
Natural resonance-theoretic conceptions of extreme electronic delocalization in soft materials†
Frank
Weinhold
 *a and
Eric D.
Glendening
b
*a and
Eric D.
Glendening
b
aTheoretical Chemistry Institute and Department of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: weinhold@chem.wisc.edu
bDepartment of Chemistry and Physics, Indiana State University, Terre Haute, IN 47809, USA. E-mail: eric.glendening@indstate.edu
First published on 28th December 2023
Abstract
In the broad context of Dalton's atomic hypothesis and subsequent classical vs. quantum understanding of macroscopic materials, we show how Pauling's resonance-type conceptions, as quantified in natural resonance theory (NRT) analysis of modern wavefunctions, can be modified to unify description of interatomic interactions from the Lewis-like limit of localized e-pair covalency in molecules to the extreme delocalized limit of supramolecular “soft matter” aggregation. Such “NRT-centric” integration of NRT bond orders for hard- and soft-matter interactions is illustrated with application to a long-predicted and recently synthesized organometallic sandwich-type complex (“diberyllocene”) that exhibits bond orders ranging from the soft limit (bBeC ≈ 0.01) to the typical values (bCC ≈ 1.35) of molecular resonance-covalency in the organic domain, with intermediate value (bBeBe ≈ 0.86) for intermetallic Be⋯Be interaction.
Introduction: quantal vs. classical conceptions of soft matter interactions
In reconsidering prevailing general conceptions concerning molecules and materials, it is appropriate to briefly reflect on the historical sequence by which present “physical” conceptions of chemical behavior were achieved.Speculations on the atomic-level nature of matter trace back to antiquity,1 but firm scientific foundations for atomistic conceptions, building on the systematic weight measurements of Lavoisier2 and Dalton3 and rational theoretical formulations of Gibbs,4 offered ever-increasing support for Boltzmann's boldly literal conjectures concerning the “moving material points” (atoms and molecules) that underlie macroscopic chemical, electrical, and thermal phenomena.5
In the early 20th century, more detailed electronic (e-level) conceptions of chemical bonding between atoms were spurred particularly by Lewis's dot-diagrams depicting localized e-pair-sharing (“covalency”) between the valence-shell electrons of the Z-charged nucleus of each neutral atom.6 Planck's discovery7 of the quantum of action (ℏ) was subsequently recognized by Bohr8 as providing quantitative explanation for the spectral lines of hydrogen (Z = 1) as well as compelling rationale for the periodicity patterns in Mendeleev's table of chemical elements for higher Z.9
These revolutionary developments culminated in the 1925–1926 papers of Heisenberg,10 Schrödinger,11 and Dirac.12 All available results from precision comparisons of theory and experiment13 support the claim that the properties of all chemical species (implicitly including those of biological, agricultural, and geological interest, and many yet to be discovered) are correctly described by the equations of quantum mechanics, both in non-relativistic10,11 and relativistic12 limits. The quantum mechanical equations thus supersede all classical–mechanical frameworks as proper foundation for the deepest conceptions of the materials domain.
Contrary to the above statement, some workers continue to argue that “quantum effects” extend only to the covalent e-pairing interactions of molecule formation, whereas the soft “noncovalent” attractions (e.g., of ligation or complexation) are to be rationalized in terms of classical electrostatic conceptions of dipole–dipole or higher multipole type. In this view, quantum equations are reserved for the “hard” interactions of molecular single-, double-, triple-bond formation, with intermediate fractional values attributable to weaker resonance-type corrections, whereas interactions of the intermolecular domain can be adequately “simulated” with molecular dynamics (MD) equations of purely classical form, thereby avoiding the complexities of exchange, superposition, and related orbital-type features of the molecular regime.
However, any such presumption of separable “covalent vs. noncovalent” (or “intramolecular vs. intermolecular,” “covalent vs. ionic,” etc.) bonding represents a false dichotomy in the logical sense. Such dichotomy would erroneously suggest that disjoint aspects of chemical behavior can be evaluated by mutually inconsistent mathematical assumptions and simply added together (in the style of “energy decomposition analysis” schemes14) to obtain successful theoretical predictions of material properties.
According to Bohr's correspondence principle,15 proper quantum equations must automatically reduce to their classical counterpart in the limit of large quantum numbers or vanishing quantum of action (ℏ → 0). This implies that the quantum equations already include all relevant contributions of classical type, rather than requiring addition of separate “quantal” and “classical” contributions to total energy.
