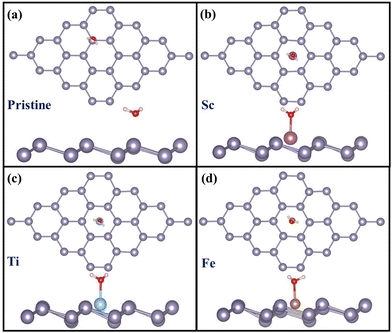 |
| | Fig. 5 H2O adsorption on (a) pristine and (b) Sc- (c) Ti- and (d) Fe-doped stanene: top view and side view. | |
Table 5 TM–O distance, H–O–H angle, adsorption energies (Eads) and free energies (ΔG) for H2O adsorption on TM@Sn. TM magnetization is also listed
| Pure stanene |
d
TM–O (Å) |
∠H–O–H |
E
ads (eV) |
ΔG (eV) |
μ
d
|
| 2.95 |
105.0 |
−0.16 |
0.43 |
0.00 |
| TM@Sn |
| Sc |
2.22 |
108.3 |
−1.18 |
−0.59 |
0.07 |
| Ti |
2.16 |
108.0 |
−0.96 |
−0.37 |
0.00 |
| V |
2.14 |
107.1 |
−0.69 |
−0.10 |
0.57 |
| Cr |
2.21 |
106.9 |
−0.67 |
−0.08 |
3.73 |
| Mn |
2.20 |
106.6 |
−0.72 |
−0.13 |
3.73 |
| Fe |
2.17 |
106.8 |
−0.61 |
−0.02 |
2.80 |
| Co |
2.10 |
106.8 |
−0.58 |
0.01 |
1.47 |
| Ni |
2.15 |
106.1 |
−0.50 |
0.09 |
0.00 |
| Cu |
2.22 |
106.0 |
−0.45 |
0.14 |
0.02 |
| Zn |
2.76 |
105.7 |
−0.12 |
0.47 |
0.00 |
It is apparent that H2O binds weakly on pure and Zn-doped stanene with Eads of −0.16 eV and −0.12 eV, respectively. However, in all the TM-doped stanenes (except Zn), water binds strongly on the catalyst with the strongest adsorption observed in Sc-doped stanene (−1.18 eV). To explore the effect of water on CO2 binding and activation on the catalyst, we further studied CO2 adsorption on TM@Sn in the presence of a water molecule as described below.
3.3.2 Co-adsorption of CO2 and H2O on TM@Sn.
For simulating adsorption of CO2 in the presence of H2O, the 2D symmetry of the catalyst can be exploited considering CO2 and H2O located on the same side as well as on the opposite sides of the stanene monolayer.52 In the present study, we have considered adsorption of one H2O molecule (i) on the same side and (ii) on the opposite side of CO2 on the catalyst. The detailed structural geometry, the adsorption energies (Eads) and the adsorption free energies for co-adsorption of CO2 and H2O are listed in Table 6. The adsorption energies for CO2 are calculated considering the catalyst with pre-adsorbed H2O as a reference. For CO2 and H2O adsorption on the opposite sides, both the molecules move away from a pure stanene monolayer with no activation of the CO2 molecule. In Sc-doped stanene, the Sc atom moves closer to the H2O molecule, in line with its ability to bind H2O strongly. Furthermore, similar to dry Sc@Sn, there is no activation of CO2 in the presence of H2O. A similar trend is observed when both the molecules lie on the same side of the Sc-doped stanene: the CO2 and H2O molecules move away from each other and there is no activation of CO2. For Ti-doped stanene as well, the Ti atom moves very close to the H2O molecule as expected from its strong adsorption of H2O. We observe that the CO2 molecule does not get activated when H2O is added on the opposite side of the catalyst. However, when adsorbed on the same side of the catalyst, the Ti atom moves closer to the CO2 molecule and the latter gets activated on the catalyst with an adsorption energy of −0.39 eV. In Fe-doped stanene, on the other hand, CO2 gets activated in both cases and the H2O molecule moves away from the Fe atom. A strong binding and subsequent activation of CO2 is evident from the O–C–O bond angle of 149° (141°) and an elongated C–O bond of 1.22 Å (1.29 Å) when placed on the opposite (same) side of H2O on Fe-doped stanene.
