
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure, bonding and stability of fumarate, maleate, and succinate dianions from X-ray spectroscopy†
Viktoriia
Savchenko
 *a,
Sebastian
Eckert
a,
Mattis
Fondell
a,
Rolf
Mitzner
a,
Vincius
Vaz da Cruz
a and
Alexander
Föhlisch
ab
*a,
Sebastian
Eckert
a,
Mattis
Fondell
a,
Rolf
Mitzner
a,
Vincius
Vaz da Cruz
a and
Alexander
Föhlisch
ab
aHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Institute for Methods and Instrumentation for Synchrotron Radiation Research, 12489 Berlin, Germany. E-mail: viktoriia.savchenko@helmholtz-berlin.de
bInstitut für Physik und Astronomie, Universität Potsdam, 14476 Potsdam, Germany
First published on 18th December 2023
Abstract
The electronic structure of the fumarate, maleate, and succinate dianions in the context of their stability is determined in a joint experimental and computational study with X-ray absorption spectroscopy and resonant inelastic X-ray scattering at the O K-edge. The study reveals differences in the electronic states and molecular orbitals of the three molecules. In particular, maleate has a non-degenerate oxygen core-orbital with an energy difference of approximately 0.15 eV, visible in a two peak structure in XAS. Polarization-dependent RIXS provides information on the orientation of the occupied valence molecular orbitals with respect to the carboxylate group plane and shows a gradually increasing energy gap between the HOMO and excited π* LUMO from fumarate to maleate to succinate. We also demonstrate the energy excitation dependence of the RIXS spectra of maleate, with the total inelastic RIXS profile shifting towards higher energy loss as the detuning is increased from negative to positive values. Our findings show that maleate is less stable than fumarate and succinate due to the presence of electronic density on its HOMO orbital on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between carboxylate groups, which can lead to weaker bonding of maleate with molecules or ions.
C bond between carboxylate groups, which can lead to weaker bonding of maleate with molecules or ions.
1 Introduction
Fumarate, maleate, and succinate dianions (Fig. 1) are common organic compounds that play a key role in coordination chemistry, often serving as bridging ligands that link metal centers in complex structures.1–4 The inherent structural differences between fumarate, maleate, and succinate influence their roles in biological pathways and their chemical behaviors in the formation, stability, and efficacy of coordination polymers.5–7 The trans isomer of C4H2O42−, fumarate, and its hydrogenated version, succinate (C4H4O42−), which are essential intermediates in the Krebs cycle, participate in metabolic reactions within the cell.8 Compounds associated with these metabolites can be biodegradable, potentially reducing the impact on the environment. In contrast, the cis isomer of C4H2O42−, maleate, does not typically form in natural biological processes, and its compounds are noted for their stability, making them suitable for various applications that demand durable materials.9,10 | ||
| Fig. 1 Molecular geometry structures of the trans- and cis-isomers fumarate C4H4O42− (a) and maleate C4H4O42− (b) together with their hydrogenated molecule, succinate C4H2O42− (c) dianions. | ||
Under identical external conditions, the stability of fumarate, maleate, and succinate dianions is influenced by their unique molecular geometries and electronic configurations. Previous studies have examined the geometries of fumarate, maleate, and succinate using various experimental and theoretical methods.11–15 Specifically, trans configuration of fumarate often leads to extended and open structures, while cis configuration of maleate results in more compact topologies.5,16–20 In the case of succinate, the absence of a CC bond allows the molecule to more freely rotate. As a result, this flexibility in rotation contributes to a variety of succinate-based coordination polymer structures.12,21,22
In addition to the effects of geometry, the electronic structure of molecules and their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) also have a significant influence on molecular properties.23–25 Previous studies examining the electronic structure have predominantly focused on elucidating the conformational stability of fumarate, maleate, and succinate dianions in liquid or gas phases.11–13 However, the impact of the electronic density distribution across the HOMO orbital on their stability remains unexplored.
In this study we aim to elucidate the influence of the electronic structure on the stability of fumarate, maleate and succinate dianions. For this purpose, we employ X-ray absorption spectroscopy (XAS) and polarization-dependent resonant inelastic X-ray scattering (RIXS) at the O K-edge. XAS and RIXS are powerful techniques for characterizing the electronic structure of molecules, which is influenced by various types of intra-molecular and inter-molecular interactions in different environments.26–33 In particular, XAS allows us to probe the unoccupied electronic states of a system, giving insight into the electronic structure of the LUMO in these dianions. On the other hand, the sensitivity of RIXS to both the occupied and unoccupied states, offers a unique perspective on HOMO–LUMO interactions and electronic transitions, which are crucial for understanding the molecular stability. Our spectroscopic study of the dianions at the O K-edge enables us to probe the relative stability of potential bonding between fumarate, maleate, succinate and metal ions or clusters in coordination polymers. Therefore, the combination of XAS and RIXS spectroscopic data with theoretical simulations will contribute to a fundamental understanding of the structure and properties of fumarate, maleate, and succinate dianions, which is pivotal for optimizing their use in coordination chemistry applications.
The paper is organized as follows: first, we describe experimental and theoretical methods used in the study. We then present and discuss the electronic structure properties of the three dicarboxylic acid dianions (fumarate, maleate, and succinate), the experimental and theoretical polarization dependent RIXS at the O K-edge of the three molecules and finally NEXAFS, comparing their electronic states and molecular orbitals. A detailed study of the photon energy dependence of RIXS gives further insight into local symmetry and bonding properties. In our final result, the excitation dependence of the RIXS for maleate is discussed, demonstrating a strong agreement between the experimental RIXS data and our theoretical spectra simulations, which are based on the localized approximation. In conclusion (Section 4) we summarize the key findings from the XAS and RIXS studies. It emphasizes the differences in electronic states and molecular orbitals among fumarate, maleate, and succinate. We find that maleate is seen as less stable than fumarate and succinate due to the presence of electronic density on its HOMO orbital on the CC bond. Fumarate and succinate, on the other hand, are more stable because their HOMO orbitals are equally delocalized.
