
Electrostatically tunable interaction of CO2 with MgO surfaces and chemical switching: first-principles theory†
Arpita
Sen
a,
Ayush K.
Narsaria
 *b,
Meghna A.
Manae
a,
Sharan
Shetty
b and
Umesh V.
Waghmare
*a
*b,
Meghna A.
Manae
a,
Sharan
Shetty
b and
Umesh V.
Waghmare
*a
aTheoretical Sciences Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India. E-mail: waghmare@jncasr.ac.in
bShell India Markets Pvt. Ltd, Mahadeva Kodigehalli, Bengaluru, Karnataka 562149, India. E-mail: ayush-kumar.narsaria@shell.com
First published on 3rd January 2024
Abstract
Electric field-assisted CO2 capture using solid adsorbents based on basic oxides can immensely reduce the required energy consumption compared to the conventional processes of temperature or pressure swing adsorption. In this work, we present first-principles density functional theoretical calculations to investigate the effects of an applied external electric field (AEEF) within the range from −1 to 1 V Å−1 on the CO2 adsorption behavior on various high and low-index facets of MgO. When CO2 is strongly adsorbed on MgO surfaces to form carbonate species, the coupling of electric fields with the resulting intrinsic dipole moment induces a ‘switch’ from a strongly chemisorbed state to a weakly chemisorbed or physisorbed state at a critical value of AEEF. We demonstrate that such ‘switching’ enables access to different metastable states with variations in the AEEF. On polar MgO(111) surfaces, we find a distinct feature of the adsorptive dissociation of CO2 towards the formation of CO in contrast to that on the non-polar MgO(100) and MgO(110) surfaces. In some cases, we observe broken inversion symmetry because of the AEEF that results in induced polarity at the interaction site of CO2 on MgO surfaces. Our results provide fundamental insights into the possibility of using AEEFs in novel solid adsorbent systems for CO2 capture and reduction.
1. Introduction
Carbon capture and storage (CCS) is one of the most important technological steps in reducing the carbon footprint to achieve environmentally benign chemical processes.1 The major contributions to the CO2 released into the atmosphere are from power plants and transportation.2 Carbon capture from pre- and post-combustion processes is commercially practiced using the amine solvent process where CO2 is selectively absorbed by amines.3,4 This method is energetically demanding due to regeneration cycles and the degradation of amines leading to environmentally unsafe processes.5,6 In recent years, materials based on solid adsorbents, such as those based on metal oxides, metal organic framework (MOF), and zeolites, have been proposed that have certain advantages over solvent-based technologies.7,8 The efficiency of solid adsorbents critically depends on the thermodynamics of the adsorption–desorption process. Pressure swing adsorption (PSA) and temperature swing adsorption (TSA) technologies are typically employed for the CO2 adsorption process for industrial applications.9–11 However, these technologies can be energy intensive based on the scale of the process. The availability of ‘green’ electricity produced from renewable sources, such as solar or wind energy, and electric swing adsorption (ESA) could be an attractive technology for CO2 separation and capture.12,13 The main advantage of this process is the ambient operation conditions that can avoid large pressure and temperature variations experienced by the solid adsorbents. Another CCS technology that has been gaining traction is external electric field (EEF) swing adsorption.14–19 EEF can have a significant effect on the adsorption behavior of the molecules on the metal surface due to the dipole change at the interface of the surface+molecule system.20–24 McEwen and co-workers combined experimental and DFT calculations to demonstrate the applicability of EEFs for steam methane reforming on Ni catalysts.25,26 They were able to show that in the presence of EEF, coke formation is reduced and the process can operate at lower temperatures. In another study, they discussed the effect of negative and positive EEFs on the reaction towards water dehydrogenation on a Ni surface.27Shaik et al. discussed their views on the application of oriented EEFs on the reaction kinetics.28,29 They proposed that the orientation of the EEF was very critical in changing the reaction pathways. Chuah et al. showed that CO and H2 could be desorbed by high electric field pulses to achieve steady-state conditions in the methanol decomposition reaction on rhodium.30 Keller et al., in a recent study, demonstrated the use of ESA in capturing CO2 in hollow fibers that have Joule heating properties.31 In an interesting study on the oxidative coupling of methane reaction, Sugiura et al. proved that Ce2(WO4)3 supported on CeO2 exhibited higher selectivity toward C2 in the presence of an electric field as compared to the conventional process.32 Shetty et al., in a recent work, combined electric field-based DFT calculations with machine learning algorithms to predict the field-dependent adsorption energies on the Pt(111) surface.33 These studies provide evidence of the importance of studying electric field-assisted chemical reactions, specifically in the ESA-driven adsorption–desorption processes.
MgO has been recognized as a prototypical basic oxide material for CO2 capture and storage, mostly due to the abundance of MgO in nature.34,35 Moreover, due to the presence of the various crystallographic orientations that can be differentiated based on the polarity of the surfaces, MgO can play an important role in its reactivity towards CO2.36–43 In a recent computational study, Manae et al. showed that CO2 interactions with various MgO surfaces depend on the local electronic and structural properties of the active site.35 They proposed that CO2 interacts weakly with the (100) and oxygen-terminated (111) surface due to the distinct properties of the active sites. It has been proposed in other studies that the reactivity of surface sites towards CO2 adsorption can be altered by promoters.44–46 Motivated by this and earlier work on the effects of an applied external electric field (AEEF) on the catalytic properties of molecular adsorption, herein, we investigate the effects of AEEFs on the adsorption behavior of CO2 on different MgO surfaces.