Table 6 TM–C (C of CO2 molecule) and TM–O (O of H2O molecule) distances, C–O–C bond angles, adsorption energies and free energies for CO2 adsorption in the presence of one water molecule
| TM |
H2O on the opposite side |
H2O on the same side |
|
d
TM–C (Å) |
d
TM–O (Å) |
∠O–C–O |
C–O (Å) |

(eV) |
ΔGCO2 (eV) |
d
TM–C (Å) |
d
TM–O (Å) |
∠O–C–O |
C–O (Å) |

(eV) |
ΔGCO2 (eV) |
| Sc |
4.82 |
2.23 |
180 |
1.18 |
−0.15 |
0.53 |
4.78 |
5.64 |
180 |
1.18 |
0.67 |
1.35 |
| Ti |
4.61 |
2.19 |
179 |
1.18 |
−0.12 |
0.56 |
2.04 |
3.90 |
127 |
1.40 |
−0.39 |
0.29 |
| Fe |
2.06 |
4.20 |
149 |
1.22 |
0.28 |
0.96 |
1.94 |
3.87 |
141 |
1.29 |
−0.28 |
0.40 |
Our calculations revealed that the catalysts that activated CO2 in the dry phase also activate the molecule in the presence of water. Surprisingly, the presence of water in close vicinity seems to enhance the strength of CO2 adsorption on the catalyst: and the adsorption energy decreases by 0.48 and 0.16 eV for Ti and Fe, respectively, a behaviour opposite to what has recently been observed in N-doped graphene.52
To explore the origin of the enhanced CO2 adsorption on the catalysts in the presence of water, the interaction energies between activated CO2 and the nearby H2O molecules have been calculated as per the super-molecular approach.73,74 The negative interaction energies resulted for Ti and Fe-doped stanene (ESI†) indicate the formation of H-bonds between the molecules. The H-bond lengths for Ti@Sn and Fe@Sn are 1.89 and 1.95 Å, respectively, similar to the H-bond lengths reported previously for various oxygen containing functional groups.75 In addition, the distances between the donor and acceptor atoms in Ti@Sn and Fe@Sn are much shorter than the sum of van der Waals radii of the donor and acceptor atoms and the O–H bond length in H2O (ESI†). Furthermore, our Bader charge analysis indicates the shift in electron density: charge on the O atom of water increases while on the C atom of the CO2 molecule decreases (Table S3, ESI†). The above two factors collectively point towards the formation of H-bonds between the activated CO2 and the nearby H2O molecules resulting in the enhanced adsorption strength of CO2 on the catalysts in the presence of water.75,76
Our calculations pertaining to CO2 activation on Ti- and Fe-doped stanene in the presence of two H2O molecules simultaneously adsorbed on both sides of the stanene monolayer (Fig. 6) revealed a trend similar to same side co-adsorption of the molecules. For Ti@Sn, the CO2 activation is well sustained with ∠O–C–O 129° and C–O bond 1.36 Å, while the same for Fe@Sn are measured as 140° and 1.29 Å, respectively. In this case as well, we observe an increased adsorption strength of CO2 in the presence of two water molecules compared to dry catalysts: Eads is −0.52 eV for Ti@Sn and −0.26 eV for Fe@Sn, similar to co-adsorption of CO2 and H2O on the same side of the catalysts as discussed above.
 |
| | Fig. 6 Co-adsorption of CO2 and H2O on Sc-, Ti- and Fe-doped stanene. CO2 and one H2O on the opposite side (1st panel) and same side (2nd panel) of the catalysts, CO2 and two H2O molecules, on both sides of the catalysts (3rd panel). | |
