
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The energetics of N2 reduction by vanadium containing nitrogenase†
Per E. M.
Siegbahn
 *a and
Wen-Jie
Wei
ab
*a and
Wen-Jie
Wei
ab
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91, Stockholm, Sweden. E-mail: per.siegbahn@su.se
bKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, Hubei Key Laboratory of Materials Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 14th December 2023
Abstract
The main class of nitrogenases has a molybdenum in its cofactor. A mechanism for Mo-nitrogenase has recently been described. In the present study, another class of nitrogenases has been studied, the one with a vanadium instead of a molybdenum in its cofactor. It is generally believed that these classes use the same general mechanism to activate nitrogen. The same methodology has been used here as the one used for Mo-nitrogenase. N2 activation is known to occur after four reductions in the catalytic cycle, in the E4 state. The main features of the mechanism for Mo-nitrogenase found in the previous study are an activation process in four steps prior to catalysis, the release of a sulfide during the activation steps and the formation of H2 from two hydrides in E4, just before N2 is activated. The same features have been found here for V-nitrogenase. A difference is that five steps are needed in the activation process, which explains why the ground state of V-nitrogenase is a triplet (even number) and the one for Mo-nitrogenase is a quartet (odd number). The reason an additional step is needed for V-nitrogenase is that V3+ can be reduced to V2+, in contrast to the case for Mo3+ in Mo-nitrogenase. The fact that V3+ is Jahn–Teller active has important consequences. N2H2 is formed in E4 with reasonably small barriers.
I. Introduction
In nature, the reduction of N2 is performed by enzymes termed nitrogenases. The by far dominating nitrogenase is molybdenum-containing nitrogenase, which has a cofactor (FeMoco) including one molybdenum and seven irons bound together by sulfides. A cysteine and a histidine are ligands of the cofactor, as well as a homocitrate, which is unusual in biology.1 Almost twenty years after the first X-ray structure, it was discovered that there is also a central carbide in the structure, only seen at a very high resolution of 1.0 Å.2 A less common nitrogenase employs vanadium instead of molybdenum. An X-ray structure with a resolution of 1.35 Å has been determined.3 The active site is shown in Fig. 1. The structure of the cofactor (FeVco) is very similar to FeMoco. A notable difference is that, instead of one of the sulfides (S3A) in FeMoco, there is a carbonate bridging two irons (Fe4 and Fe5).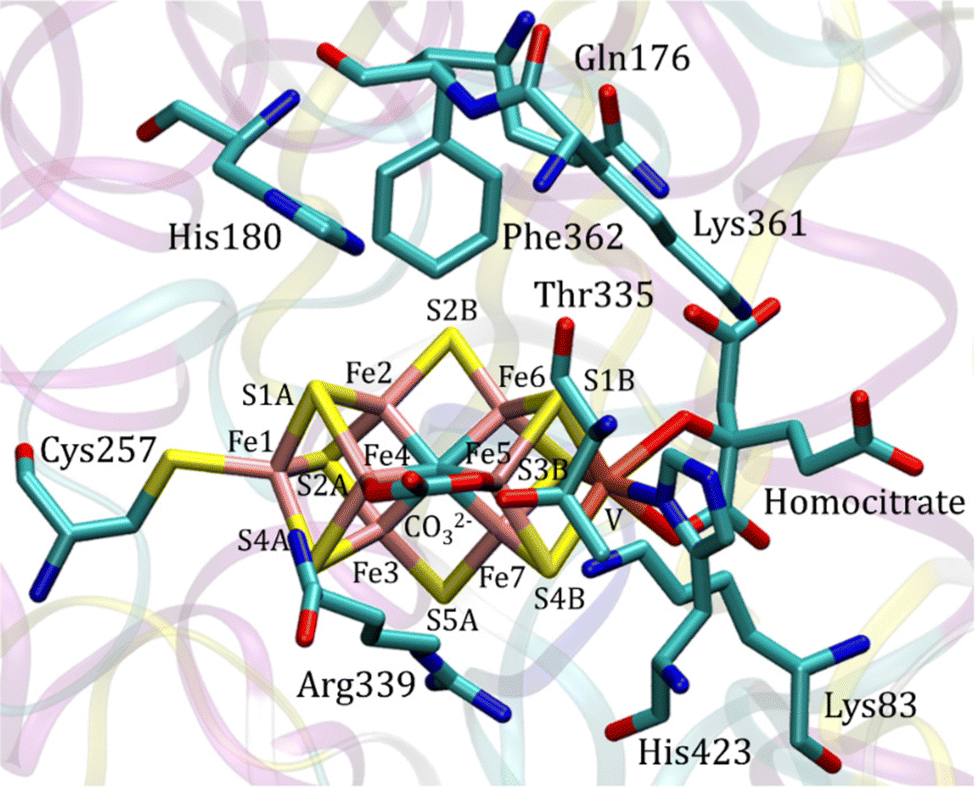
The mechanism for Mo-nitrogenase has been extensively studied for decades, but the understanding has still major unresolved issues. It has long been known that the state of Mo-nitrogenase that activates N2 is reached after four reductions in the catalytic cycle.4,5 The states involved are termed E0–E4 and, therefore, the state that activates N2 is E4. The understanding of the mechanism for Mo-nitrogenase has seen significant progress during the past decade. In a set of EPR studies, it has been shown that, just before N2 activation, two hydrides leave the cofactor as H2.6–8 The release of H2 was considered obligatory for N2 activation, and explained a long-standing mystery that the activation of N2 in Mo-nitrogenase is always accompanied by a loss of one H2. Suggestions that there should be a pre-activation before catalysis starts have been made in a series of theoretical modeling papers.9–12 However, the presence of a pre-activation has been questioned in other studies.6–8 The best experimental evidence for the presence of a pre-activation is that two different assignments of the E1 state have been made in two experiments, where the preparations of the states have been different.13,14 The reason for that difference has been analyzed recently by a modeling study and it was concluded that a likely explanation for the results is that there is an activation prior to catalysis.15 Several experimental studies have furthermore led to the conclusion that catalysis is preceded by an activation.16–19
In the present study, the mechanism of V-nitrogenase has been investigated using a theoretical modeling. It has been shown experimentally that Mo- and V-nitrogenase in the EPR active E4 state use the same general mechanism for the activation of N2.20 While Mo-nitrogenase has been intensively studied during the past decades by theoretical methods, there are only a few such studies of V-nitrogenase. A significant problem has been that the electronic ground state of the cofactor has not been safely determined until very recently.21 It was found that the oxidation state for the ground state should be (V3+ 4Fe3+ 3Fe 2+), which is an S = 0 (or integer spin non-Kramers) state. For many years it was assumed that the ground state instead was (V3+ 3Fe3+ 4Fe 2+) in contrast to FeMoco, which has been determined to be (Mo3+ 4Fe3+ 3Fe 2+). In a recent theoretical study, performed before the new assignment of the ground state of FeVco was determined, it was for that reason assumed that the ground state for FeVco was more similar to the E1 state of FeMoco, obtained after one reduction.22 That difference was also suggested to be an explanation for the experimentally observed differences in reactivities, such as the ones towards CO and CO2. The present study is the first one built on the new assignment of the ground state.21 The nature of the four-atom bridging ligand in FeVco, not found in FeMoco, was also investigated in those studies. Analysis of the X-ray structure for FeVco has led to the conclusion that the bridging four atom ligand should be an unprotonated CO32−.22 The same conclusion concerning that ligand was drawn in another theoretical study, which also suggested that the sulfide ligands in FeVco are unprotonated in the X-ray structure.23
V-nitrogenase is a much less common enzyme than Mo-nitrogenase. The importance of studying V-nitrogenase is the generally accepted similarity of the mechanisms of these enzymes. It is of high interest to investigate if calculations give a mechanism for V-nitrogenase following the same general principles as the one suggested for Mo-nitrogenase.10 If not, it casts a doubt on the mechanism reached for Mo-nitrogenase.