2. Computational details
We performed first-principles calculations within the framework of plane wave density functional theory (DFT) as implemented in the Quantum ESPRESSO package.47 A kinetic energy cutoff of 60 Ry was used for the plane wave basis set in the representation of wave functions and a cutoff of 400 Ry was taken to represent the charge density. Interactions between the valence electrons and the core electrons were modelled with the projector augmented wave (PAW) potentials.48 We have used the revised exchange–correlation energy of the Perdew–Burke–Ernzerhof (PBEsol) functional within a generalized gradient approximation (GGA).49 The occupation numbers of electronic states were smeared with the Fermi–Dirac distribution with a smearing width (kBT) of 0.04 eV. We have included van der Waals (vdW) interactions using the Grimme scheme.50 Equilibrium structures were obtained through the minimization of energy until the Hellmann–Feynman forces on each atom were smaller than 7 × 10−6 eV Å−1 in magnitude. The various surfaces of MgO were modelled by periodic supercells, including a vacuum layer of 15 Å thickness parallel to the slab separating its adjacent periodic images. Each supercell contains a slab of 5 atomic planes, with each plane of the surface containing 3 × 3 in-plane units. In bare surface calculations, all 5 atomic planes were optimized. Adsorption calculations were performed by freezing the bottom 2 atomic planes while relaxing only the top 3 planes with the CO2 adsorbate (please check the Computational details section in the ESI,† for further details). Brillouin zone integrations were performed with a uniform grid of 4 × 4 × 1 k-points for all surfaces. We optimized the structures of bare MgO surfaces, the surfaces with CO2 molecules, and the isolated CO2 molecule, including an external electric field ranging from −1.0 to 1.0 V Å−1. Although such high electric fields could be difficult to realize in controlled experiments, there are a few good reasons for us to choose such a magnitude of electric fields. Che et al. showed in a combined computational and experimental study that large electric fields are necessary for altering the adsorption energy of molecular species such as H2O and OH. A high electric field is needed to change the electronic states of the adsorbates and the surface for field-induced chemisorption.51,52 Large electric fields can also be observed in the electrochemical cells at the electrode and the electrolyte interface.51,53 Moreover, AEEFs used in DFT simulations are typically large because they correspond to low temperatures (∼0 K) and in the absence of any pressure. A high AEEF is needed to cross the barrier separating one metastable state from another. Under experimental conditions, thermal fluctuations facilitate such a crossover at a lower electric field. To simulate the response of MgO to an electric field, we added a saw-tooth potential as a function of z (perpendicular to the surface). The electric field was applied using a saw-tooth potential with a sharp, short step in the middle of the vacuum. The slope of the saw-tooth potential is the electric field. Dipole corrections were included to eliminate the effects of a polar field arising from the continuity and periodicity of the electrostatic potential.54We have calculated the adsorption energy (Ead) of CO2-adsorbed MgO surfaces with varying electric fields, i.e., ranging from −1 to 1 V Å−1 and an interval of 0.1 V Å−1, unless otherwise mentioned. The Ead is defined as follows:
| Ead = Esurface+molecule − Esurface − Emolecule | (1) |
 | ||
| Scheme 1 A schematic diagram depicting the electric field AEEF along the z-axis of the MgO slab (side view in left figure) and the different adsorption configurations of CO2 on MgO surfaces (right figure) at the bridge site (1), Mg-top site (2), O-top site (3), and hollow site (4). | ||
3. Results and discussion
3.1 MgO(100) surface
The MgO(100) surface has the lowest surface energy among the four MgO surfaces considered here and hence is the most stable and dominant surface of the rocksalt crystals of MgO.35Fig. 1 describes the change in the adsorption energy of CO2 on the (001) surface with respect to the AEEF. In the absence of the AEEF, CO2 is adsorbed on the bridge site (Fig. 1(b)) with an Ead of −1.0 eV, where the CO2 is bent with an O–C–O angle of 133°. The C atom of CO2 interacts strongly with the surface O atom, indicating the formation of carbonate species where the more electronegative O atoms of CO2 interact with Mg atoms on the surface. Starting with strong adsorption at a negative AEEF, the adsorption energy increases linearly and gradually with the AEEF until it reaches 0.202 V Å−1. Such a linear variation of energy with the AEEF confirms that the inversion symmetry of the adsorption site is broken due to the AEEF giving rise to an induced dipole moment. Interestingly, at 0.203 V Å−1 there is a sudden step change in the adsorption energy from −0.79 to −0.4 eV. Examination of the structural evolution with the AEEF reveals that the CO2 molecule remains bent from −1.0 till 0.202 V Å−1 and essentially detaches from the bridge site at 0.203 V Å−1, changing to a weakly adsorbed state (Fig. 1(d) and Fig. S2, ESI†). The O–C–O bond angle changes from 134° to 169° and the O of the MgO slab and the C of the CO2 bond length increase from 1.45 to 2.38 Å (see Fig. S1 and S2, ESI†). The adsorption energy attains a plateau beyond AEEF of 0.203 V Å−1. This shows that the CO2 molecule is in a weakly bound state on the MgO surface beyond an AEEF of 0.203 V Å−1.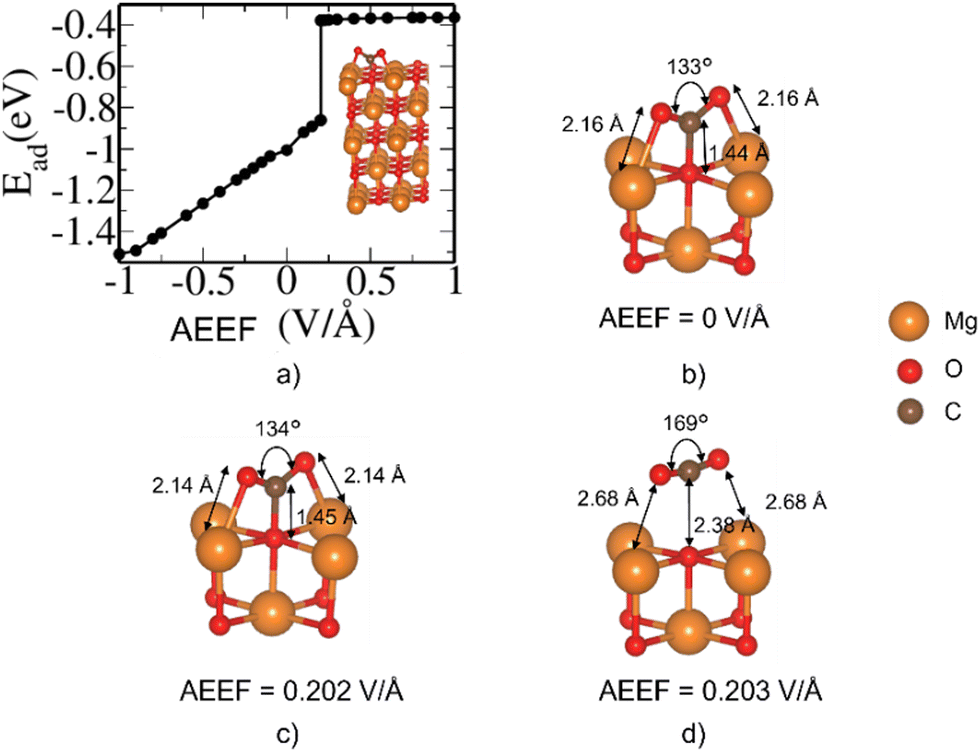 | ||
| Fig. 1 (a) Variation of adsorption energy (eV) with respect to the AEEF (V Å−1). (b)–(d) The optimized structures of CO2 adsorbed on the bridge site of Mg(100) at AEEFs of 0, 0.202 and 0.203 V Å−1, respectively. | ||
The E − E0 defined in Fig. 2 is the total energy difference with AEEF (E) and without AEEF (E0). We can see that the energy of the bare surface (Fig. 2(a)) and of the molecule (Fig. 2(b)) is a quadratic function of AEEF (symmetric parabola), while the CO2 adsorbed on the surface state has a discontinuity at 0.203 V Å−1. To gain further insight into this discontinuity, we examined the behavior of Esurface+molecule with positive and negative electric fields separately, as shown in Fig. 2(d) and (e). The linear term (b.E in eqn (2)) corresponds to the energy of the electric field interacting with the dipole moment (b in b.E or the dipolar energy). The dipolar energy changes significantly in these two regions (see Fig. 2(d) and (e)). The noticeable change in the Esurface+molecule value at 0.203 V Å−1 therefore originates from the change in dipole moment that arises from the bending of CO2. It should be noted that the quadratic term (c.E2 in eqn (2)) that accounts for polarizability (c in c.E2) is about the same in these two regions.
