
On the dual behaviour of water in octanol-rich aqueous n-octanol mixtures: an X-ray scattering and computer simulation study†
Martina
Požar
 a,
Jennifer
Bolle
b,
Susanne
Dogan-Surmeier
b,
Eric
Schneider
b,
Michael
Paulus
b,
Christian
Sternemann
*b and
Aurélien
Perera
c
a,
Jennifer
Bolle
b,
Susanne
Dogan-Surmeier
b,
Eric
Schneider
b,
Michael
Paulus
b,
Christian
Sternemann
*b and
Aurélien
Perera
c
aFaculty of Science, University of Split, Ru era Boškovic'a 33, 21000 Split, Croatia. E-mail: christian.sternemann@tu-dortmund.de
bFakultät Physik/DELTA, Technische Universität Dortmund, D-44221 Dortmund, Germany
cLaboratoire de Physique Théorique de la Matière Condensée (UMR CNRS 7600), Sorbonne Université, 4 Place Jussieu, F75252, Paris cedex 05, France. E-mail: aup@lptmc.jussieu.fr
First published on 3rd January 2024
Abstract
Aqueous n-octanol (n = 1, 2, 3, and 4) mixtures from the octanol rich side are studied by X-ray scattering and computer simulation, with a focus on structural changes, particularly in what concerns the hydration of the hydroxyl-group aggregated chain-like structures, under the influence of various branching of the alkyl tails. Previous studies have indicated that hydroxyl-group chain-cluster formation is hindered in proportion to the branching number. Here, water mole fractions up to x = 0.2 are examined, i.e. up to the miscibility limit. It is found that water molecules within the hydroxyl-chain domains participate in the chain formations in a different manner for 1-octanol and the branched octanols. The hydration of the octanol hydroxyl chains is confirmed by the shifting of the scattering pre-peak position kPP to smaller values, both from measured and simulated X-ray scattering intensities, which corresponds to an increased size of the clusters. Experimental pre-peak amplitudes are seen to increase with increasing water content for 1-octanol, while this trend is reversed in all branched octanols, with the amplitudes decreasing with the increase of the branching number. Conjecturing that the amplitudes of pre-peaks are related to the density of the corresponding aggregates, these results are interpreted as water breaking large OH hydroxyl chains in 1-octanol, hence increasing the density of aggregates, while enhancing hydroxyl aggregates in branched alcohols by inserting itself into the OH chains. The analysis of the cluster distributions from computer simulations provide more details on the role of water. For cluster sizes smaller than dc = 2π/kPP, water is found to always play the role of a structure enforcer for all n-octanols, while for clusters of size dc water is always a destructor. For cluster sizes larger than dc, the role of water differs from 1-octanol and the branched ones: it acts as a structure maker or breaker in inverse proportion to the hindering of OH hydroxyl chain structures arising from the topology of the alkyl tails (branched or not).
1 Introduction
It is well known that alcohols are characterized by the chain-like association of the OH hydroxyl groups, which give rise to a scattering pre-peak in radiation scattering experiments,1–14 and also from scattering intensities calculated from computer simulations.15–23 What is perhaps less known is the influence of the alkyl tail parts, both in their length and their branching possibilities. n-Octanols are an ideal case for this study, because the alkyl tails are reasonably long, and 4 branching conditions are possible, as illustrated in Fig. 1. | ||
| Fig. 1 Octanol molecule branching. The molecules were visualised using ChemDraw (Chem3D; PerkinElmer Informatics). | ||
Linear alcohols beyond propanol are no longer fully miscible in water, indicating that linear alkyl chain lengths beyond 4 are already too hydrophobic for water to accommodate. Indeed, the volume ratio of charged (hydroxyl head groups) to uncharged (alkyl tails) decreases as 1/m, where m is the alkyl tail length in terms of methyl/methylene units. For octanol, the volume of hydrophobic units is 8-fold that of the hydrophilic units, twice that of 1-propanol. As water is added to neat alcohols, it is initially expected that it would associate with the hydroxyl groups, which are already grouped in chains. However, this Coulomb empathetic association is not expected to hold when the water content is increased. For small alcohols, micro-segregation of the alcohol and water is observed, with the help of water–hydroxyl group proximity.24 Also, chaining of the hydroxyl groups is suppressed at high water content. This crossover from monomeric to chain formation is conditioned by the alkyl chain length and bulkiness, and cannot occur without a full phase separation beyond m = 4.25 This implies that the micro-segregation picture of miscible small alcohols is lost with longer ones. Therefore, it is interesting to examine how water affects the OH chain formation before the breaking point, and also how the alkyl tail “bath” influences water location.
The experimental X-ray spectra can be intuitively interpreted in terms of underlying structure formation, but only to some extent. Computer simulations provide a microscopic picture through the statistical analysis of many possible molecular configurations. However, simulations come with their own load of problems, the most important being the use of model molecular force fields, such that the systems studied do not quite correspond to the experimental ones. Interpreting differences in fine details at par with the intuitive ideas provided by the experimental spectra is a difficult exercise, possibly there is no guarantee that a unique interpretation is possible. This is confirmed by previous studies of 1-octanol–water mixtures.
The 1993 study of Franks et al.9 used a modified hard sphere model to fit the hydroxyl group aggregates of neat 1-octanol to a spherical core, thus providing a perfect fit of the pre-peak shape. The 2002 study by McCallum and Tieleman26 concerns computer simulations of neat and hydrated 1-octanol. The snapshots from computer simulations show very clearly that the OH hydroxyl groups form chains and not spheres, hence proving the model of Franks and coworkers incorrect, despite the impeccable fit of the pre-peak. The same simulation shows that water tends to insert within the hydroxyl chains, and that the pre-peak amplitude increases with water concentration. Finally, the 2006 paper by Chen and Siepmann18 is a Monte Carlo simulation study of dry and wet octanol with different force fields. The authors confirm the formation of OH chains, with the insertion of water inside the chains. In addition, they report that water tends to distribute itself more on very long neat 1-octanol chains than short ones, hence diminishing the number of pure 1-octanol long chains. This last trend is equally observed by us, but with the added interpretation that water tends in fact to break pure octanol OH chains, thus acting as a structure breaker, a conclusion that is not reached by this earlier study. Other experimental studies of aqueous n-octanol, are mostly limited to 1-octanol, and do not provide very accurate microscopic pictures. Several types of studies have been performed, such as thermodynamic studies,27,28 spectroscopic studies,29,30 or interfacial descriptions through simulation methods.31
The pathway we choose in this paper is as follows. Various octanol alcohols are systems ordered at the microscopic level, in the sense that hydroxyl groups form various types of chains. Adding water to these alcohols will eventually lead to full demixing already at water concentrations as low as x ≈ 0.25. Hence, studying the mixing of water with these alcohols is akin to studying precursor demixing conditions. On the other hand, it seems intuitively clear that water molecules will preferably stay in the vicinity of the hydrophilic hydroxyl chain groups, and tend to perturb these chains. Eventually, this perturbation will break the stability of the system, which will prefer to phase separate by segregating the water molecules into pockets separated from the ordered octanols. Computer simulation can provide some support to how this happens. What we find is that water plays both the role of a structure maker and breaker, depending on if the octanols are branched or not, and depending on the cluster sizes relative to the pre-peak positions. In what follows, we try to explain how the various microscopic indicators from the simulation can help support this picture.
2 Experimental, simulation and theoretical details
2.1 Experimental
Pure octanol isomers and octanol/water mixtures were studied by X-ray diffraction at 293 K. We purchased 1-octanol (purity >99.7%), 2-octanol (purity >99.5%), 3-octanol (purity >99.5%) and 4-octanol (purity >97%) from Sigma Aldrich and used them without further treatment. Mixtures were produced using MilliQ water. The solubility of water in 1-octanol and 2-octanol is low and even lower for highly branched octanols. Hence we were able to study 1- and 2-octanol for water molar fractions x up to 0.2 while 3- and 4-octanol were investigated up to x = 0.1 to avoid demixing. The pure alcohols and water-alcohol mixtures were filled into borosilicate capillaries with 0.01 mm wall thickness for the diffraction study.The X-ray diffraction measurements were performed at the bending magnet beamline BL2 of the synchrotron radiation source DELTA (Dortmund, Germany).32 We used a multilayer monochromator providing an incident energy of 10.87 keV with a bandpass of about 10−2 and a beamsize of 0.5 × 0.5 mm2. The diffraction images were measured with a MAR345 image plate detector. LaB6 was used as a calibration standard. The 2D diffraction images were converted to diffraction patterns with the software package Fit2D.33 Finally, the air scattering and scattering from capillary was subtracted and the data were normalized to the integrated intensity of the calculated diffraction patterns of the pure octanols in the wave-vector transfer k-range between 0.2 and 2.3 Å−1.