3.4 Charge density difference and Bader charge analysis
3.4.1 Bare catalysts.
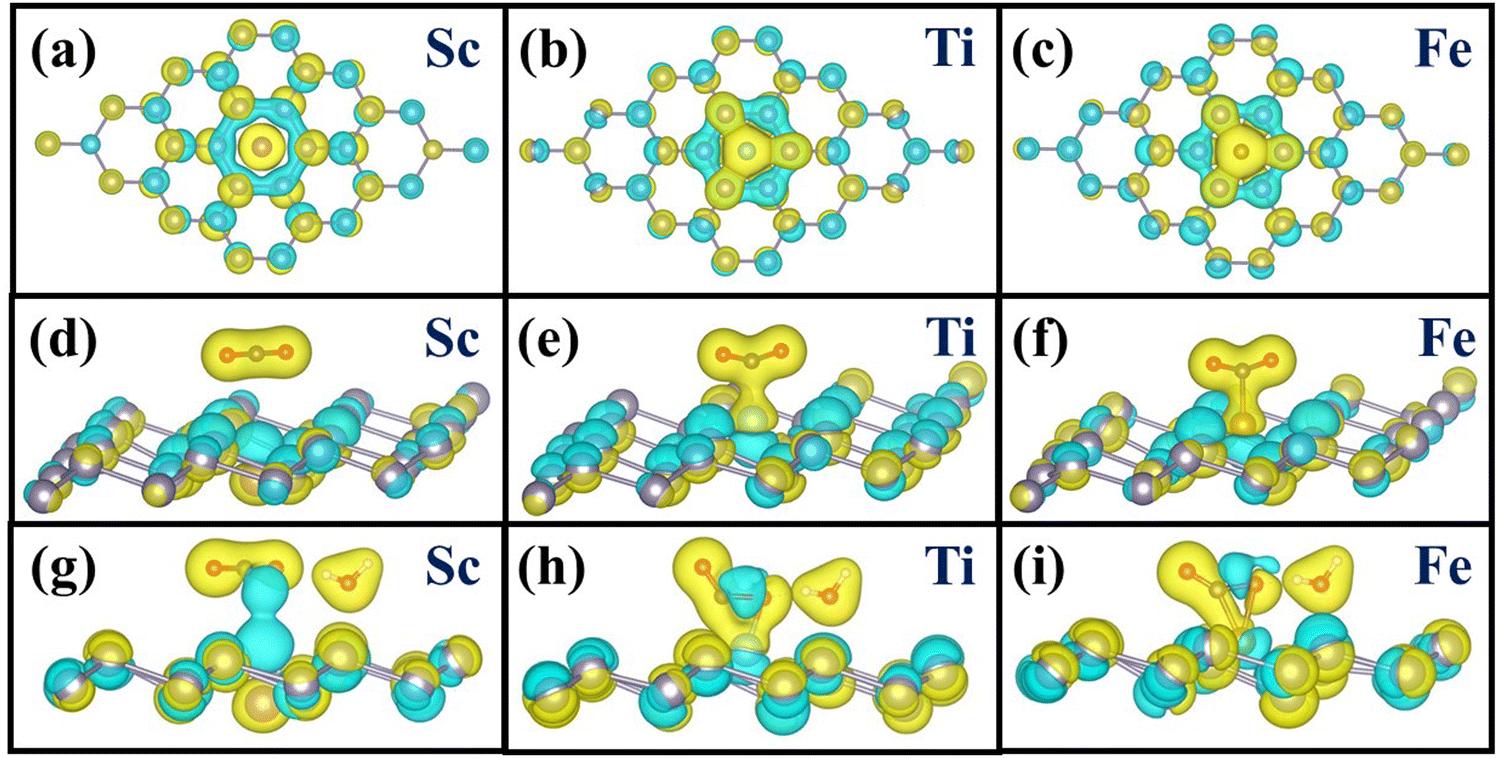
Finally, to understand the observed activity of a few TM SA and the inertness of the remaining TMs, we have performed charge density difference (CDD) calculations and Bader charge analysis77 for Sc, Ti, and Fe-doped stanene. Fig. 7(a)–(c) depicts the CDD plots for TM-doped stanene without the adsorbed molecules. For Ti and Fe, charge accumulation (yellow region) is clearly visible near the TM and the neighboring Sn atom, which is not present in Sc@Sn. The CDD plots also indicate strong binding of Ti and Fe in the stanene monolayer and hence the stability of the catalysts. The Bader charge calculations revealed that while a charge transfer of 1.30e occurs from Sc to Sn, the amount of charges transferred between the TM and neighboring Sn atoms are 0.99e and 0.13e for Ti and Fe-doped stanene, respectively. The decrease in charge transfer from Sc to Ti and Fe may indicate that Ti and Fe retain excess charge accumulated around them, which can later be transferred to a molecule when adsorbed on the catalyst.