II. Methods
The methods used here to study V-nitrogenase are the same as those used in other recent studies of nitrogenase.9–12,24 The starting point is DFT with the standard hybrid functional B3LYP.25 The geometries were optimized with a medium size basis (LACVP*). Single point energies in the optimized geometries using B3LYP with 15% were calculated using a large basis with cc-pvtz(-f) for the first-row atoms, and with LACV3P* for the metals. For the large basis set, the energies had to be computed without the pseudo-spectral approach. In the optimized points, dielectric effects were added, obtained using a Poisson–Boltzmann solver,26 with a dielectric constant of 4.0. D2 dispersion27 and zero-point effects were finally added. An empirical value of 14 kcal mol−1 was used for the cost of taking a water molecule from bulk water and placing it on the cofactor. That value can be approximately derived from the experimental free energy for the binding of one water molecule to bulk water of −6.3 kcal mol−1.35 A large entropy effect is obtained in that case since the water is moved from the gas phase. In the present case, essentially no such entropy effect should be present. The programs used were Jaguar26 and Gaussian.28The methods used here have been carefully tested.29,30 In the most recent of these studies, redox mechanisms have been tested for Photosystem II, cytochrome c Oxidase, NiFe and FeFe hydrogenases, NiFe–CO dehydrogenase, multi-copper oxidases and acetyl–CoA synthase. The results are in all cases in very good agreement with experimental findings.30 There are no counter-examples found for redox enzymes, which are all stable at room temperature, which is an important reason for the high accuracy. It is also important to note that the cases mentioned are relevant for the present study since they deal with energies not higher than 20 kcal mol−1 from their ground states, Failures of the methods for excited states and structures 30 kcal mol−1 or higher in energy are not relevant for the present study. In the literature, there are many such cases of failures. At higher energies, multireference effects start to appear which cannot be handled by the present single determinant approach. Unsaturated systems, not present in the enzymes mentioned, may also be cases of failures.
A cluster model was used for the active site.31 Besides the ligands of the cofactor, homocitrate, Cys257 and His423, the amino acids Lys83, Gln176, His180, Thr335, Arg339, Lys361 and Phe362 were included in the model, see Fig. 1. Some coordinates, marked with red circles in Fig. S1 (ESI†), were frozen from the X-ray structure, in order to avoid artificial movements. The model is of the same size as the most recent models used for Mo-nitrogenase.11,12 Since a positive region outside the homocitrate was left out of the model the homocitrate was additionally protonated, which is a common procedure in the cluster modeling. The necessity for including Phe362 was realized at a late stage of the present investigation. The results for the mechanism for the two models, with and without Phe362, were found to be quite similar and, therefore, some qualitative conclusions from the tests of the mechanisms were taken from the earlier investigations of the model without Phe362. The energetic results reported below are all for the model with Phe362. The total charge of the model of the active site is −1, which is different from the charge of −2 for Mo-nitrogenase.
Concerning the size of the model, it is of high importance to emphasize that the calculated reduction energies are all (H+,e−) additions, which do not have long-range effects, since the additions are neutral. For that purpose, models with about 200 atoms are quite sufficient. pKa values and redox potentials require larger models than the one used here, but no such values are reported.
There are many possible spin-couplings for each of the structures investigated. One of them is a coupling (+ −−+ + + −) with a numbering of the iron atoms from the X-ray structure. That coupling is here written as (−2,−3,−7) to conform with a convention used earlier, indicating which irons have negative spins. The list of different spin-states that were investigated were taken from a thorough spin-investigation for the ground state for Mo-nitrogenase.32 However, it should be noted that at the time of that study, it was believed that the spin on molybdenum was zero. It was later discovered that there are three unpaired spins on molybdenum and two on vanadium. Those spins interact strongly with the iron spins and make new investigations necessary. Clusters for an odd number of reductions were here found to be doublets (except for the ground state) and for an even number triplets.
In order to obtain the energetics for the different reductions, an energy value for the addition of a (H+, e−) is needed. The same value as in our previous studies for Mo-nitrogenase of 348.6 kcal mol−1 was used.9–11 This value is based on a redox potential for the reductant of −1.6 V and a solvation energy for a proton in water of 279.8 kcal mol−1 at pH = 7.33 It also includes a driving force of 3 kcal mol−1 for the electron transfer process.
III. Results
An interesting difference between V- and Mo-nitrogenase is that they do not have the same spin for their ground states. Assuming the activation of N2 in E4 to be the same,20 the E4 state should probably be a doublet in both cases. In our previous studies on Mo-nitrogenase, an activation prior to catalysis consisting of four reduction steps was suggested,.9–12 Therefore, for V-nitrogenase, there should be an odd number of steps in the activation process to reach a doublet E4 state. As will be discussed below, the calculations indicate an activation consisting of five steps.Another difference between the nitrogenases is the limiting overall reaction. For V-nitrogenase it is usually written:21
| N2 + 12H+ +24MgATP +12e− → 2NH3 +3H2 +24MgADP +24Pi | (1) |
In a study of the present type, where there are many reduction steps, a decision on the nature of the reductions may significantly simplify the calculations. In the case of Mo-nitrogenase, all reductions were assumed to be proton coupled. With an addition of a (H+·e−) couple, the charge of the model does not change, which leads to a technical simplification. It means that inhomogeneous long range electrostatic effects from regions outside the model can be neglected, which is not the case when the charge is changed as in cases of reductions that are not proton coupled. The homogeneous long-range effects are well covered by the dielectric cavity model. The assumption of proton coupled reductions is in general a very good approximation for models of the present size, as shown in previous studies.29,30 In the present case of V-nitrogenase, it is quite clear that all reductions are proton coupled, since for each electronic reduction, the calculated pKa values are much higher than 7.
An important difference between Mo- and V-nitrogenase is that the charges of the models for the active sites differ by one unit. The reason is that for V-nitrogenase there are three positive residues outside the cofactor, two lysines and one arginine, while for Mo-nitrogenase there are only two, both arginines. That leads to a model of the active site with a charge of −1 for V-nitrogenase and −2 for Mo-nitrogenase.
There are many possible spin-couplings for each structure in the mechanism. Many of them were tried here, but far from all of them. The choice of spin-coupling makes most difference for the first redox states, where there are still Fe3+ atoms (with five unpaired spins) present. As the Fe3+ atoms are reduced to Fe2+ (with four unpaired spins), the couplings between the irons decrease in strength, and the coupling to the spins on vanadium becomes the most important factor. A surprising finding, from the many investigations of spin states performed here, was that one spin-coupling was favorable for all E-states in the catalytic cycle except E0. That spin-coupling is (−2,−3,−7). Interestingly, a similar result was found in our study on Mo-nitrogenase. In that case, another spin-coupling dominates for the more reduced states, the one with (−2,−3,−4). That difference between V- and Mo-nitrogenase is due to the higher spin for Mo3+.
The activation steps A0–A5
The recent experimental assignment for the oxidation state of the ground state (A0) of FeVco with (V3+, 3Fe2+, 4Fe3+) is used in the present study.21 This is the same redox level as the one for FeMoco. Ten different spin-couplings were investigated for A0 and the best one found was (2-,3-,5-), which is the same as the one found for A0 in a previous study of V-nitrogenase.22 It should be noted that even though the protonation state is the same for A0 in the two studies, there is a different assignment of A0 with (V3+, 4Fe2+, 3Fe3+) in the previous study. The energy for spin-coupling (1-,5-,6-) is +2.8 kcal mol−1 higher and for (2-,3-,4) +3.9 kcal mol−1 higher (lacvp*). The calculations show that the ground state is a triplet, with the singlet +18.5 kcal mol−1 higher, using the same spin-coupling. The optimized (triplet) structure, shown in Fig. S1 (ESI†), is very close to the X-ray structure shown in Fig. 1.For the first proton-coupled reduction step from A0 to A1, the belt sulfide S2B becomes protonated. That is the same sulfide which was most easily protonated for Mo-nitrogenase. The oxidation state of the cofactor is now (V3+, 4Fe2+, 3Fe3+). The best spin-coupling found for A1 is (2-,3-,5-). This is the same as the one found for A0 in the previous study,22 which is logical since the oxidation state for A0 used in that study is the same as the one for A1 here. The transition from A0 to A1 was here found to be exergonic by −13.9 kcal mol−1. The energy for a protonated carbonate is only +1.2 kcal mol−1 higher.