2 Methods
2.1 Experiment
The dicarboxylate anions (aq) were prepared by dissolving fumaric-, maleic- and succinic-acid, supplied by Sigma Aldrich, in deionized water. The pH of the solutions was adjusted using sodiumhydroxide (NaOH) to ∼6, ∼9 and ∼8, respectively, to fully deprotonate the systems. The concentrations of the dicarboxylate anions were 0.5 M, 0.3 M and 0.3 M. The samples were injected into the experimental vacuum chamber of the EDAX experiment34 at the UE49 SGM beamline35 of the synchrotron BESSY II using a cylindrical liquid jet sample environment. Linear horizontally and vertically polarized radiation with a bandwidth of ∼0.35 eV was used to resonantly excite the sample at different energies at the oxygen 1s absorption edge. The emitted photons were detected under a 90° scattering geometry using an adapted Scienta XES 350 spherical grating spectrometer (1200 lines per mm) with an MCP detector in the second diffraction order. The experimental energy scale was calibrated by aligning the pre-edge 4a1 resonance in a partial fluorescence yield measurement of a water sample at 535 eV.2.2 Theory
All theoretical simulations were performed with the ORCA36 package (version 5.0.1) and the Multiwfn37 analysis suite. The geometry optimization of fumarate (C2h symmetry), maleate (Cs symmetry) and succinate (C2 symmetry) was performed at the DFT/PBE0 level of theory with the d3bj-correction. The def2-TZVPPD basis set was used together with the def2/J auxiliary basis sets and the RIJCOSX approximation. The water solvation was included implicitly by using a continuum solvation model (SMD) which is based on the polarized continuous quantum mechanical charge density of the solute.38 The spectral calculations (XAS and RIXS) were performed using the TD-DFT method where the nuclear motion was neglected. The core-valence separation (CVS) approximation was applied for the XAS calculation. For the simulated XAS spectra the Gaussian broadening of Γ = 0.5 eV was used. The energies of the calculated XAS for each molecule were shifted by +13 eV to match the calculated O 1s → π* succinate peak maximum with the experimental O K-edge peak maximum. The RSA-TD-DFT scheme was used for the electronic RIXS calculations.39 RIXS spectra were simulated in the localized approximation, where a single oxygen 1s core-orbital is included in TD-DFT donor space at a time, and the total spectrum is the sum of the partial spectra.3 Results and discussion
3.1 Electronic structure and X-ray absorption
Fumarate (C4H4O42−) and maleate (C4H4O42−) are molecules with two carboxylate groups, which are connected by a CC bond. Succinate (C4H6O42−), on the other hand, has two carboxylate groups connected by a single C–C bond, with two additional hydrogen atoms present in the molecule. Here we consider how the electronic structure is modified in the dicarboxylic acid dianions, depending on the type of isomer and the presence or absence of a double bond between carboxylate groups.
Fig. 2(a) presents the molecular orbital diagram for fumarate, maleate and succinate, illustrating the electronic transitions in O K-edge XAS and the O 1s core-orbitals from which core electrons are excited into unoccupied π* orbital(s). The corresponding experimental and theoretical XAS profiles are shown in the upper and lower panel of Fig. 2(b), respectively.
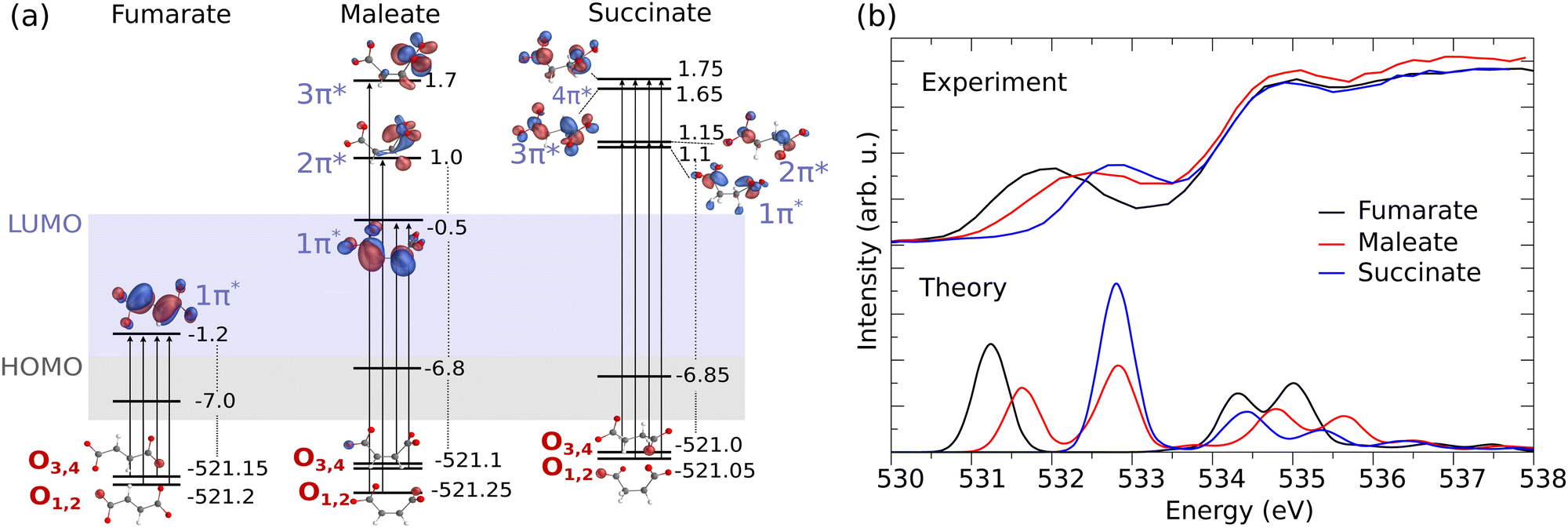 | ||
| Fig. 2 (a) Electronic structure of the dicarboxylic acid dianions (orbital energy in eV). The illustration highlights the O 1s core-orbitals for fumarate, maleate, and succinate. O1,2, associated with one carboxylate group, and O3,4, associated with the other, are labeled in accordance with the atom number in Fig. 1. The transitions to the excited π* unoccupied orbitals are also depicted. (b) The corresponding measured in partial fluorescence yield mode (liquid, upper panel) and theoretical XAS spectra at the O K-edge (monomer, lower panel) for fumarate (black line), maleate (red line) and succinate (blue line). | ||
From the orbital diagram in Fig. 2(a) we can see that the four O 1s core-orbitals of fumarate are degenerate (calculated orbital energy levels of O1 1s and O2 1s are merged at −521.2 eV, O3 1s and O4 1s at −521.15 eV) or almost degenerate with energy differences of only 0.05 eV. Therefore, due to the excitations to the single unoccupied π* the core-excited states O 1s → π* (O1 1s → π*, O2 1s → π*, O3 1s → π* and O4 1s → π*) are closely spaced (see detailed XAS in Fig. S4(a), ESI†) and form one peak in the XAS spectrum at around 531.2 eV for fumarate (Fig. 2(b)). It is important to note that, since we employ hybrid functionals in our simulations, the energy differences between Kohn–Sham orbitals are no longer a good approximation of the true transition energies. This discrepancy leads to a mismatch between the calculated orbital energy differences and the actual core-excitation energies.