Aside from these formal considerations, one can look at the historical record of atomistic-level classical-MD methods in attempts to solve substantive scientific problems. Perhaps most famous (or infamous) in this respect is the protein-folding problem, formalized over nearly 30 years in the biennial CASP (“Critical Assessment of Structure Prediction”) competition co-founded by computational biologist John Mount.16 Each new CASP event featured a newly chosen protein whose native folded structure had been recently determined by X-ray crystallography, but with structural coordinates withheld from public distribution. Competing MD specialists were provided with the primary amino acid sequence, and all have access to the many previously determined native protein structures. Each competitor then employs a favored MD force field to computationally “predict” the correctly folded protein structure, usually with profoundly disappointing results for all concerned.
However, as recently reported,16 a new artificial-intelligence (AI) competitor AlphaFold swept the 2020 competition with essentially perfect results, even though it has nothing in common with atomistic-level MD simulations! Magically, AlphaFold was able to solve the problem simply by using AI pattern-recognition algorithms on the widely available crystallographic database of previously solved structures. The combined efforts of 30+ years of classical-MD simulations therefore appear to have contributed less than “nothing” to the eventual successful predictions of protein-folding. “In some sense the problem is solved,” said Moult. However, the same classical-based MD conceptions that failed to properly predict protein folding can hardly be expected to correctly predict the chemical functionality of the folded structures as now provided by AlphaFold or experiment. Human scientists have no more insight than before about how and why proteins fold and function as they do.
Our goal here is to urge precedence for theoretical studies of materials that are firmly based on authentic quantum chemical methods and the conceptual insights they provide through the lens of natural bond orbital (NBO)17 and natural resonance theory (NRT)18 analysis. Throughout this discussion, we focus primarily on the neutral closed-shell ground states of common laboratory substances. All NBO/NRT methods rest on Löwdin's “natural” orbital conceptions,19 which in turn originate in von Neumann's density operator (![[capital Gamma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e132.gif) ) formulation of quantum mechanics20 for a specific state Ψ of the N-electron system. In Dirac notation, is expressed as the integral operator
) formulation of quantum mechanics20 for a specific state Ψ of the N-electron system. In Dirac notation, is expressed as the integral operator
| = |Ψ〉〈Ψ| | (1) |
| Γ(1,2,…,N|1′,2′,…,N′) = Ψ(1,2,…,N)Ψ*(1′,2′,…,N) | (2) |
(1), with kernel | (3) |
(2), etc.).
In a chosen spin-orbital basis {χk}, the “density matrix” representation (D) of (1) has elements
(D)jk = 〈χj|![[capital Gamma, Greek, circumflex]](https://www.rsc.org/images/entities/char_e132.gif) (1)|χk〉 (1)|χk〉 | (4) |
NBO/NRT methods are implemented in a widely used computer program (currently NBO 7.0)22 that can be interactively linked with a wide variety of popular electronic structure systems,23 insuring strict consistency of theoretical descriptors across a broad selection of quantum chemical methods [semi-empirical, HF, DFT, MP(n), CI, CC(SDT…), CASSCF,…] and basis sets [Gaussian, Slater, or implicit (semi-empirical) AOs] in common usage.24
In the ensuing discussion, we first outline the extremes of localization and delocalization that are found in NBO/NRT studies across the periodic table. We then focus on the characteristic features of NRT description in the case of species of weak vibrational rigidity, dissociation energy, or thermal stability (evidence of mechanical “softness”) or high electrical and thermal conductivity (evidence of electronic “delocalization”). In each case we employ NRT interatomic bond orders {bAB} as primary theoretical descriptors of localized bonding strength, expected to serve as principal theoretical correlators with experimentally measurable bond properties such as bond length (RAB), bond dissociation energy (ΔEA⋯B), bond vibrational frequency (νAB), NMR coupling (1JAB), and the like. Our goal is thereby to further broaden the NBO/NRT framework to deal with the more extreme delocalization effects in materials that lack conventional “molecule-like” connectivity.