In the transition from A1 to A2, the proton ends up on the carbonate (Fig. S2, ESI†). The exergonicity is again quite large with −13.2 kcal mol−1. The best spin-coupling found was (2-,4-,5-) and the oxidation state is (V3+, 5Fe2+,2Fe3+). In the A2 to A3 transition, the second (and final) belt sulfide S5A is protonated. The optimized structure of A3 is shown in Fig. S3 (ESI†). Six different spin-couplings were tried with the best one (2-,3-,7-) with an exergonicity of −9.7 kcal mol−1. The oxidation state for A3 is (V3+, 6Fe2+, 1Fe3+). An interesting finding is that the spin-population on vanadium increases from 2.1 to 2.4 in the direction towards V2+, which has an ideal spin of 3.0.
In the next transition from A3 to A4, an interesting and important effect appears. When the proton on S5A was moved to S3B, the V-S3B distance increased very much from 2.54 Å in A3 to nearly 4 Å in A4, leading to a 5-coordination for vanadium. That is due to a Jahn–Teller effect, since V3+ has an octahedral geometry and a high-spin d2 configuration. The effect only becomes strong when S3B is protonated and, therefore, the effect was not seen in A3, where S3B was un-protonated. Since there is now an open site on vanadium, a hydride can be placed on vanadium. In the optimization of that structure (Fig. S4, ESI†), the hydride formed a short bond both to vanadium and Fe6. Placing a hydride between Fe2 and Fe6 and moving S2B over to a terminal position on Fe6 has a much higher energy. The transition from A3 to this A4 structure is exergonic by only −1.0 kcal mol−1. The protonated sulfides are now S2B and S3B, and the carbonate remains protonated. The best spin-coupling is (2-,3-,5-) and the oxidation state is (V3+, 5Fe2+, 2Fe3+).
There is a second important structure of A4, here termed  (Fig. S5, ESI†). In that structure a sulfide has been released. The sulfide that is most easily released is S3B. The release involves a proton being transferred from S2B to S3B, and after that a H2S can be released. At the TS, the H-S2B distance is 1.68 Å and the H-S3B distance 1.64 Å. The Fe6 to S3B distance is increased to 3.10 Å from 2.40 Å and shows that S3B is about to be released. The barrier is +17.2 kcal mol−1, which is feasible. It is exergonic by −5.9 kcal mol−1, using an energy for a bound H2S in the enzyme of 14 kcal mol−1 as for a water. The argument for that is that H2S should be in equilibrium with H2S in the surrounding water. H2S in water should have approximately the same type of hydrogen bonds as a water molecule. The exact value is not important in the present case. The TS is shown in Fig. 2. The total exergonicity from A3 to
(Fig. S5, ESI†). In that structure a sulfide has been released. The sulfide that is most easily released is S3B. The release involves a proton being transferred from S2B to S3B, and after that a H2S can be released. At the TS, the H-S2B distance is 1.68 Å and the H-S3B distance 1.64 Å. The Fe6 to S3B distance is increased to 3.10 Å from 2.40 Å and shows that S3B is about to be released. The barrier is +17.2 kcal mol−1, which is feasible. It is exergonic by −5.9 kcal mol−1, using an energy for a bound H2S in the enzyme of 14 kcal mol−1 as for a water. The argument for that is that H2S should be in equilibrium with H2S in the surrounding water. H2S in water should have approximately the same type of hydrogen bonds as a water molecule. The exact value is not important in the present case. The TS is shown in Fig. 2. The total exergonicity from A3 to  is −6.9 kcal mol−1, which fulfills the requirement for a driving force of at least 3 kcal mol−1 to secure catalytic progress. Without the sulfide release, that requirement would not be fulfilled. Finally, it can be noted that a sulfide release was also found in A4 for Mo-nitrogenase.15
is −6.9 kcal mol−1, which fulfills the requirement for a driving force of at least 3 kcal mol−1 to secure catalytic progress. Without the sulfide release, that requirement would not be fulfilled. Finally, it can be noted that a sulfide release was also found in A4 for Mo-nitrogenase.15
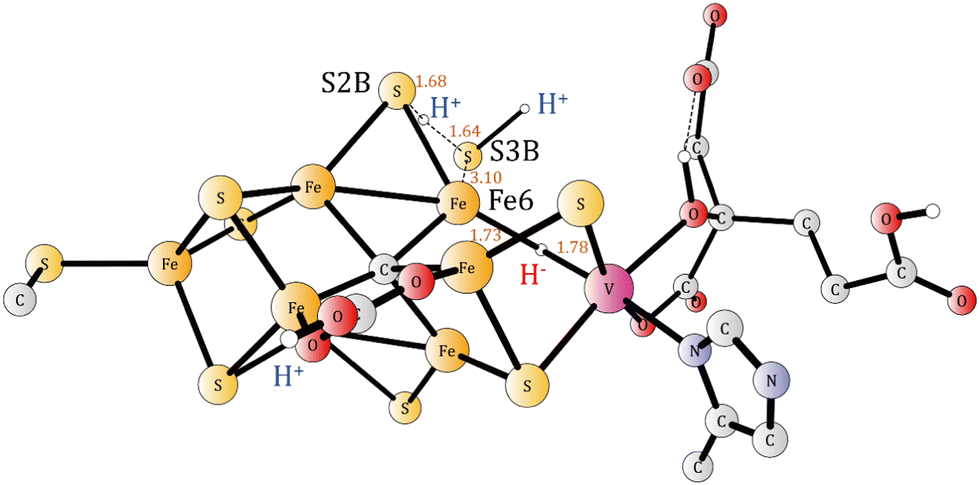 | ||
| Fig. 2 The optimized transition state for the release of H2S in A4. | ||
The preferred protonation site in A5 (Fig. 3) is S2B, which is exergonic by −16.3 kcal mol−1.
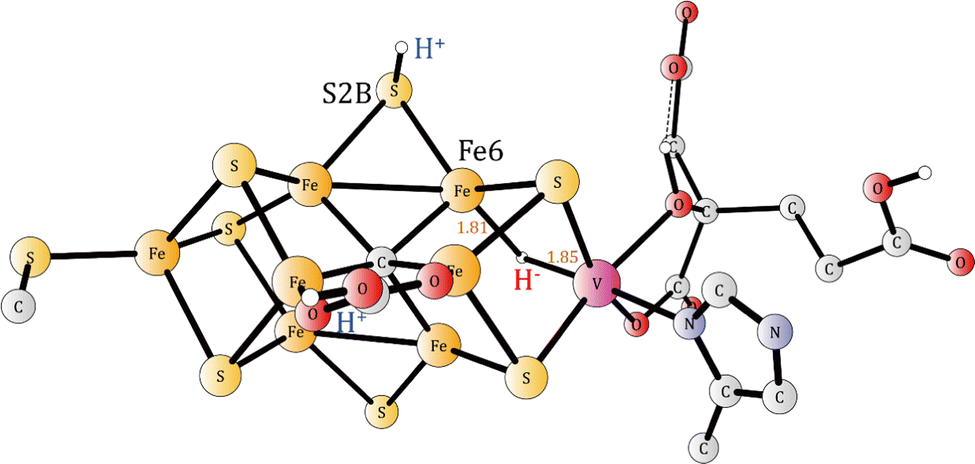 | ||
| Fig. 3 The E0 (A5) structure formed after the five activation steps. The carbonate and S2B are protonated and there is one hydride. | ||
However, the most favorable one is the carbide but to reach the carbide, the proton has to pass a TS, which turned out to be too high. The fifth reduction step completes the activation process to reach E0. This process is done only once before the catalytic cycling starts. It is important to note that the release of the sulfide occurs before catalysis and, therefore, it does not need to be replaced during catalysis. The oxidation state is (V3+, 6Fe2+, 1Fe3+) and the best spin-coupling is (2-,3-,5-). The reason the activation process contains five steps for V-nitrogenase, and only four for Mo- nitrogenase will become evident below. What has been achieved for V-nitrogenase in the activation process is that the cluster has been reduced and only one Fe3+ remains. A sulfide has been released and a hydride has been placed between Fe6 and vanadium, and will remain that way to E4 where N2 will be activated. In Mo-nitrogenase, the first hydride is instead placed in the region between Fe1, Fe2, Fe4, which has consequences for where the activation of N2 occurs, see further below. The oxidation state after the pre-activation can be compared to the corresponding one after the four-step pre-activation process for Mo-nitrogenase, which leads to (Mo3+,5Fe2+, 2Fe3+) with two Fe3+.