| Ead = a + b.E + c.E2 | (2) |
 | ||
| Fig. 2 Contribution of each term (see eqn (1)) to the energy of adsorption at different AEEF: Variation of electronic energy of (a) the bare MgO(100) surface, (b) isolated CO2 molecule and (c) CO2 adsorbed on the bridge site of the MgO(100) slab with AEEF relative to the case with zero AEEF. Quadratic fit of energy vs. AEEF for the CO2 adsorbed on the MgO surface with (d) negative and (e) positive AEEF (see eqn (2)). | ||
The adsorption of CO2 on the Mg site of the Mg(100) surface shows rather contrasting behavior as evidenced in Fig. 3(a) when compared to the adsorption on the bridge site discussed above. The large fluctuations or the jumps in adsorption energy, as seen in Fig. 3a, is because there are three metastable structures, labeled C, Ah and BMg, which switch from one to another with the AEEF. For a particular metastable structure (for example C), the adsorption energy varies linearly with the AEEF. This linear component in the variation of adsorption energy with the AEEF is associated with the interaction of the dipole moment with the AEEF. This is true for all three metastable structures (please see the dashed lines in Fig. 3a). With the AEEF, one can transition or switch from one metastable state to another. The first region is designated as Ah, which corresponds to the Ead around −0.4 eV and is an intermediate adsorbed state; the second region (BMg) is a weakly bound state around Ead ∼ −0.1 eV, and the third region (C) is a strongly adsorbed state corresponding to Ead < −0.8 eV. The most stable state at AEEF = 0.0 V Å−1 belongs to the first kind, i.e., Ah, where CO2 gets adsorbed at the hollow site. As the AEEF shifts to the negative field (from ∼−0.1 to −0.5 V Å−1), the Ead proceeds through a transition from the Ah region to BMg to Ah. We also observed that the CO2 molecule shifts from a hollow site (Fig. 3(c)) to the Mg top site (Fig. 3(d)) where the CO2 is displaced vertically upwards minimizing the interaction with the surface. At an AEEF of around −0.5 V Å−1 to −0.8 V Å−1, the magnitude of Ead increases (more negative) in the range of −1.0 eV to −1.2 eV (Region C). The CO2 adsorbed state in Region C indicates carbonate formation, where CO2 bends and forms a strong bond with the surface O (Fig. 3(e)). Surprisingly, at an AEEF of around −1.0 V Å−1, we noticed that the state of adsorption switched to the BMg type, i.e., a weakly adsorbed state (Fig. 3(f)). At the positive AEEF field, we observed only BMg and Ah regions. However, at AEEF > 0.5 V Å−1, CO2 preferentially remained in the relatively weakly bound Ah state. The structures C and BMg switched at AEEF = −1.0 V Å−1. Relaxation of the structure type C at AEEF = −1.0 V Å−1 leads to no qualitative change in the structure (preserving the C-type structure, see the details given in the ESI†).
 | ||
| Fig. 3 (a) The dependence of Ead (in eV) on the AEEF of MgO(100)/CO2 on the Mg top site. (b) The initial structure; the relaxed structures at AEEFs of (c) 0.0 V Å−1 (the set of structures marked as Ah), (d) −0.2 V Å−1 (set of structures marked as BMg), (e) −0.8 V Å−1 (set of structures marked as C), and (f) at −1.0 V Å−1 (set of structures marked as C). | ||
The states of CO2 adsorption at the O site of the Mg(100) surface (Fig. 4) can be classified into two types of states, viz., Region I: Ead < −0.6 eV, and Region II: Ead > −0.6 eV. In the former, Ead has a rich behavior where it passes through several energy maxima/minima (Fig. 4(a)), particularly at a negative AEEF, and attains a stable state at around AEEF = −1 V Å−1, corresponding to Ead of ∼−1.2 eV. These several adsorption states are separated by an Ead of ∼0.4 eV (see Fig. 4(a)). From these results, we infer that the CO2 molecule experiences different low-lying minima at negative AEEFs (accessible with AEEF and small perturbations). The structures (Fig. 4(e) and (f)) correspond to Region I where the C of the CO2 interacts with the O of the MgO surface, and the CO2 is bent to enable a favorable interaction with MgO. In Region II, Ead becomes less stabilizing and reaches a constant value of −0.3 eV for AEEF > 0.3 V Å−1. Consequently, the O–C–O bond angle shifts from a bent configuration (130°) at AEEF = 0 V Å−1 to linear (175°–177°) as the AEEF becomes positive. Broadly, we find here two types of metastable structures, the MgO slab with linear CO2 and the MgO slab with bent CO2. The details are given in the ESI.†
 | ||