2.2 Simulations
Molecular dynamics simulations of octanol–water mixtures were conducted using the Gromacs program package.34 The initial configurations of 2048 molecules for all systems were created with Packmol.35 These initial configurations were first energy minimized and then equilibrated for 5 ns. Finally, production runs of 5 ns were performed during which at least 2000 configurations were gathered. The simulations were done in the NpT ensemble at T = 300 K and p = 1 bar. The temperature was maintained with the v-rescale thermostat,36 whereas the Parrinello–Rahman barostat37,38 was utilized to keep the pressure constant. The temperature algorithm had a time constant of 0.2 ps and the pressure algorithm was set at 2 ps.The leap-frog algorithm39 was used as the integration algorithm, at every time-step of 2 fs. The short-range interactions were calculated within the 1.5 nm cut-off radius. The long-range electrostatics were handled using the PME method40 and the constraints with the LINCS algorithm.41 The forcefield of choice for the octanols was the OPLS-UA,42 while the SPC/e model was used for water.43
2.3 Theory
In order to compute the scattering intensities I(k) we require all of the atom–atom pair correlation functions gaibj(r) for every pair of atoms ai and bj in the mixtures, belonging respectively to species i and j. These are obtained directly from Gromacs trajectory files, where we typically consider runs of 5 ns, sampled every picosecond, which gurantees smooth and noiseless correlation functions. The atom–atom structure factors Saibj(k) are then computed from the Fourier transform spectra of the gaibj(r) using expression22 | (1) |
 51
51 | (2) |
From the total structure factors, one can finally compute the X-ray scattering intensities I(k) from the Debye expression:22,44,45
 | (3) |
In order to investigate the OH hydroxyl group cluster structure, the probability of finding a cluster of size s is defined as46
 | (4) |
 and N(s,k) represents the number of clusters of size s in a given simulation box k. For the oxygen atom clusters we are interested in, the O–O distance is taken as the hydrogen bonding distance between 2 oxygen atoms.
and N(s,k) represents the number of clusters of size s in a given simulation box k. For the oxygen atom clusters we are interested in, the O–O distance is taken as the hydrogen bonding distance between 2 oxygen atoms.
3 Comparison of experimental and calculated X-ray intensities
3.1 The experimental data and two conjectures
The X-ray scattering experiments are summarized in Fig. 2, for different branching of octanol and different water mole fractions of x = 0.07, 0.1, and 0.2 for 1-octanol, x = 0.045, 0.15, and 0.2 for 2-octanol, x = 0.07 and 0.1 for 3-octanol, and x = 0.06 and 0.1 for 4-octanol.In the discussion below, we pay attention to two principal features: the amplitudes and the k-positions of the main peak (around k ≈ 1.4 Å−1) and the pre-peak (in the k-range 0.4–0.6 Å−1) and their changes with water insertion. We use two conjectures: the k-positions are related to objects size and the amplitudes are related to the density of the objects. By object, we mean the atoms or atomic groups (such as the methyl group) inside molecules and the clusters of the hydroxyl head groups when it concerns the main peak and pre-peak, respectively. By object size is defined through the inter-object contact, for instance defined by the minimum of the inter-particle interaction.
In a previous work on octanols,23 we have shown that both conjectures were fully consistent with the temperature dependence of I(k), both for the main peak and the pre-peak. Essentially, we found that both the amplitudes of the main peak and pre-peak increased with the decrease of temperature, a trend which is consistent with the densification of liquids upon cooling since objects tend to occupy less volume. Similarly, the increase of peaks' k-values are shown to be consistent with a tightening of contact due to increased interactions due to the Boltzmann factor kBT. In the present case, because of the very small effects of water insertion on I(k), the conjecture is less obvious, particularly for the pre-peak.
These conjectures make sense for the main peak, since it concerns atomic groups which are well defined by the interaction force field. For the octanols considered here, the methylene groups of size σm ≈ 3.75 Å are dominating the system in a ratio 8/10. Hence, the scattering main peak is positioned at k = 2π/σm ≈ 1.67 Å−1, which confirms part of the stated conjecture. However, the experimental main peak positions at around 1.4 Å−1 indicate a somehow larger effective diameter of about 4.5 Å. Concerning the amplitudes of the main peak, they are found to be nearly insensitive to the addition of water, since the density change is so small.
For the pre-peak, however, the conjectures are important to explain the observed changes, highlighted by the arrows in Fig. 2, particularly in what concerns the amplitudes. This is because clusters are entities with a distribution of sizes and densities. While all isomers exhibit a decreasing pre-peak position kPP with increasing water addition, their amplitudes vary in a very different way. For 1-octanol the pre-peak intensity increases with higher water content. For 3- and 4-octanol this trend is reversed. Latter is observed also for 2-octanol but to a lesser extent.
 | ||
| Fig. 2 X-ray scattering intensities of n-octanol–water mixtures with n = 1, 2, 3, and 4 with different mole fractions of water. The data are normalized to the integrated scattering intensities of the pure octanols from the simulations. The arrows are guides to the eyes. | ||
3.2 Data from simulations
The calculated diffraction intensities based on the molecular dynamics simulations are presented in Fig. 3. We simulated octanol–water mixtures for molar fractions of x = 0.02, 0.05, 0.1, and 0.2 for 1 and 2-octanol and x = 0.02, 0.05, 0.1,0.1 and 5 for 3 and 4-octanol. The most notable facts are the positions of the main peaks and pre-peaks, as well as both their amplitudes, which follow the same patterns as those of the experimental data in Fig. 2, except for pure 3-octanol and to a lesser extent pure 4-octanol. Similarly to the experimental data, the main peak does not change significantly with the addition of water. The OPLS force field gives the methylene CH2 atom size of around σ ≈ 3.75 Å which corresponds to the main peak positions k(calculated)MP ≈ 1.67 Å−1, enforcing the interpretation of the peaks in terms of mean scattering object size. | ||
| Fig. 3 X-ray scattering intensities of n-octanol–water mixtures with n = 1, 2, 3, 4 as calculated from computer simulations. | ||
Interestingly, the pre-peak behavior of the simulated intensities resembles surprisingly well the findings from the experiment. Except for dry 3-octanol and 4-octanol, we observe in the simulation a shift of the pre-peak to lower k. The variation of the pre-peak intensity is most obvious if the lowest (2%) and highest (15–20%) water concentrations are considered: for 1-octanol the intensity increases while it decreases for all branched octanols with the addition of water. Overall we see a rather good agreement between experiment and simulation, as can be witnessed in Fig. SI-1 and SI.2 of the ESI,† which compare the pre-peak intensities and positions between experimental and simulation data. This agreement allows us to look deeper into the structural details analyzing the simulated snapshots, atom–atom structure factors, and correlation functions. The case of 4-octanol is also examined below. There are obvious cases of disagreements, such as the intensity of the pre-peak of dry 3-octanol being much higher than those of the wet ones. Latter is observed in opposite but to a lesser extent for 4-octanol. Surprisingly, these trends are reproducible from one run to another, even when started from different initial conditions. We get back to this later, when examining microscopic details from simulations.
The interpretation of the intensity of the pre-peak in terms of aggregate density is not obvious to justify from theoretical grounds. Indeed, the pre-peak is the result of cancellations of positive atom–atom correlation pre-peaks and negative atom–atom correlation anti-peaks. Fig. SI-3 in the ESI,† shows all the structure factors for 1-octanol and highlights typical ones, where the pre-peaks and anti-peaks are visible. The origin of these atom–atom pre-peaks and atomic peaks is related to charge order, and ultimately refer to the local arrangements of the charged molecular groups with respect to the uncharged ones. Indeed, the correlations between the charged groups tend to produce positive atom–atom pre-peaks, while the negative anti-peaks come from the charged–uncharged cross correlations. For example, the pre-peak for the case of methanol is weaker (almost a shoulder), while that of ethanol and higher alcohols is more pronounced and higher in amplitude.22 From these information, we can conclude that the intensity of the pre-peak reflect the density of the charged groups in the midst of uncharged ones, namely that of the hydroxyl group clusters in the “bath” of alkyl tails. This interpretation had been tested in the case of pure branched octanols23 and could be put to the test in the present case.
