
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production and transport of plasma-generated hydrogen peroxide from gas to liquid
Steffen
Schüttler
 a,
Anna Lena
Schöne
b,
Emanuel
Jeß
a,
Andrew R.
Gibson
b and
Judith
Golda
a
a,
Anna Lena
Schöne
b,
Emanuel
Jeß
a,
Andrew R.
Gibson
b and
Judith
Golda
a
aPlasma Interface Physics, Ruhr University Bochum, Bochum, Germany. E-mail: steffen.schuettler@rub.de
bResearch Group for Biomedical Plasma Technology, Ruhr University Bochum, Bochum, Germany
First published on 15th February 2024
Abstract
In this work, the transport of hydroxyl radicals and hydrogen peroxide from a humid atmospheric pressure plasma jet into plasma-treated liquids is analysed. The concentration of H2O2 was measured by a spectrophotometric approach using the reagent ammonium metavanadate. OH was measured by the terephthalic acid dosimeter and the chemiluminescence of luminol. The plasma jet used is based on the design of the well-investigated COST reference jet and is extended by a capillary between the two electrodes. In addition to the experiments, the 0-dimensional plasma-chemical kinetics code GlobalKin was used to analyse the plasma chemistry in the gas phase in more detail. After 5 min plasma treatment, a maximum H2O2 concentration of 1 mM was found in the liquid, while the OH concentration was a factor 50 lower. The concentrations of both species in the liquid increased with plasma power, and the H2O2 concentration also increased with the humidity concentration of the feed gas, while the OH concentration first increased with humidity admixture and then decreased. The transport of both species could be controlled by the treatment distance, the gas flow rate and low frequency pulsing of the RF jet in such a way that the selectivity towards the long-lived species H2O2 was increased. Qualitative trends in the simulated number densities of gas phase H2O2 and OH at the location of the gas–liquid interface fit relatively well to the experimental measurements in the liquid.
1 Introduction
Atmospheric pressure plasmas offer opportunities for various applications such as nitrogen fixation,1,2 plasma medicine,3,4 bacteria inactivation5,6 and plasma-driven biocatalysis.7,8 These applications have the common feature that, plasma–liquid interactions play an important role.9,10 One class of atmospheric pressure plasma source with strong potential for application in these areas is the plasma jet. In these sources the plasma itself does not need to be in direct contact with the liquid with reactivity transfer occurring via the effluent gas. Here, reactive oxygen and nitrogen species (RONS) generated in the plasma and transported into the liquid are the main driver for the desired reactions within the liquid.11 For example, in plasma-driven biocatalysis, enzymes are located in a liquid treated by a plasma jet and are activated by the reactive species. One class of enzymes that work in plasma-driven biocatalysis are peroxygenases.7 Peroxygenases utilize hydrogen peroxide and perform oxyfunctionalization reactions such as enantioselective hydroxylations and epoxidations.12 As shown by Yayci et al., atmospheric pressure plasma jets offer a promising plasma source for plasma-driven biocatalysis using peroxygenases as enzymes.7 There, the H2O2 is produced by the plasma jet and is transported to the enzymes located in the liquid.The production of H2O2 in atmospheric pressure plasmas containing water has been extensively studied by several research groups in experiments,13–16 modelling17,18 and the combination of both.19–24 In the gas phase, the main production reaction for H2O2 is typically the three-body reaction of two OH radicals and a collision partner M, which can, in principle, be any atom or molecule:19,21
| OH + OH + M → H2O2 + M | (1) |
| OH + H2O2 → H2O + HO2 | (2) |
| H2O + e− → H + OH + e− | (3) |
H2O2 can be produced by an atmospheric pressure plasma and due to its stable character, can be transported into a liquid. In plasma-treated liquid systems, the origin of H2O2 may not be clear, as it can also be produced ex situ in the liquid environment. Gorbanev et al. investigated the origin of OH and H2O2 by using isotopically labeled water. In their plasma systems, they found that the origin of these reactive species was within the plasma region, with small amounts being formed from the humidity in the effluent region.21 Thus, in their plasma–liquid configuration, H2O2 is produced before it enters the liquid.
In this work, we use an extension of the well-investigated COST reference plasma jet,25,26 namely the capillary plasma jet.27 The features of this device mean that a wider range of plasma parameters (e.g. power and molecular gas admixture) can be used in comparison to the COST reference jet, allowing for a wider range of plasma properties that, in turn, lead to a wider range of possible properties of liquids treated by the source. For example, the COST reference plasma jet is limited to a plasma power of 2 W (power density of 67 W cm−3), and other plasma jets that have been studied for H2O2 formation have been similarly limited in their range of plasma powers (maximal power density of 14 W cm−3 in ref. 19). Using the capillary plasma jet, new regimes of plasma power (in this case up to 12 W with a power density of 300 W cm−3) can be accessed and their influence on the production of H2O2 and its delivery to the liquid can be investigated.
The concentration of aqueous H2O2 in the plasma-treated liquid is measured by a spectrophotometric approach using ammonium metavanadate. The concentration of OH is investigated by the terephthalic acid dosimeter and its distribution in the liquid is visualised by the chemiluminescence of luminol. In addition to experiments, 0-D plasma-chemical kinetics simulations are used to investigate the most important production and consumption rates and spatial profiles of the species along the gas flow channel in the plasma jet and its effluent. In addition, the distribution of OH in the liquid is visualised in a liquid treated by a plasma jet, leading to an advanced understanding of the transport processes. Furthermore, by combining the concentration measurements of H2O2 and OH with the simulation results, a comprehensive overview of the production and consumption of H2O2 and OH from the plasma to the liquid surface is obtained.