Localized and delocalized extremes of chemical bonding
Electronic structure theory advanced rapidly in the years immediately following discovery of the Schrödinger equation. Early numerical applications of molecular orbital (MO) theory to the hydrogen molecular ion25 and valence bond (VB) theory to the hydrogen molecule26 soon led to Pauling's masterful “nature of the chemical bond” exposition of hybridization and resonance concepts, initially in a series of papers27 and subsequently in the famous book28 that “brought quantum mechanics into practical chemistry” and has been compared with Newton's Principia and Darwin's Origin of Species for its iconic status in the development of scientific thought.29Pauling's VB-style empiricism was chosen to map directly onto G. N. Lewis's dot-diagrams6 depicting the shared e-pairs that underlie the localized interatomic “sticks” of covalent bonding to form molecules. In what came to be identified as the HLSP-PP-VB (Heitler–London–Slater–Pauling perfect-pairing valence bond) formulation,30 each localized σAB bond of the skeletal framework is composed from overlapping spn-type directed hybrid orbitals (hA, hB) on the bonding atoms. In VB-representation, the spatial factor of each 2-center/2-electron (2c/2e) wavefunction is taken to be of the form
| σ(VB)AB(1,2) = 2−1/2[hA(1)hB(2) + hB(1)hA(2)] | (5) |
| σ(BO)AB(1,2) = [cAhA(1) + cBhB(1)][cAhA(2) + cBhB(2)] | (6a) |
| |cA|2 + |cB|2 = 1 | (6b) |
Pauling's resonance concept was introduced to deal with benzene and similar exceptional cases where the Lewis electron-dot mnemonic requires a pattern of alternating single- and double-bonds, but the experimental structure is of higher symmetry (e.g., D6h for actual benzene vs. D3h for Lewis-structural “cyclohexatriene”). In such cases, the actual many-electron wavefunction Ψ is envisioned as a “resonance hybrid” of the two alternative localized Lewis-structural patterns ΦI, ΦII, which can be expressed in von Neumann's density-operator notation (1) as
| NRT = wI|ΦI〉〈ΦI| + wII|ΦII〉〈ΦII| | (7a) |
| wI + wII = 1 | (7b) |
| 〈Ψ|NRT|Ψ〉 = 〈Ψ|[wI|ΦI〉〈ΦI| + wII|ΦII〉〈ΦII|]|Ψ〉 = max | (8) |
and NRT. The convex form (7a) for density-based optimizations may be contrasted with the corresponding configuration interaction (CI) form for energy-based optimizations, with superposition coefficients of either sign and multiple cross-terms contributing to the target variational functional.
In benzene or other strongly conjugated molecules, the weightings wI, wII approach the symmetry-enforced limit (wI ≈ wII ≈ 0.5), but in the weaker resonance of amides or the still weaker cases of hyperconjugation,32 the weightings shift increasingly toward the near-Lewis-structure limit (wI ≈ 1, wII ≈ 0) of resonance-free bonding. Because each such resonance effect involves a type of “mixing” (delocalization) of the original 2c e-pair with another e-pair situated (partially or wholly) at a 3rd center, it is commonly identified as “3c/4e” (partially delocalized) bonding, thereby distinguished from conventional 2c/2e bonding of a fully localized Lewis-structural bonding pattern.
Note that NRT methods can also be applied to open-shell radicals or excited-state species to study the delocalized spin distributions and related magnetic properties. However, the NRT bonding patterns of a given system and state are only “precursors” to possible low-barrier reactive pathways for forming bond-shifted products, or envisioned multi-state “quasi-particle” excitation phenomena of condensed phases. Furthermore, current NBO/NRT methodology applies only to fixed-N species, rather than infinitely extended models for condensed phases.
In most respects, NRT results closely mirror Pauling's original resonance concepts,33 but with two important exceptions:
(i) The numerical NRT weightings {wr} are determined from a rigorous “maximum density” criterion (analogous to that used to determine the hybridization and polarization properties of NBOs) rather than the empirical fitting to measured structural parameters as adopted by Pauling;
(ii) NRT bond orders can in principle acquire non-negative fractional values anywhere from zero upward, whereas Pauling's original resonance-mixing applied only to intramolecular interactions, thereby yielding fractional values only between positive integer (single, double, triple,…) bonds of the molecular skeleton (as judged from empirical covalent radii for each atom).