 |
| | Fig. 7 Charge density difference plots for TM@Sn (1st panel), CO2 adsorption on TM@Sn (2nd panel) and CO2 and H2O co-adsorption on TM@Sn (3rd panel) with isosurface values 0.005 se Å−3, 0.04 e Å−3 and 0.03 e Å−3 respectively. Yellow and cyan regions indicate electron accumulation and depletion, respectively. | |
3.4.2 CO2 adsorption in the dry phase.
The CDD plots of the complexes with adsorbed CO2 molecules are shown in Fig. 7(d)–(f). Our Bader charge calculations revealed that there is almost no charge transfer (0.05e) from Sc to the CO2 molecule, also evident from the charge density difference plot (Fig. 7d); instead, a charge depletion region is noticed just above the Sc atom. On the other hand, for the Ti doped system, a total of 0.57e has been transferred to CO2 from the surface, which is further confirmed by the CDD plot in Fig. 7(e). The charge depletion region (in cyan) just beneath the Ti atom further indicates a charge transfer from the Ti atom to a CO2 molecule. In Fe doped stanene, 0.46e charge has been transferred to CO2 from the surface. Fig. 7(f) shows that there exists a charge depletion zone below the Fe atom, similar to Ti, indicating that electrons have been transferred from Fe to CO2 molecules.
3.4.3 CO2 adsorption in the presence of water.
Charge density difference and Bader charge calculations have also been carried out for CO2 adsorption on the catalysts in the presence of H2O to investigate if the charge transfers from the catalyst to CO2 as observed in the dry phase still persist in the presence of water. Fig. 7(g)–(i) exhibits the CDD plots for CO2 adsorption in the presence of water on the catalysts. Our calculation revealed that for Sc-doped stanene, there is almost no charge transfer (0.04e) to the CO2 molecule from the catalyst in the presence of water. On the other hand, similar to the dry catalyst, a net charge of 1.02e has been transferred to the CO2 molecule from Ti, even in the presence of water, as evident from the CDD plot (Fig. 7h). In Fe doped stanene as well, the CO2 molecule gains 0.63e charge from the surface, clearly revealed from the CDD plot (Fig. 7i). Hence, from the CDD and Bader charge calculations it is apparent that it is the transfer of excess charges from the TM (Ti, Fe) to CO2 which is responsible for the binding and subsequent activation of the CO2 molecule, and the observed activity of the catalysts.
4 Conclusion
Employing density functional theory, this present work investigates the efficiency of pure and transition metal-doped stanene as catalysts for the CO2 reduction reaction, investigating the first two electron transfer steps, i.e., reduction of CO2 to CO. As activation of CO2 is crucial for the CO2RR, we have studied the relative ability of pure and TM-doped stanene to bind and activate CO2. Our calculations revealed that only Ti- and Fe-doped stanene bind and activate CO2 while pristine stanene and the rest of the TM@Sn do not activate the molecule, indicating that activating CO2 is difficult because of the inert nature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. Our DFT results pertaining to the next hydrogenation steps revealed that the formation of OCHO as a reaction intermediate is preferred over COOH, during the 1st hydrogenation step, leading to HCOOH as the end product for the two-electron-transfer CO2RR pathway. This trend is observed in both Ti- and Fe-doped stanene. The selectivity of the catalysts for the CO2RR over the competitive HER has also been observed. As the reaction takes place in an aqueous environment, water adsorption on the catalysts and activation of CO2 in the presence of a water molecule has also been investigated in detail with a particular emphasis on the role of water in simulating CO2 adsorption on the catalysts. It has been observed that in presence of water, CO2 adsorption is promoted on the catalyst. The enhanced strength of CO2 adsorption on the catalysts is attributed to the potential formation of the hydrogen bond between the activated CO2 and neighboring H2O molecule. Finally, to understand the reason behind the activation of CO2 on Ti- and Fe-doped stanene and the inertness of the rest of the TMs studied, charge density difference and Bader charge analysis have been carried out. While in Ti- and Fe@Sn a substantial charge transfer has been observed from the catalyst to CO2, there is almost no charge transfer from the rest of the TM@Sn studied (such as Sc) contributing to their inertness in CO2 activation. It is evident, hence, from our calculation that the observed activity of a few TMs can be attributed to the charge transfer from the catalyst to the CO2. Our results obtained within the DFT framework can be extended to similar materials beyond graphene with improved functionals, for screening efficient 2D catalysts for reduction of CO2 to CO and beyond.