3.3 The behaviour of water inferred from the I(k) data
The main peak is seen to be quite insensitive to the addition of water, which is fully consistent with the picture that water essentially contributes to the aggregates through the cluster pre-peak. Indeed, the main peak reflects the mean atom size, which is dominantly that of the methyl/methylene groups (around 4 Å for the OPLS united atom model42).The behaviour of the pre-peak allows making precise conclusions about the role of water in octanol-rich mixtures, precisely because the pre-peak contains information about the cluster structures, and particularly their sizes.23
We first note that the addition of water in all types of octanol shifts the pre-peak positions to smaller k-values, as shown by the arrows in Fig. 2. This trend is equally reproduced in the calculated results in Fig. 3. According to the first conjecture, this feature indicates an increase in the mean size of the cluster in the presence of water. This is more or less obvious, since water is likely to insert into the charged OH hydroxyl chains, hence making them larger. However, this information does not indicate which types of aggregates are affected, pure hydroxyl chains or water mixed chains. We equally note that the pre-peak positions kPP varies from kPP ≈ 0.4 Å−1 for 1-octanol to kPP ≈ 0.6 Å−1 for 4-octanol, indicating corresponding cluster sizes dc = 2π/kPP varying from dc ≈ 15 Å for 1-octanol to dc ≈ 10 Å for 4-octanol. With a water molecule of diameter σW ≈ 3 Å, these dc represent clusters of 5 to 3 units.
Turning to the pre-peak intensities, we note that the change in the intensities is different from 1-octanol and all the branched ones, indicating that water plays a special role in 1-octanol, and emphazising the difference in the role of water when the alkyl tails are branched. The intensity increases for 1-octanol, while it decreases for all branched octanols. Both trends are equally reproduced by the simulations, albeit not in the same marked way. According to the second conjecture, an increase in amplitude should indicate an increase in the density of clusters. Since, in the simulations, the total number of oxygen atoms for a given water concentration is the same in all cases, an increase in the number of oxygen aggregates necessarily implies a decrease in their size. Hence, water plays the role of an aggregate destructor in the case of 1-octanol. Conversely, a decrease in the pre-peak amplitude, as observed for the branched octanols, necessarily implies an increase in the aggregate size (since the number of O is fixed), which is compatible with the shift of the position of the pre-peaks to lower k-values. In these cases, water appears to play the role of a cluster structure enforcer.
The case of 1-octanol presents an apparent contradiction, since we both concluded that the mean cluster size increased and that the clusters were shrunk. This contradiction is lifted with the analysis of the different cluster distributions in the next section, whether they contain only hydroxyl groups or mixed water-OH clusters, and even pure water clusters. This distinction cannot be made from the experimental X-ray scattering only, and necessitates the help of computer simulations.
4 Microscopic details from computer simulations
The most straightforward approach is to analyse the cluster distributions obtained from the computer simulations, which we provide in the next subsection. The analysis of the atom–atom pair correlation functions and related structure factors in a second subsection, helps better understand what happens at the atomic level.4.1 Cluster analysis
We examine the detailed evolution with water inclusion of the 1-octanol cluster distribution in Fig. 4 and that of 4-octanol in Fig. 5. Similar distributions for 2-octanol and 3-octanol are provided in the ESI,† Fig. SI-4 and SI-5. For each octanol, the upper panel shows the cross water–octanol oxygen cluster distribution as a function of cluster size, while the lower left and right panels show the octanol oxygen and water oxygen cluster distributions, respectively.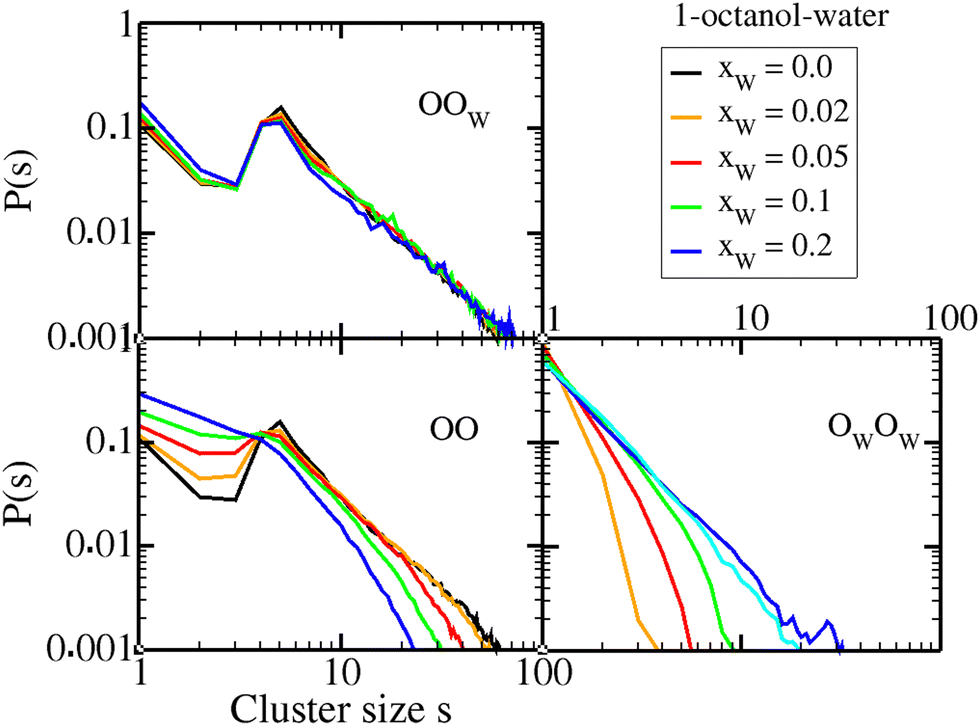 | ||
| Fig. 4 Oxygen atom cluster size distributions for 1-octanol and for different water contents. Neat water cluster curve is shown in cyan in the lower right panel. | ||
 | ||
| Fig. 5 Oxygen atom clusters size distributions for 4-octanol and for different water content. Neat water cluster curve is shown in cyan in the lower right panel. | ||
The first important information provided by these figures is that the cluster size under the peak position and that under the pre-peak position are narrowly related. For the 1-octanol system, the oxygen cluster size is 5 units, which for a diameter of the oxygen atom of about 3 Å gives a cluster size of sc ≈ 15 Å, which corresponds quite well to the pre-peak positions kPP ≈ 0.4 Å−1 related to cluster size dc = 2π/kPP ≈ 15 Å. This correspondence underlies that the aggregates are mostly linear. This is not true with branched octanols, which have both smaller and non-linear OH aggregates.8 Indeed, for 4-octanol we find that the cluster peak is 4.5 units, which is about sc ≈ 13 Å, while the pre-peak position kPP ≈ 0.6 Å−1 relates to cluster size dc ≈ 10 Å < sc. This discrepancy is compatible with the OH clusters being more globular instead of cylindrical.8 With the addition of water, the pre-peak and cluster position peaks move oppositely, indicating that the geometry of the hydrated clusters enter into the picture. How so is what we analyze in the following.
The second information concerns the small OO cluster sizes between dimers and trimers in all cases: the probability of their size increases with the addition of water. In this case, water plays the role of hydroxyl cluster enforcer in all cases. Similarly, the OO cluster peak, namely pentamers, always decreases with water addition, indicating that water is a destructor of hydroxyl pentamers in all cases. But for higher clusters above pentamers, there is a difference between 1-octanol and branched ones, which we analyze below.
We first focus on the upper panel of Fig. 4, and notice that these cross water–octanol oxygen cluster distribution, for all water concentrations, are nearly identical to their respective pure octanol oxygen distributions (the black curves for xW = 0). The first thing to notice is that all the distributions look more or less alike, and more importantly look like the neat 1-octanol cluster distribution. This is a confirmation that the mixed oxygen clusters are similar to that of neat 1-octanol clusters. Clearly, this implies that the 1-octanol oxygen clusters will be reduced in number and size. This is exactly what we observe in the left lower panel: while large OO clusters are in a smaller number compared to that of pure 1-octanol, including the cluster-peak region, the number of smaller OO clusters are increased, and in both cases with a monotonous trend in water concentration increase. The lower right panel shows water oxygen clusters, which strikes by the fact that there is no cluster peak, just like for pure water.46 This is partly because water forms polydispersed globular clusters instead of chain-like ones, since it tends to form tetrahedrally extended space filling patterns.