However, from the many short contact distances that were found between non-bonded atoms in the 40-plus years since his original resonance conceptions, Pauling later recognized34 that resonance-type stabilization must extend to “no-bond resonance” of the intermolecular domain in certain metallic cases. This concurs with the general NBO/NRT perception that quantal resonance-covalency (3c/4e donor–acceptor stabilization) extends into the regime of “soft” intermolecular interactions (with bond orders in the range 0 ≤ bA⋯B ≤ 1, smoothly connecting with known intramolecular bond-order variations). In this respect, Pauling's readjusted position on supramolecular resonance is closer to the early viewpoint of Sidgwick35 and many subsequent workers, particularly for the H-bonding phenomenon.36
In outline, the NRT program employs an iterative algorithm to generate a “tree” of candidate resonance structures from the initial NBO structure, as well as any other user-suggested structures that may be provided in $NRTSTR keylist input.18 Successive iterative cycles then lead to many such candidate bonding patterns (possibly tens of thousands!) that might seem to present an insuperable numerical task for the weightings-optimizer. However, the intrinsic “convex programming”37 mathematical structure of the optimization problem ensures that the final weightings are generated in robustly stable and efficient fashion with full numerical accuracy.18d
Resonance primacy: the other side of the localization coin
Historically, the development of electronic bonding concepts, as well as the original NBO program itself, was dominated by G. N. Lewis's seminal recognition of localized 2c/2e “pairing” as the primary feature of chemical bonding. In this limit, the iconic ball-and-stick picture of molecular connectivity in some “primary” bonding pattern becomes the starting point for perturbative evaluation of “secondary” resonance-type delocalization effects. In the present NBO7 program, the primacy of the natural Lewis structure (NLS) bonding pattern is implicitly assumed, and indeed, the combined sum of secondary NRT-type corrections is commonly less than that of the primary NLS contribution in organic main-group applications. [Note that the corrupted and obsolete “NBO 3.1” program distributed with Gaussian-16 has no authentic NRT capability, even though it contains code that apparently reads NRT-related keyword input.]However, with a robustly general NRT search algorithm in hand, it becomes feasible to envision an alternative NRT-centric analysis designed for an entirely different domain of electronic delocalization effects, as well as smooth reduction to the NLS-centric limit. In such NRT-centric formulation, the formal NLS bonding pattern may play little or no role as “parent” for generation of trial structures to be included in variational optimization of NRT weightings. Far from being “primary” or “dominant,” the formal NLS may have negligible weighting in the final NRT expansion, which instead represents an extended delocalization network with no single bonding pattern achieving the significant majority weighting to warrant ball-and-stick (molecule-like) preferential status. In some respects, this is analogous to the way in which a particular free-atom valence-shell NAO may become a small minority contributor to the more general natural hybrid orbital (NHO) that better serves as conceptual building-block for the network of covalent bonds in a Lewis-structural bonding pattern.
For specificity, we identify the leading (highest weighted) structure of the NRT expansion as the “natural resonance-type Lewis-structure” (NRLS) to distinguish it (if necessary) from the NBO-centric NLS. In the extreme NRT-centric limit, only the final bond orders (many or all in the 0 < bAB < 1 range) remain as useful “local” descriptors of resonance-type stabilization that correlate with structural bond lengths or other measurable interatomic properties. Of course, many intermediate cases can occur between the envisioned extreme limits of NLS-centric (“localized Lewis-like”) and NRT-centric (“completely delocalized”) electronic behavior, but a properly generalized NBO/NRT analysis framework should be able to range freely between these “hard” (bAB > 1) and “soft” (bAB → 0) limits of material behavior.
In the present work, we employ a simple “intermediate” example of resonance delocalization in a sandwich-type organometallic complex (diberyllocene) to illustrate how the present NBO 7.0 algorithms can be extended toward the NRT-centric limit of “extreme” delocalization in metallic or soft-matter species. In this intermediate-delocalization case, the default NRT search is found to be adequate, but the illustrated protocol serves to ensure “completeness” of the NRT search for contributing structures. In future NBO program versions, the illustrated strategy (involving use of $CHOOSE and $NRTSTR keylists) will be automated to ensure consistency between the final NRLS bonding pattern of highest NRT weighting and the starting point for the recursive “tree” of NRT-guess structures for which optimized weightings are computed.