O bond. Our DFT results pertaining to the next hydrogenation steps revealed that the formation of OCHO as a reaction intermediate is preferred over COOH, during the 1st hydrogenation step, leading to HCOOH as the end product for the two-electron-transfer CO2RR pathway. This trend is observed in both Ti- and Fe-doped stanene. The selectivity of the catalysts for the CO2RR over the competitive HER has also been observed. As the reaction takes place in an aqueous environment, water adsorption on the catalysts and activation of CO2 in the presence of a water molecule has also been investigated in detail with a particular emphasis on the role of water in simulating CO2 adsorption on the catalysts. It has been observed that in presence of water, CO2 adsorption is promoted on the catalyst. The enhanced strength of CO2 adsorption on the catalysts is attributed to the potential formation of the hydrogen bond between the activated CO2 and neighboring H2O molecule. Finally, to understand the reason behind the activation of CO2 on Ti- and Fe-doped stanene and the inertness of the rest of the TMs studied, charge density difference and Bader charge analysis have been carried out. While in Ti- and Fe@Sn a substantial charge transfer has been observed from the catalyst to CO2, there is almost no charge transfer from the rest of the TM@Sn studied (such as Sc) contributing to their inertness in CO2 activation. It is evident, hence, from our calculation that the observed activity of a few TMs can be attributed to the charge transfer from the catalyst to the CO2. Our results obtained within the DFT framework can be extended to similar materials beyond graphene with improved functionals, for screening efficient 2D catalysts for reduction of CO2 to CO and beyond.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SG acknowledges the support from IIITDM Kancheepuram through research assistantship. DM thanks IIITDM Kancheepuram for financial support through institute SEED grant (No: IIITDM/ISG/PHY/7).
References
- N.-N. Vu, S. Kaliaguine and T.-O. Do, Critical Aspects and Recent Advances in Structural Engineering of Photocatalysts for Sunlight-Driven Photocatalytic Reduction of CO2 into Fuels, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C. B. Park, Photoelectrochemical Reduction of Carbon Dioxide to Methanol through a Highly Efficient Enzyme Cascade, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed.
- Z. Zhang, S.-Y. Pan, H. Li, J. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Recent advances in carbon dioxide utilization, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS.
- S. C. Peter, Reduction of CO2 to Chemicals and Fuels: A Solution to Global Warming and Energy Crisis, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- A. Saravanan, P. Senthil kumar, D.-V. N. Vo, S. Jeevanantham, V. Bhuvaneswari, V. Anantha Narayanan, P. R. Yaashikaa, S. Swetha and B. Reshma, A comprehensive review on different approaches for CO2 utilization and conversion pathways, Chem. Eng. Sci., 2021, 236, 116515 CrossRef CAS.
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. Wang and H. Tan, Current technology development for CO2 utilization into solar fuels and chemicals: a review, J. Energy Chem., 2020, 49, 96–123 CrossRef.
- S. Samanta and R. Srivastava, Catalytic conversion of CO2 to chemicals and fuels: the collective thermocatalytic/photocatalytic/electrocatalytic approach with graphitic carbon nitride, Mater. Adv., 2020, 1, 1506–1545 RSC.
- R. Daiyan, W. H. Saputera, H. Masood, J. Leverett, X. Lu and R. Amal, A Disquisition on the Active Sites of Heterogeneous Catalysts for Electrochemical Reduction of CO2 to Value-Added Chemicals and Fuel, Adv. Energy Mater., 2020, 10, 1902106 CrossRef CAS.
- D. Sharma, R. Sharma, D. Chand and A. Chaudhary, Nanocatalysts as potential candidates in transforming CO2 into valuable fuels and chemicals: a review, Environ. Nanotechnol. Monit. Manage., 2022, 18, 100671 CAS.
- H. S. Whang, J. Lim, M. S. Choi, J. Lee and H. Lee, Heterogeneous catalysts for catalytic CO2 conversion into value-added chemicals, BMC Chem. Eng., 2019, 1, 9 CrossRef.
- S. A. Mazari, N. Hossain, W. J. Basirun, N. M. Mubarak, R. Abro, N. Sabzoi and A. Shah, An overview of catalytic conversion of CO2 into fuels and chemicals using metal organic frameworks, Process Saf. Environ. Prot., 2021, 149, 67–92 CrossRef CAS.
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Heterogeneous Catalysis for CO2 Conversion into Chemicals and Fuels, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS.
- Y. Ling, Q. Ma, Y. Yu and B. Zhang, Optimization Strategies for Selective CO2 Electroreduction to Fuels, Trans. Tianjin Univ., 2021, 27, 180–200 CrossRef CAS.
- Q. Cui, G. Qin, W. Wang, K. R. Geethalakshmi, A. Du and Q. Sun, Novel two-dimensional MOF as a promising single-atom electrocatalyst for CO2 reduction: a theoretical study, Appl. Surf. Sci., 2020, 500, 143993 CrossRef CAS.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Organic–Inorganic Hybrid Nanomaterials for Electrocatalytic CO2 Reduction, Small, 2020, 16, 2001847 CrossRef CAS PubMed.
