
Early bird or night owl? Controlling the ultrafast photodynamics of triphenylamine substituted 2,2′:6′,2′′-terpyridine†
Anna Maria
Maroń
 *a,
Oliviero
Cannelli
*bc,
Etienne Christophe
Socie
b,
Piotr
Lodowski
a,
Malte
Oppermann
bd,
Barbara
Machura
a and
Majed
Chergui
be
*a,
Oliviero
Cannelli
*bc,
Etienne Christophe
Socie
b,
Piotr
Lodowski
a,
Malte
Oppermann
bd,
Barbara
Machura
a and
Majed
Chergui
be
aInstitute of Chemistry, University of Silesia, Szkolna 9, 40-007 Katowice, Poland. E-mail: anna.maron@us.edu.pl
bLaboratory of Ultrafast Spectroscopy (LSU) and Lausanne Centre for Ultrafast Science (LACUS), École Polytechnique Fédérale de Lausanne, ISIC CH H1 625, Station 6, CH-1015, Lausanne, Switzerland
cCenter for Free-Electron Laser Science, DESY, Notkestraße 85, 22607 Hamburg, Germany. E-mail: oliviero.cannelli@cfel.de
dDepartment of Chemistry, University of Basel, Klingelbergstrasse 80, 4056 Basel, Switzerland
eElettra – Sincrotrone Trieste S.C.p.A., S.S.14 Km.163, 5 in Area Science Park, I – 34149, Trieste, Italy
First published on 24th January 2024
Abstract
Controlling the ultrafast photodynamics of metal-free organic molecules has great potential for technological applications. In this work, we use solvent polarity and viscosity as “external knobs” to govern the photodynamics of an electron-donating derivative of 2,2′:6′,2′′-terpyridine (terpy), namely 4′-(4-(di(4-tert-butylphenyl)amine)phenyl)-2,2′:6′,2′′-terpyridine (tBuTPAterpy). We combine femtosecond fluorescence upconversion (FlUC), transient absorption (TA) and quantum mechanical calculations to provide a comprehensive description of the tBuTPAterpy's photodynamics. Our results demonstrate that, by changing the solvent, the time scale of light-induced conformational changes of the system can be tuned over two orders of magnitude, controlling the tBuTPAterpy fluorescence spectral region and yield. As a result, depending on the local environment, tBuTPAterpy can act either as an “early bird” or a “night owl”, with a tunability that makes it a promising candidate for metal-free sensors.
Introduction
Intramolecular charge transfer (ICT) is a fundamental phenomenon responsible for a large number of chemical and biological processes in organic and inorganic systems.1 Several investigations were devoted to ICT, which is exploited in applications for molecular electronics, solar energy-conversion, quantum optics and chemical sensors.2–7 In addition, the fundamental understanding of ICT has fascinated scientists over the past few decades.8–20 In ICT systems, the early photodynamics upon optical transitions to the lowest electronic states is associated with the population transfer from the locally-excited (LE) Franck–Condon (FC) region to the relaxed geometry of the ICT state.21 These conformational changes include twisting,9,22–27 planarization,23,24,27–30 bending,31 rehybridization of molecular fragments within the ICT moiety,13,32 and/or intersystem crossing (ISC) to the triplet manifold.33–37 Such structural and electronic changes are strongly influenced by environment effects. As a consequence, solvent parameters like polarity and viscosity can be used as experimental knobs for the external control of the ICT photodynamics.3 A rational design of ICT devices requires characterizing their ultrafast photodynamics and quantifying the impact of solvent parameters on their photoresponse.21Herein, we focus on the donor–acceptor molecule 4′-(4-(di(4-tert-butylphenyl)amine)phenyl)-2,2′:6′,2′′-terpyridine (tBuTPAterpy) shown in Fig. 1. Derivatives of 2,2′:6′,2′′-terpyridine (terpy) are among the most important organic ligands in coordination chemistry.38–46 Several spectroscopic investigations described the photophysical properties of transition metal complexes with terpy ligands,43,46–51 showing that electron-donating substituents lengthen the photoluminescence lifetime of the compounds. This is generally due to an energy barrier increase between the emissive metal-to-ligand charge transfer triplet (3MLCT) state and the metal-centred triplet (3MC) state,52–55 or to the equilibration between the 3MLCT state and the intraligand triplet (3ILCT/3IL) state.47,51,56,57 Despite the widespread use of terpys and their derivatives, the photophysics of the isolated ligands was to a great extent overlooked. Only a limited number of studies described the ultrafast photodynamics of these organic compounds,58,59 underestimating the great potential of these metal-free systems for various applications.
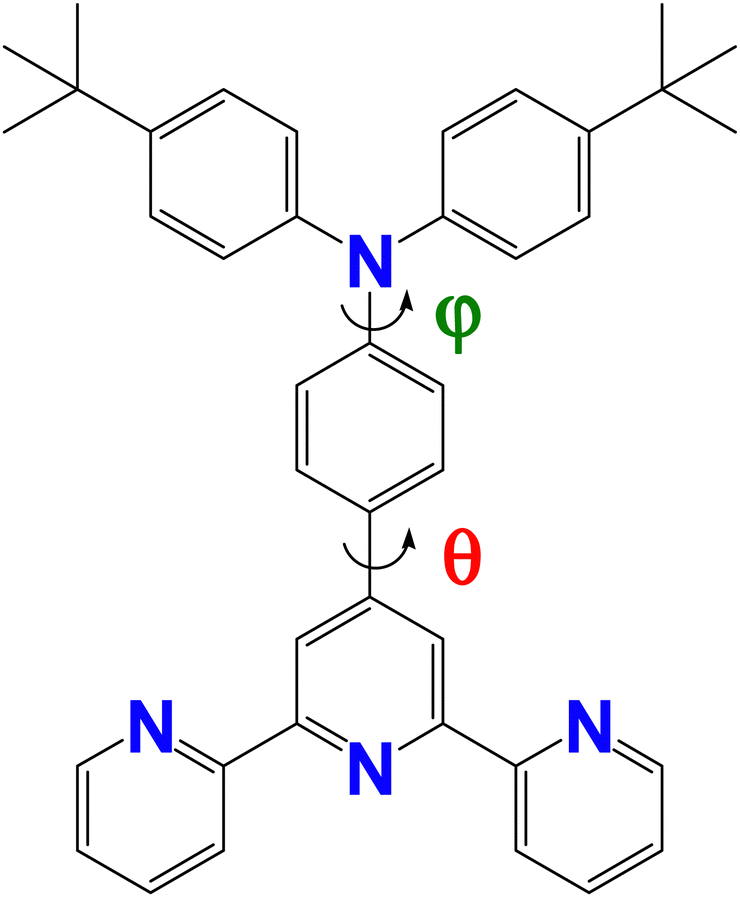 | ||
| Fig. 1 Structure of 4′-(4-(di(4-tert-butylphenyl)amine)phenyl)-2,2′:6′,2′′-terpyridine (tBuTPAterpy). The dihedral angles θ and φ correspond to the torsions of the terpyridine and phenyl units, and of the phenyl and amide units, respectively. | ||
In a recent study,60 we investigated the steady-state properties of tBuTPAterpy, showing that it is highly sensitive to solvent polarity, viscosity and pH. These features, together with its high fluorescence quantum yield (ΦPL = 0.48–0.83 and τPL = 1.53–6.56 ns), make it a potential candidate as a metal-free environment sensor. Our previous joint experimental and computational work provided a picture of tBuTPAterpy's energy diagram, showing that the absorption process is dominated by an ICT S0 → S1 transition, while the near-lying S0 → S2 transition has an oscillator strength several orders of magnitude lower. However, fully exploiting the applicative potentials of this system requires a better understanding of how the environment influences its early photodynamics, which governs its final emission properties. In this work, we performed a detailed study of the tBuTPAterpy ultrafast photoresponse using femtosecond fluorescence upconversion (FlUC), femtosecond transient absorption (TA) spectroscopy, and quantum chemical calculations, rationalizing solvent polarity and viscosity effects on the system's photophysics. In chloroform, we show that at least three conformers participate in the deactivation pathways of the molecule. Increasing the solvent polarity leads to a barrier-less motion that activates a conformational change to the lowest energy excited state on sub-ps time scales, while higher solvent viscosities slow down the process up to hundreds of ps. Finally, in the absence of strong polarity and viscosity effects, the ISC to the triplet manifold becomes the dominant process.