In the XAS of succinate at the O K-edge, we observe a single peak at around 532.8 eV (Fig. 2(b)), just as observed with fumarate. This can be attributed to the two factors: firstly, succinate has four equivalent oxygen atoms, which exhibit 1s orbital energy differences of up to 0.05 eV. Secondly, the excitations occur to four unoccupied π* molecular orbitals (1π*, 2π*, 3π*, 4π* in the Fig. 2(a)) and the maximal orbital energy difference between them is about 0.65 eV. As a result of these excitations, four core-excited states are close-lying (see detailed XAS in Fig. S4(c), ESI†) and form the single peak. Due to the dipole selection rules the transitions to σ orbitals LUMO and LUMO+1 are weak and do not make significant contribution to the XAS profile at the O K-edge of succinate. Therefore LUMO and LUMO+1 were eliminated from the orbital diagram.
The different orientation of carboxylate groups with respect to the CC bond in the cis isomer of fumarate, maleate, leads to a removal of degeneracy between O 1s orbitals of two carboxylate groups. Thus, the maleate dianion has an energy difference between non-degenerate oxygen core-orbitals (O1,2 and O3,4), which is approximately ≈0.15 eV, as seen in Fig. 2(a). We can observe two peaks in calculated XAS for maleate (Fig. 2(b) lower panel). One can notice that the energy difference between the two XAS features of maleate is bigger than 0.15 eV. The increased energy shift is influenced not only by the energy difference between O 1s orbitals but most dominantly by the orbital energy difference between the unoccupied π* orbitals, which are populated within the X-ray absorption. The delocalized LUMO 1π* spans across the CC bond has therefore lower energy than the unoccupied 2π* and 3π* orbitals, which are localized only on one carboxylate group. Therefore the first peak is formed by two core-excitations from O3,4 1s orbitals into the LUMO 1π* orbital at 531.65 eV and the second peak results from excitations from two lowest O1,2 1s orbitals to the higher LUMOs (2π* and 3π*) at 532.85 eV. The energy separation between the core-excited states may be overestimated due to the limitations of the density functional theory (DFT) method and could potentially be further broadened by considering vibrational and environmental effects. As a result, the corresponding measured XAS spectrum for maleate, as shown in Fig. 2(b) in the upper panel, exhibits a broadened peak rather than distinct peaks.
The comparative analysis of XAS spectra across fumarate, maleate, and succinate offers valuable insights into their respective electronic structures. We can see how the electronic structure changes from the trans isomer (fumarate) to the cis isomer (maleate) and leads to a shift of the resonance to higher energies. In the case of fumarate, the molecular geometry allows for more effective π* orbital overlap between the two carboxylate groups. This enhanced overlap results in a greater extent of delocalization of the π* orbital. As a consequence, the energy of these π* orbital is lower in fumarate compared to maleate. The maleate is exhibiting less delocalization as one carboxylate group is perpendicularly oriented to the remaining π system. This reduced delocalization of the π* orbital increases its energy relative to fumarate. Therefore, the observed higher energy shift of the CO π* peak in maleate in the XAS spectra can be attributed to these intramolecular interactions between two carboxylate groups. In succinate, the presence of 2 additional hydrogen atoms instead of the CC bond causes its LUMO orbital to have σ* character while the LUMO of fumarate and maleate is π*. The lowest lying π* orbital of succinate is nonconjugated and lying higher in energy than the LUMO of fumarate and maleate. Consequently, the first O 1s to π* resonance in XAS spectrum of succinate is at a higher energy compared to fumarate and maleate.
This analysis highlights the capability of XAS to detect changes in electronic structures arising from molecular variations. However, it is essential to acknowledge the limitations of XAS in determining molecular stability. While the orbital diagrams and XAS profiles provide detailed insights into the electronic transitions and energy levels, particularly involving the oxygen 1s core-orbitals and unoccupied π* orbitals, they do not fully reflect the effects of the factors influencing molecular stability. For example, the overall stability of a molecule involves its entire electronic structure, including occupied molecular orbitals, and can be significantly affected by dynamic processes and interactions with the environment, which are not captured in XAS measurements. In this context, RIXS is a more comprehensive tool which is able to probe deeper into the electronic structure. In the next section we show detailed analysis of occupied molecular orbitals through RIXS which therefore will contribute to our understanding of stability of fumarate, maleate and succinate.