Based on the knowledge of the influence of plasma parameters on the production and delivery of H2O2 and OH, the effect of important treatment parameters such as treatment distance and gas flow rate on the species concentrations in the liquid and their evolution in the effluent are investigated. It is shown that the selectivity towards the long-lived species H2O2 can be increased remarkably by choosing appropriate treatment parameters, which is necessary for application purposes such as plasma-assisted biocatalysis, as explained above.
At the end, the effect of pulsing the RF-jet with low frequencies on the species concentrations in the liquid is discussed. This low frequency pulsing of an RF plasma jet to trigger the production of H2O2 and its delivery in a liquid was previously investigated by Vasko et al.19 They found the pulsing to have only minor effect on the H2O2 production. However, in that work only one pulsing frequency at various duty cycles was used. Here, it is shown that lower pulsing frequencies can trigger plasma chemistry and that both frequency and duty cycle can be used to control reactive species production.
2 Experiments
2.1 Atmospheric pressure plasma jet
The atmospheric pressure plasma jet used is based on the design of the well-investigated COST Jet25,28 and was previously investigated by Winzer et al.27 Between two plane parallel stainless-steel electrodes, a borosilicate glass capillary (CM Scientific) is placed with an inner square cross-section of 1 mm2 and a wall thickness of 0.2 mm. The length of the electrodes is 40 mm leading to a plasma volume of 1 mm × 1 mm × 40 mm with a distance of 1.4 mm between the two electrodes. The power was provided by an RF power generator (Coaxial Power Systems RFG 150) operating at a frequency of 13.56 MHz. It was connected via a matching network (Coaxial Power Systems MMN 150) to one electrode while the other electrode was grounded. The matching network provides an adjustable tuning of the impedance of the plasma source to avoid reflected power. The signal of the RF generator can be modulated by a second waveform generator (Tektronix AFG 3011). The modulation pulse used was a square wave with frequencies in the order of tens to thousands of Hz and duty cycles of 10% and 50%. Power measurements of the dissipated plasma power were performed as described by Golda et al.28 Briefly, the voltage was measured by a miniaturised voltage probe which is placed in parallel behind to the powered electrode. The calibration of the voltage probe was performed by a commercial high voltage probe (Tektronix P5100A). The calibration factor lay between 100 and 200. The current was measured via a 4.7 Ω resistor positioned between the grounded electrode and ground. The current is proportional to the voltage drop across the resistor by Ohm's law. The signal of the voltage probe and the voltage drop across the resistor were measured by a fast 10 GS s−1 oscilloscope (Teledyne LeCroy HDO6104A). The dissipated plasma power can then be calculated by P = U·I·cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Φ with the measured voltage U, the current I and the phase shift Φ between the voltage and the current, corrected by the reference phase shift of the system without plasma.
Φ with the measured voltage U, the current I and the phase shift Φ between the voltage and the current, corrected by the reference phase shift of the system without plasma.
The capillary plasma jet was operated in helium (purity 5.0). Mass flow controllers (Analyt MTC) were used to control the gas flow. If not otherwise noted 1 slm was used as gas flow rate. By use of an ice-cooled bubbler system filled with distilled water (Fisher Scientific), water vapor was added to the helium gas flow.13,20,21,29 The amount of water vapor in the feed gas could be adjusted by passing a certain amount of the gas flow through the bubbler vessel. The temperature in the bubbler vessel was measured to 1.40 °C ± 0.22 °C and was controlled for each experiment. Willems et al. measured the water vapor concentration in the feed gas of an atmospheric pressure plasma jet in operation with a similar bubbler system by mass spectrometry.20 Good agreement between the measured water vapour concentration and theoretically calculated at the measured temperature was found.20 Here, the water vapour concentration was calculated. The maximum water concentration in the feed gas was 6400 ppm ± 140 ppm.
2.2 Liquid treatment
The treatment of liquid samples was performed in UV cuvettes (Sarstedt polystyrene) that were placed beneath the plasma source. As liquid medium a 100 mM potassium phosphate buffer was used to ensure a stable pH value of 7.0 during the plasma treatments. 3 mL of buffer was filled in the cuvettes using a pipette (Eppendorf). To check that the same amount of buffer was filled in each cuvette, the weight of the cuvette was measured. Ten measurements showed a standard deviation of only 1%. Thus, it can be assumed that the amount of buffer was the same for each treatment. The distance from plasma end (end of the electrodes) to liquid surface was 24 mm. Between the end of the electrodes and the capillary end a gap of 10 mm was present. Thus, the plasma effluent was guided for 10 mm through the capillary, protecting it from ambient air. On the last 14 mm the effluent passed through ambient air to the liquid surface. The treatment time for the measurements was set to 5 min. To measure the evaporation of the liquid during treatment, the weight of ten cuvettes was measured. The difference in weight was only 3.3%, so that evaporation can be neglected.2.3 H2O2 concentration
To measure the H2O2 concentration, a spectrophotometric approach utilising an ammonium metavanadate (NH4VO3) solution was used. The ammonium metavanadate solution was prepared as described by Nogueira et al.30 and has already been used to measure the H2O2 concentration in a plasma treated liquid.19 The lower detection limit of this spectrophotometric method was found to be 143 μM.30 3.109 mL sulfuric acid (Fisher Chemical, >95%) was used to dissolve 725.26 mg ammonium metavanadate (therma scientific, 99.5%), which was then diluted with distilled water. The final solution had a pH of less than 3. The reaction of ammonium metavanadate with H2O2 results in a red-orange peroxovanadium cation solution with an absorption peak at 450 nm. The setup of the absorption measurement is part of Fig. 1. To measure the absorption, light from a laser-stabilised broadband light source (Energetiq EQ-99 LDLS) was used, which passed through the absorption medium in the cuvette during the plasma treatment. The measurement depth was 8 mm beneath the liquid surface to ensure that no disturbance from the liquid surface occurs in the beam path. The beam had a width of 1 mm, so the measurement was taken in a liquid volume of 1 mm × 10 mm × 10 mm. Spectra were recorded every 30 s using a spectrometer (Avantes Avaspec-ULS 2049×64 TEC-EVO) and the first spectrum was taken as a reference spectrum. The absorption is proportional to the H2O2 concentration c and follows the Beer–Lambert Law with the intensity I, the reference intensity I0, the absorptivity
with the intensity I, the reference intensity I0, the absorptivity  and the absorption length d. Calibration of the setup was performed using 30% H2O2 stock solution (Fisher Chemical). Calibration solutions of H2O2 with concentrations up to 5 mM were used and linear behaviour of the absorption with increasing H2O2 concentration was found.