Otherwise, the nitrogenases are very similar at E0. Most notably, a sulfide has been released and only one sulfide (S2B) is protonated in both cases. The suggested mechanism for the activation steps from A0 to A5 is shown in Scheme 1 and the energies are summarized in a diagram in Fig. 4.
 | ||
| Scheme 1 Suggested mechanism for the activation steps from A0 to A5. | ||
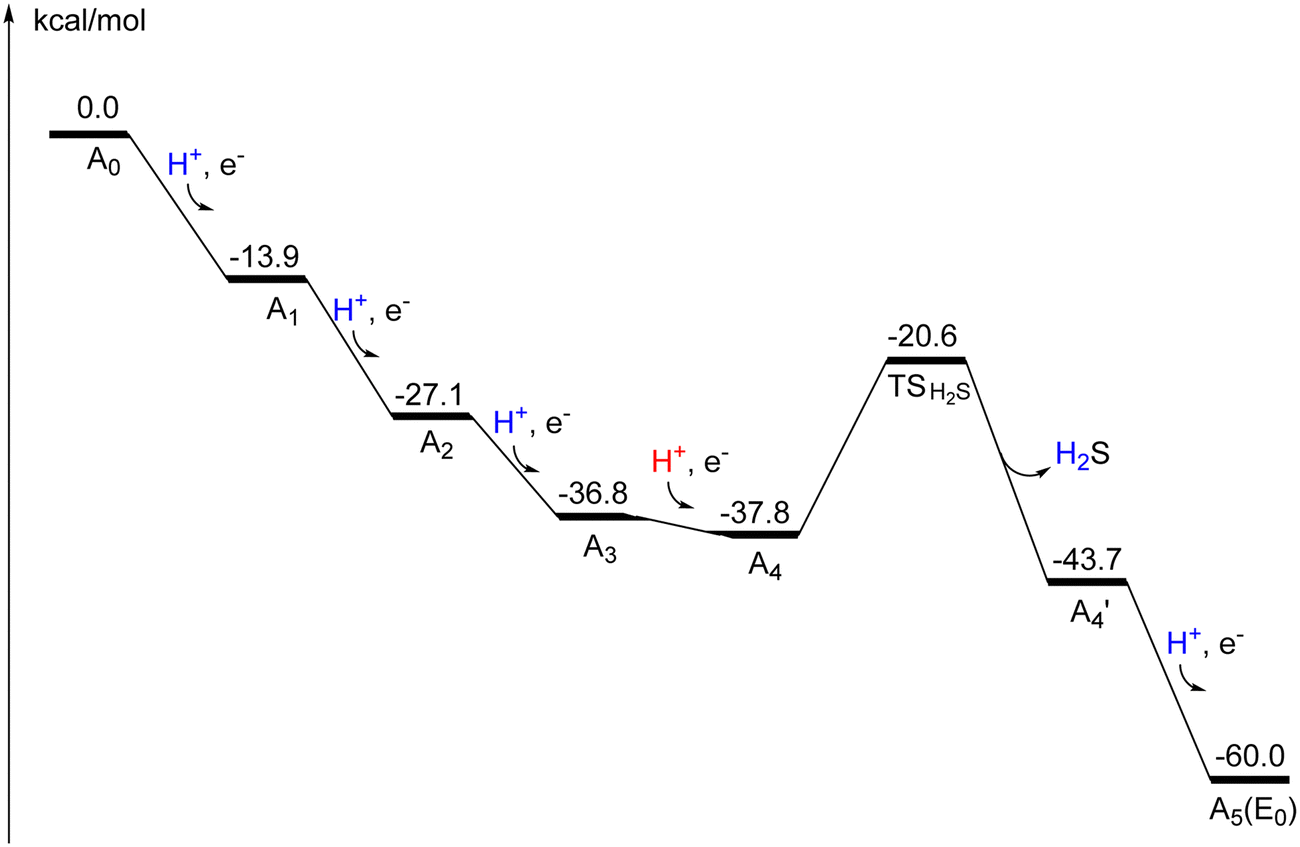 | ||
| Fig. 4 Energy diagram for the activation steps from A0 to A5. | ||
E0–E4 in the catalytic cycle
At E0 the catalytic cycling starts. According to experiments, N2 should be activated after four additional reductions. In the reduction from E0 to E1, there is a protonation of the carbide. The incoming proton is first bound as a hydride between Fe4 and Fe5 and then moves to the carbide. The optimized TS is shown in Fig. 5. The proton has a short bond of 1.63 Å to Fe5 and to the carbide the distance is 2.07 Å. The local barrier is +5.3 kcal mol−1, but since the resting state has a proton on S5A, which is bound by −4.3 kcal mol−1, the actual barrier becomes (4.3 + 5.3) = +9.6 kcal mol−1. Since the barrier is so low, the protonation of the carbide can probably not be avoided. That is unlike the situation for Mo-nitrogenase,12 where the corresponding barrier (in A4) is +17.9 kcal mol−1, which is higher than for the release of a sulfide. For Mo-nitrogenase, the avoidance of the carbide protonation is important for the mechanism. For V-nitrogenase, there is no corresponding advantage of avoiding carbide protonation, see further below. The protonation of the carbide might actually occur already in A4, with a barrier of +16.3 kcal mol−1. The calculations give a small endergonicity of +0.1 kcal mol−1, but an exergonicity is possible considering the uncertainty of the methods. That scenario would lead to the avoidance of carbide protonation and deprotonation in the catalytic cycling. At the present stage, it is not clear if that avoidance is an advantage. The exergonicity of the reduction from E0 to E1 (Fig. S6, ESI†) is −20.8 kcal mol−1 for V-nitrogenase. The irons are now all reduced to Fe2+ and the oxidation state is (V3+, 7Fe2+). The spin-coupling is (2-,3-,7-), which will actually remain the best one all the way until N2 is activated.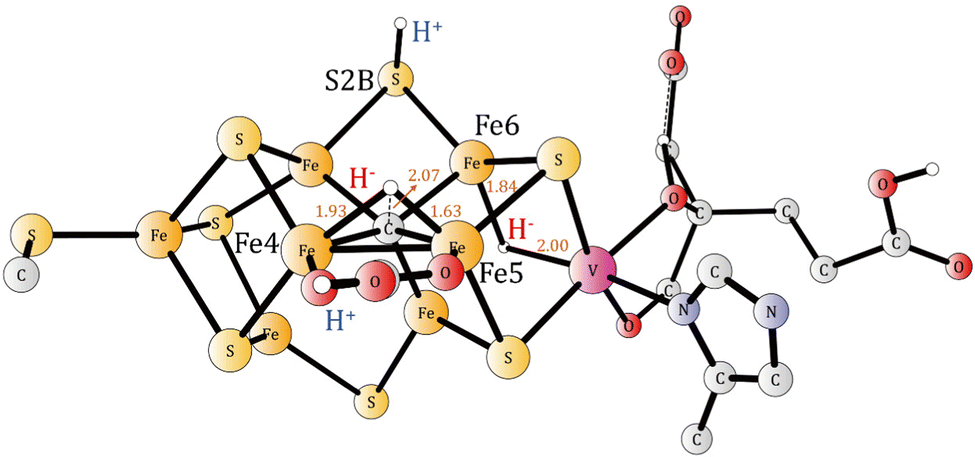 | ||
| Fig. 5 The optimized transition state for formation of a protonated carbide in E1. | ||
The presence of two hydrides is necessary for the mechanism of N2 activation in E4, as suggested experimentally.6–8 In E2, the second hydride becomes bound, bridging between Fe6 and Fe7 (Fig. S7, ESI†), with a bond-distance of 1.82 Å to both of them. The reason the second hydride can bind in that region is that the carbide is protonated and moves away somewhat from its central position, which leaves enough space for the second hydride to bind rather close to the first one. In A4, where there is an un-protonated carbide, there is not enough space and the hydride that entered in that state was instead bound between Fe6 and vanadium, see above. Since the second hydride takes an electron from the irons, the oxidation state in E2 becomes (V3+, 6Fe2+, 1Fe3+), the same as in E0. The exergonicity of the reduction from E1 to E2 is −9.6 kcal mol−1. The spin-coupling is the same as in E1. In E3, S5A becomes protonated (Fig. S8, ESI†) and the oxidation state is back to (V3+, 7Fe2+). The best spin-coupling is again the same as in E1. The exergonicity of the E2 to E3 transition is −6.9 kcal mol−1.