| Fig. 4 (a) The variation in Ead (in eV) of MgO(100)/CO2 on the O top site with AEEF. The relaxed structure at AEEFs of (b) 0.0 V Å−1, (c) 0.2 V Å−1, (d) 0.3 V Å−1, (e) −0.5 V Å−1 and (f) −0.8 V Å−1. Ead has been classified into two regions, Region I (Ead < −0.6 eV, more stabilizing) and Region II (Ead > −0.6 eV, less stabilizing), respectively. | ||
3.2 MgO(110) surface
We now consider the MgO(110) surface that is less stable (higher surface energy by 0.4 eV Å−12) than the MgO(100) surface and is highly reactive towards CO2 adsorption compared to the (100) surface.42 We obtained optimized structures starting with CO2 on top of the O site, Mg site, and bridge site. At AEEF = 0 V Å−1, we find that the CO2 molecule has an adsorption energy of −3.2 eV and forms CO32− species (Fig. 5(b)) when relaxed from the O top site. We see a linear relationship between the AEEF and Ead (Fig. 5(a)) in contrast to the switching seen in the case of the MgO(100) surface. As there are no accessible metastable states in our analysis presented in Fig. 5, there are no fluctuations or jumps in Ead with AEEF. The quadratic variation or a slight deviation from the linear variation in the adsorption energy with AEEF is attributed to polarizability (or dielectric constant of MgO), which causes a dipole moment induced by AEEF. The total energy difference with and without AEEF, i.e., E − E0, of the pristine MgO(110) slab (Fig. 5(c)) and CO2 adsorbed on the slab on the O of MgO (Fig. 5(d)) is a quadratic function for the (110) surface similar to the (100) surface. However, there is no discontinuous change in Ead for CO2 adsorption on the (110) surface in contrast to that on the (100) surface (Fig. 2(c)). The structure of CO2 in the adsorbed state at AEEF in the range from −1 to 1 V Å−1 does not change and qualitatively remains in the same CO32− state, as described in Fig. 5(b). It is clear that the changes in Ead seen in Fig. 5(a) without many structural changes are due to the difference in the E − E0 of the bare MgO(110) surface (Fig. 5(c)) and the CO2-adsorbed MgO(110) surface (Fig. 5(d)). One should note that the CO2 contribution to Ead is relatively weaker, as shown in Fig. 2(b). To understand the orbitals involved in the interaction of the CO2 molecule with the MgO surface, we considered a configuration of CO2 interacting with the MgO(110) surface adsorbed on the O site at an AEEF of 0.1 V Å−1. From the partial density of states, the specific occupied state (overlapping with the valence band) that has contributions from orbitals of the CO2 molecule was identified. Visualization of an isosurface of charge density associated with this state at the Y-point shows that the HOMO of CO2, which is essentially the lone pair p-orbitals of its O atoms, interacts with Mg atoms of the MgO(110) surface, and the p-orbitals of O atoms on the MgO surface interact with the C atom of the adsorbed CO2 molecule, leading to the formation of the CO32− state (see Fig. S5, ESI†).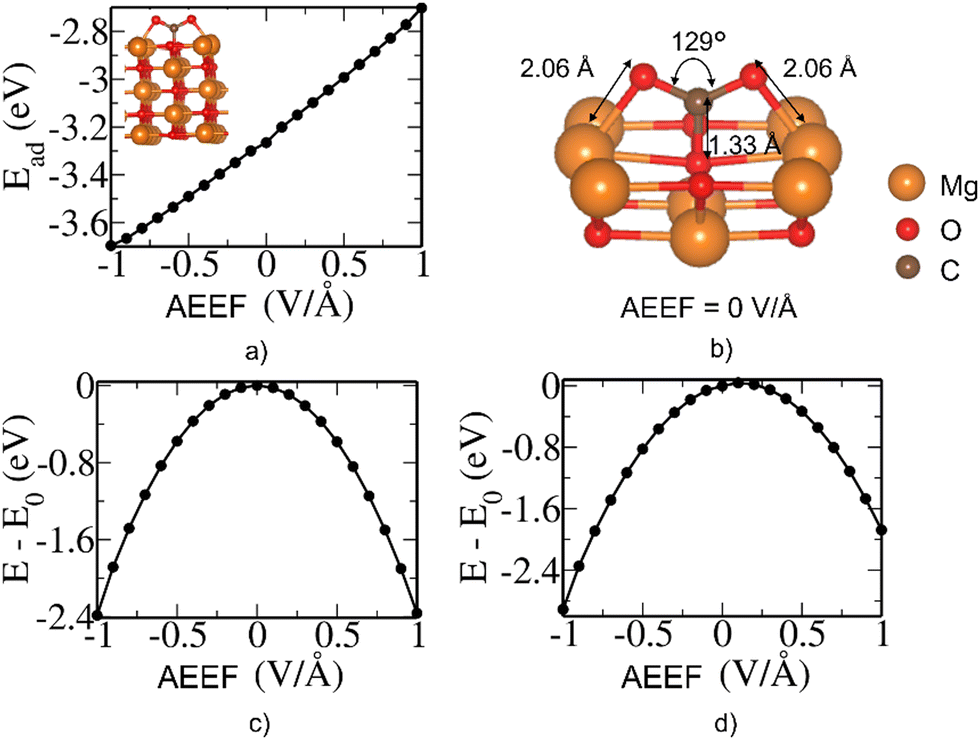 | ||
| Fig. 5 (a) The variation of Ead (in eV) with the AEEF for MgO(110)/CO2 on the O top site. (b) The relaxed structure at zero AEEF. The variation of the electronic energy (in eV) of the (c) bare (110) surface and (d) CO2 adsorbed on the O site of the (110) MgO surface with the AEEF relative to the case with zero AEEF. | ||
We also explored the adsorption of CO2 on the Mg site (Fig. 6(b)) of the MgO(110) surface. The CO2 molecule shifts to the O site in the relaxed structure at AEEF = 0 V Å−1 resulting in the formation of a CO32−-like structure (see Fig. 5(b)). At an AEEF of −0.1 V Å−1, CO2 desorbs (Fig. 6(d)) and remains in that state till −0.4 V Å−1, and the desorbed state switches back to the adsorbed state at AEEF = −0.5 V Å−1 with Ead of −3.5 eV. Surprisingly, at AEEF = −0.8 V Å−1, the structures switch back to the desorbed state. While there are oscillations in the adsorption energy behavior at negative AEEF, it exhibits a linear variation at positive AEEF, remaining in the CO32− state (Fig. 6(a)). Oscillation in the CO2 adsorbed state (at the Mg site (Fig. 6)) is intriguing and probably reflects local energy minima, in contrast to adsorption at the O site (Fig. 5), while the two states are structurally similar. Thus, we found three metastable states, and their switching and structural transformations are discussed in detail in ESI.†
 | ||
| Fig. 6 (a) The variation of Ead (in eV) with the AEEF for MgO(110)/CO2 on the Mg top site. (b) The initial structure with CO2 on the Mg site. The relaxed structures of the configurations at AEEFs of (c) 0.0 V Å−1, (d) −0.1 V Å−1, (e) −0.5 V Å−1, and (f) −1.0 V Å−1. Small adsorption energies in (a) represent weakly adsorbed states with long distances between CO2 and the surface (e.g. (d) and (f)). | ||