This analysis clearly demonstrates that water tends to destroy OO clusters, either by inserting into them, or by breaking them apart into smaller ones. We conclude that water acts as a structure breaker in the case of 1-octanol, since it really breaks the original neat 1-octanol ordering. It is important to note here that by structure breaker we do not merely mean to say that water inserts itself in octanol oxygen chains, but that it really make long OO chains unfavourable.
This situation changes when branched octanols are concerned. In Fig. 5, we see from the upper panel that the mixed OOw clusters are still more or less similar to those of pure 4-octanol OO clusters (shown in black). We note that the mid range mixed cluster size 7 < s < 11 tends to be somewhat larger than OO clusters. However, the large OO clusters remain similar to that of neat 4-octanol, as can be seen in the lower left panel. Even though the increase in small OO clusters with water content is similar to that observed in Fig. 4 for 1-octanol (lower left panel), it is really the intermediate/large cluster distribution which is strikingly different. This analysis shows that water acts as a structure maker for 4-octanol, in the sense that it preserves the original OO cluster structure, and promotes larger OOw mixed clusters, a trend also observed for 2-octanol and 3-octanol, as shown in the ESI.†
4.2 Correlation function analysis
Herein, we present a comparative analysis of structure functions for 1-octanol and 4-octanol, mostly from the perspective of supporting the dual role of water. The corresponding data for 2-octanol and 3-octanol can be found in the ESI.†The various oxygen–oxygen pair correlation functions are shown in Fig. 6, 8 and 10, and the corresponding structure factors in Fig. 7, 9 and 11. We focus here on comparing the aqueous 1-octanol mixtures with the aqueous 4-octanol mixtures, respectively, shown in the right and left panels of the figures. Since the pair correlations are essentially governed by the strong hydrogen bonding interactions, the first peak is unusually high, thus the logarithmic scale has been used. All these figures show features typical of alcohols, with a narrow and high first peak, which witnesses the strong first neighbour correlations due to strong hydrogen binding (represented here by Coulomb interactions), followed by second and higher neighbour depletion correlations, which witness the chaining of the hydroxyl groups. It is these two mechanisms that produce respectively positive and negative contributions to the oxygen–oxygen structure factors, which cancel into forming the pre-peak feature. We note in Fig. 7, 9, and 11, that the pre-peak positions correspond to that in the calculated scattering intensities in Fig. 3.
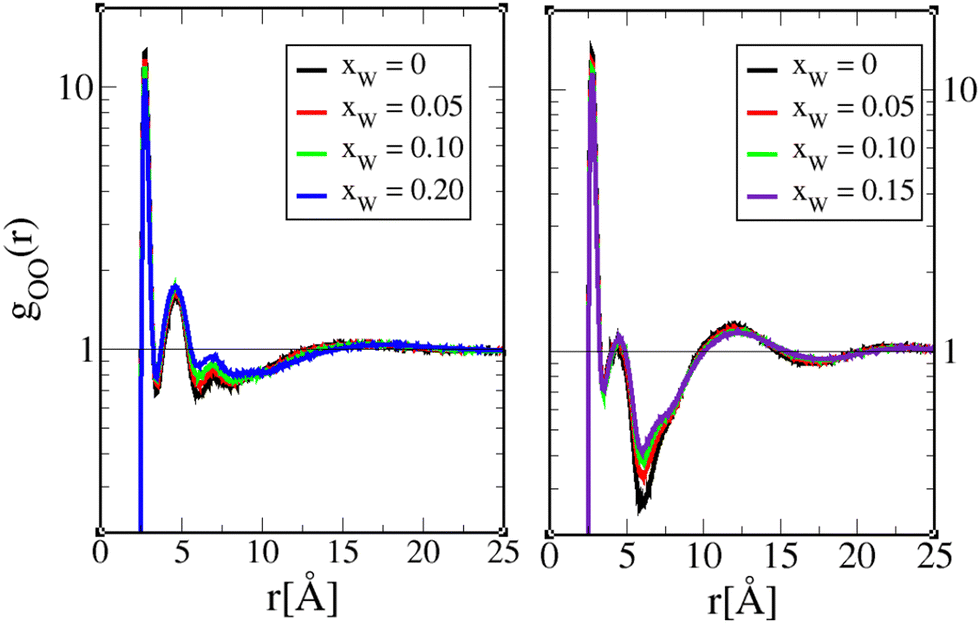 | ||
| Fig. 6 Octanol oxygen–oxygen pair correlation function gOO(r) for 1-octanol (left) and 4-octanol (right), and for various water mole fractions as shown in the legend panels. The pure octanol curves (xw = 0) are presented in black. Note that the vertical scale for the gOO(r) is logarithmic. | ||
 | ||
| Fig. 7 Octanol oxygen–oxygen pair structure factor SOO(k) for 1-octanol (left) and 4-octanol (right), and for various water mole fractions as shown in Fig. 6. The pure octanol curves (xW = 0) are presented in black. | ||
Fig. 6 illustrates the features described above, between two octanol oxygens. In addition, we see that adding water in small concentrations leads to small changes in the correlations for both 1-octanol and 4-octanol, which is the general diminution of correlations. Only the minimum of the depletion well shows marked changes around r ≈ 7.5 Å and r ≈ 6 Å for 1-octanol and 4-octanol, respectively, indicating that water plays its most important role in this distance range, by reducing the depletion. The shape of the depletion is not the same between 1-octanol and 4-octanol, as previously analyzed in ref. 23, and provides important information about the related cluster shapes, which we recall here. As noted in ref. 22, the sharp O–O contact first peak in gOO(r), followed by the depletion correlations in the range 5 Å−15 Å are descriptive of O–O chain clusters. For 1-octanol, in addition to the first peak, the second peak is also quite marked, which is not the case for 4-octanol. Similarly, the depletion range clearly extends more for 1-octanol than for 4-octanol. These 2 features suggest that 1-octanol OH chains are in average longer than those of 4-octanol. In addition, the fact that the domain correlations, as witnessed by the large secondary large oscillations in 4-octanol gOO(r), of period ≈5 Å, indicate that the chain domains are rather short, looking more globular than cylindrical. However, the features discussed above in Fig. 6 are in the visible distance ranges below 20 Å, which corresponds to small clusters of 4–6 oxygen atom units.
In order to gather information about larger clusters, we need to examine the atom–atom structure factors in Fig. 7. We recall that the prominent peak observed in the structure factors at kpp = 2π/dc in the range 0.4 Å−1 < kpp < 0.6 Å−1, corresponds to the OH clusters of size dc, which contribute to the scattering pre-peak observed in Fig. 2 and 3. Once again, we note that there are only little changes in the general shape of these functions with small water addition. Notable features are the marked decrease of the pre-peak amplitude for 4-octanol, as compared to 1-octanol, but also the lesser dependence of overall features with water inclusion for 4-octanol than for 1-octanol, which suggests that OO clusters are not as much affected for 4-octanol than for 1-octanol, except for their numbers, as suggested by the pre-peak amplitude decrease. This is consistent with the cluster analysis results.
Concerning the mixed OOW correlations in Fig. 8, we observe the same features as in Fig. 6, namely, that the depletion well is the most affected by water addition, as the depletion correlations are decreased. We note, however, a clear shift of the peak positions at r ≈ 15–17 Å for 1-octanol, suggesting the existence of larger mixed clusters with the increase of water content. This is consistent with the cluster analysis about large mixed clusters being more prominent than large OO clusters for wet 1-octanol while in the case of wet 4-octanol mixed clusters of average size increase.