E4 is according to experiments, the state which activates N2.4–8 Since this is the most important state in the nitrogenase mechanism, a thorough investigation of spin-couplings, for 10 different cases, was made again. The lowest energy was again found for (2-,3-,7-) but with (2-,3-,4-) only +1.8 kcal mol−1 higher with all effects included. It might have been expected that it should be difficult to reduce the cofactor further from (V3+, 7Fe2+) in E3, since all irons are now Fe2+. However, it turns out that also vanadium can be reduced, from V3+ to V2+. After the reduction, the spin on vanadium is −2.93, close to the ideal spin of −3.0 for V2+. In E3 the spin on vanadium is −2.52, in between the ideal spin of −3.0 for V2+ and −2.0 for V3+. The sulfide most easily protonated in E4 was found to be S1A. The exergonicity of the E3 to E4 transition is −11.9 kcal mol−1. This means that for V-nitrogenase with the ground state (V3+, 3Fe2+, 4Fe3+) there are five easily reduced metal atoms, while for Mo-nitrogenase with the ground state (Mo3+, 3Fe2+, 4Fe3+) there are only four. That explains the necessity of one more activation step in V-nitrogenase. Apart from the five reductions of the metals, four reductions are needed to produce two hydrides, and still keep a low oxidation state of the cofactor. That means altogether 9 reduction steps from A0, 5 prior activation steps and 4 steps in the catalytic cycle. In E4 there are now two hydrides, a protonated carbide and three protonated sulfides, S2B, S5A and S1A, see Fig. 6. In Mo-nitrogenase there are four protonated sulfides, since the carbide is not protonated in that case.12
 | ||
| Fig. 6 The E4 structure where the activation of N2 occurs. There are two hydrides, a protonated carbonate, a protonated carbide and three protonated sulfides, S2B, S5A and S1A. | ||
At this stage, H2 can be formed from the two hydrides, as required for the activation of N2. The purpose of the formation of H2 from the two hydrides is to reduce the (formal) oxidation state of the cofactor by two units to become very low with (V2+, 5Fe2+, 2Fe1+), which is needed for electron transfer to N2. A straightforward reduction from Fe2+ to Fe1+ without the release of the hydrides is not energetically possible with the redox potential of −1.6 V. The barrier for H2 formation is +15.0 kcal mol−1. The transition state is shown in Fig. 7. In the TS there are two short bonds of the hydrides to Fe6 with 1.65 Å and 1.71 Å, and the distance between the hydrides is 1.26 Å. The distance from one of the hydrides to vanadium is 2.05 Å, and the distance from the other hydride to Fe7 is 2.03 Å. The distance to Fe2 is much longer with 2.74 Å. The release of H2 is endergonic by +3.0 kcal mol−1, which is quite different from the situation in Mo-nitrogenase, where this release was found to be endergonic by +8.7 kcal mol−1.12 There is a large gain of entropy of −8.4 kcal mol−1 as H2 is released. After H2 release, the irons with the lowest spins are Fe2, Fe5 and Fe6 with 3.3 and the one with highest is Fe1 with 3.66.
 | ||
| Fig. 7 The transition state for H2 formation. | ||
After H2 is released, N2 can bind. Several positions for N2 were tried and one of them has a much lower energy than the other ones. The structure is shown in Fig. 8. N2 is bound end-on to Fe6, with a short distance of 1.91 Å. The N–N distance is 1.14 Å, elongated from 1.11 Å for free N2. These distances indicate that N2 is clearly activated. The charge transfer from the cofactor to N2 is −0.26 and the spin on N2 is −0.33. The iron with the lowest spin is Fe6 with 3.20 and the one with highest Fe1 with 3.68. The enthalpic binding energy for N2 compared to the structure after H2 release is as large as −15.5 kcal mol−1, which with the entropy penalty of +9.9 kcal mol−1 is reduced to −5.6 kcal mol−1. Since the release of H2 is endergonic by +3.0 kcal mol−1, see above, the release of H2 and binding of N2 is altogether exergonic by −2.6 kcal mol−1. For Mo-nitrogenase, the corresponding energy is +5.9 kcal mol−1, where the difference comes from the much less favorable release of H2 in that case.
 | ||
| Fig. 8 The optimized structure after N2 binding. | ||
It is interesting, and very important, to note that the position for N2 binding on Fe6 is different from the one of Mo-nitrogenase, where N2 is bound on Fe4.12 The preference compared to other positions is very large in both cases. The origin of that difference is clearly connected to the preferred positions of the hydrides. For V-nitrogenase, the preferred positions for the hydrides are close to V and Fe6, because of the Jahn–Teller distortion of V3+, see above, while for Mo-nitrogenase the preferred positions are close to Fe4. The conclusion drawn here is that when the hydrides are released as H2, the two electrons which stay on the cofactor and are donated to N2, remain in the region where the hydrides were bound. That is a very surprising result, since with a look at the spin-populations, the electrons on the cofactor appear to be very delocalized. In contrast to the present conclusion, there is no obvious conclusion concerning the position for N2 binding that can be drawn from the positions of the amino acids surrounding the cofactor.
The mechanism from E0 to E4 is shown in Scheme 2 and the energies are summarized in a diagram in Fig. 9. E0 is the same state as A5 in Fig. 3. The transition energies are all reasonably large, ranging from −6.9 kcal mol−1 for E2 to E3, to −20.8 kcal mol−1 for E0 to E1. The requirement that the transition energy should be larger than −3 kcal mol−1 to drive catalysis forward is fulfilled for all of them. The oxidation state starts with (V3+, 1Fe3+, 6Fe2+) for E0. The reason there is still one Fe3+ present is that there is a hydride which has taken an electron from one of the irons in E0. The second hydride, necessary for the activation of N2, enters in E2. There are three protonated sulfides at the start of E4. All E-states lack one sulfide, which was released in A4 prior to catalysis. The first step in E4 is to form H2 from the two hydrides. In the TS, both hydrides are bound to Fe6. The barrier for H-H formation is +15.0 kcal mol−1, which can easily be overcome. The barrier will not be seen on the kinetics, since the barrier for electron transfer is +18 kcal mol−1. The reaction energy for the release of H2 is slightly endergonic by +3.0 kcal mol−1. The oxidation state is now (V2+, 5Fe2+, 2Fe1+), which is exceptionally low. The hydrides leave an empty site on Fe6, where N2 can bind end-on and electrons can flow over from the cofactor. The binding of N2 is exergonic by −5.9 kcal mol−1.