and the absorption length d. Calibration of the setup was performed using 30% H2O2 stock solution (Fisher Chemical). Calibration solutions of H2O2 with concentrations up to 5 mM were used and linear behaviour of the absorption with increasing H2O2 concentration was found.
 | ||
| Fig. 1 Sketch of the plasma setup (upper part) and the setups of the liquid diagnostics (lower part). The involved devices of the different liquid diagnostics are marked by different borders: solid: spectrophotometric approach using ammonium metavanadate; dashed: TA dosimeter; dotted: detection of chemiluminescence of luminol. | ||
To validate the trends and the absolute values of the H2O2 measurements, the spectrophotometric approach was compared to measurements by electrochemical H2O2 sensing and was published in another work.31 Excellent agreement was found between the two diagnostics, allowing only the spectrophotometric approach to be used in the work presented here.
2.4 OH concentration
The hydroxyl radical OH is a precursor of H2O2 in the plasma chemistry from H2O to H2O2. Thus, knowledge of the OH production is crucial in order to optimize the H2O2 production. Two methods are explained in the following, one for measuring the OH concentration in the liquid and one for visualising the OH distribution at the liquid surface.The setup for detecting the CL is part of Fig. 1. Luminol (Serva Electrophoresis) was diluted in buffer solution to a concentration of 5 mM. The CL was measured using an iCCD camera (Andor iStar CCD DH334T-18U-E3). Since the CL spectrum peaks at 425 nm,41,42 a filter at 424 nm with a FWHM of 12 nm was used in front of the camera. The exposure time was set to 10 s, so the CL signal was accumulated during the first 10 s of the plasma treatment. An image of the buffer solution without addition of luminol under plasma treatment was taken as a background image for each plasma condition to exclude possible light reflection at the liquid surface by the plasma emission.
In another work, both diagnostics for measuring OH were directly compared to ensure a high selectivity towards OH of the diagnostics.31 A very good agreement between the two diagnostics was found for this plasma–liquid system. Thus, although luminol is mainly excited by O2−, it can be used to visualise the distribution of OH in the liquid.
2.5 Uncertainty estimation
Several sources of uncertainties exist in the plasma treatment of the liquid and the liquid diagnostics. Regarding liquid diagnostics, the calibration of the spectrophotometric method and the TA dosimeter might be prone to uncertainties. Fits of the calibration curves and a repetition of the calibrations showed uncertainties of less than 1%. The reproducibility of the measurements for five consecutive measurements also showed a low uncertainty of less than 1%. The setup for detecting the chemiluminescence of luminol also showed stable reproducibility with daily repetitions. The biggest source of uncertainty was given by the arrangement of the plasma source. By changing the setup, the plasma source was rearranged so that the capillary was parallel between the two electrodes. A small tilt of the capillary resulted in a different ignition behaviour of the plasma. In the best parallel alignment, the plasma ignites homogeneously distributed and the main emission occurs in the center of the discharge gap. If the alignment was not perfect, the strongest emission occurs at the gas inlet or gas outlet of the discharge channel, which negatively influenced the behaviour of the plasma. The alignment of the plasma source was attempted to be as parallel as possible so that the strongest plasma emission occurred in the centre of the discharge gap. Measuring the plasma power ensured that the same treatment conditions prevailed after the plasma source was arranged. The reproducibility of the measurements when the plasma source was rearranged showed an uncertainty of 15% for five consecutive measurements. In order to compare the results, this uncertainty was chosen for the results of the spectrophotometric approach shown in this paper using ammonium metavanadate and the TA dosimeter.2.6 Plasma-chemical kinetics model
The plasma-chemical kinetics in the capillary plasma jet are simulated using the zero-dimensional plasma-chemical kinetics code GlobalKin44 to get deeper insights into reactions and species evolution in the system. GlobalKin solves the mass continuity equation, including gas-phase and surface loss processes for charged and neutral particles. The electron temperature is calculated using the electron energy conservation equation considering elastic and inelastic collisions and the power deposited in the plasma. Additionally, the electron energy distribution function and electron impact rate and transport coefficients are calculated internally using the two-term approximation of the Boltzmann equation. The energy balance equation for the neutral gas is also solved to determine the gas temperature. As an input for the simulation, the wall temperature of the capillary at the end of the discharge channel was measured using a thermocouple (Voltcraft, K-type). The gas temperature was measured as described in ref. 27 with the same thermocouple and was compared to the gas temperature obtained from the model. A good agreement between measured and simulated gas temperature was found (not shown here). The gas temperature increased linearly with plasma power (not shown here) and varied from 330 K at 1 W plasma power up to 500 K at 12 W.In this work, spatial changes of species concentrations along the direction of the gas flow are of interest to compare the experimentally measured results in the liquid with the gas-phase simulations. Hence, a pseudo-1D plug flow model is used to relate the temporal variation of species concentrations to their spatial variation along the gas flow using the gas flow velocity. In the figures following, a negative position corresponds to the plasma area located inside the jet, from the end of the electrodes to the gas inlet. Positive values represent the effluent region, meaning the distance between the end of the electrodes and the liquid surface. Unless otherwise mentioned, in this work, the last value of the gas-phase simulation, directly at the liquid surface, is compared to the experimental results in the liquid. The simulation includes charged and neutral species. Since a cross-field plasma jet was used, charged particles are mainly present in the discharge channel and not in the effluent. The simulation showed (not shown here) that only neutral species reach the distance of 24 mm where the liquid surface is present. Thus, only neutral species were analysed at this position. It should be emphasised that the liquid itself is not included in the simulations. In addition, any reduction in the gas flow velocity as the gas exits the capillary and any mixing with the ambient air are also not included. In reality, the gas velocity will decrease upon exiting the capillary, and a certain degree of mixing with the ambient air will occur. Therefore, these simplifications in the model could lead to an overestimation of the concentration of highly reactive species like OH at the liquid surface as the time required for reactive species to reach the liquid surface will be underestimated and any potential reactions with molecules in the ambient air are omitted. However, given the relatively high gas velocities and the proximity of the capillary nozzle to the liquid surface, we do not expect these effects to have a major impact on the trends observed in this work.