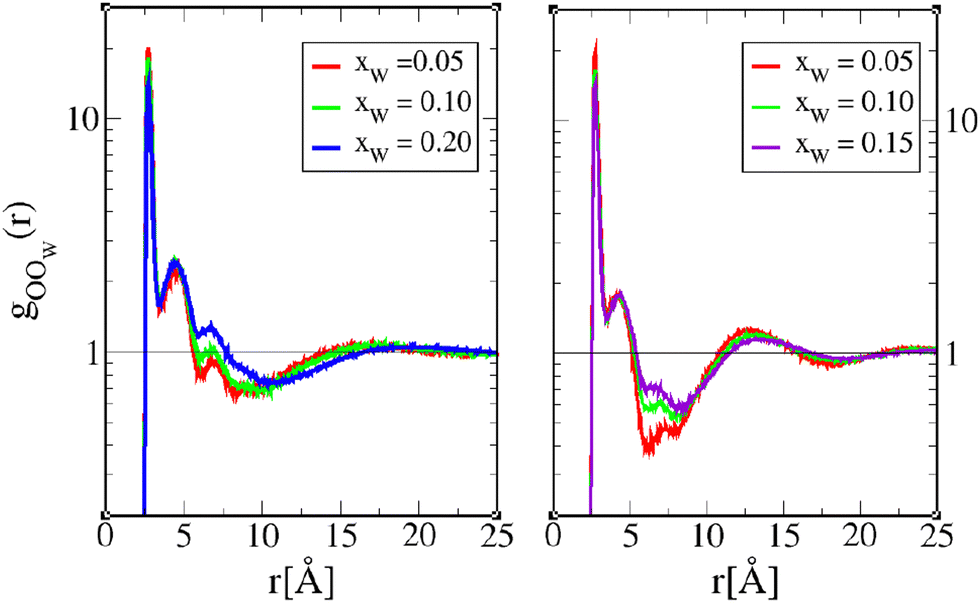 | ||
| Fig. 8 Octanol oxygen–water–oxygen pair correlation function gOOW(r) for 1-octanol (left) and 4-octanol (right). The lines are as in Fig. 6. | ||
The corresponding structure factors shown in Fig. 9 show much more marked features than in the case of OO correlations. The 1-octanol SOOW(k) structure factors have a higher pre-peak amplitude than those for 4-octanol, and the changes with water content are more prominent. This is fully consistent with the scenario suggested by the cluster analysis, that mixed oxygen clusters are more abundant in 1-octanol than in 4-octanol, confirming the intruder role of water in 1-octanol and to a much lesser extent in 4-octanol.
 | ||
| Fig. 9 Octanol oxygen–water–oxygen structure factor SOOW(k) for 1-octanol (left) and 4-octanol (right). The lines are as in Fig. 6. | ||
The water oxygen correlations in Fig. 10 and 11 show very peculiar features, especially when compared with the corresponding correlations in pure water (black curves). The gOWOW(r) clearly suggests the existence of tiny water droplet clusters of 2–3 members, which is witnessed by the fact that the second and third neighbour correlations are well above 1. The large oscillatory features represent droplet–droplet correlations, most probably across alkyl tails, and not within the water-hydroxyl chain clusters. The marked self-water correlations are also consistent with the water cluster analysis not presenting a cluster peak.
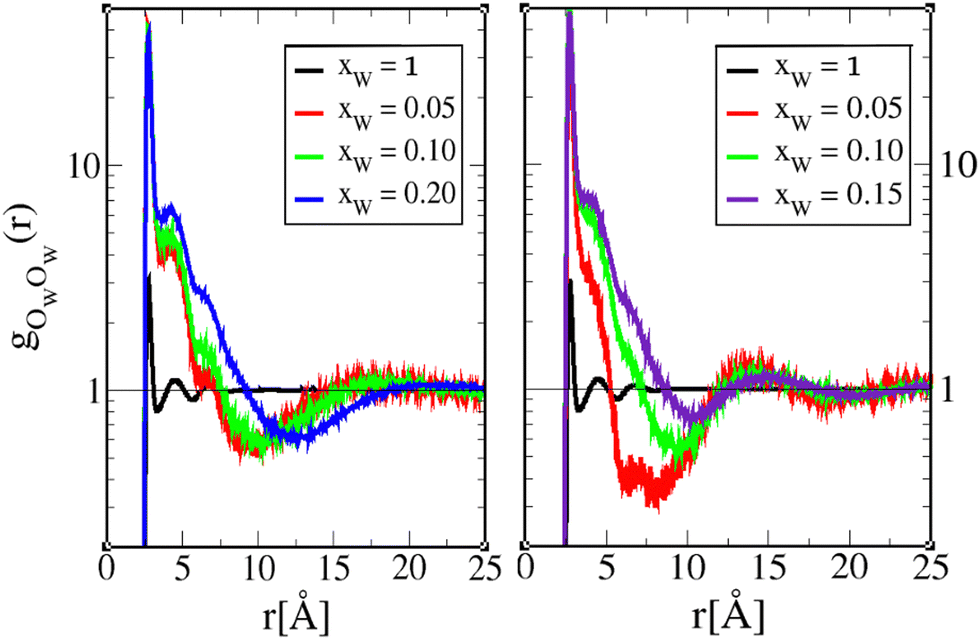 | ||
| Fig. 10 Water–oxygen–water–oxygen pair correlation function gOWOW(r) for 1-octanol (left) and 4-octanol (right). The lines are as in Fig. 6, and the pure water correlations are shown in black curves. | ||
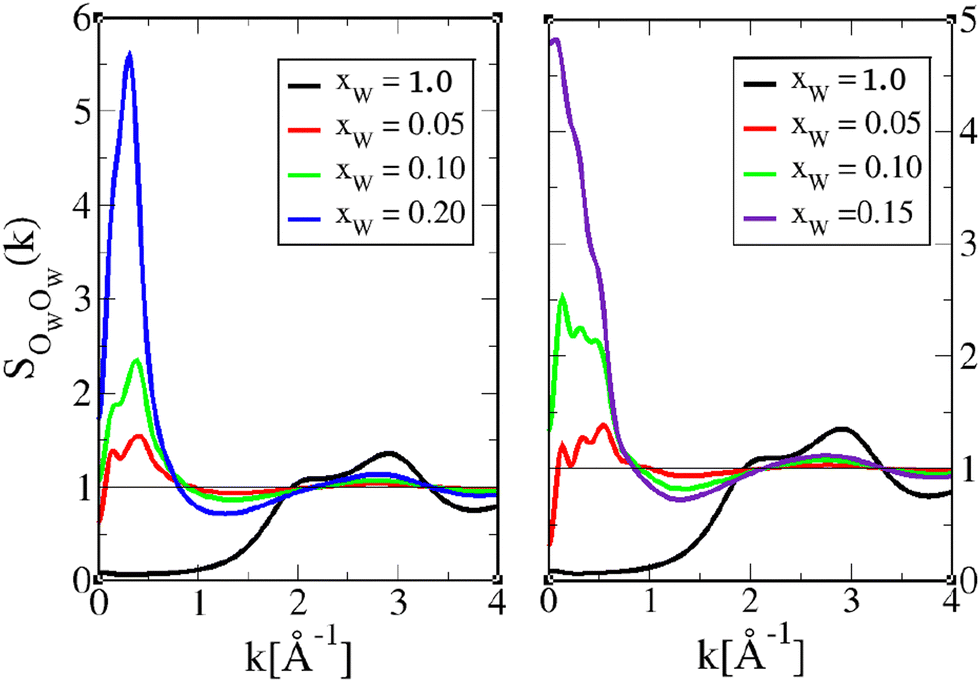 | ||
| Fig. 11 Water–oxygen–water–oxygen structure factors SOWOW(k) for 1-octanol (left) and 4-octanol (right). The lines are as shown in Fig. 6, except that the pure water correlations are shown in black curves. | ||
The structure factors in Fig. 11 tend to confirm the above analysis for gOWOW(r). There is a marked pre-peak at around k ≈ 0.4 Å−1, which is in line with that of the octanol pre-peak and X-ray pre-peak, suggesting that water is part of the hydroxyl clusters, and not unexpectedly so. We note also that this pre-peak tends to become a k = 0 concentration fluctuation peak in the case of 4-octanol, which is markedly visible for xW = 0.15. This is consistent with the earlier demixing of 4-octanol with water content.
We note the enormous difference between pure water correlations (black curves) and those for very small water concentrations, suggesting the radical change in the microscopic structure. The main peak in SOWOW(k) for pure water, which is in fact the shoulder peak at around k ≈ 2 Å−1 (which corresponds to water diameter σ ≈ 3 Å), disappears for the aqueous octanol mixtures, indicating that the bulk water structure does not exist anymore, and is replaced by water aggregated structures.