- W. da Silva Freitas, A. D’Epifanio and B. Mecheri, Electrocatalytic CO2 reduction on nanostructured metal-based materials: challenges and constraints for a sustainable pathway to decarbonization, J. CO2 Util., 2021, 50, 101579 CrossRef CAS.
- M. N. Hossain, J. Wen and A. Chen, Unique copper and reduced graphene oxide nanocomposite toward the efficient electrochemical reduction of carbon dioxide, Sci. Rep., 2017, 7, 3184 CrossRef PubMed.
- S. Lu, F. Lou and Z. Yu, Recent Progress in Two-Dimensional Materials for Electrocatalytic CO2 Reduction, Catalysts, 2022, 12, 228 CrossRef CAS.
- Y. Wang, Y. Li, J. Liu, C. Dong, C. Xiao, L. Cheng, H. Jiang, H. Jiang and C. Li, BiPO4-Derived 2D Nanosheets for Efficient Electrocatalytic Reduction of CO2 to Liquid Fuel, Angew. Chem., Int. Ed., 2021, 60, 7681–7685 CrossRef CAS PubMed.
- S. Payra, S. Shenoy, C. Chakraborty, K. Tarafder and S. Roy, Structure-Sensitive Electrocatalytic Reduction of CO2 to Methanol over Carbon-Supported Intermetallic PtZn Nano-Alloys, ACS Appl. Mater. Interfaces, 2020, 12, 19402–19414 CrossRef CAS PubMed.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Metal-Free Carbon Materials for CO2 Electrochemical Reduction, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, Understanding Trends in the Electrocatalytic Activity of Metals and Enzymes for CO2 Reduction to CO, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- R. Küngas, Review—Electrochemical CO2 Reduction for CO Production: Comparison of Low- and High-Temperature Electrolysis Technologies, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- X. Liu and L. Dai, Carbon-based metal-free catalysts, Nat. Rev. Mater., 2016, 1, 1–12 CrossRef.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Hierarchical Mesoporous SnO2 Nanosheets on Carbon Cloth: A Robust and Flexible Electrocatalyst for CO2 Reduction with High Efficiency and Selectivity, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Recent Advances in Carbon-Based Metal-Free Electrocatalysts, Adv. Mater., 2019, 31, 1806403 CrossRef PubMed.
- D. Misra and S. K. Yadav, Nb-Implanted BaO as a Support for Gold Single Atoms, J. Phys. Chem. C, 2021, 125, 28059–28066 CrossRef CAS.
- W. Kong, J. Deng and L. Li, Recent advances in noble metal MXene-based catalysts for electrocatalysis, J. Mater. Chem. A, 2022, 10, 14674–14691 RSC.
- C. Anichini, W. Czepa, D. Pakulski, A. Aliprandi, A. Ciesielski and P. Samorì, Chemical sensing with 2D materials, Chem. Soc. Rev., 2018, 47, 4860–4908 RSC.
- D. Akinwande, C. J. Brennan, J. S. Bunch, P. Egberts, J. R. Felts, H. Gao, R. Huang, J.-S. Kim, T. Li, Y. Li, K. M. Liechti, N. Lu, H. S. Park, E. J. Reed, P. Wang, B. I. Yakobson, T. Zhang, Y.-W. Zhang, Y. Zhou and Y. Zhu, A review on mechanics and mechanical properties of 2D materials—Graphene and beyond, Extreme Mech. Lett., 2017, 13, 42–77 CrossRef.
- R. Hu, G. Liao, Z. Huang, H. Qiao, H. Liu, Y. Shu, B. Wang and X. Qi, Recent advances of monoelemental 2D materials for photocatalytic applications, J. Hazard. Mater., 2021, 405, 124179 CrossRef CAS PubMed.
- T. Wang, H. Wang, Z. Kou, W. Liang, X. Luo, F. Verpoort, Y.-J. Zeng and H. Zhang, Xenes as an Emerging 2D Monoelemental Family: Fundamental Electrochemistry and Energy Applications, Adv. Funct. Mater., 2020, 30, 2002885 CrossRef CAS.
- H. Qiao, H. Liu, Z. Huang, R. Hu, Q. Ma, J. Zhong and X. Qi, Tunable Electronic and Optical Properties of 2D Monoelemental Materials Beyond Graphene for Promising Applications, Energy Environ. Mater., 2021, 4, 522–543 CrossRef CAS.