Experimental and computations
Materials
tBuTPAterpy was obtained according to the procedure described in ref. 51. Analytical data for tBuTPAterpy (1H and 13C NMR spectroscopy and elemental analysis) are reported in the ESI,† and are in good agreement with those discussed in ref. 51. All solvents were of spectroscopic grade (chloroform, n-hexane, and glyceryl triacetate) or HPLC grade (acetonitrile, water impurities <0.02%) and were commercially available (Merck, Karl–Fischer).Measurement techniques, sample preparation and method of analysis
FlUC and TA spectroscopy studies were conducted using the setups described in ref. 61 and 62, and additional details are reported in the ESI.† Quartz cuvettes of 2 mm (TA) and 1 mm (FlUC) thicknesses were used for sample solutions having concentrations of 125 μM and 250 μM, respectively. The stability of tBuTPAterpy in chloroform, acetonitrile, glycerol triacetate and n-hexane was spectrophotometrically monitored over 48 hours prior to the analysis (Fig. S1, ESI†). Photostability experiments and fluence dependence characterizations are reported in Fig. S2 and S3 (ESI†), respectively.The FlUC setup consists of a Ti:sapphire laser (Libra, Coherent, 800 nm central wavelength, 45 fs pulse duration, and 1 kHz repetition rate) and a FlUC spectrofluorimeter (LIOP- TEC). 400 nm excitation pulses were produced by doubling the 800 nm light with a I type BBO crystal (d = 0.5 mm). The sample solution was excited in the magic angle geometry (54.7°). The type II sum-frequency generation was obtained from the horizontally polarized gate beam and the vertically polarized fluorescence. The upconverted light was collected using an unfolded Czerny–Turner spectrograph and detected using a CCD camera (Newton 920, Andor). The instrument response function (IRF) of the measurements was estimated from the full widths at half maximum (FWHM) of the cross-correlation signal of the pump and probe pulses, corresponding to 0.140 ps and 0.150 ps, respectively for CHCl3 and acetonitrile solutions.
The TA spectra were measured using a Helios setup (Ultrafast Systems) equipped with a femtosecond Ti:sapphire regenerative amplified laser system (Astrella, Coherent, 800 nm central wavelength, 1 kHz repetition rate). A CaF2 crystal and an optical parametric amplifier (Light Conversion, TOPAS prime) were used for the generation of the white light probe and the 405 nm pump beam, respectively. A CCD detector was used to collect the TA signal in transmission geometry. The IRF was measured in pure solvents (FWHM of about 160 fs, 150 fs, 150 fs, 150 fs were measured in CHCl3, glyceryl triacetate, acetonitrile, n-hexane, respectively).
The Surface Xplorer (Ultrafast Systems) software was employed for the data processing of the TA data sets. The analysis of FlUC and TA data sets was performed using the OptimusTM software.63 A detailed description of the data analysis procedure is given in the ESI.†
Computational details
All calculations were performed at the density-functional theory (DFT)64 and time-dependent density-functional theory (TD-DFT)65 level, using the hybrid Perdew–Burke–Ernzerhof (PBE0) functional66,67 and the def2-SVP basis set.68 Solvation effects on the system's geometry and electronic structure were included by using a polarizable continuum model (PCM).69 The geometry of the ground state (S0) was fully optimized without any structural parameter constraints. The geometries of the excited states were determined using TD-DFT without (S1) and with (S2) structural constraints on the dihedral terpy-phenil angle (θ).Results and discussion
FlUC and TA of tBuTPAterpy were measured in both CHCl3 (ε = 4.81 and η = 0.51 cP) and acetonitrile (ε = 37.5 and η = 0.34 cP), while TA data were further collected varying the solvent polarity and viscosity, respectively, measuring the system in n-hexane (ε = 1.88, η = 0.28 cP), and glyceryl triacetate (triacetine, ε = 7.01, η = 25 cP).70–72 The results are summarized in Table 1, in Fig. 2 (FlUC in CHCl3) and in Fig. 3–6 (TA in all solvents), and are discussed in more detail hereafter. Additional information is reported in Fig. S4–S14 in the ESI.†| FlUC | TA | ||||||
|---|---|---|---|---|---|---|---|
| CHCl3 | CH3CN | CHCl3 | CH3CN | n-Hexane | Triacetine | ||
| τ 1 | 0.31 | 0.23 | τ 1 | 0.54 | 0.20 | — | 0.67 |
| τ 2 | 3.7 | 1.2 | τ 2 | 3.1 | 1.4 | — | 10 |
| τ 3 | 9.8 | — | τ 3 | 8.9 | — | — | 110 |
| τ 4 | — | — | τ 4 | 210 | 110 | 72 | 560 |
| τ 5 | Infinite | Infinite | τ 5 | 4400 | 5800 | 1600 | 4800 |
| τ 6 | — | — | τ 6 | — | — | Infinite | — |
 | ||
| Fig. 2 FlUC data for tBuTPAterpy in chloroform solution (λexc = 400 nm): 2D time-wavenumber plot (A); FlUC spectra at selected time delays (B); and TRANES (C). The time axis break at 1 ps separates linear and logarithmic ranges. | ||
 | ||
| Fig. 3 TA 2D maps (panel A), TA spectra at selected time delays (panel B), selected time traces with their fits (panel C) for tBuTPAterpy in CHCl3 (λexc = 405 nm). The time axis break at 1 ps separates linear and logarithmic ranges. The arrow in the color bar of panel A indicates the 0 OD value of the transient absorption. | ||
 | ||
Fig. 4 TA 2D maps (panel A), TA spectra at selected time delays (panel B), selected time traces with their fits (panel C) for tBuTPAterpy in acetonitrile (λexc = 405 nm). The time axis break at 1 ps separates linear and logarithmic ranges, whereas the axis break at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 316–24390 cm−1 (395–410 nm) corresponds to the photon energy of the Raman scattering line of the acetonitrile solvent and was excluded from the analysis. The arrow in the color bar of panel A indicates the 0 OD value of the transient absorption. 316–24390 cm−1 (395–410 nm) corresponds to the photon energy of the Raman scattering line of the acetonitrile solvent and was excluded from the analysis. The arrow in the color bar of panel A indicates the 0 OD value of the transient absorption. | ||
 | ||
| Fig. 5 TA 2D maps (panel A), TA spectra at selected time delays (panel B), selected time traces with their fits (panel C) for tBuTPAterpy in n-hexane (λexc = 405 nm). The time axis break at 1 ps separates linear and logarithmic ranges. The arrow in the color bar of panel A indicates the 0 OD value of the transient absorption. | ||
 | ||
| Fig. 6 TA 2D maps (panel A), TA spectra at selected time delays (panel B), selected time traces with their fits (panel C) for tBuTPAterpy in glyceryl acetate (λexc = 405 nm). The time axis break at 1 ps separates linear and logarithmic ranges. The arrow in the color bar of panel A indicates the 0 OD value of the transient absorption. | ||
Femtosecond studies in chloroform.