Illustrative application to diberyllocene
As an example of current research interest, we illustrate the NRT-centric approach to analyzing the delocalization in diberyllocene, (BeC5H5)2, a surprising sandwich-like organometallic complex with cyclopentadienyl groups encapsulating what is nominally a dimer of beryllium (Z = 4), a notoriously poisonous element with known aversion to forming dimetallic Be–Be bonds. The structural details of diberyllocene (including the curiously eclipsed C5h geometry) were first predicted theoretically by the Schaefer group in 2005,38 but a successful synthesis and X-ray characterization was achieved only recently,39 heralded as “A big breakthrough for beryllium.”40A variety of ab initio and DFT methods and basis sets are found to yield satisfactory agreement with the experimental X-ray structure for diberyllocene. For present purposes we employ a simple B3LYP/6-311++G** level of hybrid density functional theory, which allows direct comparison with NBO/NRT results for many other chemical species.36Table 1 compares calculated B3LYP/6-311++G** structural parameters with those of the earlier theoretical prediction,38 both in close agreement with the recent X-ray measurements.37
| Geometry | Ref. 38 | Present |
|---|---|---|
| R C(1)Be(21) | 1.968 | 1.952 |
| R C(1)C(2) | 1.425 | 1.418 |
| R C(1)H(6) | 1.085 | 1.079 |
| R Be(21)Be(22) | 2.057 | 2.041 |
| ∡x–C(1)–H(6) | 3.4° | 3.2° |
Diberyllocene exemplifies a dilemma of the original NRT implementation18 that occurs whenever the “leading” (NRLS) structure of the NRT expansion is inconsistent with the supposedly “best” (NLS) single Lewis structure (according to the variational maximum-density criterion by which NBO algorithms are governed17). This inconsistency is uncommon in the vast majority of stable chemical compounds to be found in the chemical laboratory and discussed in chemistry classrooms. However, examples of such NLS/NRLS mismatch become increasingly common in the “soft-matter” domain of weak supramolecular interactions, where no interatomic bond order may reach the threshold (bAB ≥ 1) considered necessary for “molecule” formation.
Given the NLS vs. NRLS distinction, we can recognize that the choice of NLS as starting point for NRT-based studies of resonance phenomena is a rather arbitrary consequence of the bias introduced by historical sequence, with general recognition of “Lewis structure” preceding that of multi-structural “resonance” by more than a decade. The variational NRT algorithm (7) is the appropriate generalization of the self-same maximum-density criterion that governs all “natural” orbital concepts.19 As indicated above, multi-reference NRT is manifestly more general than single-NLS description. Accordingly, both logical and numerical consistency demand that the former NRT algorithm be iteratively extended (if NLS and NRLS fail to coincide) with the NRLS (as obtained from initial NLS-based iteration) chosen as the starting point for full NRT re-optimization. This added step ensures self-consistency between initial and final assessment of which resonance structure is considered “best” in the more general NRLS-based framework. Such NRLS-based bond orders can be obtained by simply inserting the NRLS structure found in an initial NLS-based NRT search as $CHOOSE keylist input (replacing the NLS) in a subsequent NRLS-based NRT search.
Fig. 1(a)–(e) shows further details of how NLRS-based NRT analysis is performed for diberyllocene. Fig. 1(a) shows details of the NLS bonding pattern that initiates the default NRT search [with LONE pairs on atoms 1, 14 and BOND pairs “S 1 2” (single bond between atoms 1, 2), “S 1 5” (single bond between atoms 1, 5), and so forth]. Fig. 1(b) shows the brief output summary of iterative cycles for the NLS-based search, which successively reduces overall D(w) “error” by about 34% (from initial 0.09257 in cycle 1 to final 0.06078 in cycle 6) and leads to a final 223-term NRT expansion (out of 4737 trial structures considered). Fig. 1(c) shows details of the resulting NRLS bonding pattern that is highest-weighted (w1 = 2.67%) in the initial NRT search [with LONE pairs on atoms 5, 15 and BOND pairs “D 1 2” (double bond between atoms 1 and 2), “S 1 5” (single bond between atoms 1 and 5), and so forth], all differing from the NLS bonding pattern in Fig. 1(a). Fig. 1(d) shows how the NRLS bonding pattern is inserted in a $CHOOSE…$END keylist as input (to replace the NLS) in a new NRLS-based NRT search. Finally, Fig. 1(e) presents the summary of the alternative NRLS-based NRT search, which may be compared with the NLS-based search in Fig. 1(b).
 | ||
| Fig. 1 Details of NRLS-based NRT analysis for diberyllocene (see text), showing alternative starting structures [NLS (a) vs. NRLS (c)], $CHOOSE input for the latter [(d)], and respective NRT search summaries [(b) and (e)]. | ||
In the present case, the NLS and NRLS are merely symmetry-equivalent alternatives, so the final NRLS-based NRT bond orders are identical to those of the default NLS-based search. However, this extra step gives insurance that no untapped reservoir of contributing resonance structures has been overlooked. It also justifies confidence that the calculated NRT bond orders are well converged and suitable for the intended correlations with measurable properties, a unique feature of bond-order measures compared to theoretical descriptors from other analysis methodologies.