Similar analysis for 2-octanol and 3-octanol can be made from the figures in the ESI† (Fig. SI-6–SI-8 for 2-octanol and Fig. SI-9–SI-11 for 3-octanol).
4.3 Alkyl tail correlations
If the role of water appears as important, one should not forget that this role is dependent on the topology of the alkyl tails. We have already noticed in ref. 23 that the role of the branching was to reduce the aggregation of the OH hydroxyl groups. This is apparent in Fig. 2 and 3 through the position of the pre-peak positions, as well as their heights. One may question the presence of pre-peaks on alkyl tail correlations between carbon atoms. Our interpretation is that these pre-peak reflect the global formation of octanol aggregates, as induced by the OH hydroxyl head aggregation. Therefore, the pre-peaks in SC8C8(k) should reflect this molecular aggregation, and in particular the “intra” aggregate alkyl tail correlations. This is the reason to select the last carbon atoms, far from the OH head group.The influence of the water content on the alkyl tail carbon atoms is illustrated in the case of the last carbon atom C8, for 1-octanol and 4-octanol, in Fig. 12 and 13, respectively.
 | ||
| Fig. 12 Last carbon atom C8 pair correlation function gC8C8(r) (left panel) and corresponding structure factor SC8C8(k) (right panel) for 1-octanol and for different water contents. The inset in the right panel is a zoom over the pre-peak. | ||
 | ||
| Fig. 13 Same as 12 but for 4-octanol. | ||
The most remarkable feature is the relative insensitivity of these functions for water inclusion for gC8C8(r), except for the pre-peak amplitudes. 1-Octanol appears to be appreciably more sensitive to water inclusion than 4-octanol in the large distances part of the correlation function. This is directly translated into a larger dependence on water inclusion in the structure factor pre-peak, as can be seen from the right panels of Fig. 12 and 13, and particularly visible in the insets.
For 1-octanol, the pre-peak increases with the addition of water and moves to smaller k-values, while it is the opposite for 4-octanol. These trends are exactly those observed in the X-ray scattering intensities in Fig. 2. We have interpreted them in favour of the dual role of water in the n-octanol mixtures. The alkyl tail correlations are therefore in line with water being a structure breaker for 1-octanol and a structure maker for 4-octanol.
4.4 Details from snapshots
It is instructive to compare the aggregate structures formed by the hydroxyl groups and water molecules. In Fig. 14 we provide such a comparison between aqueous 1-octanol (left) and aqueous 4-octanol (right) for a low water content of xw = 0.05, and in Fig. 15 for the higher water content xw = 0.15. For the low water content, we observe mostly the differences in chaining of the hydroxyl groups. For 1-octanol the longer chaining is quite apparent, while smaller chains and and also monomers are visible on the right panel for 4-octanol. It is equally seen that small water clusters are part of larger octanol clusters, in both panels.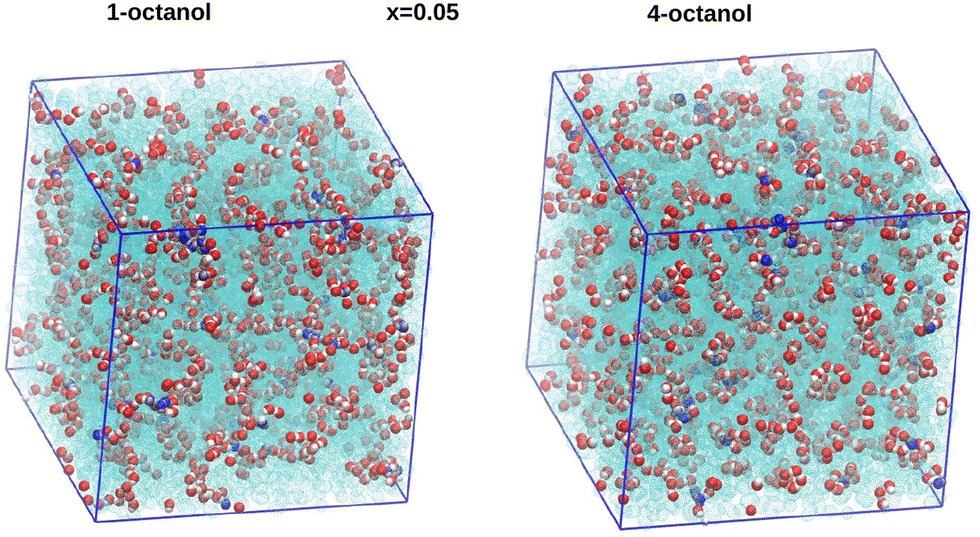 | ||
| Fig. 14 Comparison of snapshots for the xw = 0.05 low water mole fraction, between 1-octanol (left) and 4-octanol (right). Only the hydroxyl groups (oxygen in red and hydrogen in white) and water (in blue) are shown. Snapshots are made with VMD.47 | ||
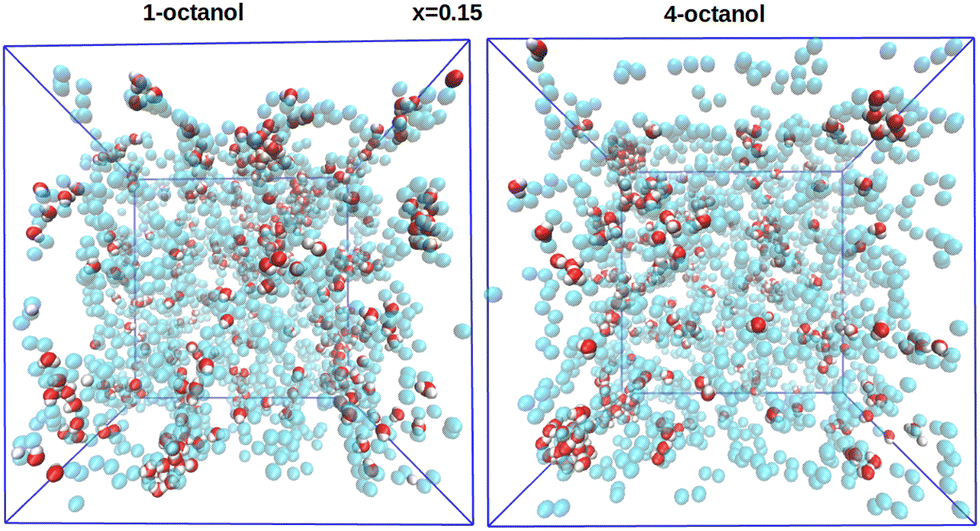 | ||
| Fig. 15 Comparison of snapshots for the xw = 0.15 higher water mole fraction, between 1-octanol (left) and 4-octanol (right). Only the water molecules are shown (oxygen in red and hydrogen in white). The oxygen molecules of the octanols are shown in the cyan ghost mode. Snapshots are made with VMD.47 | ||
In Fig. 15, for xw = 0.15, we see a marked difference in clustering between 1-octanol and 4-octanol, the latter being more clustered and fewer monomers than for 1-octanol are observed. This is consistent with water increasing the number of chains in 1-octanol (since all of the water is always part of the octanol chain clusters), hence with water acting as an OO chain breaker (there is a clear illusion of “more” water in the left panel, even the total number of water molecules is the same in both panels).
5 Discussion and conclusion
In this work, we have highlighted the special role of small concentrations of water in the self-assembled structures of the n-octanol hydroxyl groups, through the analysis of the scattering pre-peaks, which witness these structures. Indeed, scattering pre-peaks are important indicators of the microscopic aggregation in a variety of systems, such as micelles and micro-emulsions,48 room temperature ionic liquids49 or branched and star polymers.50 Such aggregative structures can be experimentally detected using dynamical light scattering (DLS) techniques as well, mainly through the measure of the diffusion coefficient which gives information about the size of the structures. Such studies have been performed on particular aqueous alcohol mixtures such as tert-butanol or butoxytethanol,51–53 and can detect structures in the 20 Å range. It would be particularly interesting to have similar experiments in water–octanol mixtures.We have shown that computer simulations can be used to interpret experimental X-ray spectra of aqueous alcohol n-octanol mixtures in the water poor region, not only by reproducing the changes in the shape of the diffraction intensities with water concentrations, but additionally by providing support to the intuitive interpretations of the pre-peak positions and amplitude displacements with water inclusion. More importantly, this study has highlighted the dual role played by water, which depends indirectly on the topology (i.e. branching) of the alkyl tails. It is generally believed that water has a H-bond structure which is so complex that it leads to many anomalies found in neat water,54 but also that the nature of different solutes perturb this complex structure, either by destroying or enforcing it. Such solutes are called chaotropes and cosmotropes.55 Many interesting effects in aqueous mixtures are often described or attributed in terms of this vocabulary.56–60
In the present paper, we show that water itself can act as a structure maker/preserver or breaker, depending on the nature of the order in non-aqueous systems, herein n-octanols. More importantly, this dual role depends on the hydrophobic sub-molecular component of the solute. Since water demixes from long alcohols, it is not surprising that it would act as a structure breaker for 1-octanol. On the other hand, the role of water as a structure preserver in branched octanol is intriguing. It is not the first example of water acting as a structure maker, since direct micelle formation is promoted by cooperation of water molecules in the outer corona region. Similarly, the role of water in the biological environment is also believed to play a structuring role. The structural details provided herein may help better understand the role of water, and pave the way for further investigations.