The FlUC results of tBuTPAterpy in CHCl3 are shown in Fig. 2 (see also Fig. S4 and S5 in the ESI†). Within the first 10 ps of the dynamics, the fluorescence signal grows and shifts towards lower energies and decays at longer time scales (Fig. 2A and B). The emission band centred at ∼21740 cm−1 (460 nm), which promptly appears after the pump pulse, largely overlaps with the steady-state emission spectrum of tBuTPAterpy at 77 K (see Fig. S4, panel D, ESI†). Instead, the band after 1000 ps time delay, which is centred at ∼20410 cm−1 (490 nm), matches well with the steady-state emission spectrum at room temperature (Fig. S4, panel D, ESI†). Time-resolved area normalized emission spectra (TRANES, Fig. 2C) show a quasi-isoemissive point at ∼21000 cm−1 (475 nm) for time delays longer than 10 ps, pointing to the presence of two transient emitting states.20,73–78
To obtain further insights into the relaxation dynamics of tBuTPAterpy, a global lifetime analysis of the FlUC data was performed, with the best fit achieved using a four-component exponential decay model. Three-component fits were attempted without reaching a satisfactory agreement with the experiment, whereas adding a fifth component to the four-component fit did not provide significant improvements, as shown in Fig. S15 in the ESI.† The two shortest components, τ1 and τ2, respectively of 0.31 ps and 3.7 ps, agree well with the inertial (0.285 ps) and diffusional (4.15 ps) solvation processes in chloroform for coumarin153,79 while the longer time constants τ3 and τ4 are ascribed to solute kinetics. These results are complemented and confirmed by the TA measurements presented in the following.
Fig. 3A shows the TA signal of tBuTPAterpy in CHCl3. On sub-ps time scales, it consists of two negative and two positive alternating regions. The negative features correspond to a ground-state bleaching (GSB) in the energy range 28600–25000 cm−1 (350–400 nm) and to a stimulated emission band (SE1) lying between 22700 cm−1 (441 nm) and 20800 cm−1 (481 nm). The latter overlaps well with the fluorescence spectra at short time scales of Fig. 2. The two positive bands are related to excited state absorption (ESA) processes and lie at 25000–22700 cm−1 (400–441 nm, ESA1) and 20800–16700 cm−1 (481–599 nm, ESA2). Within 1 ps after excitation, a third band (ESA3) appears between 16700 cm−1 (599 nm) and 14900 cm−1 (671 nm). The SE1 band grows during the first 2 ps and red-shifts into a new band (SE2) at longer time delays. The presence of an isoemissive point at 21050 cm−1 (475 nm) supports the assignment of these emission bands to two different transient species. The ESA2 band grows within the IRF and starts decaying on a sub-ps time scale. As reported for similar π-conjugate structures,58,59 we assign it to S1 → Sn transitions within the terpy moiety. The narrow and intense ESA1 band is typical of polypyridine anion radicals80 in free organic ligands and their metal coordination compounds.81–84 Instead, the ESA3 band is ascribed to the cation radical species of the donor unit.85 The coexistence of ESA1 and ESA3 transient signals respectively corresponding to anion and cation radicals is consistent with a photoinduced ICT process. The experimental results are satisfactorily reproduced using a five-component global target analysis (see Table 1, and also Fig. S6 and S17 in the ESI† for a complete set of time traces and for the map of residuals). The obtained time constants are in good agreement with the FlUC analysis, with the notable exception for the presence of an additional time constant of 210 ps in the TA fit (Table 1). Based on the analysis of the TA spectra at different time delays, we conclude the following:
(i) The GSB and all ESA features start growing within the IRF of the experiment;
(ii) The first component with a lifetime of τ1 = 0.54 ps is related to the formation of SE1 and a partial growth (decay) of ESA1 (ESA2). These signals reflect the formation of an S1 excited state having a moderate ICT character and populating the lowest unoccupied molecular orbital (LUMO) centred on the terpy moiety.60 At these time scales, the SE1 band and the FlUC signal are blue-shifted with respect to the room temperature emission spectrum. As discussed in ref. 60, this is typical of the tBuTPAterpy system in the absence of structural changes with respect to the FC geometry;
(iii) The time constant τ2 (3.1 ps) is associated with a progressive red shift and intensity drop of the SE1 and ESA2 bands, and a further growth of the ESA1 band;
(iv) During τ3 (8.9 ps), the ESA1 band keeps growing, whereas the SE1 band red shifts towards the newly formed SE2 band, giving rise to the isoemissive point at 21050 cm−1 (475 nm). During τ2 and τ3, the system undergoes a relaxation process towards a molecular configuration having an emission spectrum compatible with room temperature fluorescence (see Fig. S4, panel D, ESI†). This observation, together with the presence of the ESA1 and ESA3 bands, is consistent with the formation of an ICT state upon conformational changes of the molecular moieties;60
(v) The τ4 time component (210 ps) is mostly associated with the decay of the ESA1 and SE2 features;
(vi) The τ5 time constant of 4.4 ns agrees with the photoluminescence decay times measured in Time-correlated Single Photon Counting (TCSPC, 4.3 ns)60 and therefore corresponds to the radiative decay of the final excited state.
Fig. S5 and S6 in the ESI† show the comparison of absorption, emission and excitation steady-state spectra of tBuTPAterpy in CHCl3 with FlUC and TA energy traces at multiple time delays, and TA time traces with their fits, respectively.
Femtosecond studies in acetonitrile and n-hexane.
The comparison of TA experiments in solvents of higher (acetonitrile) and lower (n-hexane) polarity with respect to the moderately polar CHCl3 provides a better understanding of how the solvent polarity affects the photodynamics of the push–pull tBuTPAterpy molecule (Fig. 4 and 5).In acetonitrile (Fig. 4), the TA spectral shapes resemble those in chloroform, showing only a minor hypsochromic shift of the ESA1 band and a bathochromic shift of the SE2 band. However, differently from chloroform, the best fit of the TA global target analysis was achieved with a four-component model (see Fig. S8 and S18, ESI†). In this case, the spectral changes from the SE1 to the SE2 band occur almost immediately after photoexcitation, giving rise to an isoemissive point at 20040 cm−1 (499 nm) between the traces at 0.2 and 0.5 ps (Fig. 4B). The τ1 and τ2 components approximately agree with the time constants previously reported for the solvation dynamics of coumarin153 in acetonitrile.79 Thus, if a population transfer occurs between two different emitting states, it overlaps with the sub-ps inertial solvation process, explaining the absence of a fifth time component in the fit with respect to the chloroform data set. This observation agrees with the TRANES results of the FlUC measurements in acetonitrile, where no isoemissive point is observed (Fig. S9, ESI†) and where the transient spectral shifts are typical of solvation dynamics time scales. Similar to chloroform, in acetonitrile the TA fit of tBuTPAterpy requires one additional component (four) with respect to the FlUC data (three), as reported in Table 1, and the longest time component matches well the decay time of the TCSPC measurements.60
In the non-polar n-hexane, the transient signal intensity is significantly lower than that in the polar solvents under similar pump fluences. This is a consequence of the hypsochromic shift of the steady-state tBuTPAterpy absorption onset, which leads to a lower sample absorbance at 24690 cm−1 (405 nm). Compared to chloroform, both ESA1 and ESA2 bands are bathochromically shifted by about 500–1300 cm−1, while a hypsochromic shift of ∼950 cm−1 is observed in the case of ESA3 band. The SE1 band at 24570 cm−1 (407 nm)60 partially overlaps with the pump signal and is buried under the stronger ESA1 band, resulting in a positive feature of the TA map (see Fig. S11 for the comparison of TA spectra with the steady-state spectra in n-hexane, ESI†). The global lifetime analysis of the n-hexane data was performed using three components (see Table 1, Fig. 5C, and Fig. S12 in the ESI,† which show the details of the global analysis in n-hexane). Both the first and second time constants correspond to a decay of the positive signal, with the latter closely resembling the PL decay time in n-hexane (1.53 ns).60 By comparing the results in n-hexane and CHCl3, a remarkable difference is the presence of a persisting signal up to the longest measured delays in n-hexane. This “slow” component is characteristic of the ESA1 and ESA3 bands only, which have maxima respectively at 23810 cm−1 (420 nm) and <15400 cm−1 (>649 nm). Instead, the ESA2 band disappears at about 5000 ps time delay. In the ns time delay range, the transients agree with the triplet–triplet ESA bands of the TA spectra of TPA-substituted benzimidazole derivatives, as reported in ref. 37.