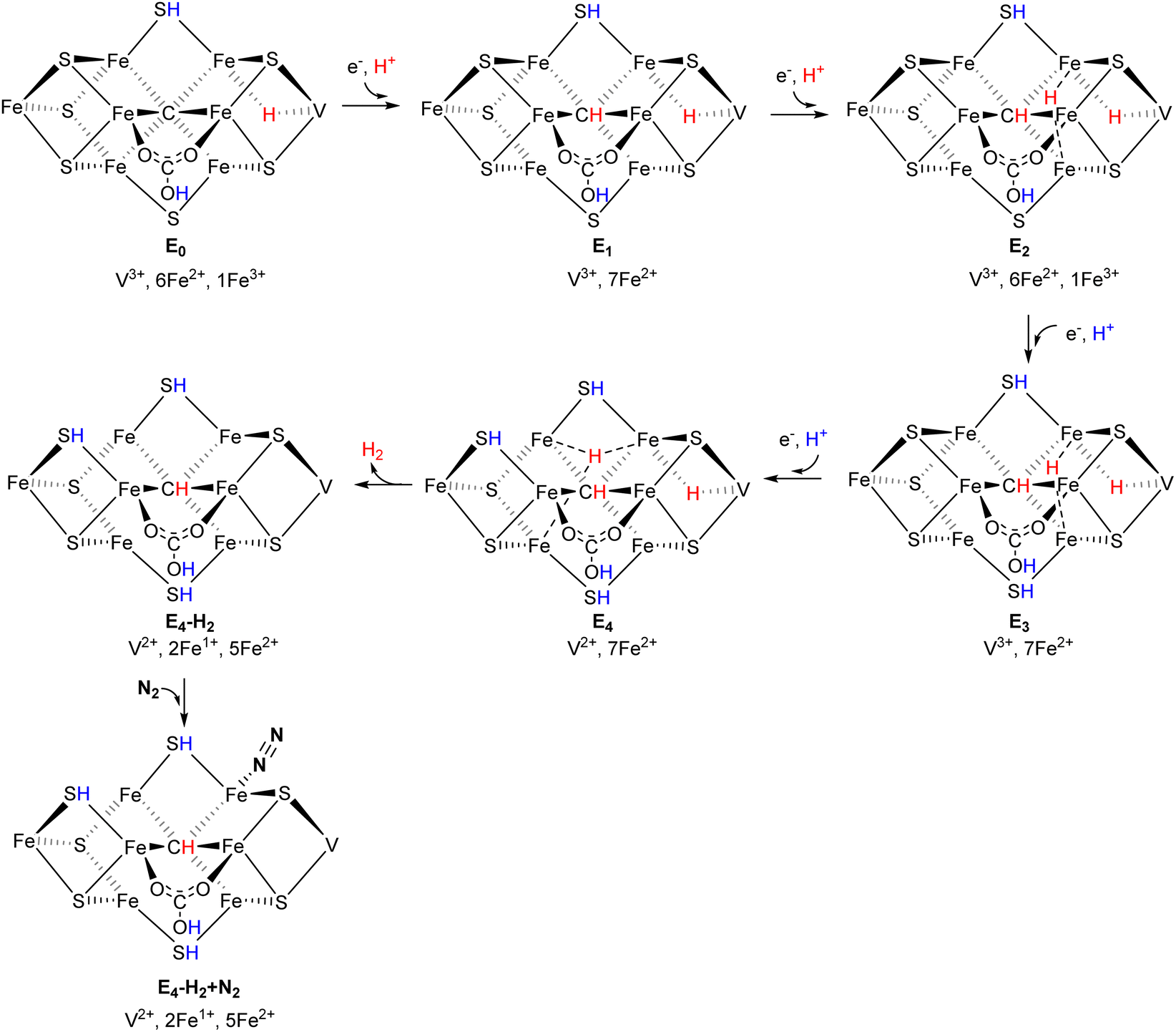 | ||
| Scheme 2 Suggested mechanism for the catalytic cycle from E0 to E4–H2 + N2. | ||
 | ||
| Fig. 9 Energy diagram for the catalytic cycle from E0 to E4–H2 + N2. | ||
H2 release and N2 binding in E4 has also been studied for the spin-coupling (2-,3-,4-), which is the second lowest one in energy. The comparison was made for the model without Phe362. In the starting E4 state, the energy for (2-,3-,7-) is lower by −1.8 kcal mol−1. The barrier for H2 formation is higher by +2.5 kcal mol−1 for (2-,3-,7-), and the endergonicity is larger by +2.7 kcal mol−1. The most significant difference between the spin-couplings occurs for the binding of N2, which is more exergonic by −7.3 kcal mol−1 for (2-,3-,7-). This means that the overall process of releasing H2 and binding N2 becomes more exergonic for (2-,3-,7-) by −4.6 kcal mol−1. A similar effect of changing spin-coupling in E4 was found also for Mo-nitrogenase. Besides having slightly lower energies, the results for (2-,3-,7-) are in better agreement with experiments.
Some tests of the accuracy of the results in E4 were also obtained in the usual way by varying the fraction of exact change with 10%, 15% and 20%. The model without Phe362 was used. The barrier for H2 formation has an uncertainty with a variation of 0.9 kcal mol−1 for each percent. The endergonicity for H2 release has a variation of 0.5 kcal mol−1 for each percent and the binding of N2 a similar uncertainty of also 0.5 kcal mol−1. The variation of the energies with the fraction of exchange showed as usual a nearly linear behavior. Based on previous experience, considering the results with 15% as most reliable, an estimate of the accuracy of the results with +/− 3 kcal mol−1 appears reasonable.
The next step in E4, is a protonation of N2. The proton in the best position for the protonation is the one on S2B. However, this is not the sulphide with the lowest pKa. Following the order in which the sulphides were protonated, the sulfide with the lowest pKa is the one on S1A, see further below. The TS for the protonation of N2 is shown in Fig. 10, and the product in Fig. 11. It is very important for lowering the barrier and stability of the product that N2 is stabilized by a bond to vanadium. After the protonation, all the irons have similar spins between 3.4 and 3.6. The spin on vanadium is hardly changed with 3.9 indicating a redox state with V2+. The calculated barrier is 19.3 kcal mol−1 and the proton transfer is endergonic by +9.2 kcal mol−1. The values are similar to the ones for Mo-nitrogenase with 17.6 kcal mol−1 and +4.0 kcal mol−1. For V-nitrogenase the resting state for the proton transfer is the state where N2 is bound, but that is not the case for Mo-nitrogenase, where the protonation starts at an energy of +5.9 kcal mol−1 above the resting state in E4. The calculated effective barrier for Mo-nitrogenase is therefore +23.5 kcal mol−1, which is too high to be allowed. It was therefore concluded that the calculated barrier for this complicated case, where two electrons and one proton are transferred, is overestimated by 4–5 kcal mol−1. There is probably an overestimation also for V-nitrogenase. For Mo-nitrogenase this step is rate-limiting, and that could be the case also for V-nitrogenase, see further below.
 | ||
| Fig. 10 The transition state for N2H formation. | ||
 | ||
| Fig. 11 The optimized structure for N2H with S1A protonated. | ||
Before the second proton binds to N2H, the proton on S1A needs to be transferred to S2B, in a step which is exergonic by 2.5 kcal mol−1. That, apparently trivial, step turned out to be more complicated than expected. Moving protons from one sulphide to another one has recently been shown to be trivial in most cases, with low barriers, when one or two water molecules could be used in the transfer.34 However, in the present case there is a complicating factor. In the reactant, there is a long distance of 4.0 Å between S1A and Fe2, but in the product the distance is only 2.4 Å. Without moving the sulphide towards Fe2, the barrier is quite high, but with the sulphide moved much closer to Fe2 in the starting structure for the TS (Fig. 12) search, the optimized barrier is only 6.7 kcal mol−1. Since the N2H reactant is not the resting state, but +9.2 kcal mol−1 higher, the effective barrier becomes as high 15.9 kcal mol−1. It can be added that the two water molecules are strongly bound in the reactant, so there is no cost for taking them from the bulk water.
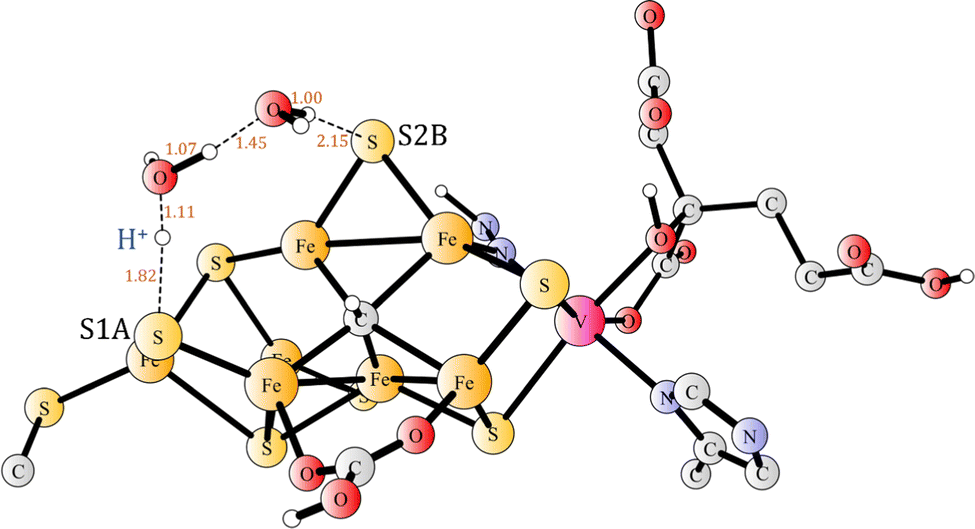 | ||
| Fig. 12 The transition state for the transfer of the proton between S1A and S2B using two water molecules. | ||
The final step in the activation of N2 in E4 is to form N2H2. This is done by moving the proton on S5A over to N2H. The TS is shown in Fig. 13. Since the transfer (Fig. 14) is strongly exergonic by −17.4 kcal mol−1, an early TS was found. The S–H distance is 1.41 Å and the N–H distance 1.97 Å. The local barrier is 13.3 kcal mol−1, but since the reactant N2H is 6.7 kcal mol−1 higher than the resting state, the overall barrier becomes 20.0 kcal mol−1. That means a barrier somewhat higher than the one for the first protonation of N2. Attempts to reduce the barrier by the use of water molecules were not done but would probably reduce the barrier by a few kcal mol−1.