While the computer simulation study of water rich binary mixtures is plagued with spurious demixing issues in many cases,61–66 in the present study we demonstrate that this is not the case for water poor mixtures. In contrast, it is apparent that the study provides results that are in good agreement with experimental scattering intensities, indicating that it is not a model issue, as is often formulated when considering problematic water rich simulations.63,67,68 It is quite tempting to deduce that the apparent spurious water segregation and demixing could be a genuine physical effect, in the very short time (less than the microsecond) and small sizes (box sizes below 50 nm). Water may first tend to induce large segregated solute domains in a very short period, before such domains melt and lead to final equilibrium with less segregated mixture. This problem may not be perceptible at the experimental level (unless explicitly looked for). From these remarks, it could be interesting to look at water nucleation problems in binary mixtures of aqueous organic molecules from the water rich side. As stated above, the very good agreement observed between the experimental and calculated X-ray scattering intensities, and for very different systems, tends to indicate that subtle self-assembly issues in aqueous poor conditions can be properly studied by model computer simulations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank DELTA for providing synchrotron radiation at beamline BL2 and technical support. Martina Požar gratefully acknowledges funding from the Croatian Science Foundation under project no. UIP-2017-05-1863 Dynamics in Micro-segregated Systems and the computational resources of the server UniST-Phy at the University of Split. This work was supported by the BMBF via DAAD (PROCOPE 2022–2023, Project-ID: 57560563, and PROCOPE 2024–2025, Project-ID 57704875) within the French-German collaborations PROCOPE (46644XK), Supra-molecular order in complex liquids: experiment and theory and PROCOPE (50951YA), Analysis of the molecular coherence in the self-assembly process: experiment and theory.References
- W. Pierce and D. MacMillan, X-ray studies on liquids: the inner peak for alcohols and acids, J. Am. Chem. Soc., 1938, 60, 779–783 CrossRef CAS.
- B. Warren, X-ray diffraction in long chain liquids, Phys. Rev., 1933, 44, 969–973 CrossRef CAS.
- M. Magini, G. Paschina and G. Piccaluga, On the structure of methyl alcohol at room temperature, J. Chem. Phys., 1982, 77, 2051–2056 CrossRef CAS.
- A. Narten and A. Habenschuss, Hydrogen bonding in liquid methanol and ethanol determined by X-ray diffraction, J. Chem. Phys., 1984, 80, 3387–3391 CrossRef CAS.
- S. Sarkar and R. N. Joarder, Molecular clusters and correlations in liquid methanol at room temperature, J. Chem. Phys., 1993, 99, 2032–2039 CrossRef CAS.
- A. Sahoo, P. Nath, V. Bhagat, P. Krishna and R. Joarder, Effect of temperature on the molecular association in liquid D-methanol using neutron diffraction data, Phys. Chem. Liq., 2010, 48, 546–559 CrossRef CAS.
- A. Karmakar, P. Krishna and R. Joarder, On the structure function of liquid alcohols at small wave numbers and signature of hydrogen-bonded clusters in the liquid state, Phys. Lett. A, 1999, 253, 207–210 CrossRef CAS.
- S. Sarkar and R. N. Joarder, Molecular clusters in liquid ethanol at room temperature, J. Chem. Phys., 1994, 100, 5118–5122 CrossRef CAS.
- N. P. Franks, M. H. Abraham and W. R. Lieb, Molecular organization of liquid n-octanol: an X-ray diffraction analysis, J. Pharm. Sci., 1993, 82(5), 466–470 CrossRef CAS PubMed.
- K. S. Vahvaselkä, R. Serimaa and M. Torkkeli, Determination of liquid structures of the primary alcohols methanol, ethanol, 1-propanol, 1-butanol and 1-octanol by X-ray scattering, J. Appl. Crystallogr., 1995, 28, 189–195 CrossRef.
- A. Sahoo, S. Sarkar, V. Bhagat and R. N. Joarder, The probable molecular association in liquid D-1-propanol through neutron diffraction, J. Phys. Chem. A, 2009, 113, 5160–5162 CrossRef CAS PubMed.
- A. K. Karmakar, S. Sarkar and R. N. Joarder, Molecular clusters in liquid tert-butyl alcohol at room temperature, J. Phys. Chem., 1995, 99, 16501–16503 CrossRef CAS.
- M. Tomšič, A. Jamnik, G. Fritz-Popovski, O. Glatter and L. Vlček, Structural properties of pure simple alcohols from ethanol, propanol, butanol, pentanol, to hexanol: comparing monte carlo simulations with experimental saxs data, J. Phys. Chem. B, 2007, 111, 1738–1751 CrossRef PubMed.
- T. Yamaguchi, K. Hidaka and A. Soper, The structure of liquid methanol revisited: a neutron diffraction experiment at −80C and +25C, Mol. Phys., 1999, 96, 1159 CrossRef CAS.
- C. Benmore and Y. Loh, The structure of liquid ethanol: a neutron diffraction and molecular dynamics study, J. Chem. Phys., 2000, 112, 5877–5883 CrossRef CAS.
- I. Akiyama, M. Ogawa, K. Takase, T. Takamuku, T. Yamaguchi and N. Ohtori, Liquid structure of 1-propanol by molecular dynamics simulations and X-ray scattering, J. Solution Chem., 2004, 33, 797–809 CrossRef CAS.
- P. Sillrén, J. Swenson, J. Mattsson, D. Bowron and A. Matic, The temperature dependent structure of liquid 1-propanol as studied by neutron diffraction and epsr simulations, J. Chem. Phys., 2013, 138, 214501 CrossRef PubMed.
- B. Chen and J. I. Siepmann, Microscopic structure and solvation in dry and wet octanol, J. Phys. Chem. B, 2006, 110(8), 3555–3563 CrossRef CAS PubMed . PMID: 16494411.
- A. Vrhovšek, O. Gereben, A. Jamnik and L. Pusztai, Hydrogen bonding and molecular aggregates in liquid methanol, ethanol, and 1-propanol, J. Phys. Chem. B, 2011, 115, 13473–13488 CrossRef PubMed.
- J. Cerar, A. Lajovic, A. Jamnik and M. Tomšič, Performance of various models in structural characterization of n-butanol: molecular dynamics and X-ray scattering studies, J. Mol. Liq., 2017, 229, 346–357 CrossRef CAS.
- J. Lehtola, M. Hakala and K. Hämäläinen, Structure of liquid linear alcohols, J. Phys. Chem. B, 2010, 114, 6426–6436 CrossRef CAS PubMed.
- M. Požar, J. Bolle, C. Sternemann and A. Perera, On the X-ray scattering pre-peak of linear mono-ols and the related microstructure from computer simulations, J. Phys. Chem. B, 2020, 124(38), 8358–8371 CrossRef PubMed.
- J. Bolle, S. P. Bierwirth, M. Požar, A. Perera, M. Paulus, P. Münzner, C. Albers, S. Dogan, M. Elbers, R. Sakrowski, G. Surmeier, R. Böhmer, M. Tolan and C. Sternemann, Isomeric effects in structure formation and dielectric dynamics of different octanols, Phys. Chem. Chem. Phys., 2021, 23, 24211–24221 RSC.