Femtosecond studies on glyceryl acetate
The TA 2D map of tBuTPAterpy in the viscous triacetine solvent is reported in Fig. 6 (panel A). The spectral profiles resemble the TA spectra in CHCl3: the ESA bands are in the same energy range, and the SE1 and SE2 bands undergo similar spectral changes. The position of the SE2 band agrees with the steady-state emission spectrum of tBuTPAterpy in the triacetine solvent (see Fig. S13 in the ESI†), and the 2D map is satisfactorily fitted using a five-component model. However, with respect to CHCl3, the system's photodynamics significantly slows down upon the increase of solvent viscosity (see Table 1 and Fig. S14, S20 in the ESI† for details about the global analysis). The highest increase concerns τ3, which is one order of magnitude larger in triacetine than in chloroform, suggesting that this time constant corresponds to a conformational change of the solute. The isoemissive point at 20880 cm−1 (479 nm) further suggests the presence of two emissive states. Instead, the τ5 component matches the decay time observed in the TCSPC measurement.60 Minor differences between the two solvents are also present, as the absence of an initial blue shift of the ESA1 band and a fast growth of the SE1 band within τ1 (not within τ2, as it happens in chloroform).
Excited state kinetics of tBuTPAterpy
Combining the experimental results reported in the previous sections with DFT calculations, we propose in Fig. 7 the energy diagram describing the tBuTPAterpy's photodynamics in CHCl3. In the ground state, tBuTPAterpy is characterized by an equilibrium structure where its building blocks lie in different planes forming dihedral angles of θ = 26° (terpy-phenyl angle) and ϕ = 33° (phenyl-amide angle).60 The excitation at 405 nm initially populates the S1 excited state, which in the FC geometry has a moderate intramolecular charge transfer character (ICT1).60 Our experimental evidences show that the inertial and diffusive solvation processes, which are responsible for the first two time components of the photodynamics, lead to a Stokes shift of the emissive band, consistently with an energy stabilization of the ICT1 state. The third relaxation process, which is strongly affected by the solvent viscosity (see triacetine results), is ascribed to conformational changes of the system towards geometries at lower energy with respect to the FC region.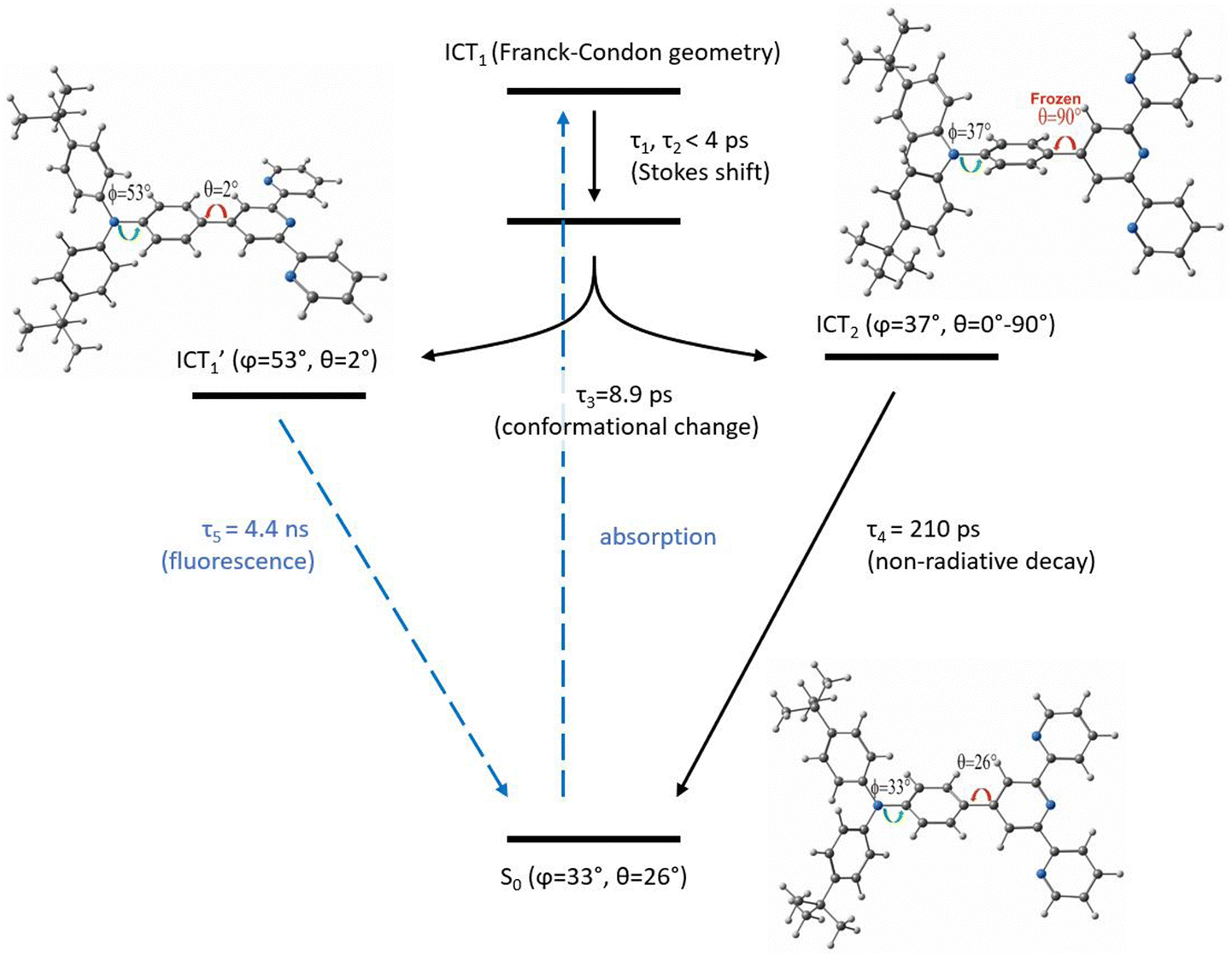 | ||
Fig. 7 Schematic of the excited state dynamics of tBuTPAterpy in CHCl3. The dihedral angles θ and φ correspond to the rotations of the bonds connecting the terpy-phenyl and phenyl-amide, respectively. The S0 and 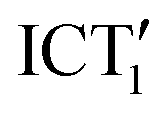 structures correspond to the optimized geometries of the ground and first excited state, respectively. The ICT2 structure was obtained by optimising the geometry of the excited state under the constraint of θ = 90°. structures correspond to the optimized geometries of the ground and first excited state, respectively. The ICT2 structure was obtained by optimising the geometry of the excited state under the constraint of θ = 90°. | ||
We elucidated these structural modifications by performing excited-state optimisations at the TD-DFT level. The calculations highlight that the torsions along the θ and φ dihedral angles have high influence on the system's energy.60 In particular, the energy minimum of the S1 state  is reached for φ and θ approaching 53° and 2°, respectively (Fig. 7, left structure). This is due to the fact that the planarization of the θ angle stabilizes the LUMO, which was shown to span over the terpy and central phenyl units.60 For the same reason, increasing θ destabilizes the first excited state, while it marginally affects the second non-emissive excited state (ICT2), whose LUMO+1 occupied orbital was reported to be solely localised on the terpy substituent.60 As a consequence, the torsion along θ causes a change in the relative energy of the first bright and second dark excited states, with an energy barrier of only few kcal mol−1 between them.60 This energy separation is of the same order of magnitude of the energy dissipated by the system in the relaxation dynamics, as observed through the red shift of the tBuTPAterpy transient emission spectra within the first 10 ps. Therefore, it is reasonable to assume that during the third relaxation process (τ3) two different excited-state conformers,
is reached for φ and θ approaching 53° and 2°, respectively (Fig. 7, left structure). This is due to the fact that the planarization of the θ angle stabilizes the LUMO, which was shown to span over the terpy and central phenyl units.60 For the same reason, increasing θ destabilizes the first excited state, while it marginally affects the second non-emissive excited state (ICT2), whose LUMO+1 occupied orbital was reported to be solely localised on the terpy substituent.60 As a consequence, the torsion along θ causes a change in the relative energy of the first bright and second dark excited states, with an energy barrier of only few kcal mol−1 between them.60 This energy separation is of the same order of magnitude of the energy dissipated by the system in the relaxation dynamics, as observed through the red shift of the tBuTPAterpy transient emission spectra within the first 10 ps. Therefore, it is reasonable to assume that during the third relaxation process (τ3) two different excited-state conformers,  and ICT2, are populated (Fig. 7). Considering that the potential energy surface of the ICT2 along the θ coordinate is approximately flat,60 the energy minimum of this state is mostly defined by the torsion along the φ dihedral angle. Thus, even though unconstrained structural optimisations of the ICT2 state cannot be performed at the TD-DFT level, we could estimate its minimum-energy geometry along the φ coordinate in the case of θ frozen at 90° (Fig. 7, right structure), a value at which the first two excited states change in relative order.60
and ICT2, are populated (Fig. 7). Considering that the potential energy surface of the ICT2 along the θ coordinate is approximately flat,60 the energy minimum of this state is mostly defined by the torsion along the φ dihedral angle. Thus, even though unconstrained structural optimisations of the ICT2 state cannot be performed at the TD-DFT level, we could estimate its minimum-energy geometry along the φ coordinate in the case of θ frozen at 90° (Fig. 7, right structure), a value at which the first two excited states change in relative order.60
A population transfer towards a new conformer is consistent with the presence of an isoemissive point in both TRANES and SE bands. We ascribe the  state to the long living emissive state of tBuTPAterpy, as confirmed by the overlap of the late FlUC transient emission spectrum and the room temperature fluorescence signal (see Fig. S4, ESI†). Furthermore, the