3.2 Oxygen K-edge RIXS of dicarboxylate dianions
In the previous section we discussed the XAS spectroscopy of fumarate, maleate and succinate dianions, which constitutes the initial step in the two-step RIXS process. During the absorption, a preferred orientation of molecules is established. In the current research we focus on π* resonance of O K-edge energy region where absorption is maximum under horizontal polarization. The RIXS cross-section depends on the angle (χ) between incoming photon polarization (e) and the wave vector of the emitted photon (k′) as cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) χ = e·k′. Thus, from the oriented sub-ensemble RIXS anisotropy allows to determine the orientation of the valence (occupied) molecular orbitals due to different intensity ratios of the emission from σ and π orbitals. Fumarate, maleate and succinate have different point group symmetry which leads to higher diversity of their molecular orbital symmetry than just σ and π (see orbitals in Fig. S1–S3, ESI†). Therefore, the comparison of electronic states and molecular orbitals in different symmetry of these molecules would be inefficient. RIXS scattering anisotropy allow us not only to distinguish but also systematize and compare emitting occupied valence orbitals in terms of in-plane and out-of-plane with respect to their carboxylate groups.
χ = e·k′. Thus, from the oriented sub-ensemble RIXS anisotropy allows to determine the orientation of the valence (occupied) molecular orbitals due to different intensity ratios of the emission from σ and π orbitals. Fumarate, maleate and succinate have different point group symmetry which leads to higher diversity of their molecular orbital symmetry than just σ and π (see orbitals in Fig. S1–S3, ESI†). Therefore, the comparison of electronic states and molecular orbitals in different symmetry of these molecules would be inefficient. RIXS scattering anisotropy allow us not only to distinguish but also systematize and compare emitting occupied valence orbitals in terms of in-plane and out-of-plane with respect to their carboxylate groups.
The O K-edge RIXS spectra of fumarate, maleate and succinate excited under horizontally (χ = 0°) and vertically (χ = 90°) polarized light are shown in the Fig. 3(a), (b), (d), (e), (g) and (h) from theoretical simulations and experiment. In our comparison of RIXS spectra we include the RIXS of maleate for the first O3,4 1s → 1π* and the second O1,2 1s → 2π*, 3π* resonances in order from lower to higher excitation energy which were considered in the previous Section 3.1. In the RIXS spectra of all three molecules, we observe a prominent double feature between 3.0–8.0 eV on the energy loss scale, and indicated it by the green shaded area in the theoretical spectra. Additionally, the energies of the orbitals responsible for this RIXS double feature are highlighted by green shading in Fig. 3(c), (f) and (k). The energies of excited unoccupied π* and of some representative occupied orbitals are depicted as sticks (blue and red) positioned above and below the energy axis. The position above and below corresponds to their orientation (in-plane and out-of-plane) in relation to a carboxylate group. The rest remaining significant orbitals are shown in Fig. S1–S3, ESI.† Due to the different orbital orientation of excited π* LUMOs of dianions and their in-plane lone-pair occupied orbitals (12Bu, 22A′, 8A′′, 16A in Fig. 3(c), (f) and (k)), we observe a more intense signal of the first inelastic peak of the RIXS double feature under horizontally polarized excitation as seen in Fig. 3(a), (d) and (g). In contrast, in the case of second inelastic peak of the RIXS double feature the decay dominantly occurring from out-of-plane occupied orbitals (like 3Au for fumarate, 6A′′ for maleate (both resonances) and 13B for succinate) which have the same orientation as the excited π* LUMO orbitals. According to the selection rules in RIXS processes, the symmetry and orientation of these orbitals with respect to the polarization direction of the incident light play a crucial role. The vertical polarization aligns with the orbital plane of the out-of-plane orbitals, enhancing the interaction and thus the intensity of the RIXS signal. Therefore, the second inelastic RIXS peak has stronger intensity under the vertically polarized excitation. Generally, the RIXS spectra of the dianions show resemblance to the RIXS spectra at the O K-edge and their anisotropy behavior of other systems which contain dicarboxylate groups.40,41
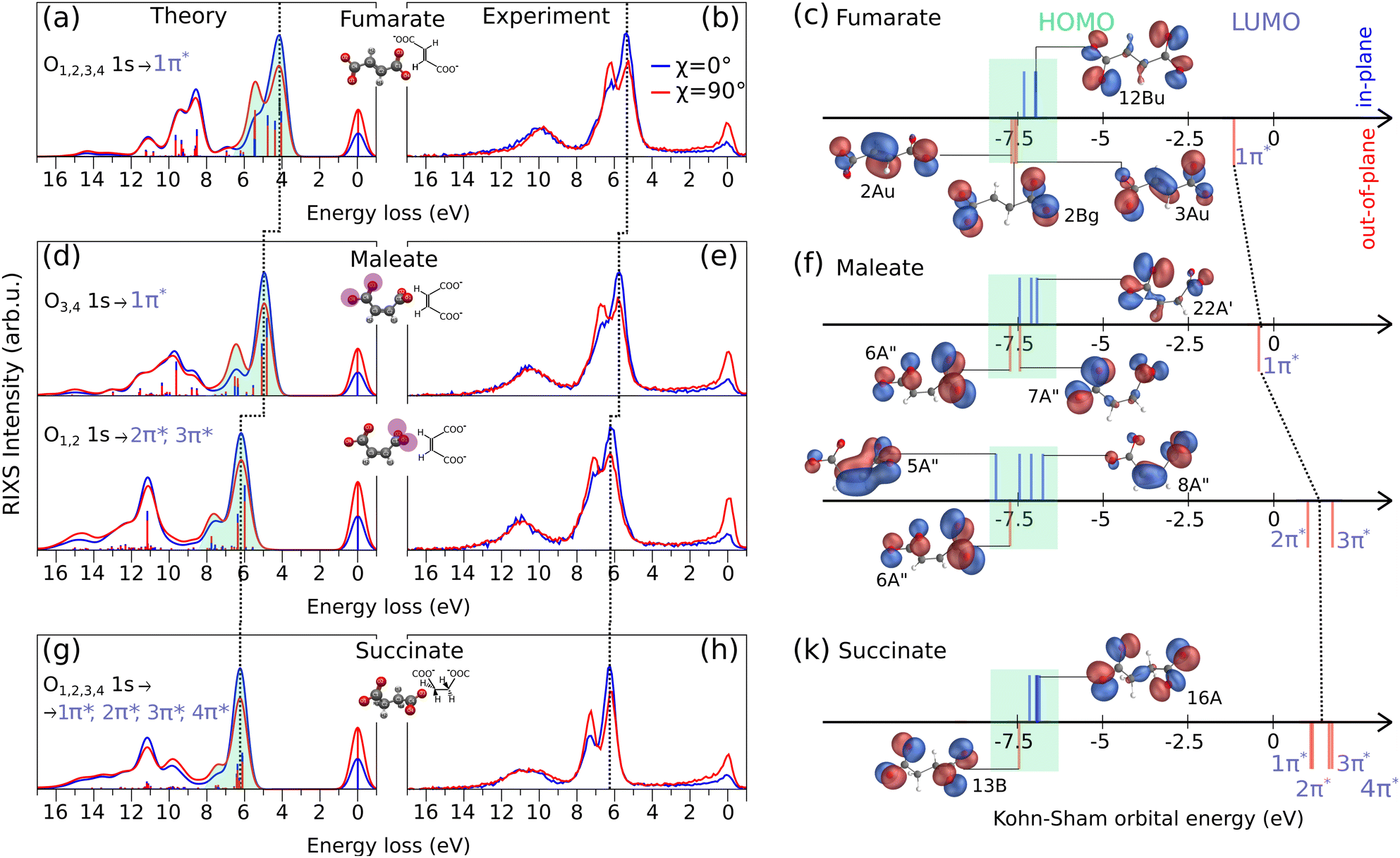 | ||