Fig. 2 displays the optimized structure and NRLS-based NRT bond orders of diberyllocene at the B3LYP/6-311++G** level. The structure shows the striking D5h symmetry of the complex, the curious eclipsed conformation of the two opposed Cp rings, and the barely perceptible inward “tilt” of CH bonds on each Cp ring toward the opposite face of the sandwich, all calling for electronic explanation.
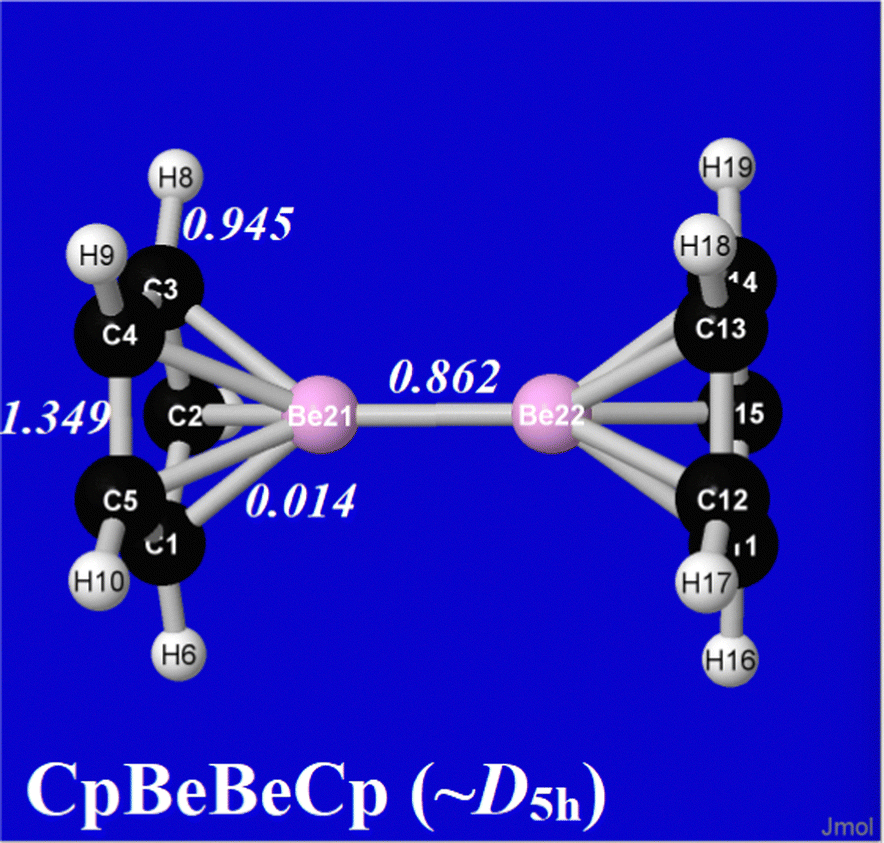 | ||
| Fig. 2 NRT bond orders for the optimized B3LYP/6-311++G** structure of (C5H5Be)2 of the present work. | ||
Noteworthy in the present context is the distribution of NRT bond orders in Fig. 2, with “molecule-like” values (bAB ≥ 1) restricted to CC bonds of the capping cyclopendienyl groups, whereas each Be atom engages only in distinctly sub-molecular bonding [bBeBe = 0.862, bBeC = 0.014] to adjacent atoms of the sandwich complex. Quantitative NBO/NRT descriptors also show sharp departures from ionic-type Cp−⋯Be+–Be+⋯Cp− depiction. The natural population analysis (NPA) atomic charge (Q(NPA)Be = 0.84) falls significantly below that of the formal “Be(I)” oxidation state of an ionic description. However, the authors of ref. 39 attribute this discrepancy to the fact that NPA charges “do not take into account the presence of the non-nuclear attractor” of the corresponding Bader-type “quantum theory of atoms in molecules” (QTAIM) value (Q(QTAIM)Be = 1.39). We believe that NBO/NRT charges and bond orders offer a more accurate and nuanced picture of bonding in diberyllocene than concepts based on the “non-nuclear attractor” artifacts of QTAIM partitioning.41
Concluding summary
We have sketched the outlines of an alternative “NRT-centric” formulation of NBO/NRT analysis methods that better ensures formal continuity of resonance-type bond-order descriptors that can range from the strongly localized Lewis-like limit of molecule formation (“hard” matter) to the strongly delocalized metallic-like limit of supramolecular (“soft” matter) interactions. Specific technical details of how such hard- → soft-matter transition can be accomplished in the framework of the current NBO 7.0 analysis program are described and illustrated with application to a recently synthesized “diberyllocene” complex that promotes intermediate-type supramolecular Be⋯Be bonding in the sub-molecular (bBeBe < 1) domain. Details of NBO/NRT analysis of diberyllocene exhibit the significant deviations from ionic-type [Cp−⋯Be+⋯Be+⋯Cp−] conceptions, as well as their alleged support from QTAIM-type descriptors.The present application illustrates the case where no resonance structure acquires more than a small minority weighting (all wi < 3%), demonstrates the procedure to be followed whenever the leading structure of the NRT expansion differs from that of the starting NLS, and exhibits a variety of bond orders in the weak (bCBe), intermediate (bBeBe), and multiple-bonding (bCC) range. However, the beryllocene sandwich-complex, although interesting in other respects, is found to remain safely within the localized “molecule-like” range of the current NLS-based NRT search. Further exploration of the extreme delocalization limit therefore awaits NRT applications to transition-metal species with more characteristic metallic conduction and magnetic properties, as are expected to follow.