state to the long living emissive state of tBuTPAterpy, as confirmed by the overlap of the late FlUC transient emission spectrum and the room temperature fluorescence signal (see Fig. S4, ESI†). Furthermore, the  emission decays in few ns, in agreement with the time scales observed in the TCSPC experiment.60 The hypothesis of a small fraction of photoexcited tBuTPAterpy in a non-radiative state (ICT2), which is populated alternatively to the
emission decays in few ns, in agreement with the time scales observed in the TCSPC experiment.60 The hypothesis of a small fraction of photoexcited tBuTPAterpy in a non-radiative state (ICT2), which is populated alternatively to the  conformer during τ3, is supported by the presence of the second-to-last time constant of the TA experiments. We interpret this 210 ps (560 ps) decay component in CHCl3 (triacetine) as a non-radiative decay from the ICT2 to the ground state, compatibly with its absence in the FlUC experiment.
conformer during τ3, is supported by the presence of the second-to-last time constant of the TA experiments. We interpret this 210 ps (560 ps) decay component in CHCl3 (triacetine) as a non-radiative decay from the ICT2 to the ground state, compatibly with its absence in the FlUC experiment.
Changes in the solvent lead to modifications of the early photodynamics of tBuTPAterpy. For instance, the rate of the conformational change of the system is modulated by changing the viscosity while keeping the same polarity. Hence, in the viscous triacetine we observe a lengthening of the time components ascribed to population transfers between ICT1 and the excited-state minima  and ICT2, and to the relaxation of the ICT2 to the ground state. More polar environments facilitate the formation of the
and ICT2, and to the relaxation of the ICT2 to the ground state. More polar environments facilitate the formation of the  state, stabilizing it and lowering the conformational energy barrier with the initially populated ICT1 state, similarly to what was reported for push–pull pyridium salts by Carlotti et al.86 Thus, in the highly polar acetonitrile, the tBuTPAterpy photodynamics is solvation-controlled, while in the moderately polar CHCl3 it is controlled by the slower conformational changes.
state, stabilizing it and lowering the conformational energy barrier with the initially populated ICT1 state, similarly to what was reported for push–pull pyridium salts by Carlotti et al.86 Thus, in the highly polar acetonitrile, the tBuTPAterpy photodynamics is solvation-controlled, while in the moderately polar CHCl3 it is controlled by the slower conformational changes.
Finally, since a pronounced solvent polarity is required for the stabilization of the CT states, the apolar n-hexane favours a different relaxation pathway over the population transfer towards new conformers, namely an ISC process to the triplet manifold. The ICT1 (S1) → LE (Tn) process, which is allowed by the El-Sayed rule, was previously observed in apolar solvents in the case of 2,6-bis(diphenylamino)anthraquinone.36 In the specific case of the push–pull tBuTPAterpy system, the population of the initially formed ICT1 (S1) singlet state in the FC geometry is directly transferred to the LE (T1) triplet state localized on the 4-(di(4-tert-butylphenyl)amine)phenyl moiety before the structural change to the  conformer occurs, a process which is possibly slowed down by a high conformational energy barrier.
conformer occurs, a process which is possibly slowed down by a high conformational energy barrier.
Conclusions
In this work, we characterized the ultrafast photodynamics of the push–pull molecule tBuTPAterpy, rationalizing the solvent polarity- and viscosity-dependence of its steady-state properties that we analysed in our previous study.60 Based on DFT calculations, and FlUC and TA measurements as a function of solvent properties, we proposed a diagram describing the system's relaxation dynamics in CHCl3, showing the presence of multiple transient ICT conformers. Upon photoexcitation, the tBuTPAterpy populates a singlet state (ICT1) which undergoes energy stabilization due to solvent relaxation processes. Within 10 ps, a structural change involving the torsions of the terpy, phenyl and amide units along the θ and φ dihedral angles leads to a population transfer to a second emitting conformer, ICT1′, which decays to the ground state through a fluorescence channel occurring in the ns time scales. We also determined the presence of a second “dark” ICT conformer (ICT2), populated alternatively to the ICT1 state and undergoing non-radiative decay to the ground state.By combining ultrafast measurements as a function of solvent polarity and viscosity, we corroborated the interpretation proposed in the energy diagram and explained how the environment influences the tBuTPAterpy photodynamics. Specifically, increasing the solvent viscosity slows down the ICT1 →  conformational change of about one order of magnitude, as observed in tBuTPAterpy in glyceryl triacetate. Conversely, a solvent polarity increase reduces the energy barrier of the ICT1 →
conformational change of about one order of magnitude, as observed in tBuTPAterpy in glyceryl triacetate. Conversely, a solvent polarity increase reduces the energy barrier of the ICT1 →  conformational change, speeding up a population transfer process which occurs on sub-ps time scales in acetonitrile. Instead, by minimizing the solvent polarity effects, as in the case of apolar n-hexane, a parallel pathway involving ISC is favoured. In this solvent, the ICT1 (S1) → LE (Tn) decay is the dominant relaxation channel and it occurs on shorter time scales than the conformational change.
conformational change, speeding up a population transfer process which occurs on sub-ps time scales in acetonitrile. Instead, by minimizing the solvent polarity effects, as in the case of apolar n-hexane, a parallel pathway involving ISC is favoured. In this solvent, the ICT1 (S1) → LE (Tn) decay is the dominant relaxation channel and it occurs on shorter time scales than the conformational change.
Overall, this study reveals the complexity of tBuTPAterpy photodynamic as a representative example of push–pull derivatives of 2,2′:6′,2′′-terpyridine. Even though these ligands are mostly used as building blocks in coordination chemistry for the enhancement of photoluminescence yields in metal complexes, our work stresses the potential of terpy derivatives as metal-free environment probes whose photodynamics can be easily controlled through external solvent parameters. The reported ultrafast investigation confirms that tBuTPAterpy is a promising candidate for sensing applications, and it provides a fundamental understanding of its excited state dynamics, laying the foundations for chemical and technological optimisations of metal-free sensors.