| Fig. 3 The normalized theoretical RIXS spectra (a), (d), (g) and experimental RIXS spectra (b), (e), (h) through oxygen K-edge of fumarate (C4H4O42−), maleate (C4H4O42−), and succinate (C4H2O42−), respectively. The horizontal polarization is represented by the blue curve and the vertical polarization by the red curve. The simplified Kohn–Sham orbital-energy diagrams (c), (f), (k) display significant orbitals for the RIXS process for each molecule. Blue sticks positioned above the orbital energy axis indicate the in-plane orientation of orbitals, while red sticks positioned below the orbital energy axis indicate the out-of-plane orientation of orbitals, relative to the plane of the carboxylate group. | ||
From the RIXS spectra of all three molecules and from the orbital-energy diagram we can conclude that fumarate, maleate and succinate mainly have the same order of in-plane and out-of-plane occupied orbitals involved in RIXS double feature with the exception for the RIXS of maleate at the second resonance (O1,2 1s → 2π*, 3π*). The in-plane 5A′′ orbital of maleate contributes to the emission band under horizontal polarization in the region of the second inelastic peak at around 7.5 eV energy loss in the theoretical spectrum in Fig. 3(d) at O1,2 1s → 2π*, 3π* resonance. Therefore, we observe a broader emission band under horizontal polarization and a minimal dependence on anisotropy for the second inelastic peak in the case of the second resonance of maleate. In addition, it is worthwhile to note that only in the case of fumarate which has planar structure a symmetry character of orbitals can be unambiguously probed by RIXS since they are in agreement with the orbital orientation. Thus, in-plane orbitals of fumarate correspond to the σ orbitals and out-of-plane to the π orbitals (Fig. 3(c)).
In Fig. 3(a), (d) and (g), we have indicated with a dashed line the emission band shift from fumarate to maleate, and from maleate to succinate, towards higher energy loss. This trend is attributed to the gradually increasing energy gap between the HOMO and the excited LUMO from molecule to molecule (see Fig. 3(c), (f) and (k)). The HOMO orbitals of all three molecules are in approximately the same orbital energy region (from −7.0 eV to −6.8 eV) and therefore the cause of increasing energy gap is the increasing energy of the excited LUMO orbitals (marked by dashed line through Fig. 3(c), (f) and (k)) which was described in previous section for XAS. It is also observed that the RIXS spectra evolve depending on the internal interaction between the two carboxylate groups and how they are twisted with respect to each other, as is the case with the XAS spectra. Since we focus on the most prominent spectral features of fumarate, maleate and succinate in the experimental RIXS (Fig. 3(b), (e) and (h)), we omit a description of energy loss spectral lines beyond first two inelastic peaks which are formed by the decays associated mostly with in-plane σ occupied orbitals (Fig. S1–S3, ESI†).
Even though the experimental RIXS spectra in the Fig. 3(b), (e) and (h) are affected by environmental and vibrational broadening we observe that first two inelastic peaks of succinate (in Fig. 3(h) at 6 eV and 7.5 eV) are more separated than in the case of fumarate (Fig. 3(b) at 5.3 eV and 6.5 eV) or maleate (Fig. 3(d) at 6 eV and 7.1 for the first resonance, at 6.1 eV and 7.3 eV for the second resonance). A cause of their separation is the absence of a CC bond in molecular structure of succinate. The two carboxylate groups of succinate are linked only by σ bonds while fumarate and maleate have also π bonding along CC bond, see e.g. orbitals 2Au and 3Au of fumarate on Fig. 3(c). Therefore, the highest occupied out-of-plane orbital of succinate 13B (Fig. 3(k)) is fully localized on the oxygen atoms and provides maximal transition dipole moment which leads to the high intensity of the second inelastic peak. The in-plane orbitals of succinate are almost degenerate, leading to the valence-excited states being energetically close. Thus, the valence-excited states of succinate which correspond to the first two inelastic peaks are well separated in energy between two features while in the case of fumarate and maleate the valence-excited states are also located between two inelastic peaks as we can see in theoretical spectra in the Fig. 3(a), (d) and (g).
It is worthwhile to note that for the second maleate resonance the out-of-plane orbital 6A′′ contributes to the second inelastic peak and it is located only on the oxygen atoms. Therefore, we would expect narrow peak as in the case of corresponding peak in RIXS of succinate, but the peak is broader due to the presence of valence-excited states between the two features. These states are also present in the RIXS through the first maleate resonance, but the intensity ratios are different due to excitation energy dependence, which will be discussed in the next section.