- M. Požar, B. Lovrinčević, L. Zoranić, T. Primorac, F. Sokolić and A. Perera, Micro-heterogeneity versus clustering in binary mixtures of ethanol with water or alkanes, Phys. Chem. Chem. Phys., 2016, 18, 23971 RSC.
- A. E. Hill and W. M. Malisoff, The mutual solubility of liquids. iii. the mutual solubility of phenol and water. iv. the mutual solubility of normal butyl alcohol and water, J. Am. Chem. Soc., 1926, 48(4), 918–927 CrossRef CAS.
- J. L. MacCallum and D. P. Tieleman, Structures of neat and hydrated 1-octanol from computer simulations, J. Am. Chem. Soc., 2002, 124(50), 15085–15093 CrossRef CAS PubMed . PMID: 12475354.
- Y. Marcus, Structural aspects of water in 1-octanol, J. Solution Chem., 1990, 19, 507–517 CrossRef CAS.
- K. Liltorp, P. Westh and Y. Koga, Thermodynamic properties of water in the water-poor region of binary water + alcohol mixtures, Can. J. Chem., 2005, 83(5), 420–429 CrossRef CAS.
- F. Palombo, T. Tassaing, M. Paolantoni, P. Sassi and A. Morresi, Elucidating the association of water in wet 1-octanol from normal to high temperature by near- and mid-infrared spectroscopy, J. Phys. Chem. B, 2010, 114(28), 9085–9093 CrossRef CAS PubMed.
- P. Sassi, M. Paolantoni, R. S. Cataliotti, F. Palombo and A. Morresi, Water/alcohol mixtures: a spectroscopic study of the water-saturated 1-octanol solution, J. Phys. Chem. B, 2004, 108(50), 19557–19565 CrossRef CAS.
- R. L. Napoleon and P. B. Moore, Structural characterization of interfacial n-octanol and 3-octanol using molecular dynamic simulations, J. Phys. Chem. B, 2006, 110(8), 3666–3673 CrossRef CAS PubMed.
- M. Dargasz, J. Bolle, A. Faulstich, E. Schneider, M. Kowalski, C. Sternemann, J. Savelkouls, B. Murphy and M. Paulus, X-ray scattering at beamline bl2 of delta: studies of lysozyme-lysozyme interaction in heavy water and structure formation in 1-hexanol, J. Phys.: Conf. Ser., 2022, 2380(1), 012031 CrossRef.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, Two-dimensional detector software: from real detector to idealised image or two-theta scan, High Press. Res., 1996, 14(4–6), 235–248 CrossRef.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. Shirts, J. Smith, P. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Gromacs 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- J. Martínez and L. Martínez, Packing optimization for automated generation of complex system's initial configurations for molecular dynamics and docking, J. Comput. Chem., 2003, 24, 819 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, Crystal structure and pair potentials: a molecular-dynamics study, Phys. Rev. Lett., 1980, 45, 1196 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182 CrossRef CAS.
- R. Hockney, Methods in computational physics, The potential calculation and some applications, Orlando Academic Press, 1970, vol. 9, pp. 135–221 Search PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh ewald: an n
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) log(n) method for ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
log(n) method for ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS. - B. Hess, H. Bekker, H. Berendsen and J. Fraaije, Lincs: a linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- W. Jorgensen, Optimized intermolecular potential functions for liquid alcohols, J. Phys. Chem., 1986, 90, 1276 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 1987, 91(24), 6269–6271 CrossRef CAS.
- P. Debye, Zerstreuung von röntgenstrahlen, Ann. Phys., 1915, 351, 809–823 CrossRef.
- P. Debye, Scattering of X-rays. The collected papers of Peter J.W. Debye, Interscience Publishers, 1954 Search PubMed.
- L. Zoranić, F. Sokolić and A. Perera, Microstructure of neat alcohols: a molecular dynamics study, J. Chem. Phys., 2007, 127, 024502 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, Vmd: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- M. Teubner and R. Strey, Origin of the scattering peak in microemulsions, J. Chem. Phys., 1987, 87(5), 3195 CrossRef CAS.
- H. Annapureddy, H. Kashyap, P. De Biase and C. Margulis, What is the origin of the prepeak in the X-ray scattering of imidazolium-based room-temperature ionic liquids?, J. Phys. Chem. B, 2010, 114(50), 16838–16846 CrossRef CAS PubMed . PMID: 21077649.
- S. Jin, K. S. Jin, J. Yoon, K. Heo, J. Kim, K.-W. Kim, M. Ree, T. Higashihara, T. Watanabe and A. Hirao, X-ray scattering studies on molecular structures of star and dendritic polymers, Macromol. Res., 2008, 16(8), 686–694 CrossRef CAS.
- G. W. Euliss and C. M. Sorensen, Dynamic light scattering studies of concentration fluctuations in aqueous t-butyl alcohol solutions, J. Chem. Phys., 1984, 80(10), 4767–4773 CrossRef CAS.
- T. M. Bender and R. Pecora, Dynamic light scattering measurements of mutual diffusion coefficients of water-rich 2-butoxyethanol/water systems, J. Phys. Chem., 1988, 92(6), 1675–1677 CrossRef CAS.
- J. Troncoso, K. Zemánková and A. Jover, Dynamic light scattering study of aggregation in aqueous solutions of five amphiphiles, J. Mol. Liq., 2017, 241, 525–529 CrossRef CAS.
- M. Chaplin, water structure and science (website).
- P. Ball and J. E. Hallsworth, Water structure and chaotropicity: their uses, abuses and biological implications, Phys. Chem. Chem. Phys., 2015, 17, 8297–8305 RSC.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Ions in water: characterizing the forces that control chemical processes and biological structure, Biophys. Chem., 2007, 128(2), 95–104 CrossRef CAS PubMed.
- B. Hribar, N. T. Southall, V. Vlachy and K. A. Dill, How ions affect the structure of water, J. Am. Chem. Soc., 2002, 124(41), 12302–12311 CrossRef CAS PubMed.
- Y. Marcus, Effect of ions on the structure of water: structure making and breaking, Chem. Rev., 2009, 109(3), 1346–1370 CrossRef CAS PubMed.
- H. Laurent, A. K. Soper and L. Dougan, Trimethylamine n-oxide (tmao) resists the compression of water structure by magnesium perchlorate: terrestrial kosmotrope vs. martian chaotrope, Phys. Chem. Chem. Phys., 2020, 22, 4924–4937 RSC.
- M. Vraneš, A. Tot, S. Papović, J. Panić and S. Gadžurić, Is choline kosmotrope or chaotrope?, J. Chem. Thermodyn., 2018, 124, 65–73 CrossRef.
- A. Perera and F. Sokolić, Modeling nonionic aqueous solutions: the acetone–water mixture, J. Chem. Phys., 2004, 121, 11272 CrossRef CAS PubMed.
- B. Kežić and A. Perera, Revisiting aqueous-acetone mixtures through the concept of molecular emulsions, J. Chem. Phys., 2012, 137, 134502 CrossRef PubMed.
- M. Lee and N. van der Vegt, A new force field for atomistic simulations of aqueous tertiary butanol solutions, J. Chem. Phys., 2005, 122(11), 114509 CrossRef PubMed.
- R. Gupta and G. N. Patey, Aggregation in dilute aqueous tert-butyl alcohol solutions: insights from large-scale simulations, J. Chem. Phys., 2012, 137(3), 034509 CrossRef PubMed.
- S. D. Overduin, A. Perera and G. N. Patey, Structural behavior of aqueous t-butanol solutions from large-scale molecular dynamics simulations, J. Chem. Phys., 2019, 150(18), 184504 CrossRef CAS PubMed.
- S. Banerjee, J. Furtado and B. Bagchi, Fluctuating micro-heterogeneity in water-tert-butyl alcohol mixtures and lambda-type divergence of the mean cluster size with phase transition-like multiple anomalies, J. Chem. Phys., 2014, 140(19), 194502 CrossRef.
- R. Chitra and P. E. Smith, A comparison of the properties of 2,2,2-trifluoroethanol and 2,2,2-trifluoroethanol/water mixtures using different force fields, J. Chem. Phys., 2001, 115(12), 5521–5530 CrossRef CAS.
- S. D. Overduin and G. N. Patey, Comparison of simulation and experimental results for a model aqueous tert-butanol solution, J. Chem. Phys., 2017, 147(2), 024503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04651f |
| This journal is © the Owner Societies 2024 |