Author contributions
Conceptualisation: A. M. M., O. C. and M. C.; methodology: A. M. M., O. C., P. L. and M. O.; software: A. M. M. and P. L.; validation: A. M. M., O. C. and B. M.; formal analysis: A. M. M., O. C., P. L. and E. C. S.; investigation: A. M. M. and E. C. S.; resources: A. M. M., B. M. and M. C.; data curation: A. M. M., O. C., P. L. and E. C. S.; writing—original draft preparation: A. M. M., O. C. and B. M.; writing—review and editing: A. M. M., O. C., M. O., B. M. and M. C.; visualisation: A. M. M., O. C., P. L. and M. C.; supervision: A. M. M. and M. C.; project administration: A. M. M.; funding acquisition: A. M. M, B. M. and M. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Science Centre of Poland, SONATA grant no. 2020/39/D/ST4/00286. The research activities were co-financed by the funds granted under the Research Excellence Initiative of the University of Silesia in Katowice. This work was supported by the ERC via the DYNAMOX and CHIRAX advanced grants, and the Swiss SNF via the NCCR:MUST. AMM thanks the Polish National Agency for Academic Exchange (PPN/BEK/2018/1/00275/U/00001).Notes and references
- M. Hao, W. Chi, C. Wang, Z. Xu, Z. Li and X. Liu, Molecular Origins of Photoinduced Backward Intramolecular Charge Transfer, J. Phys. Chem. C, 2020, 124, 16820–16826 CrossRef CAS.
- M. Ratner, A brief history of molecular electronics, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- S. Sasaki, G. P. C. Drummen and G. Konishi, Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- V. Malytskyi, J.-J. Simon, L. Patrone and J.-M. Raimundo, Thiophene-based push–pull chromophores for small molecule organic solar cells (SMOSCs), RSC Adv., 2015, 5, 354–397 RSC.
- A. M. El-Zohry and M. Karlsson, Gigantic Relevance of Twisted Intramolecular Charge Transfer for Organic Dyes Used in Solar Cells, J. Phys. Chem. C, 2018, 122, 23998–24003 CrossRef CAS.
- K. Nakayama, T. Okura, Y. Okuda, J. Matsui, A. Masuhara, T. Yoshida, M. S. White, C. Yumusak, P. Stadler, M. Scharber and N. S. Sariciftci, Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption, Materials, 2021, 14, 1200 CrossRef CAS PubMed.
- H. Y. Chung, J. Oh, J.-H. Park, I. Cho, W. S. Yoon, J. E. Kwon, D. Kim and S. Y. Park, Spectroscopic Studies on Intramolecular Charge-Transfer Characteristics in Small-Molecule Organic Solar Cell Donors: A Case Study on ADA and DAD Triad Donors, J. Phys. Chem. C, 2020, 124, 18502–18512 CrossRef CAS.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826–58851 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- G. L. Closs and J. R. Miller, Intramolecular Long-Distance Electron Transfer in Organic Molecules, Science, 1988, 240, 440–447 CrossRef CAS PubMed.
- M. Park, C. H. Kim and T. Joo, Multifaceted Ultrafast Intramolecular Charge Transfer Dynamics of 4-(Dimethylamino)benzonitrile (DMABN), J. Phys. Chem. A, 2013, 117, 370–377 CrossRef CAS.
- P. B. Coto, L. Serrano-Andrés, T. Gustavsson, T. Fujiwara and E. C. Lim, Intramolecular charge transfer and dual fluorescence of 4-(dimethylamino)benzonitrile: ultrafast branching followed by a two-fold decay mechanism, Phys. Chem. Chem. Phys., 2011, 13, 15182 RSC.
- K. A. Zachariasse, S. I. Druzhinin, S. A. Kovalenko and T. Senyushkina, Intramolecular charge transfer of 4-(dimethylamino)benzonitrile probed by time-resolved fluorescence and transient absorption: No evidence for two ICT states and a πσ* reaction intermediate, J. Chem. Phys., 2009, 131, 224313 CrossRef PubMed.
- S. I. Druzhinin, N. P. Ernsting, S. A. Kovalenko, L. P. Lustres, T. A. Senyushkina and K. A. Zachariasse, Dynamics of Ultrafast Intramolecular Charge Transfer with 4-(Dimethylamino)benzonitrile in Acetonitrile, J. Phys. Chem. A, 2006, 110, 2955–2969 CrossRef CAS PubMed.
- M. A. Kochman, B. Durbeej and A. Kubas, Simulation and Analysis of the Transient Absorption Spectrum of 4-(N, N -Dimethylamino)benzonitrile (DMABN) in Acetonitrile, J. Phys. Chem. A, 2021, 125, 8635–8648 CrossRef CAS PubMed.
- M. A. Kochman and B. Durbeej, Simulating the Nonadiabatic Relaxation Dynamics of 4-(N, N -Dimethylamino)benzonitrile (DMABN) in Polar Solution, J. Phys. Chem. A, 2020, 124, 2193–2206 CrossRef CAS.
- I. Gómez, P. J. Castro and M. Reguero, Insight into the Mechanisms of Luminescence of Aminobenzonitrile and Dimethylaminobenzonitrile in Polar Solvents. An ab Initio Study, J. Phys. Chem. A, 2015, 119, 1983–1995 CrossRef PubMed.
- I. Georgieva, A. J. A. Aquino, F. Plasser, N. Trendafilova, A. Köhn and H. Lischka, Intramolecular Charge-Transfer Excited-State Processes in 4-(N, N -Dimethylamino)benzonitrile: The Role of Twisting and the πσ* State, J. Phys. Chem. A, 2015, 119, 6232–6243 CrossRef CAS PubMed.
- A. Perveaux, P. J. Castro, D. Lauvergnat, M. Reguero and B. Lasorne, Intramolecular Charge Transfer in 4-Aminobenzonitrile Does Not Need the Twist and May Not Need the Bend, J. Phys. Chem. Lett., 2015, 6, 1316–1320 CrossRef CAS PubMed.
- A. S. R. Koti and N. Periasamy, Time resolved area normalized emission spectroscopy (TRANES) of DMABN confirms emission from two states, Res. Chem. Intermed., 2002, 28, 831–836 CrossRef CAS.
- B. Patrizi, C. Cozza, A. Pietropaolo, P. Foggi and M. Siciliani de Cumis, Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules, Molecules, 2020, 25, 430 CrossRef CAS PubMed.
- Z. Szakács, S. Rousseva, M. Bojtár, D. Hessz, I. Bitter, M. Kállay, M. Hilbers, H. Zhang and M. Kubinyi, Experimental evidence of TICT state in 4-piperidinyl-1,8-naphthalimide – a kinetic and mechanistic study, Phys. Chem. Chem. Phys., 2018, 20, 10155–10164 RSC.
- G. Haberhauer, Planarized and Twisted Intramolecular Charge Transfer: A Concept for Fluorophores Showing Two Independent Rotations in Excited State, Chem. – Eur. J., 2017, 23, 9288–9296 CrossRef CAS.
- G. Haberhauer, R. Gleiter and C. Burkhart, Planarized Intramolecular Charge Transfer: A Concept for Fluorophores with both Large Stokes Shifts and High Fluorescence Quantum Yields, Chem. – Eur. J., 2016, 22, 971–978 CrossRef CAS PubMed.
- R. Ghosh, A. Nandi and D. K. Palit, Solvent sensitive intramolecular charge transfer dynamics in the excited states of 4-N,N-dimethylamino-4′-nitrobiphenyl, Phys. Chem. Chem. Phys., 2016, 18, 7661–7671 RSC.
- E. Benassi, B. Carlotti, M. Segado, A. Cesaretti, A. Spalletti, F. Elisei and V. Barone, Presence of Two Emissive Minima in the Lowest Excited State of a Push–Pull Cationic Dye Unequivocally Proved by Femtosecond Up-Conversion Spectroscopy and Vibronic Quantum-Mechanical Computations, J. Phys. Chem. B, 2015, 119, 6035–6040 CrossRef CAS PubMed.
- Z. Zhang, G. Zhang, J. Wang, S. Sun and Z. Zhang, The mechanisms of Large Stokes Shift and Fluorescence Quantum Yields in anilino substituted Rhodamine analogue: TICT and PICT, Comput. Theor. Chem., 2016, 1095, 44–53 CrossRef CAS.