Based on the information about occupied orbitals which we get with the RIXS spectra simulation we can consider how the electronic density distribution over these orbitals affect the stability of fumarate, maleate and succinate. In maleate, a significant part of the electronic density of HOMO orbital 8A′′ localized on the CC bond. This electronic density distribution makes the electrons of O1,2 less available for interaction with electrons of other molecules or ions. Contrarily, the high electronic density on the CC bond indicates that there are more electrons available for bonding, which makes it as a reaction center for such a reaction like isomerization. As an example, isomerisation reaction can occur via a radical chain reaction mechanism15 where at the initial step, a bromine radical adds to one of the carbon atoms of the CC bond in the maleate structure, leading to the formation of a carbon-centered radical complex. In contrast, the HOMO orbitals of succinate (16A) and fumarate (12Bu) are delocalized over both carboxylate groups, which potentially would lead to a strong bonding between them and other molecules. Therefore, the delocalization of HOMO can contribute to the stability of fumarate and succinate, as the electrons can be shared between the two carboxylate groups and maintain the bonding. In addition to the orbital analysis of fumarate, maleate and succinate, the Mulliken analysis from our DFT simulation, (see Fig. S5, ESI†) shows that fumarate and succinate are likely to be more stable than maleate due to their symmetrical charge distribution. In contrast, maleate exhibits greater variation in the charges on the carbon atoms, with one carbon atom having a higher positive charge than the other and negative charge on the CC bond. This could make the molecule less stable than fumarate and succinate, as it might have a higher reactivity due to the imbalance in charge distribution.
3.3 Detuning dependence of the RIXS of maleate
In the analysis of RIXS, changing the excitation energy (detuning) from the top energy of XAS resonance plays an important role.42 Detuning helps in reducing the impact of vibrational nuclear dynamics, however, in cases where detuning is not significant, vibronic coupling between electronic states can also affect the RIXS spectra of symmetric systems. This occurs due to the degeneracy of delocalized orbitals of equivalent atoms in the system which results in degenerate or quasi-degenerate core-excited states.29 Upon core-excitation, the symmetry of the molecule is broken and the initially delocalized core-orbitals become localized within the core-hole lifetime, making the selection rules embedded in RIXS simulation inapplicable and can lead to a disagreement between theoretical simulation and experiment.In the Fig. 4(b) and (c) we show theoretical and experimental RIXS spectra of maleate depending on the different detuning, denoted by Ω = ω − ωTOPXAS, where ω is excitation energy and ωTOPXAS is the maximum of X-ray absorption band. In the studied range from Ω1 = −1.2 eV to Ω2 = +0.4 eV we observe the same anisotropy behaviour of double RIXS feature which is the result of an identical orbital orientation sequence participating in the scattering. Due to the redistribution of intensities between different final states we observe the shift of the total theoretical and experimental inelastic RIXS profile towards higher energy loss from negative to positive detuning. The most perceptible transformation in the electronically inelastic channel we see at the Ω2 = +0.8 eV. The first two inelastic peaks in RIXS become almost indistinguishable in the theoretical and experimental spectra as result of similar emission decay intensities for both horizontal and vertical polarization. This can be understood as we lose sensitivity to the symmetry of the LUMO orbitals when we tune far above the resonance, effectively quenching the anisotropy observed for resonant excitation. Furthermore, the agreement between simulation and experiment confirms the applicability of the localized approximation for describing RIXS in these systems, which indicates the presence of symmetry breaking effects upon core-excitation. These effects could be elucidated in future high-resolution experiments.
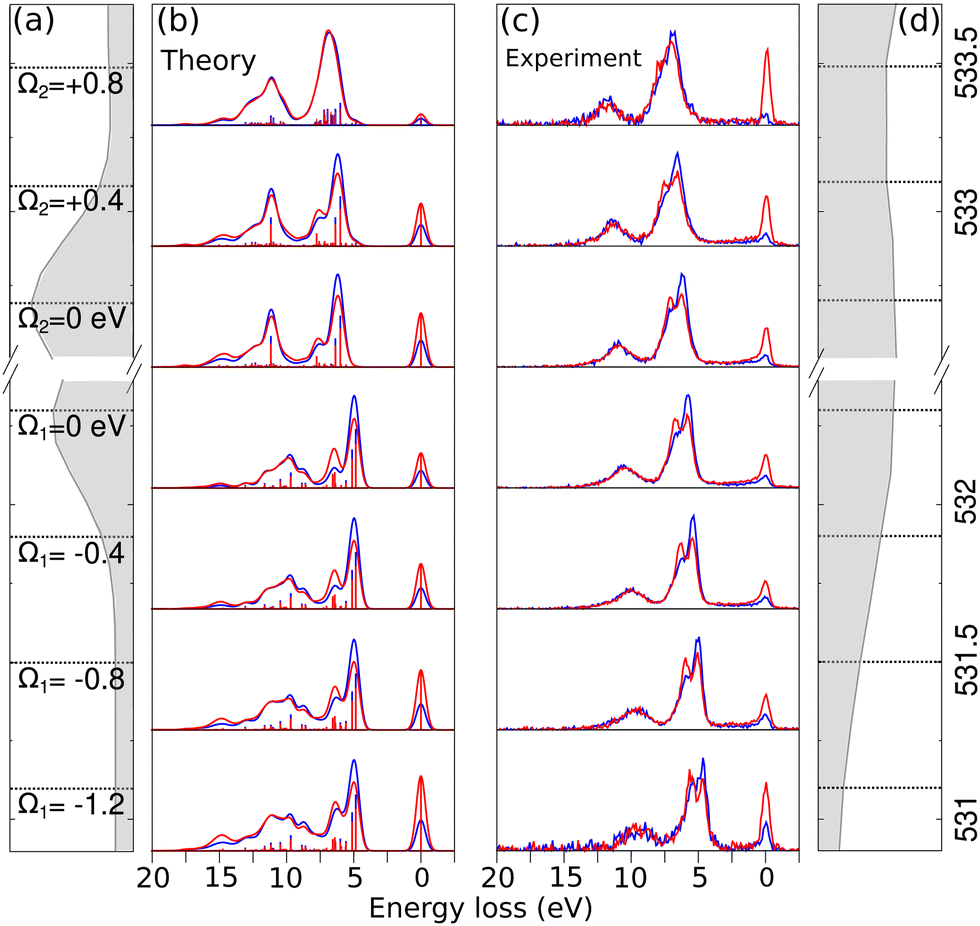 | ||
| Fig. 4 Comparison of theoretical (b) and experimental (c) normalized RIXS spectra of maleate for different excitation energy values. Panels (a) and (d) show the theoretical and experimental XAS, respectively, with dashed lines indicating the energy excitation (ω) and the corresponding detuning (Ω). The negative detuning is denoted as Ω1 from the first resonance (Ω1 = 0 eV), and the positive detuning is denoted as Ω2 from the second resonance (Ω2 = 0 eV). The position of the first resonant peak in panel (a) is shifted by +0.65 eV to the second resonance, in addition to the rigid shift specified in Section 2.2 in accordance with the experimental energy excitation values. | ||
The dependency of RIXS spectra on the detuning are also demonstrated for fumarate and succinate in ESI† (Fig. S6 and S7).