Conflicts of interest
There are no conflicts to declare.References
- Lucretius, De Rerum Natura (ca. 60 BC).
- A. Lavoisier, Traité Élémentaire de Chimie, Paris, 1789 Search PubMed.
- J. Dalton, A New System of Chemical Philosophy, S. Russell, London, 1808 Search PubMed.
- (a) J. W. Gibbs, On the Equilibrium of Heterogeneous Substances, Trans. Conn. Acad., 1874–1878, 3, 108–248 Search PubMed; (b) J. W. Gibbs, Elementary Principles in Statistical Mechanics, Charles Scribner's Sons, New York, 1902 Search PubMed.
- L. Boltzmann, Studien über das Gleichgewicht der lebendigen Kraft zwischen bewegeten materiellen Punkten, Wien. Ber., 1868, vol. 58, pp. 517–560.
- (a) G. N. Lewis, The Atom and the Molecule, J. Am. Chem. Soc., 1916, 38, 762–785 CrossRef CAS; (b) G. N. Lewis, Valence and the Nature of the Chemical Bond, Chemical Catalog Co., New York, 1923 Search PubMed.
- M. Planck, Zur Theorie des Gesetzes der Energieverteilung im Normalspectrum, Verh. Deut. Physik. Gesell., 1900, 2, 202–204 CAS.
- N. Bohr, On the Constitution of Atoms and Molecules, Phil. Mag., 1913, 26, 1–24 CAS.
- V. Mendeleev, The Periodic Law of the Chemical Elements, J. Chem. Soc., 1889, 55, 634–656 RSC.
- W. Heisenberg, Über quantentheoretische Umdeuting kinematischer und mechanischer Beziehungen, Z. Phys., 1925, 33, 879–893 CrossRef CAS.
- E. Schrödinger, Quantisierung als Eigenwertsproblem, Ann. Phys., 1926, 384, 361–376 CrossRef.
- P. A. M. Dirac, On the Theory of Quantum Mechanics, Proc. Roy. Soc. A, 1926, 112, 661–677 CAS.
- For examples, see, e.g., H. Bethe and E. E. Salpeter, Quantum Mechanics of One- and Two-Electron Atoms, Springer, Berlin, 1957 CrossRef PubMed; H. F. Schaefer III, Methylene: A Paradigm for Computational Quantum Chemistry, Science, 1986, 231, 1100–1107 CrossRef PubMed.
- F. M. Bickelhaupt, C. F. Guerra, M. Mitoraj, F. Sagan, A. Michalak, S. Pan and G. Frenking, Clarifying Notes on the Bonding Analysis Adopted by the Energy Decomposition Analysis, Phys. Chem. Chem. Phys., 2022, 24, 15726–15735 RSC.
- N. Bohr, Über die Serienspektra der Elemente, Z. Phys., 1920, 2, 423–478 CrossRef CAS.
- E. Callaway, After AlphaFold: Protein-Folding Contest Seeks Next Big Break-Through, Nature, 2023, 613, 13–14 CrossRef CAS PubMed.
- F. Weinhold, The Path to Natural Bond Orbitals, Isr. J. Chem., 2022, 62, e202100026 CrossRef CAS; F. Weinhold and C. R. Landis, Discovering Chemistry with Natural Bond Orbitals, John Wiley, Hoboken NJ, 2012 Search PubMed.