- R. Ghosh, A. Nandi, A. Kushwaha and D. Das, Ultrafast Conformational Relaxation Dynamics in Anthryl-9-benzothiazole: Dynamic Planarization Driven Delocalization and Protonation-Induced Twisting Dynamics, J. Phys. Chem. B, 2019, 123, 5307–5315 CrossRef CAS PubMed.
- T. Staněk, M. Dvořák, N. Almonasy, M. Nepraš, I. Šloufová and M. Michl, Formation of planarized intramolecular charge-transfer state in dichlorotriazinyl-pyrene fluorescent probe: TD-DFT and resonance Raman study, Dyes Pigm., 2017, 141, 121–127 CrossRef.
- K. A. Zachariasse, S. I. Druzhinin, W. Bosch and R. Machinek, Intramolecular Charge Transfer with the Planarized 4-Aminobenzonitrile 1- tert -Butyl-6-cyano-1,2,3,4-tetrahydroquinoline (NTC6), J. Am. Chem. Soc., 2004, 126, 1705–1715 CrossRef CAS.
- Q. Wu, J. Liu, Y. Li, M. M. S. Lee, L. Hu, Y. Li, P. Zhou, D. Wang and B. Z. Tang, Janus luminogens with bended intramolecular charge transfer: Toward molecular transistor and brain imaging, Matter, 2021, 4, 3286–3300 CrossRef CAS.
- M. Segado, I. Gómez and M. Reguero, Intramolecular charge transfer in aminobenzonitriles and tetrafluoro counterparts: fluorescence explained by competition between low-lying excited states and radiationless deactivation. Part I: A mechanistic overview of the parent system ABN, Phys. Chem. Chem. Phys., 2016, 18, 6861–6874 RSC.
- M. Lv, X. Wang, D. Wang, X. Li, Y. Liu, H. Pan, S. Zhang, J. Xu and J. Chen, Unravelling the role of charge transfer state during ultrafast intersystem crossing in compact organic chromophores, Phys. Chem. Chem. Phys., 2021, 23, 25455–25466 RSC.
- K. Chen, I. V. Kurganskii, X. Zhang, A. Elmali, J. Zhao, A. Karatay and M. V. Fedin, Intersystem Crossing and Electron Spin Selectivity in Anthracene-Naphthalimide Compact Electron Donor-Acceptor Dyads Showing Different Geometry and Electronic Coupling Magnitudes, Chem. – Eur. J., 2021, 27, 7572–7587 CrossRef CAS PubMed.
- Y. Hou, J. Liu, N. Zhang and J. Zhao, Long-Lived Local Triplet Excited State and Charge Transfer State of 4,4′-Dimethoxy Triphenylamine-BODIPY Compact Electron Donor/Acceptor Dyads, J. Phys. Chem. A, 2020, 124, 9360–9374 CrossRef CAS PubMed.
- J. Choi, D.-S. Ahn, K. Y. Oang, D. W. Cho and H. Ihee, Charge Transfer-Induced Torsional Dynamics in the Excited State of 2,6-Bis(diphenylamino)anthraquinone, J. Phys. Chem. C, 2017, 121, 24317–24323 CrossRef CAS.
- J. Pina, J. S. Seixas de Melo, R. M. F. Batista, S. P. G. Costa and M. M. M. Raposo, Triphenylamine–Benzimidazole Derivatives: Synthesis, Excited-State Characterization, and DFT Studies, J. Org. Chem., 2013, 78, 11389–11395 CrossRef CAS PubMed.
- M. Heller and U. S. Schubert, Syntheses of Functionalized 2,2′:6′,2′′-Terpyridines, Eur. J. Org. Chem., 2003, 947–961 CrossRef CAS.
- S. S. M. Fernandes, M. Belsley, C. Ciarrocchi, M. Licchelli and M. M. M. Raposo, Terpyridine derivatives functionalized with (hetero)aromatic groups and the corresponding Ru complexes: Synthesis and characterization as SHG chromophores, Dyes Pigm., 2018, 150, 49–58 CrossRef CAS.
- C. Wei, Y. He, X. Shi and Z. Song, Terpyridine-metal complexes: Applications in catalysis and supramolecular chemistry, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed.
- R. R. Panicker and A. Sivaramakrishna, Remarkably flexible 2,2′:6′,2′′-terpyridines and their group 8–10 transition metal complexes – Chemistry and applications, Coord. Chem. Rev., 2022, 459, 214426 CrossRef CAS.
- A. M. W. Cargill Thompson, The synthesis of 2,2′:6′,2′′-terpyridine ligands—versatile building blocks for supramolecular chemistry, Coord. Chem. Rev., 1997, 160, 1–52 CrossRef.
- E. U. Mughal, M. Mirzaei, A. Sadiq, S. Fatima, A. Naseem, N. Naeem, N. Fatima, S. Kausar, A. A. Altaf, M. N. Zafar and B. A. Khan, Terpyridine-metal complexes: effects of different substituents on their physico-chemical properties and density functional theory studies, R. Soc. Open Sci., 2020, 7, 201208 CrossRef CAS PubMed.
- H. Hofmeier and U. S. Schubert, Recent developments in the supramolecular chemistry of terpyridine–metal complexes, Chem. Soc. Rev., 2004, 33, 373–399 RSC.
- R. Musiol, P. Malecki, M. Pacholczyk and J. Mularski, Terpyridines as promising antitumor agents: an overview of their discovery and development, Expert Opin. Drug Discovery, 2022, 17, 259–271 CrossRef CAS PubMed.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Square-Planar Pd(II), Pt(II), and Au(III) Terpyridine Complexes: Their Syntheses, Physical Properties, Supramolecular Constructs, and Biomedical Activities, Chem. Rev., 2008, 108, 1834–1895 CrossRef CAS PubMed.
- L. M. Hight, M. C. McGuire, Y. Zhang, M. A. Bork, P. E. Fanwick, A. Wasserman and D. R. McMillin, π Donation and Its Effects on the Excited-State Lifetimes of Luminescent Platinum(II) Terpyridine Complexes in Solution, Inorg. Chem., 2013, 52, 8476–8482 CrossRef CAS PubMed.
- P. Pal, S. Mukherjee, D. Maity and S. Baitalik, Synthesis, Photophysics, and Switchable Luminescence Properties of a New Class of Ruthenium(II)–Terpyridine Complexes Containing Photoisomerizable Styrylbenzene Units, ACS Omega, 2018, 3, 14526–14537 CrossRef CAS PubMed.
- J. Kübel, R. Schroot, M. Wächtler, U. S. Schubert, B. Dietzek and M. Jäger, Photoredox-active Dyads Based on a Ru(II) Photosensitizer Equipped with Electron Donor or Acceptor Polymer Chains: A Spectroscopic Study of Light-Induced Processes toward Efficient Charge Separation, J. Phys. Chem. C, 2015, 119, 4742–4751 CrossRef.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Complexes of the Ruthenium(II)-2,2’:6’,2’’-terpyridine Family. Effect of Electron-Accepting and -Donating Substituents on the Photophysical and Electrochemical Properties, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- A. M. Maroń, A. Szlapa-Kula, M. Matussek, R. Kruszynski, M. Siwy, H. Janeczek, J. Grzelak, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Photoluminescence enhancement of Re(I) carbonyl complexes bearing D–A and D–π–A ligands, Dalton Trans., 2020, 49, 4441–4453 RSC.
- A. K. Pal and G. S. Hanan, Design, synthesis and excited-state properties of mononuclear Ru(II) complexes of tridentate heterocyclic ligands, Chem. Soc. Rev., 2014, 43, 6184–6197 RSC.
- E. A. Medlycott and G. S. Hanan, Designing tridentate ligands for ruthenium(II) complexes with prolonged room temperature luminescence lifetimes, Chem. Soc. Rev., 2005, 34, 133–142 RSC.
- L. Xiao, Y. Xu, M. Yan, D. Galipeau, X. Peng and X. Yan, Excitation-Dependent Fluorescence of Triphenylamine-Substituted Tridentate Pyridyl Ruthenium Complexes, J. Phys. Chem. A, 2010, 114, 9090–9097 CrossRef CAS PubMed.