4 Conclusions
In conclusion, we have used X-ray absorption spectroscopy and resonant inelastic X-ray scattering at the O K-edge to investigate the electronic structures of fumarate, maleate, and succinate dianions, which are commonly used as bridging ligands. Our results reveal differences in the electronic states and molecular orbitals of the three molecules, which can be attributed to the unique molecular geometry and electronic structure of each dianion. In particular, the electronic structure of dicarboxylic acid dianions is modified by the presence or absence of a double bond between carboxylate groups and the orientation of the carboxylate groups relative to each other. Fumarate and succinate exhibit degenerate or nearly degenerate O 1s core-orbitals and due to the excitation to the single or several close-lying π* orbitals, a single XAS peak is observed for each of these molecules. In contrast, the maleate dianion has non-degenerate oxygen core-orbitals with an energy difference of approximately 0.15 eV. Consequently, in the calculated XAS for maleate, two distinct peaks are observed at around 532.85 eV and 531.65 eV. The separation between these peaks is influenced not only by the energy difference of the O 1s orbitals but also, and more predominantly, by the energy difference between the unoccupied excited π* orbitals.The RIXS measurements provide information on the orientation of the occupied valence molecular orbitals and enable a comparison of these emitting occupied valence orbitals in terms of in-plane and out-of-plane orientations with respect to their π* orbitals. The RIXS spectra of fumarate, maleate and succinate show a prominent double feature between 3.0–8.0 eV on the energy loss scale. The first inelastic peak in the RIXS spectra is more intense under horizontal polarization, while the second inelastic peak has stronger emission under the vertically polarized excitation. We also observed a gradually increasing energy gap between the HOMO and excited π* orbitals from fumarate to maleate to succinate, resulting in a shift of the emission band towards higher energy loss. The RIXS spectra evolve depending on the internal interaction between the two carboxylate groups and how they are twisted with respect to each other.
The degeneracy of delocalized orbitals of equivalent atoms in the system can result in degenerate or quasi-degenerate core-excited states, leading to a breakdown of selection rules embedded in RIXS simulation. In this study, we have demonstrated the detuning dependence of the RIXS spectra of maleate. Our analysis shows a good agreement between our theoretical predictions and the experimental results. The total theoretical and experimental inelastic RIXS profile of maleate shifts towards higher energy loss as the detuning is increased from negative to positive values. Additionally, we observed that the first two inelastic peaks in the RIXS spectra become almost indistinguishable at a detuning of Ω2 = +0.8 eV, as a result of similar emission decay intensities for both horizontal and vertical polarization.
Based on the results of the study obtained by XAS and RIXS we analysed the stability of fumarate, maleate and succinate molecules. Maleate is expected to be less stable than fumarate and succinate due to its asymmetrical charge distribution and the presence of electronic density on its HOMO orbital on the CC bond between carboxylate groups, which can lead to weaker bounding between maleate and molecules or ions. Fumarate and succinate, on the other hand, are more stable because their delocalized HOMO over the carboxylate groups and symmetrical charge distribution. Thus, use of XAS and RIXS, in combination with theoretical analysis, has provided valuable insight into the electronic structure and properties of dicarboxylate dianions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime.References
- L.-N. Zhu, Z.-P. Deng, S. W. Ng, L.-H. Huo and S. Gao, Dalton Trans., 2019, 48, 7589–7601 RSC.
- R. Sen, A. Halder and D. Ghoshal, Polyhedron, 2020, 183, 114534 CrossRef CAS.
- H. Mao, C. Zhang, G. Li, H. Zhang, H. Hou, L. Li, Q. Wu, Y. Zhu and E. Wang, Dalton Trans., 2004, 3918–3925 RSC.
- S. Basavoju, D. Boström and S. P. Velaga, Cryst. Growth Des., 2006, 6, 2699–2708 CrossRef CAS.
- G. A. Farnum, D. P. Martin, L. K. Sposato, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2010, 363, 250–256 CrossRef CAS.
- S. Demir, G. K. Kantar, Y. Topcu and Q. Li, Transition Met. Chem., 2012, 37, 257–263 CrossRef CAS.
- F. Baig, S. Khullar, S. K. Mandal and M. Sarkar, ChemistrySelect, 2017, 2, 11677–11685 CrossRef CAS.
- D. L. Nelson, A. L. Lehninger and M. M. Cox, Lehninger principles of biochemistry, Macmillan, 2008 Search PubMed.
- H. C. Erythropel, Evaluation of maleate, fumarate, and succinate diesters as potential green plasticizers, McGill University, Canada, 2016 Search PubMed.
- J. H. Schlander and T. Turek, Ind. Eng. Chem. Res., 1999, 38, 1264–1270 CrossRef CAS.
- J. M. Herbert and J. Ortiz, J. Phys. Chem. A, 2000, 104, 11786–11795 CrossRef CAS.
- P. Skurski, J. Simons, X.-B. Wang and L.-S. Wang, J. Am. Chem. Soc., 2000, 122, 4499–4507 CrossRef CAS.