Optimization of CO2 on the bridge site of the MgO(110) results in the formation of the CO32− state (AEEF = 0) as shown in Fig. 7(c), which is accompanied by the local reconstruction of the surface near the adsorption site. This state remains unchanged at the AEEF from −1.0 V Å−1 to 0.2 V Å−1. Around AEEF = 0.3 V Å−1, the CO32− state is further stabilized due to the favorable orientation of the bent CO2 relative to the surface. Remarkably, at 1.0 V Å−1, CO2 destabilizes and desorbs from the surface (Fig. 7(f)). We thus find here two metastable states that are accessible with the AEEF ranging from –1.0 to 1.0 V Å−1, upon relaxation starting from the structure stable at 0 V Å−1. The details are given in ESI.†
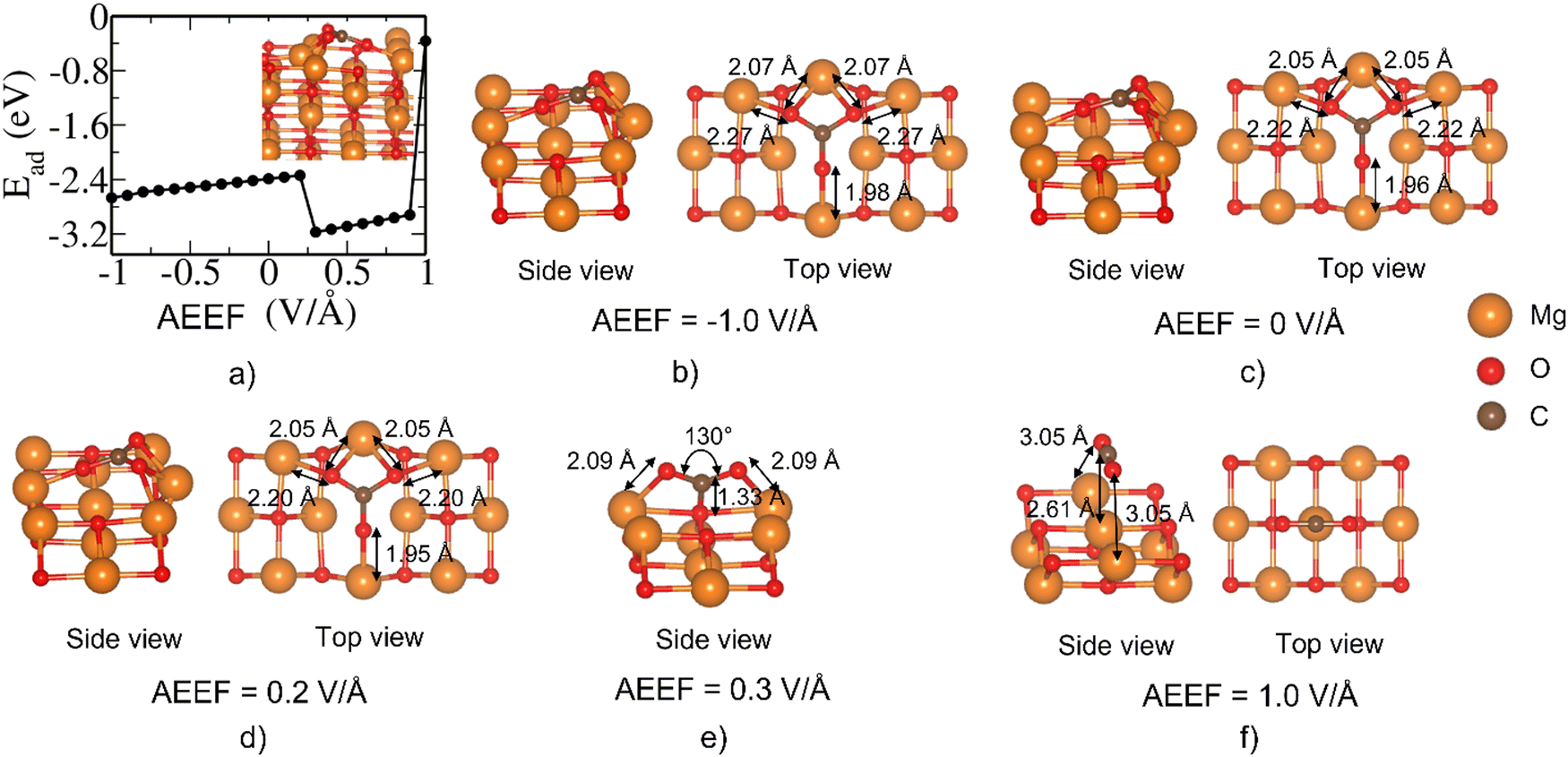 | ||
| Fig. 7 (a) The variation of Ead (in eV) with the AEEF for MgO(110)/CO2 on the bridge site. Relaxed structures of the configurations at AEEFs of (b) 0.0 V Å−1, (c) 0.2 V Å−1, (d) 0.3 V Å−1, (e) 1.0 V Å−1, and (f) -1.0 V Å−1. | ||
3.3 MgO(111) surface
The MgO(111) surface is the least stable and the most interesting in the context of the reactivity of CO2 adsorption due to the polarity of the surface, unlike its (100) and (110) surfaces.41,42 Considering the polarity of the MgO(111) surface, the reactivity of the surface depends on the surface orientation, i.e., Mg or O termination. We will discuss simulations of CO2 adsorption on these surface orientations in the presence of AEEF. Fig. 8 describes the adsorption behavior of CO2 on the Mg top site of the Mg-terminated MgO(111) surface with varying AEEF. CO2 adsorbed very weakly at AEEF from 1 V Å−1 till −0.6 V Å−1 (Fig. 8(a)). The structures of CO2 on the Mg-terminated MgO(111) surface (Fig. 8(b) and (c)) corresponding to AEEFs ranging from 1.0 to −0.6 V Å−1 confirmed its weak adsorption on the surface. At an AEEF of −0.7 V Å−1, drastic stabilization of CO2 on the surface was observed. Fig. 8(d) indicates that the CO2 forms a carbonate compound and is adsorbed on the MgO slab with the O atoms of CO2 interacting strongly with the surface Mg atoms. This state remains unchanged until an AEEF of −1 V Å−1. We found here two metastable states of CO2 adsorbed on the Mg-terminated (111) surface and a structural transformation at AEEF −0.7 V Å−1. The details are given in the ESI.† | ||
| Fig. 8 (a) The variation of Ead (in eV) with AEEF on MgO(111)-Mg-terminated/CO2 on the Mg top site. Relaxed structures of configurations at AEEFs of (b) 0.0 V Å−1, (c) −0.6 V Å−1, and (d) −0.7 V Å−1. | ||
CO2 adsorption at a bridge site of the Mg-terminated (111) surface is weak at AEEF = 0 V Å−1, similar to the adsorption over the Mg top site, as discussed above. At 0 V Å−1, CO2 is aligned horizontally (parallel to the surface) over the bridge site with its O atoms facing the Mg atoms (Fig. 9(b)), in contrast to the case where the C of the CO2 is on top of the Mg atom (Fig. 8(b)). Surprisingly, CO2 can further undergo an irreversible dissociative adsorption into CO + O (Fig. 9(c)) at AEEF = 0.5 V Å−1. The CO fragment in the dissociated state is aligned perpendicular to the surface and its O atom is bonded to the Mg atom (see Fig. 9(c)). It is interesting to note that the CO species from the dissociated state is weakly adsorbed on the surface with C–Mg bond lengths of ∼2.3 A, or, in other words, the CO formed could be easily desorbed. More details on the structural transformation can be found in the ESI.†
 | ||
| Fig. 9 (a) The variation of Ead (in eV) with the AEEF for MgO(111)-Mg-terminated/CO2 on the bridge site. Relaxed structures of configurations at AEEFs of (b) 0.0 V Å−1, (c) 0.5 V Å−1, and (d) 1.0 V Å−1. The dissociated O from CO2 in (c) is colored blue. | ||
We believe that the contribution to the higher adsorption energy is from the O adsorption on the Mg atoms rather than the interaction of the CO with the surface. With AEEF = 0.9 V Å−1, the CO2 molecule goes back to a weaker adsorption state (Fig. 9(a)), where the CO2 is linearly oriented over the MgO surface (Fig. 9(d)). We find here 3 types of structures with significant structural transformation at the AEEFs of 0.5 V Å−1 and 0.9 V Å−1. The details are given in the ESI.†
We also evaluated the adsorption of CO2 on two sites, viz., the bridge and top (Fig. 10 and 11) of the O-terminated MgO(111), i.e., the MgO(111)-O surface. It is clear that CO2 adsorption on the MgO(111)-O surface shows distinct behavior as compared to the Mg-terminated MgO(111) surface.