- X. Chen, C. Tan, Q. Yang, R. Meng, Q. Liang, M. Cai, S. Zhang and J. Jiang, Ab Initio Study of the Adsorption of Small Molecules on Stanene, J. Phys. Chem. C, 2016, 120, 13987–13994 CrossRef CAS.
- K. Ma, J. Chen, X. Dai, J. Xiao, L. Wang, L. Xu and Z. Wang, The potential of stanene with transition metal adsorbed as a promising gas sensor: a first-principles study, Results Phys., 2021, 28, 104617 CrossRef.
- A. Zhang, H. Yang, Q. Liu, W. Li and Y. Wang, DFT insights into the adsorption properties of toxic gas molecules on pure and transition metal embedded stanene monolayers: towards gas sensor devices, Synth. Met., 2020, 266, 116441 CrossRef CAS.
- J. Zhou, D. Liu, F. Wu, L. Yang, Y. Xiong and A. Abbasi, A DFT study on the possibility of embedding a single Ti atom into the perfect stanene monolayer as a highly efficient gas sensor, Theor. Chem. Acc., 2020, 139, 46 Search PubMed.
- Y.-Z. Chang, J.-N. Lin, S.-D. Li and H. Liu, Adsorption of greenhouse gases (methane and carbon dioxide) on the pure and Pd-adsorbed stanene nanosheets: a theoretical study, Surf. Interfaces, 2021, 22, 100878 Search PubMed.
- A. Abbasi and J. J. Sardroodi, Density functional theory investigation of the interactions between the buckled stanene nanosheet and XO2 gases (X = N, S, C), Comput. Theor. Chem., 2018, 1125, 15–28 Search PubMed.
- H. Vovusha, T. Hussain, M. Sajjad, H. Lee, A. Karton, R. Ahuja and U. Schwingenschlögl, Sensitivity enhancement of stanene towards toxic SO2 and H2S, Appl. Surf. Sci., 2019, 495, 143622 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- X. Li, H. Li, X. Zuo, L. Kang, D. Li, B. Cui and D. Liu, Chemically Functionalized Penta-stanene Monolayers for Light Harvesting with High Carrier Mobility, J. Phys. Chem. C, 2018, 122, 21763–21769 CrossRef CAS.
- A. Abbasi and J. J. Sardroodi, Electronic structure tuning of stanene monolayers from DFT calculations: effects of substitutional elemental doping, Appl. Surf. Sci., 2018, 456, 290–301 CrossRef CAS.
- S. R. Naqvi, T. Hussain, W. Luo and R. Ahuja, Exploring Doping Characteristics of Various Adatoms on Single-Layer Stanene, J. Phys. Chem. C, 2017, 121, 7667–7676 CrossRef CAS.
- J. Chen, Z. Wang, X. Dai, J. Xiao, M. Long and T. Chen, The effects of transition metal adatoms on the electronic properties of stanene, Phys. E, 2020, 124, 114365 CrossRef CAS.
- A. Abbasi, Theoretical Investigation of The interaction Between Noble Metals (Ag, Au, Pd, Pt) and Stanene Nanosheets: A DFT Study, J. Inorg. Organomet. Polym. Mater., 2019, 29, 1895–1915 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- D. Misra, G. Di Liberto and G. Pacchioni, CO2 electroreduction on single atom catalysts: is water just a solvent?, J. Catal., 2023, 422, 1–11 CrossRef CAS.
- S. Wang, L. Li, J. Li, C. Yuan, Y. Kang, K. S. Hui, J. Zhang, F. Bin, X. Fan, F. Chen and K. N. Hui, High-Throughput Screening of Nitrogen-Coordinated Bimetal Catalysts for Multielectron Reduction of CO2 to CH4 with High Selectivity and Low Limiting Potential, J. Phys. Chem. C, 2021, 125, 7155–7165 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- S. Zhou, W. Pei, J. Zhao and A. Du, Silicene catalysts for CO2 hydrogenation: the number of layers controls selectivity, Nanoscale, 2019, 11, 7734–7743 RSC.
- M. Wang, L. Kong, X. Lu and C.-M. L. Wu, First-row transition metal embedded pyrazine-based graphynes as high-performance single atom catalysts for the CO2 reduction reaction, J. Mater. Chem. A, 2022, 10, 9048–9058 RSC.