The chemistry is simulated using a reaction scheme for helium–water vapour mixtures that has been well-investigated for similar sources without dielectric capillary between the electrodes.18,22,23,45 The reaction scheme contains 46 species and around 600 reactions.
3 Results and discussion
3.1 H2O2 concentration in plasma-treated liquid
To optimise the H2O2 concentration in the plasma-treated liquid, the effects of feed gas humidity and dissipated plasma power on the H2O2 production were investigated. For all measurements, the H2O2 concentration shows a linear increase over time (not shown here). This is in good agreement with previously published work, as the linear temporal evolution of the H2O2 concentration in plasma-treated liquids has been observed in many experiments.14,15,19,36In Fig. 2(a) the H2O2 concentration in the liquid after plasma treatment as a function of feed gas humidity is shown. Trends at two different plasma powers, a low plasma power at 1 W and a high plasma power at 6 W, are displayed. With increasing humidity in the feed gas, the H2O2 concentration measured in the liquid rises for both investigated plasma powers. At 0 ppm humidity of the feed gas, the H2O2 concentration is less than 0.05 mM for both plasma powers. Some water impurities in the gas pipes might lead to a small H2O2 production even without additional water vapor admixture. Up to a humidity of 1500 ppm, the H2O2 concentration increases linearly to a concentration of 0.2 mM and 0.5 mM for 1 W and 6 W plasma power, respectively. At higher humidity, the concentration rise weakens and values of (0.30 ± 0.05) mM at 1 W plasma power and (0.93 ± 0.14) mM at 6 W plasma power are reached at the maximum humidity of 6400 ppm.
 | ||
| Fig. 2 H2O2 concentration for (a) varying humidity concentration at two plasma powers and (b) varying plasma power at two humidity concentrations. Dashed lines correspond to densities obtained from model results. The measurements correspond to the left axes labels and refer to concentrations in the liquid. The simulated results correspond to the right axes labels and refer to densities in the gas phase at the location of the liquid surface. | ||
Considering the main pathway in the production of H2O2 from dissociation of H2O to form OH viareaction (R3) and the recombination of OH to H2O2 according to reaction (1), the increasing trend in H2O2 concentration with increasing humidity of the feed gas is comprehensible. This trend has also been observed by other research groups in plasma-treated liquids using atmospheric pressure plasma jets.14,19,21 There, an initial linear increase and then a flattening of the H2O2 concentration was also observed, and the break of the linear correlation was explained by loss of H2O2 due to an increasing amount of OH at higher humidity, since OH is also the main consumer of H2O2viareaction (2).19,21 A lower electron density at high admixtures of reactive gases might also affect the H2O2 production.21
In Fig. 2(a) the model results are also shown. A good agreement between the trend in the measured H2O2 concentration in the liquid and the H2O2 density in the model as a function of the feed gas humidity at a plasma power of 6 W is observable. The H2O2 density obtained from the simulation reaches values of up to 0.8 × 1015 m3 at a humidity of 6400 ppm. In case of 1 W plasma power, the H2O2 density is only a few percent lower compared to the density in the 6 W case. In the experiment, on the other hand, the H2O2 concentration is lower by a factor of three for the plasma treatment with 1 W than for the treatment with 6 W. There, the simulation and experiments differ from each other. This is particularly visible when looking at the trends in H2O2 concentration or density as a function of the dissipated plasma power (Fig. 2(b)). In the experiment, with increasing plasma power the H2O2 concentration rises linearly until a plasma power of 4 W and saturates for higher plasma powers. For plasma powers higher than 6 W, a plateau in the H2O2 concentration arises. This behaviour is present both at low and high humidity of the feed gas. As the plasma power increases, the electron density in atmospheric pressure plasma jets also increases.46,47 An enhanced electron density leads to an enhanced electron impact dissociation of water to OH which in turn leads to a higher H2O2 production. The saturation at high plasma powers might be due to too high OH density in the plasma so that the destruction of H2O2 by OH might play a more important role. Furthermore, the temperature of the plasma also increases significantly for high plasma powers.27 This can negatively affect the plasma chemistry for the production of H2O2.