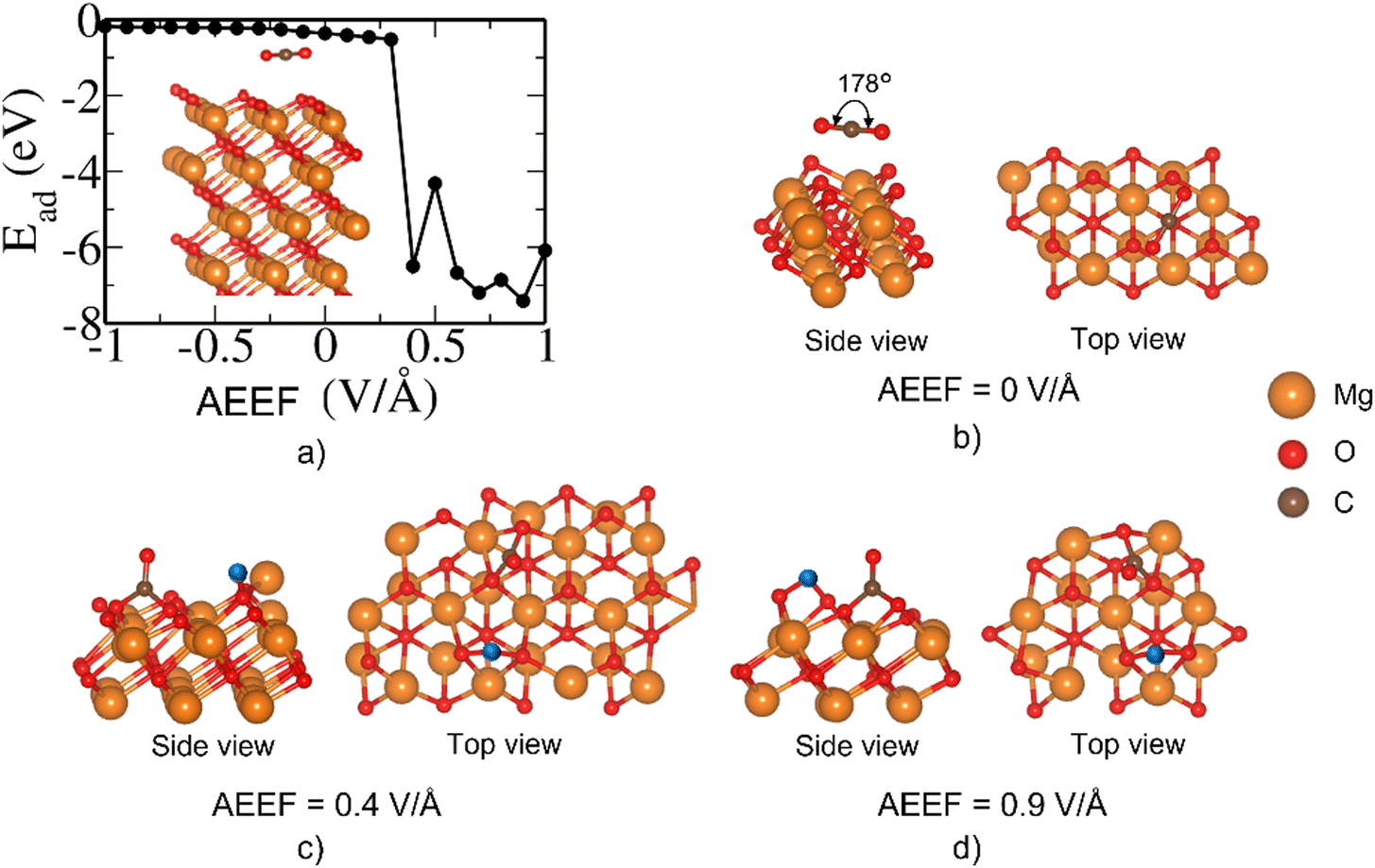 | ||
| Fig. 10 (a) The variation of Ead (in eV) with AEEF for MgO(111)–O-terminated/CO2 on the bridge site. Relaxed structures of configurations at (b) 0.0 V Å−1, (c) 0.4 V Å−1, and (d) at 0.9 V Å−1. The dissociated O from CO2 in (c) is colored blue. | ||
 | ||
| Fig. 11 (a) The variation of Ead (in eV) with AEEF for MgO(111)–O-terminated/CO2 on the O top site. Relaxed structures of configurations at (b) 0.0 V Å−1, (c) 0.2 V Å−1, (d) 0.3 V Å−1, (e) 1.0 V Å−1 and (f) −1.0 V Å−1. The dissociated O from CO2 in (c) is colored blue. | ||
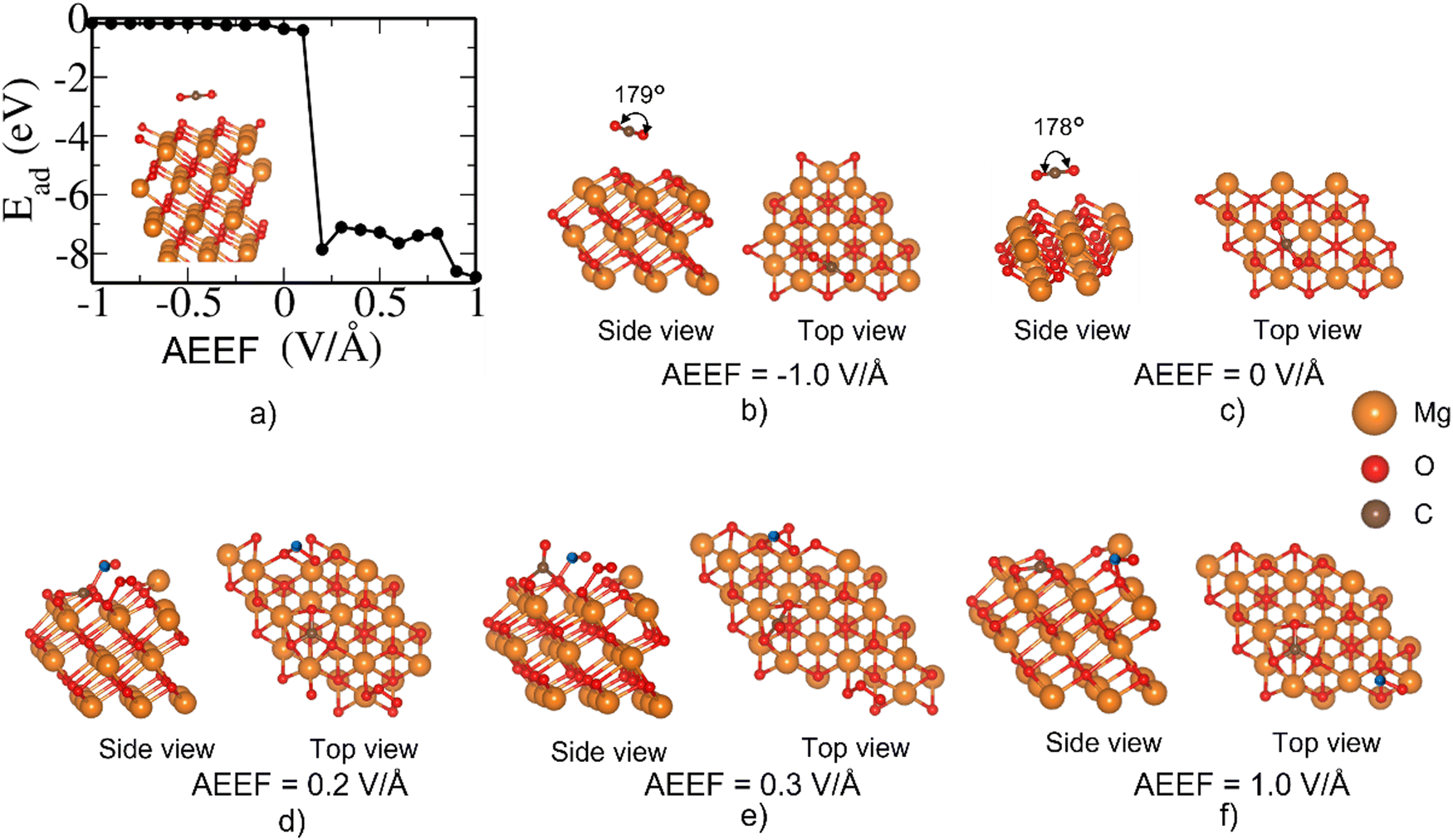
At AEEF from −1 to 0.3 V Å−1, there is a very weak adsorption of CO2 on the bridge site of the MgO(111)–O surface; the CO2 remains relatively flat (O–C–O bond angle of 178°, see Fig. 10(b)). However, at AEEF = 0.4 V Å−1, the CO2 molecule undergoes dissociation into CO + O and the adsorption energy stabilizes below −6 eV as seen in (Fig. 10(a)). CO2 remains dissociated for AEEF > 0.4 V Å−1. This drastic change in the adsorption energy is also a consequence of the large local reconstruction of the surface (see Fig. 10(c) and (d)). The strong stabilization due to CO2 dissociative adsorption is an irreversible process in the CO2 adsorption–desorption behavior.