- (a) E. D. Glendening and F. Weinhold, Natural Resonance Theory. I. General Formulation, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS; (b) E. D. Glendening and F. Weinhold, Natural Resonance Theory. II. Natural Bond Order and Valency, J. Comput. Chem., 1998, 19, 610–627 CrossRef CAS; (c) E. D. Glendening, J. K. Badenhoop and F. Weinhold, Natural Resonance Theory. III. Chemical Applications, J. Comput. Chem., 1998, 19, 628–646 CrossRef CAS; (d) E. D. Glendening, C. R. Landis and F. Weinhold, Resonance Theory Reboot, J. Am. Chem. Soc., 2019, 141, 4156–4166 CrossRef CAS PubMed.
- P.-O. Löwdin, Quantum Theory of Many–Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin Orbitals, and Convergence Problems in the Method of Configurational Interaction, Phys. Rev., 1955, 97, 1474–1489 CrossRef.
- J. von Neumann, Mathematical Foundations of Quantum Mechanics, Princeton University Press, Princeton NJ, 1955 Search PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, What is NBO Analysis and How is it Useful?, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University Wisconsin, Madison, 2018, https://nbo7.chem.wisc.edu/ Search PubMed.
- NBO “Affiliated Programs” link: https://nbo7.chem.wisc.edu/affil_css.htm.
- J. B. Foresman and Æ Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian Inc., Wallingford CT, 2015 Search PubMed.
- L. Pauling, The Application of the Quantum Mechanics to the Structure of the Hydrogen Molecule and Hydrogen Molecule-Ion and to Related Problems, Chem. Rev., 1928, 5, 173–213 CrossRef.
- W. Heitler and F. London, Wechselwirkung neutraler Atome und homopolar Bindung nach der Quantenmechanik, Z. Phys., 1927, 44, 455–472 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond. Application of Results Obtained from the Quantum Mechanics and from a Theory of Paramagnetic Susceptibility to the Structure of Molecules, J. Am. Chem. Soc., 1931, 53, 1367–1400 CrossRef CAS , and references therein.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals, Cornell University Press, Ithaca NY, 1939 Search PubMed.
- P. Ball, In Retrospect: Pauling's Primer, Nature, 2010, 468, 1036 CrossRef.
- G. A. Gallup, A Short History of Valence Bond Theory, in Valence Bond Theory, ed. D. L. Cooper, Elsevier, Amsterdam, 2002 Search PubMed.
- R. S. Mulliken, Electronic Structures of Polyatomic Molecules and Valence. II. General Considerations, Phys. Rev., 1932, 41, 49–71 CrossRef CAS.
- R. S. Mulliken, C. A. Rieke and W. G. Brown, Hyperconjugation, J. Am. Chem. Soc., 1941, 63, 41–56 CrossRef CAS.
- E. D. Glendening and F. Weinhold, Pauling's Conceptions of Hybridization and Resonance in Modern Quantum Chemistry, Molecules, 2021, 26, 4110 CrossRef CAS PubMed.
- L. Pauling, Metal-Metal Bond Lengths in Complexes of Transition Metals, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 4290–4293 CrossRef CAS PubMed.
- N. V. Sidgwick, The Electronic Theory of Valency, Oxford University Press, London, 1929 Search PubMed.
- F. Weinhold and C. R. Landis, Valency and Bonding, Cambridge University Press, Cambridge UK, 2005 Search PubMed.
- J. Nocedal and S. Wright, Numerical Optimization, Springer, New York, 2nd edn, 2006 CrossRef CAS PubMed; E. D. Glendening, S. J. Wright and F. Weinhold, Efficient Optimization of Natural Resonance Theory Weightings and Bond Orders by Gram-Based Convex Programming, J. Comput. Chem., 2019, 40, 2028–2035 CrossRef CAS PubMed.
- Y. Xie, H. F. Schaefer III and E. E. Jemnis, Characteristics of Novel Sandwiched Beryllium, Magnesium, and Calcium Dimers: C5H5BeBeC5H5, C5H5MgMgC5H5, and C5H5CaCaC5H5, Chem. Phys. Lett., 2005, 402, 414–421 CrossRef CAS.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Diberyllocene, a Stable Compound of Be(I) with a Be–Be Bond, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- J. L. Dutton, A Big Breakthrough for Beryllium: A Stable Organometallic Compound with a Be–Be Bond has been Synthesized, Science, 2023, 380, 1106–1107 CrossRef CAS PubMed.
- F. Weinhold, Natural Bond Critical Point Analysis: Quantitative Relationships between NBO-Based and QTAIM-Based Topological Descriptors of Chemical Bonding, J. Comput. Chem., 2012, 33, 2440–2449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04790c |
| This journal is © the Owner Societies 2024 |