In contrast to the experiments, the simulation shows an initial increase with a maximum H2O2 density at around 3 W and then a decreasing trend. Possible reasons for this discrepancy between experiments and simulations might be due to the rising gas temperature with increasing power. A number of the rate coefficients related to the production and consumption of H2O2 are gas temperature dependent, and the deviation in the trends as a function of plasma power may be indicative of the uncertainty in the temperature dependence of these rate coefficients.
The H2O2 production is dominated by the three-body recombination of OH (reaction (1)) as visualised in Fig. 3(a). Here, the main pathways of (a) production and (b) consumption over space are shown for a plasma power of 6.2 W and a humidity of 6400 ppm, simulated using the pseudo-1D plug flow model. The main consumption, shown in (b), occurs by reacting with OH (reaction (2) → c1), and also, but less common, with H (c3, c4) or by electron impact dissociation (c2). Overall, the production rate of H2O2 reaches a maximum value at the beginning of the jet, at around 37 mm, after a linear increase and later on decreases exponentially by reaching a constant production rate within the jet. This behaviour is caused by a longer-lasting increase of H2O2 consumption reactions, also reaching a constant value, and results in a steady state. In the effluent, the production rate decreases steeper than most consumption rates, resulting in a slightly decreasing H2O2 density.
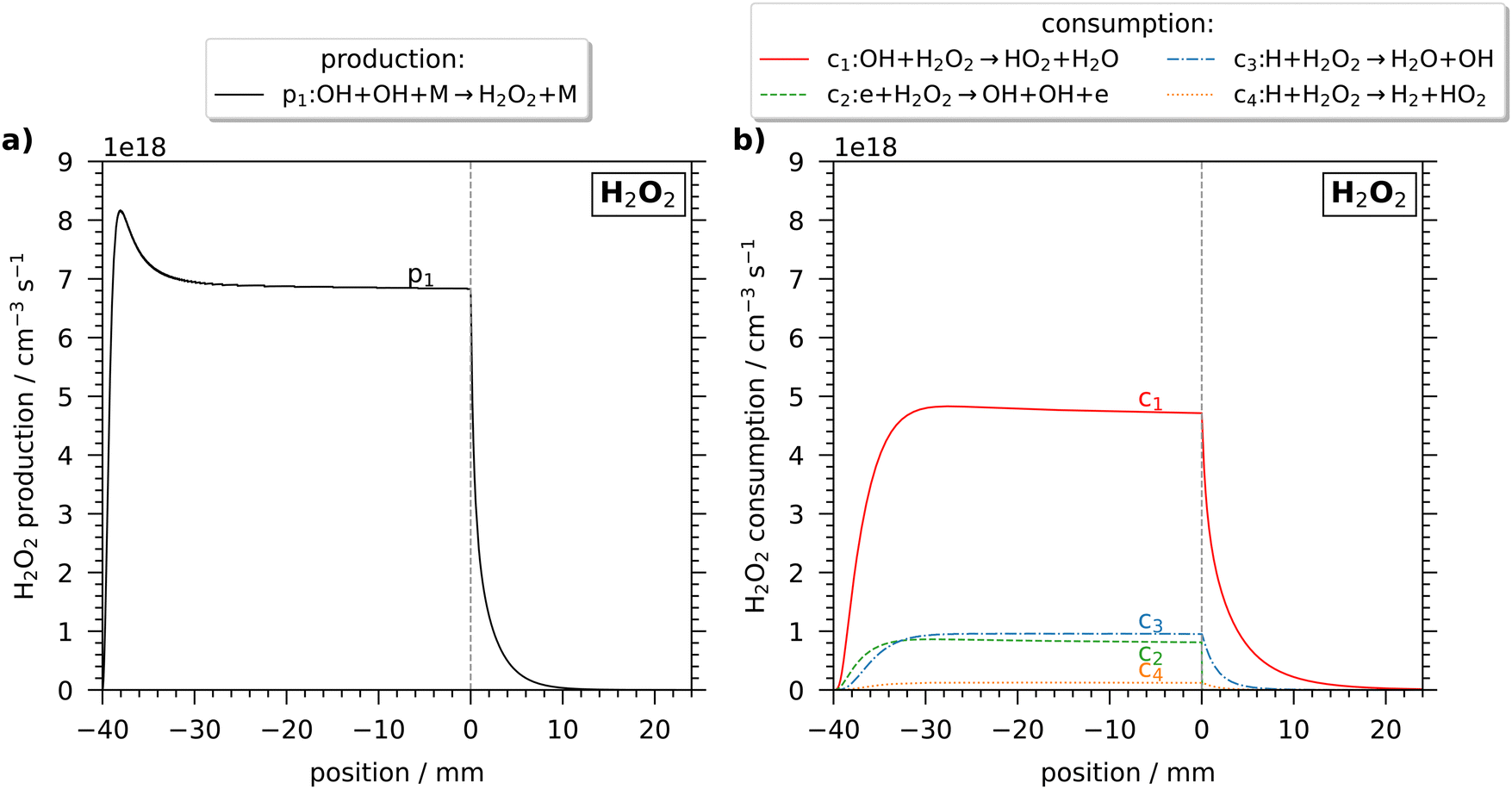 | ||
| Fig. 3 Spatial variation of the reaction rates of the main (a) production and (b) consumption reactions of H2O2 in the jet and the effluent. Simulations were performed for a humidity of 6400 ppm and a plasma power of 6.2 W using the pseudo-1D plug flow model. Negative positions correspond to positions within the discharge channel, at 0 mm the discharge channel ends and the liquid surface is placed at a distance of 24 mm. | ||
In comparison to the COST jet, Yayci et al. also found a linear relationship between the H2O2 concentration and the applied voltage.7 However, the highest applied voltage of the COST jet corresponds only to a plasma power of less than 2 W and the influence of higher plasma powers on the H2O2 production cannot be investigated using the COST jet. Using another jet, the production of H2O2 could be measured up to a power of 3 W, and this also followed a linear trend up to high powers,19 which was also observed in this work using the capillary jet.
