
A density functional theory benchmark on antioxidant-related properties of polyphenols†

Abstract
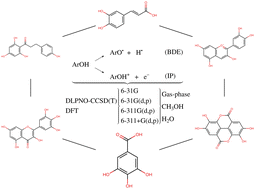
In this work, we present a density functional theory benchmark on antioxidant-related properties for a series of six polyphenols that are well-known antioxidants: caffeic acid, cyanidin, ellagic acid, gallic acid, myricetin, and phloretin. Computations on the 24 O–H bond dissociation enthalpies (BDEs) and 6 ionization potentials (IPs) were performed using twenty-three exchange–correlation functionals combined with four different basis sets in the gas-phase, water, and methanol; calibration against the Domain-based Local Pair Natural Orbital CCSD(T) (DLPNO-CCSD(T)) approach was employed. Mean absolute deviation (MAD) as well as linear fitting results suggested the LC-PBE approach as the most suitable for O–H BDEs in the gas-phase. The LC-PBE, M06-2X, and M05-2X results presented the smallest MADs for O–H BDEs when compared to the reference, in water. The LC-PBE results had the smallest MADs for IPs in the gas-phase while M05-2X, M06-2X, LC-PBE, and LC-ωPBE exhibited the best results for MAD in water. We expect the outcomes from the present work will serve as general guidance for researchers working in the field.
 Please wait while we load your content...
Please wait while we load your content...