The dissociation of CO2 into CO + O is also observed when CO2 interacts with the top site of the MgO(111)–O surface (Fig. 11). Here too, CO2 is weakly adsorbed at a low AEEF of ≤0.1 V Å−1. Surprisingly, at AEEF = 0.2 V Å−1, there is a large stabilization of Ead of about −8.0 eV. This arises primarily from the local reconstruction of the MgO(111) surface with the surface O atoms protruding out of the surface, as seen in Fig. 11(d). At all the positive AEEF values, the Ead remains below −7.0 eV, corresponding to the highly distorted structure. Such a large stabilization of the Ead is quite unusual for CO2 adsorption on MgO(111)–O surfaces. The strong dissociative adsorption could imply the irreversible capture of CO2. Furthermore, this phenomenon indicates that AEEF enhances the activity of the polar MgO(111) surfaces for CO2 reduction to CO.
4. Summary
The use of AEEF in CO2 capture on earth-abundant basic oxides such as MgO provides a new opportunity for low-cost and efficient processes for reducing anthropogenic CO2. In the present work, we have investigated CO2 adsorption–desorption behavior on various MgO surfaces under AEEFs between −1 to 1 V Å−1 using first-principles theoretical analysis. Our results show that (a) the CO2 ‘switches’ between different metastable states with varying AEEF, and (b) these metastable states are quite distinct depending on the polarity of the MgO surfaces.In the cases of the non-polar (100) and (110) surfaces of MgO, we mostly observed the formation of carbonate (CO32−) species with strong chemisorbed states at negative AEEFs, which switched to weakly adsorbed states at weakly negative or positive values of AEEF. Interestingly, on the polar MgO(111) surface, the CO2 molecule irreversibly dissociates into CO + O at positive AEEF, indicating the reduction of CO2 to CO. This manifests the role of AEEF for CO2 reduction to CO that usually requires high temperature in a thermochemical process. Therefore, different surfaces of MgO can be utilized to capture CO2, and in some cases, reduce it to CO, through an appropriate choice of AEEF (see Table 1). We have shown that in some cases, the broken inversion symmetry at the adsorption site results in an induced dipole moment, which couples linearly with the AEEF, thereby facilitating electric control of reaction mechanisms and catalytic activity.
| Surface and initial site | Final site (H: hollow; B: bridge; Mg: Mg top; and O: O top) and field (AEEF in V Å−1) | Inaccessible site | ||
|---|---|---|---|---|
| 100 B | (a) O site: (−1 ≤ AEEF ≤ 0.2, bent Mg2CO3) | (b) O site: (0.2 ≤ AEEF ≤ 1.0, comparatively linear CO2) | H | |
| 100 Mg | (a) H site: (AEEF ≤ 0, 0.5 ≤ AEEF ≤ 1.0, linear CO2) | (b) Mg site: (0.1 ≤ AEEF ≤ 0.4, AEEF = −0.2, −0.3, −0.9, −1.0, linear CO2) | (c) O site: (−0.8 ≤ AEEF ≤ −0.5, CO32− species) | (a) Mg and B, (b) H, (c) H |
| 100 O | (a) H site: (AEEF = 0.2, linear CO2) | (b) O site: (0.3 ≤ AEEF ≤ 1.0, linear CO2) | (c) O site: (−1.0 ≤ AEEF ≤ 0.1, bent CO2) | (a) Mg and O, (b) H, (c) H |
| 110 O | (a) O site: (−1.0 ≤ AEEF ≤ 1.0, bent CO2) | H | ||
| 110 Mg | (a) O site: (0.0 ≤ AEEF ≤ 1.0, −0.7 ≤ AEEF ≤ −0.5, bent CO2) | (b) Mg site: (−1.0 ≤ AEEF ≤ −0.8, −0.4 ≤ AEEF ≤ −0.1, linear CO2) | H | |
| 110 B | (a) O site: (0.3 ≤ AEEF ≤ 0.9, CO32− species with stronger adsorption) | (b) O site: (−0.1 ≤ AEEF≤ 0.2, CO32− species with weaker adsorption) | H | |
| 111-Mg-terminated Mg | (a) Mg site: (−0.6 ≤ AEEF ≤ 1.0, linear CO2) | (b) Mg site: (−0.9 ≤ AEEF ≤ 0.7, carbonate compound) | H | |
| 111-Mg-terminated B | (a) B site: (−1.0 ≤ AEEF ≤ 0.4, linear CO2) | (b) Dissociative adsorption (CO + O): 0.5 ≤ AEEF ≤ 0.8, CO on Mg site | H | |
| 111-O-terminated B | (a) B site: (−1.0 ≤ AEEF ≤ 0.3, linear CO2) | (b) Dissociative adsorption (CO + O): 0.4 ≤ AEEF ≤ 1.0 | H | |
| 111-O-terminated O | (a) Linear CO2 (−1.0 ≤ AEEF ≤ 0.1), O site | (b) Dissociative adsorption (CO + O): 0.2 ≤ AEEF ≤ 1.0 | H | |
Our specific results will stimulate experimental work to explore the influence of applied external electric fields on the adsorption–desorption and reduction of CO2 on basic oxides to develop low-energy, low-cost sustainable technologies for CO2 capture and conversion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
UVW acknowledges support from Shell India Markets Private Limited and a JC Bose National Fellowship of SERB-DST, and JNCASR. AKN and SS would like to acknowledge Sander van Bavel and Michiel Boele (Shell Global Solutions International B. V.) for the discussions.References
-
B. Metz, O. Davidson, H. de Coninck, M. Loos and L. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, Cambridge University Press, UK, 2005, p. 431 Search PubMed
.