Assuming that all H2O2 molecules enter the liquid, the gas-phase H2O2 density required to yield the concentrations measured in the liquid can be calculated from the measured H2O2 concentration. Since the Henry's law solubility constant of H2O2 is high,48 this approximation is appropriate. Multiplying the concentration with the liquid volume V and Avogadro's constant provides the total number of H2O2 molecules arriving at the liquid surface during treatment. In more general terms, this is the flux of H2O2 molecules arriving at the liquid surface during the treatment period. Dividing the total number of H2O2 molecules by the gas flow rate ϕ and the treatment time T results in the required density of H2O2nH2O2 to achieve the measured concentration.
 | (4) |
The maximum achievable H2O2 concentration in the liquid using the capillary plasma jet was 1 mM after 5 min treatment. This corresponds to a production rate of 600 nmol min−1. Compared to the COST jet, the maximum production rate of the capillary plasma jet is higher. Yayci et al. reported an H2O2 production rate ranging from 200 nmol min−1 to 300 nmol min−1.7. Other research groups also found this production rate for plasma-treated liquids by the COST jet.15,21,36 The main advantage of the capillary plasma jet over the COST jet is the possibility to apply higher plasma powers. The dissipated plasma power in the COST jet is limited by the transition to constricted mode, which occurs at a power of about 1 W to 2 W.25,49 The H2O2 production rate of the capillary jet at 1 W is also 200 nmol min−1. Thus, the capillary plasma jet is comparable to the COST jet in H2O2 production, but has the advantage of being able to operate at higher plasma powers, resulting in a higher production rate.
3.2 OH concentration in plasma-treated liquid
Since OH is also produced in the plasma and it is involved in the production and destruction of H2O2, its concentration in the plasma-treated liquid is important to consider and was measured by the TA dosimeter. It should be emphasised again that the TA dosimeter measures the time-accumulated HTA concentration due to the reaction between TA and OH. The measured HTA concentration can be viewed as proportional to the OH concentration, but does not reflect the instantaneous OH concentration. Thus, when discussing the OH concentration, the time-accumulated HTA concentration, which is corrected by the capture efficiency of TA explained in the experiments, is considered and not an instantaneous OH concentration in the liquid.Similar to the investigations of the H2O2 concentration, the OH concentration measured in the liquid after plasma treatment is shown in Fig. 4. With increasing humidity, an initial steep increase in OH concentration is visible. At a humidity of 640 ppm, the maximum concentration is reached with values of 0.01 mM and 0.025 mM for 1 W treatment and 6 W treatment, respectively. Thus, the accumulated OH concentration measured in the liquid is of the order of ten micromolar, which agrees well with previous studies using the COST reference jet.21,50 At a higher humidity, the OH concentration slightly decreases. The same general trend can be seen in the OH density obtained from the simulation. There, the OH density peaks at a slightly lower humidity of less than 500 ppm. As described above, a higher humidity leads to an enhanced production of OH radicals in the plasma due to electron impact dissociation of water, which also enter the liquid. At a higher humidity, the consumption of OH becomes dominant and the concentration in the liquid no longer increases or even decreases slightly. The described trend was also observed by Gorbanev et al. as they measured the OH concentration by DMPO spin trap in a plasma-treated liquid.21
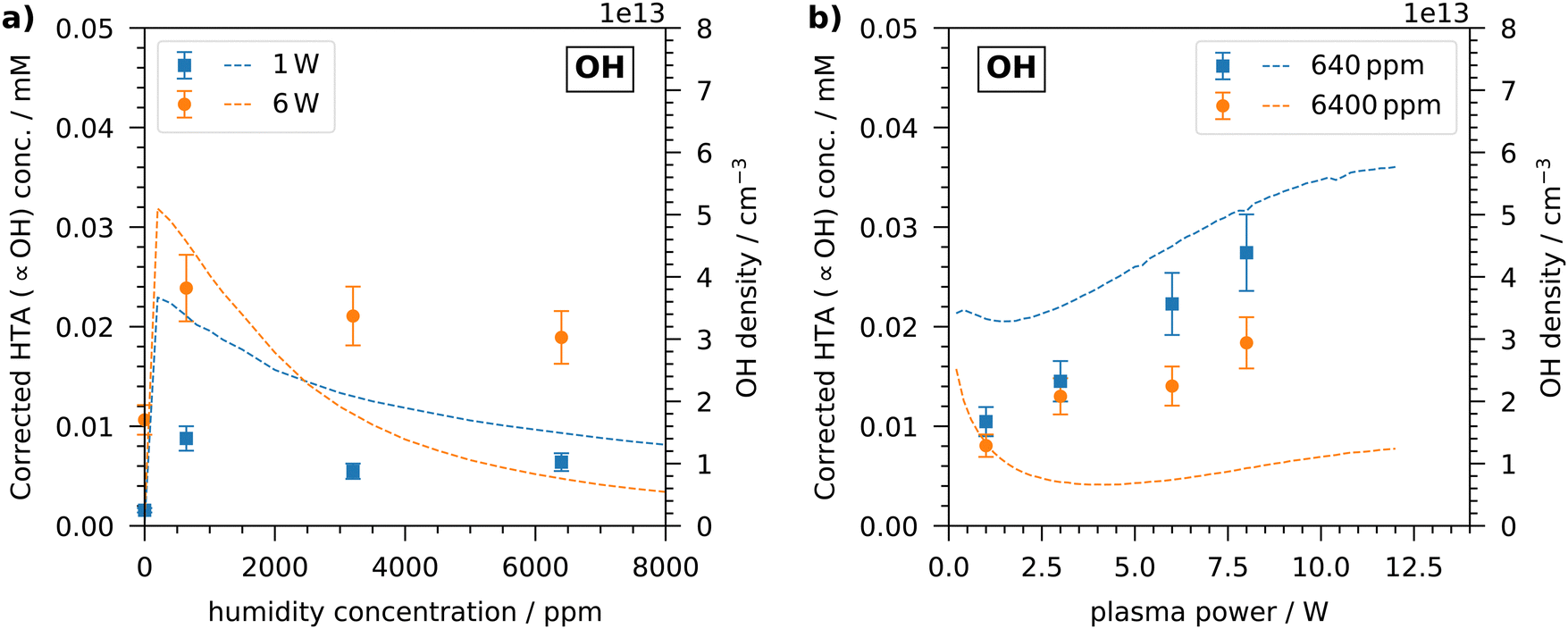 | ||
| Fig. 4 OH concentration for (a) various humidity concentrations at two plasma powers and (b) various plasma powers at two humidity concentrations. Dashed lines correspond to densities obtained from model results. The measurements correspond to the left axes labels and refer to concentrations in the liquid. The simulated results correspond to the right axes labels and refer to densities in the gas phase at the location of the liquid surface. It should be emphasised that the measured time-accumulated corrected HTA concentration in the liquid is proportional to the OH concentration, but does not reflect an instantaneous OH concentration in the liquid. | ||
At all investigated humidity concentrations, the OH concentration is higher for the treatment at 6 W plasma power than for the treatment with 1 W. In the simulation, however, this is only present for humidity concentrations up to 3000 ppm. At a higher humidity, the OH density obtained from the simulation is higher for low plasma powers. This discrepancy can be seen more clearly in Fig. 4(b). In the experiments, with increasing plasma power the OH concentration increases steadily and linearly at both low and high humidity. On the other hand, in the simulation the OH density initially decreases up to plasma powers of 2 W and increases at higher plasma powers. This is more pronounced at a high humidity of 6400 ppm, but can also be observed at a low humidity.