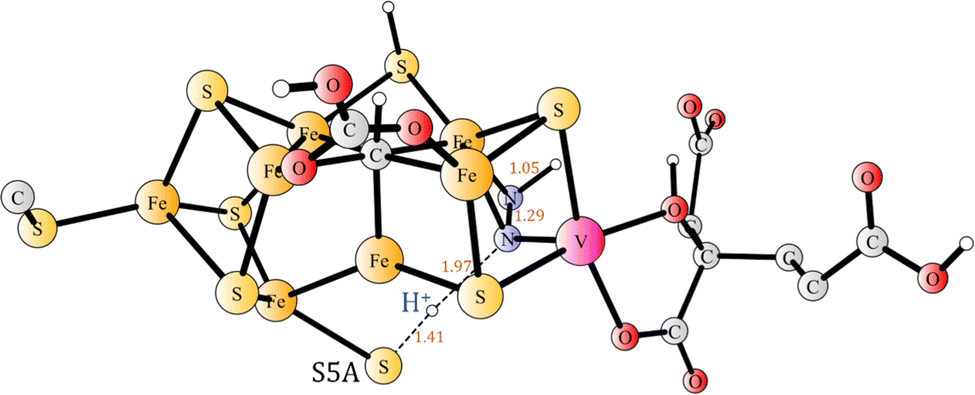 | ||
| Fig. 13 The transition state for N2H2 formation. | ||
 | ||
| Fig. 14 The optimized structure of the N2H2 product (EN2H2). | ||
The energy diagram from the point where N2 became bound to the final point with N2H2 is shown in Fig. 15. As for the case of Mo-nitrogenase, there are no observable intermediates. However, the starting point might be possible to observe in this case but not for Mo-nitrogenase. The rate-limiting barrier for Mo-nitrogenase is the first protonation of N2, and is probably also the one for the same step for V-nitrogenase. A key for the activation of N2 in V-nitrogenase is that one nitrogen of N2 forms a bond to vanadium in the TS.
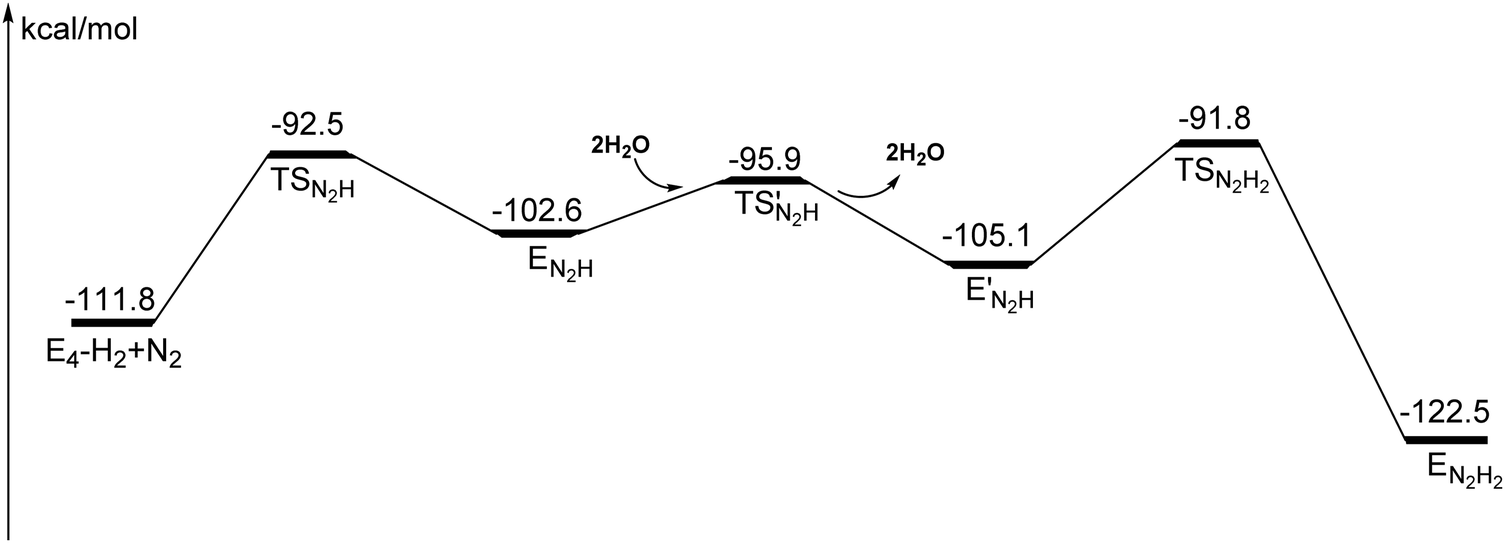 | ||
| Fig. 15 Energy diagram from E4–H2 + N2 to E4–N2H2. | ||
IV. Conclusions
The mechanism for V-nitrogenase has been studied using the same methodology as has recently been used to study Mo-nitrogenase.11,12 The studies start at the respective ground states and continues to the activation of N2 in the E4 state. The goal of the reduction processes is to achieve a state with the lowest possible stable oxidation state. This means both for V- and Mo-nitrogenase a reduction of all irons to Fe2+. An important difference in the reduction processes is that V3+ can be rather easily reduced, but Mo3+ cannot. Since there are four Fe3+ in both cases, this difference leads to a necessity for five activation steps for V-nitrogenase, but only four for Mo-nitrogenase. In turn, this leads for V-nitrogenase to different spin-states for the ground states of these enzymes, a result that has recently been demonstrated experimentally.21 The activation processes lead to a presence of a hydride in E1 in both cases, in agreement with recent experiments.14 For the present case of V-nitrogenase, the hydride states obtained after one or two reductions of the resting state are very high in energy, more than 20 kcal mol−1 higher than the ones with a protonated sulfide or bicarbonate.15There are important similarities and differences of the mechanisms in E4 for V- and Mo-nitrogenase. A similarity is that the release of a sulfide occurs already in the activation process before the catalytic cycling starts. For V-nitrogenase, there is a binding of a hydride to vanadium, which also occurs in the activation process at A4. The origin of that binding is that V3+ is Jahn–Teller active, which leads to a possibility for opening up a coordination site. That open site can later be used for hydride binding. In contrast, Mo3+ is not Jahn–Teller active and, therefore, hydride binding to molybdenum is much harder. For Mo-nitrogenase the formation of H2 has a barrier of +17.3 kcal mol−1 and is endergonic by 8.7 kcal mol−1, while for V-nitrogenase the barrier is +15.0 kcal mol−1 and endergonic by only 3.0 kcal mol−1. It should be mentioned in this context that the rate-limiting step for the nitrogenase mechanism is not N2 activation but electron transfer from the Fe-protein in both cases.4,5,7
The binding site for N2 is different in the two cases. In V-nitrogenase, N2 binds exergonically by −5.6 kcal mol−1 to Fe6 in the neighborhood of vanadium. In Mo-nitrogenase, N2 instead binds to Fe4 exergonically by −2.8 kcal mol−1 far away from molybdenum. In both cases, the enthalpic binding is larger than the large loss of entropy of +9.9 kcal mol−1. As has been demonstrated experimentally, N2 binding has to be preceded by a loss of H2 from two hydrides.6–8 The reason for the necessary loss of H2 is that an oxidation state with the presence of two Fe1+ can be formed, which would otherwise be very endergonic by straight reductions. The oxidation state of the cofactor just before N2 binding is (V2+, 5Fe2+, 2Fe1+) while for Mo-nitrogenase it is (Mo3+, 5Fe2+, 2Fe1+).