- H. Li, D. Wu, J. Wu, Y. Song, W. Lv, Z. Duan and D. Ma, Mechanistic understanding of the electrocatalytic conversion of CO into C2+ products by double-atom catalysts, Mater. Today Phys., 2023, 37, 101203 CrossRef CAS.
- D. Wu, P. Lv, J. Wu, B. He, X. Li, K. Chu, Y. Jia and D. Ma, Catalytic active centers beyond transition metals: atomically dispersed alkaline-earth metals for the electroreduction of nitrate to ammonia, J. Mater. Chem. A, 2023, 11, 1817–1828 RSC.
- J. Lin, Z. Pan and X. Wang, Photochemical Reduction of CO2 by Graphitic Carbon Nitride Polymers, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS PubMed.
- U. J. Etim, C. Zhang and Z. Zhong, Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces, Nanomaterials, 2021, 11, 3265 CrossRef CAS PubMed.
- Y. Wang, D. He, H. Chen and D. Wang, Catalysts in electro-, photo- and photoelectrocatalytic CO2 reduction reactions, J. Photochem. Photobiol., C, 2019, 40, 117–149 CrossRef.
- Y. Quan, J. Zhu and G. Zheng, Electrocatalytic Reactions for Converting CO2 to Value-Added Products, Small Sci., 2021, 1, 2100043 CrossRef CAS.
- M. Ma, F. Li and Q. Tang, Coordination environment engineering on nickel single-atom catalysts for CO2 electroreduction, Nanoscale, 2021, 13, 19133–19143 RSC.
- L. Gong, X. Wang, T. Zheng, J. Liu, J. Wang, Y.-C. Yang, J. Zhang, X. Han, L. Zhang and Z. Xia, Catalytic mechanism and design principle of coordinately unsaturated single metal atom-doped covalent triazine frameworks with high activity and selectivity for CO2 electroreduction, J. Mater. Chem. A, 2021, 9, 3555–3566 RSC.
- T. Liu, G. Wang and X. Bao, Electrochemical CO2 Reduction Reaction on 3d Transition Metal Single-Atom Catalysts Supported on Graphdiyne: A DFT Study, J. Phys. Chem. C, 2021, 125, 26013–26020 CrossRef CAS.
- D. Bohra, I. Ledezma-Yanez, G. Li, W. de Jong, E. A. Pidko and W. A. Smith, Lateral Adsorbate Interactions Inhibit HCOO− while Promoting CO Selectivity for CO2 Electrocatalysis on Silver, Angew. Chem., 2019, 131, 1359–1363 CrossRef.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements, Chem. Sci., 2017, 8, 1090–1096 RSC.
- C. Ling, Q. Li, A. Du and J. Wang, Computation-Aided Design of Single-Atom Catalysts for One-Pot CO2 Capture, Activation, and Conversion, ACS Appl. Mater. Interfaces, 2018, 10, 36866–36872 CrossRef CAS PubMed.
- S. Vijay, W. Ju, S. Brückner, S.-C. Tsang, P. Strasser and K. Chan, Unified mechanistic understanding of CO2 reduction to CO on transition metal and single atom catalysts, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- C. Zhao, X. Su, S. Wang, Y. Tian, L. Yan and Z. Su, Single-atom catalysts on supported silicomolybdic acid for CO2 electroreduction: a DFT prediction, J. Mater. Chem. A, 2022, 10, 6178–6186 RSC.
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, Machine-Learning-Accelerated Catalytic Activity Predictions of Transition Metal Phthalocyanine Dual-Metal-Site Catalysts for CO2 Reduction, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- A. K. Srivastava and N. Misra, Calculating interaction energies of hydrogen bonded dimers and complexes of HF, H2O and NH3: Super-molecular versus AIM Approach, J. Comput. Methods Mol. Des., 2014, 4, 19–23 Search PubMed.
- L. Bondesson, K. V. Mikkelsen, Y. Luo, P. Garberg and H. Ågren, Hydrogen bonding effects on infrared and Raman spectra of drug molecules, Spectrochim. Acta, Part A, 2007, 66, 213–224 CrossRef PubMed.
- D. Zhao and X. Liu, Density Functional Calculation of H2O/CO2/CH4 for Oxygen-Containing Functional Groups in Coal Molecules, ACS Omega, 2022, 7, 17330–17338 CrossRef CAS PubMed.
- Biochem 104b Lecture Notes, https://www.bio.brandeis.edu/classes/biochem104/lecture_notes.html, (accessed November 28, 2023).
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Debolina
Misra
b and
Debolina
Misra