The behaviour of production and consumption rates of OH is depicted in Fig. 5 for the case of a plasma power of 6.2 W and humidity concentration of 6400 ppm. OH has four main production mechanisms (Fig. 5(a)), in the jet dominated by the electron impact dissociation with water molecules (reaction (3) → p1) and in the effluent by a reaction between H and HO2 (p2). Because of their lower densities, water reactions with H2O+ and O* are less common. OH is mainly consumed by producing H2O2 (reaction (1) → p1), as well as O and water (p2). But also by reactions with, especially in the effluent, less common species, as O and H (c3, c4). At the end of the plasma region, the reaction with atomic oxygen becomes the most relevant, caused by the increasing number of O atoms gained in the plasma. All four consumption reactions are still relevant at the beginning of the effluent and therefore cause a decrease of OH. The strong spatial dependence of the formation of OH by reactions of H and HO2 and its consumption by O indicates that there may be the possibility to control the efficiency of OH delivery to targets through varying the residence time of the gas in the plasma channel, as the densities of HO2 and O take some time to build within the plasma, as shown later in 6.
 | ||
| Fig. 5 Spatial variation of reaction rates of the main (a) production and (b) consumption reactions of OH in the jet and the effluent. Simulations were performed for a humidity of 6400 ppm and a plasma power of 6.2 W using the pseudo-1D plug flow model. Negative positions correspond to positions within the discharge channel, at 0 mm the discharge channel ends and the liquid surface is placed at a distance of 24 mm. | ||
Following the calculations explained before, the gas-phase OH density at the liquid surface can be estimated from the measured OH concentration in in the liquid. The highest concentration measured is 0.025 mM, which corresponds to an OH density of 9 × 1012 cm−3. In the simulation, the OH density is 5 × 1013 cm−3 at the respective plasma parameters. Thus, the simulation shows a five times higher OH density. Considering the fact that the TA dosimeter does not capture all OH radicals and the low Henry's law solubility constant of OH,51 which means that not all OH radicals enter the liquid, the higher density obtained in the simulation is reasonable. Additional insight can be obtained by looking at the ratios between the H2O2 and OH densities and concentrations from both experiment and simulation. These are shown in Table 1. In the experiments, the H2O2 concentration at a humidity of 6400 ppm is about 50 times higher than the OH concentration. The densities predicted in the simulation show similar ratios, albeit with a stronger dependence on the plasma power. At a humidity of 640 ppm the ratio varies between 13 and 19 in the experiment, while the simulation shows a factor of 4. Considering all the differences between experiment and simulation, this deviation is understandable, however, the trend towards higher H2O2 selectivity with higher humidity is evident in both. A comparison with the data of Gorbanev et al. shows that a ratio of about 20 at low humidity and 40 at higher humidity was also achieved using the COST Jet.21
| c H2O [ppm] | P [W] | c H2O2 [mM] | c OH [mM] |
|
|
n OH [1013 cm−3] |
|
|---|---|---|---|---|---|---|---|
| 640 | 1 | 0.170 ± 0.024 | 0.0088 ± 0.0013 | 19 | 0.15 | 3.7 | 4 |
| 6 | 0.30 ± 0.05 | 0.024 ± 0.004 | 13 | 0.21 | 5.1 | 4 | |
| 6400 | 1 | 0.30 ± 0.05 | 0.0064 ± 0.0009 | 47 | 0.75 | 1.7 | 44 |
| 6 | 0.93 ± 0.14 | 0.0189 ± 0.0027 | 49 | 0.82 | 1.1 | 75 | |



To obtain a deeper insight into the plasma chemistry, Fig. 6 shows the simulated spatial variation of densities of neutral species related to the main OH formation and consumption reactions. The water vapour content stays almost constant for (a) 640 ppm and (b) 6400 ppm water content, within the plasma area and the effluent, respectively. O2, O and HO2 increase within the whole length of the active plasma region, steeper at the beginning, up to the end of the electrodes, while the densities of H, OH and H2O2 also increase in the first part of the plasma area, but then reach saturation. In the effluent, the H2O, O2, H2O2 and HO2 densities are nearly constant, especially compared to the more reactive species H, OH and O. The case in (b) with 6400 ppm humidity content shows steeper increases, steeper decreases and higher maximal species densities, as well as lower densities at the liquid surface for the species that decrease in the effluent, compared to the 640 ppm case in (a). The density of charged particles such as the superoxide radical O2− in front of the liquid surface was found to be negligible and accordingly not shown in the spatial profile.