- R. Siebert, A. Winter, M. Schmitt, J. Popp, U. S. Schubert and B. Dietzek, Light-Induced Dynamics in Conjugated Bis(terpyridine) Ligands – A Case Study Toward Photoactive Coordination Polymers, Macromol. Rapid Commun., 2012, 33, 481–497 CrossRef CAS PubMed.
- R. Fernández-Terán and L. Sévery, Living Long and Prosperous: Productive Intraligand Charge-Transfer States from a Rhenium(I) Terpyridine Photosensitizer with Enhanced Light Absorption, Inorg. Chem., 2021, 60, 1334–1343 CrossRef PubMed.
- R. J. Fernández-Terán and L. Sévery, Coordination Environment Prevents Access to Intraligand Charge-Transfer States through Remote Substitution in Rhenium(I) Terpyridinedicarbonyl Complexes, Inorg. Chem., 2021, 60, 1325–1333 CrossRef PubMed.
- R. Siebert, A. Winter, U. S. Schubert, B. Dietzek and J. Popp, Excited-State Planarization as Free Barrierless Motion in a π-Conjugated Terpyridine, J. Phys. Chem. C, 2010, 114, 6841–6848 CrossRef CAS.
- R. Siebert, D. Akimov, M. Schmitt, A. Winter, U. S. Schubert, B. Dietzek and J. Popp, Spectroscopic Investigation of the Ultrafast Photoinduced Dynamics in π-Conjugated Terpyridines, Chem. Phys. Chem., 2009, 10, 910–919 CrossRef CAS PubMed.
- A. M. Maroń, O. Cannelli, E. C. Socie, P. Lodowski and B. Machura, Push-Pull Effect of Terpyridine Substituted by Triphenylamine Motive—Impact of Viscosity, Polarity and Protonation on Molecular Optical Properties, Molecules, 2022, 27, 7071 CrossRef PubMed.
- E. Socie, B. R. C. Vale, A. Burgos-Caminal and J.-E. Moser, Direct Observation of Shallow Trap States in Thermal Equilibrium with Band-Edge Excitons in Strongly Confined CsPbBr3 Perovskite Nanoplatelets, Adv. Opt. Mater., 2021, 9, 2001308 CrossRef CAS.
- A. M. Maroń, K. Choroba, T. Pedzinski and B. Machura, Towards better understanding of the photophysics of platinum(II) coordination compounds with anthracene- and pyrene-substituted 2,6-bis(thiazol-2-yl)pyridines, Dalton Trans., 2020, 49, 13440–13448 RSC.
- C. Slavov, H. Hartmann and J. Wachtveitl, Implementation and Evaluation of Data Analysis Strategies for Time-Resolved Optical Spectroscopy, Anal. Chem., 2015, 87, 2328–2336 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- T. Jarusuwannapoom, W. Hongrojjanawiwat, S. Jitjaicham, L. Wannatong, M. Nithitanakul, C. Pattamaprom, P. Koombhongse, R. Rangkupan and P. Supaphol, Effect of solvents on electro-spinnability of polystyrene solutions and morphological appearance of resulting electrospun polystyrene fibers, Eur. Polym. J., 2005, 41, 409–421 CrossRef CAS.
- T. Dong, E. P. Knoshaug, P. T. Pienkos and L. M. L. Laurens, Lipid recovery from wet oleaginous microbial biomass for biofuel production: A critical review, Appl. Energy, 2016, 177, 879–895 CrossRef CAS.
- D. S. Gill and D. Rana, Preparation of Some Novel Copper(I) Complexes and their Molar Conductances in Organic Solvents, Z. Naturforsch., A: Phys. Sci., 2009, 64, 269–272 CrossRef CAS.
- A. S. R. Koti, M. M. G. Krishna and N. Periasamy, Time-Resolved Area-Normalized Emission Spectroscopy (TRANES): A Novel Method for Confirming Emission from Two Excited States, J. Phys. Chem. A, 2001, 105, 1767–1771 CrossRef CAS.
- A. S. R. Koti and N. Periasamy, Application of time resolved area normalized emission spectroscopy to multicomponent systems, J. Chem. Phys., 2001, 115, 7094–7099 CrossRef CAS.
- K. Ramamurthy, K. Ponnusamy and S. Chellappan, Excitation-resolved area-normalized emission spectroscopy: a rapid and simple steady-state technique for the analysis of heterogeneous fluorescence, RSC Adv., 2020, 10, 998–1006 RSC.
- A. Cesaretti, B. Carlotti, P. L. Gentili, R. Germani, A. Spalletti and F. Elisei, Twisting in the excited state of an N-methylpyridinium fluorescent dye modulated by nano-heterogeneous micellar systems, Photochem. Photobiol. Sci., 2016, 15, 525–535 CrossRef CAS PubMed.
- B. Carlotti, A. Cesaretti, C. G. Fortuna, A. Spalletti and F. Elisei, Experimental evidence of dual emission in a negatively solvatochromic push–pull pyridinium derivative, Phys. Chem. Chem. Phys., 2015, 17, 1877–1882 RSC.
- B. Carlotti, E. Benassi, A. Cesaretti, C. G. Fortuna, A. Spalletti, V. Barone and F. Elisei, An ultrafast spectroscopic and quantum mechanical investigation of multiple emissions in push–pull pyridinium derivatives bearing different electron donors, Phys. Chem. Chem. Phys., 2015, 17, 20981–20989 RSC.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- C. Bonaccorso, A. Cesaretti, F. Elisei, L. Mencaroni, A. Spalletti and C. G. Fortuna, New Styryl Phenanthroline Derivatives as Model D−π−A−π−D Materials for Non-Linear Optics, Chem. Phys. Chem., 2018, 19, 1917–1929 CrossRef CAS PubMed.
- A. El Nahhas, A. Cannizzo, F. van Mourik, A. M. Blanco-Rodríguez, S. Záliš, A. Vlček and M. Chergui, Ultrafast Excited-State Dynamics of [Re(L)(CO)3(bpy)] n Complexes: Involvement of the Solvent, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef CAS PubMed.
- A. El Nahhas, C. Consani, A. M. Blanco-Rodríguez, K. M. Lancaster, O. Braem, A. Cannizzo, M. Towrie, I. P. Clark, S. Záliš, M. Chergui and A. Vlček, Ultrafast Excited-State Dynamics of Rhenium(I) Photosensitizers [Re(Cl)(CO)3(N,N)] and [Re(imidazole)(CO)3(N,N)] +: Diimine Effects, Inorg. Chem., 2011, 50, 2932–2943 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vazquez, L. M. Lawson Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, Experimental Evidence of Ultrafast Quenching of the 3 MLCT Luminescence in Ruthenium(II) Tris-bipyridyl Complexes via a 3 dd State, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed.
- A. N. Tarnovsky, W. Gawelda, M. Johnson, C. Bressler and M. Chergui, Photexcitation of Aqueous Ruthenium(II)-tris-(2,2‘-bipyridine) with High-Intensity Femtosecond Laser Pulses, J. Phys. Chem. B, 2006, 110, 26497–26505 CrossRef CAS PubMed.
- M. R. Son, Y.-J. Cho, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Direct observation of the photoinduced electron transfer processes of bis(4-arylphenylamino benzo)-ortho-carborane using transient absorption spectroscopic measurements, Phys. Chem. Chem. Phys., 2017, 19, 24485–24492 RSC.
- B. Carlotti, G. Consiglio, F. Elisei, C. G. Fortuna, U. Mazzucato and A. Spalletti, Intramolecular Charge Transfer of Push–Pull Pyridinium Salts in the Singlet Manifold, J. Phys. Chem. A, 2014, 118, 3580–3592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details about experiments, photodamage and fluence dependence tests, comparison of time-resolved spectra to steady state spectra, DAS and EAS, and details about global fitting procedures. See DOI: https://doi.org/10.1039/d3cp04492k |
| This journal is © the Owner Societies 2024 |