- H.-T. Chen, J.-G. Chang, D. G. Musaev and M.-C. Lin, J. Phys. Chem. A, 2008, 112, 6621–6629 CrossRef CAS PubMed.
- M. Wanko, T. Wende, M. M. Saralegui, L. Jiang, A. Rubio and K. R. Asmis, Phys. Chem. Chem. Phys., 2013, 15, 20463–20472 RSC.
- H. Tateno, Y. Miseki, H. Kusama, S.-Y. Chen, T. Mochizuki and K. Sayama, ACS Sustainable Chem. Eng., 2021, 9, 6886–6893 CrossRef CAS.
- X. Xu, X. Zhang, X. Liu, T. Sun and E. Wang, Cryst. Growth Des., 2010, 10, 2272–2277 CrossRef CAS.
- S. Halder, A. Barma, C. Rizzoli, P. Ghosh and P. Roy, ACS Appl. Nano Mater., 2019, 2, 5469–5474 CrossRef CAS.
- Y. Gong, C. Hu, H. Li and W. Tang, Inorg. Chem. Commun., 2006, 9, 273–276 CrossRef CAS.
- L. C. Llanes, S. H. Clasen, A. T. Pires and I. P. Gross, Eur. Polym. J., 2021, 142, 110112 CrossRef CAS.
- H.-M. Ye, R.-D. Wang, J. Liu, J. Xu and B.-H. Guo, Macromolecules, 2012, 45, 5667–5675 CrossRef CAS.
- B. Bhattacharya, D. Saha, D. K. Maity, R. Dey and D. Ghoshal, CrystEngComm, 2014, 16, 4783–4795 RSC.
- A. Maiti, A. Halder, S. Dinda, G. Pahari and D. Ghoshal, Polyhedron, 2022, 227, 116144 CrossRef CAS.
- S. Datta and T. Limpanuparb, RSC Adv., 2021, 11, 20691–20700 RSC.
- S. Feuerbacher and L. S. Cederbaum, J. Am. Chem. Soc., 2003, 125, 9531–9537 CrossRef CAS PubMed.
- P. Atkins and J. De Paula, Physical chemistry for the life sciences, Oxford University Press, USA, 2011 Search PubMed.
- M. Blum, M. Odelius, L. Weinhardt, S. Pookpanratana, M. Bär, Y. Zhang, O. Fuchs, W. Yang, E. Umbach and C. Heske, J. Phys. Chem. B, 2012, 116, 13757–13764 CrossRef CAS PubMed.
- R. Golnak, K. Atak, E. Suljoti, K. F. Hodeck, K. M. Lange, M. A. Soldatov, N. Engel and E. F. Aziz, Phys. Chem. Chem. Phys., 2013, 15, 8046–8049 RSC.
- V. Savchenko, V. Ekholm, I. E. Brumboiu, P. Norman, A. Pietzsch, A. Föhlisch, J.-E. Rubensson, J. Gråsjö, O. Björneholm, C. Såthe, M. Dong, T. Schmitt, D. McNally, X. Lu, P. Krasnov, S. P. Polyutov, F. Gel’mukhanov, M. Odelius and V. Kimberg, J. Chem. Phys., 2021, 154, 214304 CrossRef CAS.
- S. Eckert, V. Vaz da Cruz, M. Ochmann, I. von Ahnen, A. Föhlisch and N. Huse, J. Phys. Chem. Lett., 2021, 12, 8637–8643 CrossRef CAS PubMed.
- F. Gel’mukhanov, M. Odelius, S. P. Polyutov, A. Föhlisch and V. Kimberg, Rev. Mod. Phys., 2021, 93, 035001 CrossRef.
- V. Vaz da Cruz, R. Büchner, M. Fondell, A. Pietzsch, S. Eckert and A. Föhlisch, J. Phys. Chem. Lett., 2022, 13, 2459–2466 CrossRef CAS PubMed.
- B. Dierker, E. Suljoti, K. Atak, K. M. Lange, N. Engel, R. Golnak, M. Dantz, K. Hodeck, M. Khan, N. Kosugi and E. F. Aziz, New J. Phys., 2013, 15, 093025 CrossRef CAS.
- E. Ertan, V. Savchenko, N. Ignatova, V. Vaz da Cruz, R. C. Couto, S. Eckert, M. Fondell, M. Dantz, B. Kennedy, T. Schmitt, A. Pietzsch, A. Föhlisch, F. Gel’mukhanov, M. Odelius and V. Kimberg, Phys. Chem. Chem. Phys., 2018, 20, 14384–14397 RSC.
- K. Kunnus, I. Rajkovic, S. Schreck, W. Quevedo, S. Eckert, M. Beye, E. Suljoti, C. Weniger, C. Kalus, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, K. J. Gaffney, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, S. Techert, P. Wernet and A. Föhlisch, Rev. Sci. Instrum., 2012, 83, 123109 CrossRef PubMed.
- A. Pietzsch and S. Eisebitt, JLSRF, 2016, 2, A54 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- V. Vaz da Cruz, S. Eckert and A. Föhlisch, Phys. Chem. Chem. Phys., 2021, 23, 1835–1848 RSC.
- S. Eckert, E. J. Mascarenhas, R. Mitzner, R. M. Jay, A. Pietzsch, M. Fondell, V. Vaz da Cruz and A. Föhlisch, Inorg. Chem., 2022, 61, 10321–10328 CrossRef CAS PubMed.
- Z. Yin, I. Rajkovic, S. T. Veedu, S. Deinert, D. Raiser, R. Jain, H. Fukuzawa, S. ichi Wada, W. Quevedo, B. Kennedy, S. Schreck, A. Pietzsch, P. Wernet, K. Ueda, A. Föhlisch and S. Techert, Z. Phys. Chem., 2015, 229, 1855–1867 CrossRef CAS.
- M. Nyberg, Y. Luo, L. Qian, J.-E. Rubensson, C. Såthe, D. Ding, J.-H. Guo, T. Käämbre and J. Nordgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 115117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures. See DOI: https://doi.org/10.1039/d3cp04348g |
| This journal is © the Owner Societies 2024 |