For both V- and Mo-nitrogenase, the end-point in E4 is formation of N2H2. The first protonation of N2 is the most difficult step. In Mo-nitrogenase, that step is rate-limiting which is probably also the case for V-nitrogenase. There are several other steps in V-nitrogenase with similar barriers and the methods are not accurate enough to conclude which step is rate-limiting. The formation of N2H is quite endergonic in both cases.
The competing reaction to nitrogen activation is formation of H2 from a proton on a sulfide and a hydride. In Mo-nitrogenase, the only H2 formed is the one from the two hydrides, which is necessary for the activation of N2.6–8 In V-nitrogenase, it has been found experimentally that there are two additional H2 formations, see eqn (1). There are no indications in the present study that these two additional H2 formations should be necessary for N2 activation. Instead, they are here concluded to occur because they cannot be avoided. For Mo- and V-nitrogenase, there are protonated sulfides that could potentially form unproductive H2 with a hydride. Interestingly, there are four protonated sulfides in Mo-nitrogenase but only three in V-nitrogenase even though more unproductive H2 is formed in V-nitrogenase. Clearly, there are also other reasons than the number of protonated sulfides that lead to a loss of H2.
After catalysis, as ATP or N2 ceases, the cluster should return to the ground state A0. Starting from the E4 state. One possible scenario is that the hydrides are lost as two H2 by combining with protons, either from solution or from the protonated sulfides. After catalysis, the P-cluster should return to its redox potential of −0.4 V and would then be a strong oxidant for FeMoco for its strongly reduced state in E4. The remaining 5 electrons should therefore leave FeMoco over the P-cluster and out of the enzyme. A0 is the most stable form of the cofactor.
There are actually not many results definitely known by experiments for V-nitrogenase. One of them is the spin and oxidation state of the ground state. The second one is the process of activating N2. The activation of N2 occurs after a preceding loss of two hydrides as H2. The process is reversible. All the results found here using the present methodology are in very good agreement with these experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at National Supercomputer Centre (NSC) partially funded by the Swedish Research Council through grant agreements no. 2022-22-955 and no. 2018-05973. This work was supported by the Swedish Research Council and the China Scholarship Council.References
- J. Kim and D. C. Rees, Structural models for the metal centers in the nitrogenase molybdenum-iron protein, Science, 1992, 257, 1677–1682 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. M. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor, Science, 2011, 334, 940 CrossRef CAS PubMed.
- D. Sippel and O. Einsle, The Structure of Vanadium Nitrogenase Reveals an Unusual Bridging Ligand, Nat. Chem. Biol., 2017, 13, 956–960 CrossRef CAS PubMed.
- R. N. F. Thorneley and D. J. Lowe, Molybdenum Enzymes, ed. T. Spiro, Wiley, N.Y., 1985, p. 221 Search PubMed.
- B. K. Burgess and D. J. Lowe, Mechanism of Molybdenum Nitrogenase, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, A Draft Mechanism, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- D. Lukoyanov, Z.-Y. Yang, N. Khadka, D. R. Dean, L. C. Seefeldt and B. J. Hoffman, Identification of a Key Catalytic Intermediate Demonstrates That Nitrogenase Is Activated by the Reversible Exchange of N2 for H2, J. Am. Chem. Soc., 2015, 137, 3610–3615 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Model Calculations Suggest that the Central Carbon in the FeMo-cofactor of Nitrogenase Becomes Protonated in the Process of Nitrogen Fixation, J. Am. Chem. Soc., 2016, 138, 10485–10495 CrossRef CAS PubMed.
- P. E. M. Siegbahn, The mechanism for nitrogenase including all steps, Phys. Chem. Chem. Phys., 2019, 21, 15747–15759 RSC.
- W.-J. Wei and P. E. M. Siegbahn, A Mechanism for Nitrogenase Including a Loss of a Sulfide, Chem. – Eur. J., 2022, e202103745 CrossRef CAS PubMed.
- P. E. M. Siegbahn, The mechanism for N2 activation in the E4 – state of nitrogenase, Phys. Chem. Chem. Phys., 2023, 25, 23602–23613 RSC.
- C. Van Stappen, A. T. Thorhallsson, L. Decamps, R. Bjornsson and S. DeBeer, Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations, Chem. Sci., 2019, 10, 9807–9821 RSC.
- D. A. Lukoyanov, D. F. Harris, Z.-Y. Yang, A. Pérez-González, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, The One-Electron Reduced Active-Site FeFe-Cofactor of Fe-Nitrogenase Contains a Hydride Bound to a Formally Oxidized Metal–Ion Core, Inorg. Chem., 2022, 61, 5459–5464 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Can the E1 state in nitrogenase tell if there is an activation process prior to catalysis?, Phys. Chem. Chem. Phys., 2023, 25, 3702–3706 RSC.
- T. Spatzal, A. Kathryn, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- D. Sippel, M. Rohde, J. Netzer, C. Trncic, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, A bound reaction inter-mediate sheds light on the mechanism of nitrogenase, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed.
- T. M. Buscagan and D. C. Rees, Rethinking the Nitrogenase Mechanism: Activating the Active Site, Joule, 2019, 3, 2662–2678 CrossRef CAS PubMed.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed.
- D. F. Harris, D. A. Lukoyanov, H. Kallas, C. Trncik, Z.-Y. Yang, P. Compton, N. Kelleher, O. Einsle, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism to Achieve N2 Reduction, Biochemistry, 2019, 58, 3293–3301 CrossRef CAS PubMed.
- Z.-Y. Yang, E. Jimenez-Vicente, H. Kallas, D. A. Lukoyanov, H. Yang, J. S. Martin del Campo, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, The electronic structure of FeV-cofactor in vanadium-dependent nitrogenase, Chem. Sci., 2021, 12, 6913–6922 RSC.
- B. Benediktsson and R. Bjornsson, Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron−Vanadium Cofactor, Inorg. Chem., 2020, 59, 11514–11527 CrossRef CAS PubMed.
- J. Bergmann, E. Oksanen and U. Ryde, Quantum-refinement studies of the bidentate ligand of V-nitrogenase and the protonation state of CO-inhibited Mo-nitrogenase, J. Inorg. Biochem., 2021, 219, 111426 CrossRef CAS PubMed.
- P. E. M. Siegbahn and M. R. A. Blomberg, A Systematic DFT Approach for Studying Mechanisms of Redox Active Enzymes, Front. Chem., 2018, 6, 644 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Jaguar, version 8.9, Schrodinger, Inc., New York, 2015 CrossRef CAS; A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. R. A. Blomberg, T. Borowski, F. Himo, R.-Z. Liao and P. E. M. Siegbahn, Quantum Chemical Studies of Mechanisms for Metalloenzymes, Chem. Rev., 2014, 114, 3601–3658 CrossRef CAS PubMed.
- P. E. M. Siegbahn, A quantum chemical approach for the mechanisms of redox-active metalloenzymes, RSC Adv., 2021, 11, 3495–3508 RSC.
- P. E. M. Siegbahn and F. Himo, The Quantum Chemical Cluster Approach for Modeling Enzyme Reactions, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 323–336 CAS.
- T. Lovell, J. Li, T. Liu, D. A. Case and L. Noodleman, FeMo Cofactor of Nitrogenase: A Density Functional Study of States MN, MOX, MR and MI, J. Am. Chem. Soc., 2001, 123, 12392–12410 CrossRef CAS PubMed.
- D. M. Camaioni and C. A. J. Schwerdtfeger, Comment on “Accurate Experimental Values for the Free Energies of Hydration of H+, OH−, and H3O+”, Phys. Chem. A, 2005, 109, 10795–10797 CrossRef CAS PubMed.
- P. E. M. Siegbahn, How protons move in enzymes – the case of nitrogenase, J. Phys. Chem. B, 2023, 127, 2156–2159 CrossRef CAS PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous Solvation Free Energies of Ions and Ion-Water Clusters Based on an Accurate Value for the Absolute Aqueous Solvation Free Energy of the Proton, J. Phys. Chem. B, 2006, 110, 16066–16081 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates for all structures discussed in the text are given as supporting information. See DOI: https://doi.org/10.1039/d3cp04698b |
| This journal is © the Owner Societies 2024 |