-
T. A. Boden, G. Marland and R. J. Andres, Global, Regional, and National Fossil-Fuel CO2 Emissions. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, Tenn., USA, 2017 Search PubMed
- H. Herzog and D. Golomb, Encyclopedia Energy, 2004, 1, 277–287 CrossRef
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427–6433 CrossRef CAS PubMed
- L. Raynal, P.-A. Bouillon, A. Gomez and P. Broutin, Chem. Eng. J., 2011, 171, 742–752 CrossRef CAS
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS
- S. E. Zanco, J.-F. Pérez-Calvo, A. Gasós, B. Cordiano, V. Becattini and M. Mazzotti, ACS Eng. Au, 2021, 1, 50–72 CrossRef CAS
- W. Y. Hong, Carbon Capture Sci. Technol., 2022, 3, 100044 CrossRef CAS
- C. Dhoke, S. Cloete, S. Krishnamurthy, H. Seo, I. Luz, M. Soukri, Y. Park, R. Blom, S. Amini and A. Zaabout, Chem. Eng. J., 2020, 380, 122201 CrossRef CAS
- L. Riboldi and O. Bolland, Energy Proc., 2017, 114, 2390–2400 CrossRef CAS
- F. Raganati, R. Chirone and P. Ammendola, Ind. Eng. Chem. Res., 2020, 59, 3593–3605 CrossRef CAS
- C. A. Grande, R. P. L. Ribeiro, E. L. G. Oliveira and A. E. Rodrigues, Energy Proc., 2009, 1, 1219–1225 CrossRef CAS
- C. A. Grande, R. P. P. L. Ribeiro and A. E. Rodrigues, Energy Fuels, 2009, 23, 2797–2803 CrossRef CAS
- Q. Sun, G. Qin, Y. Ma, W. Wang, P. Li, A. Du and Z. Li, Nanoscale, 2017, 9, 19–24 RSC
- F. Wang, P. Li, S. Wei, J. Guo, M. Dan and Y. Zhou, Appl. Surf. Sci., 2018, 445, 568–574 CrossRef CAS
- H. Xiong, H. Zhang and L. Gan, J. Mater. Sci., 2021, 56, 4341–4355 CrossRef CAS
- B. Liao, Z. Zhang, D. Wang, Y. Xu, Y. Wei, W. Bao, K. Lv, J. Wang and Y. Wang, Appl. Surf. Sci., 2022, 572, 151312 CrossRef CAS
- A. A. Khan, I. Ahmad and R. Ahmad, Chem. Phys. Lett., 2020, 742, 137155 CrossRef CAS
- H. Guo, W. Zhang, N. Lu, Z. Zhuo, X. C. Zeng, X. Wu and J. J. Yang, J. Phys. Chem. C, 2015, 119, 6912–6917 CrossRef CAS
- H. Y. Ammar, K. M. Eid and H. M. Badran, J. Phys. Chem. Solids, 2021, 153, 110033 CrossRef CAS
- M. Xiao, B. Zhang, H. Song, Y. Lv and B. Xiao, Solid State Commun., 2021, 338, 114459 CrossRef CAS
- D. Biriukov and Z. Futera, J. Phys. Chem. C, 2021, 125, 7856–7867 CrossRef CAS
- X. Zhang, H. Li, Z. Xia, S. Yu, S. Wang and G. Sun, Phys. Chem. Chem. Phys., 2021, 23, 1584–1589 RSC
- J.-G. Zhou and Q. L. Williams, J. Phys.: Condens. Matter, 2009, 21, 055008 CrossRef PubMed
- F. Che, J. T. Gray, S. Ha and J. S. McEwen, ACS Catal., 2017, 7, 551–562 CrossRef CAS
- F. Che, J. T. Gray, S. Ha and J. S. McEwen, ACS Catal., 2017, 7, 6957–6968 CrossRef CAS
- F. Che, J. T. Gray, S. Ha and J. S. McEwen, J. Catal., 2015, 332, 187–200 CrossRef CAS
- S. Shaik, D. Danovich, J. Joy, Z. Wang and T. Stuyver, J. Am. Chem. Soc., 2020, 142, 12551–12562 CrossRef CAS PubMed
- S. Shaik, R. Ramanan, D. Danovich and D. Mandal, Chem. Soc. Rev., 2018, 47, 5125–5145 RSC
- G. Chuah, N. Kruse, W. Schmidt, J. Block and G. Abend, J. Catal., 1989, 119, 342–353 CrossRef CAS
- L. Keller, T. Lohaus, L. Abduly, G. Hadler and M. Wessling, Chem. Eng. J., 2019, 371, 107–117 CrossRef CAS
- K. Sugiura, S. Ogo, K. Iwasaki, T. Yabe and Y. Sekine, Sci. Rep., 2016, 6, 25154 CrossRef CAS PubMed
- M. Shetty, M. A. Ardagh, Y. Pang, O. A. Abdelrahman and P. J. Dauenhauer, ACS Catal., 2020, 10, 12867–12880 CrossRef CAS
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, J. Mater. Chem. A, 2019, 7, 20103–20120 RSC
- M. A. Manae, L. Dheer, S. Rai, S. Shetty and U. V. Waghmare, Phys. Chem. Chem. Phys., 2022, 24, 1415–1423 RSC
- S. Arndt, G. Laugel, S. Levchenko, R. Horn, M. Baerns, M. Scheffler, R. Schlögl and R. Schomäcker, Catal. Rev., 2011, 53, 424–514 CrossRef CAS
- J. S. J. Hargreaves, G. J. Hutchings and R. W. Joyner, Catal. Today, 1990, 6, 481–488 CrossRef CAS
- H.-X. Fan, T.-Y. Cui, A. Rajendran, Q. Yang, J. Feng, X.-P. Yue and W.-Y. Li, Catal. Today, 2020, 356, 535–543 CrossRef CAS
- H. Y. Kim, H. M. Lee and J.-N. Park, J. Phys. Chem. C, 2010, 114, 7128–7131 CrossRef CAS
- M. B. Jensen, L. G. M. Pettersson, O. Swang and U. Olsbye, J. Phys. Chem. B, 2005, 109, 16774–16781 CrossRef CAS PubMed
- S. Jiang, Y. Lu, S. Wang, Y. Zhao and X. Ma, Appl. Surf. Sci., 2017, 416, 59–68 CrossRef CAS
- D. Cornu, H. Guesmi, J.-M. Krafft and H. Lauron-Pernot, J. Phys. Chem. C, 2012, 116, 6645–6654 CrossRef CAS
- G. A. Mutch, S. Shulda, A. J. McCue, M. J. Menart, C. V. Ciobanu, C. Ngo, J. A. Anderson, R. M. Richards and D. Vega-Maza, J. Am. Chem. Soc., 2018, 140, 4736–4742 CrossRef CAS PubMed
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
- W. K. Fan and M. J. Tahir, Environ. Chem. Eng., 2021, 9, 105460 CrossRef CAS
- B. Liang, H. Duan, T. Sun, J. Ma, X. Liu, J. Xu, X. Su, Y. Huang and T. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 925–932 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- F. Che, J. T. Gray, S. Ha, N. Kruse, S. L. Scott and J. S. McEwen, ACS Catal., 2018, 8, 5153–5174 CrossRef CAS
- H. J. Kreuzer, Surf. Sci., 1991, 246, 336–347 CrossRef CAS
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed
- C. Wolf, https://christoph-wolf.at/tag/dipfield/, accessed 21 August 2023.
- A. Kakekhani and S. Ismail-Beigi, ACS Catal., 2015, 5, 4537–4545 CrossRef CAS
- M. F. Sarott, M. D. Rossell, M. Fiebig and M. Trassin, Nat. Commun., 2022, 13, 3159 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04588a |
| This journal is © the Owner Societies 2024 |