 | ||
| Fig. 6 Spatial variation of the densities of neutral species related to OH production and consumption for a humidity of (a) 640 ppm and (b) 6400 ppm. Simulations were performed for a plasma power of 6.2 W using the pseudo-1D plug flow model. Negative positions correspond to positions within the discharge channel, at 0 mm the discharge channel ends and the liquid surface is placed at a distance of 24 mm. | ||
3.3 Distribution of reactive species at the liquid surface
The aforementioned measurements of OH concentration are volume-averaged measurements in the liquid. Since OH is highly reactive, its interaction at the liquid surface is of particular interest. To this end, the chemiluminescence of luminol can be used to visualise reactive species such as OH. In Fig. 7 the chemiluminescence signal during plasma treatments with various humidity concentrations at plasma powers of 1 W and 6 W is shown. In all images, a clear chemiluminescence signal is visible at the liquid surface at a height of 9 mm with a bright spot in the center at a width of 8 mm to 9 mm. The 1 mm width of the bright spot is equal to the width of the capillary and is located exactly at the position where the capillary faces the liquid surface. To the side of the bright spot, the chemiluminescence signal becomes weaker. In the center, the signal extends up to 3 mm into the liquid but weakens considerably after the first millimeter. At the sides, the signal only extends for less than 2 mm into the liquid. This profile of the chemiluminescence signal at the liquid surface is visible for all plasma treatments. Some differences in intensity are noticeable by changing the plasma parameters, while the shape remains the same. With increasing humidity, the intensity of the chemiluminescence signal initially increases, has a maximum at a humidity of 640 ppm and decreases at higher humidity concentrations. This trend is observed for both plasma powers, however, the intensity of the chemiluminescence signal is four times higher in case of the 6 W than for a plasma power of 1 W. | ||
| Fig. 7 Chemiluminescence of luminol under plasma treatment for various humidity concentrations (columns). Upper row: Treatment with plasma power of 1 W; Lower row: Treatment with plasma power of 6 W. Colour bars apply to all images in a row. Note the different scales of the colour bars per row. | ||
The increase in intensity of the chemiluminescence signal with increasing plasma power can be seen more clearly in Fig. 8. With increasing plasma power from 1 W to 8 W, the intensity of the signal increases clearly and steadily.
 | ||
| Fig. 8 Chemiluminescence of luminol under plasma treatment for various plasma powers. Measurements were taken at a humidity concentration of 640 ppm. Colour bar applies to all images. | ||
Both trends obtained by the chemiluminescence of luminol agree very well with those obtained with the TA dosimeter.
3.4 Transport phenomena
All previously shown measurements are performed at a fixed distance between plasma and liquid surface and at a constant gas flow rate. However, the transport of the reactive species is strongly dependent on the mechanisms that occur during the transport from the plasma to the liquid. The effects of a longer transport path and gas flow rate on the concentration of reactive species in the liquid are discussed in the following. | ||
| Fig. 9 Effect of treatment distance (distance to the liquid surface) on species concentration in the liquid for (a) H2O2 and (b) OH. Measurements were taken at a humidity of 6400 ppm and a plasma power of 6 W. Dashed lines correspond to densities obtained from model results in the gas-phase. It should be emphasised that the measured time-accumulated corrected HTA concentration in the liquid is proportional to the OH concentration, but does not reflect an instantaneous OH concentration in the liquid. | ||
On the other hand, the OH radical is more affected by a change in distance. Both the measured concentration and the simulated density of OH show an exponential decay with increasing distance. In the experiments, no OH can be measured at distances greater than 35 mm. A longer transport path from the plasma to the liquid leads to more time for reactive species to react in the effluent. The highly reactive OH radical is strongly affected by this longer reaction time and is consumed faster in the effluent than H2O2. The effect of diffusion might also be visible, since no OH can be measured at 42 mm in the experiment, but a low density is still visible in the simulation. However, this effect is not as clearly visible as it is the case for H2O2, since the OH distribution is more affected by chemical reactions than by diffusion.
The simulation results in Fig. 10 show the main production and consumption reactions of OH in the effluent. Fig. 10(a) shows a change from p1 to p3 as the dominant production reaction at about 30 mm and a steep decrease of p1 and p2 compared to the most consumption reactions in (b). The two main consumption reactions in Fig. 10(b), c1 and c2 also decrease, as all production and consumption reaction rates, but slower than all production rates. Overall, the total consumption rate in the effluent is larger than the production rate, leading to a strong decrease in the OH density. In general, both the production and consumption rates of OH depend on relatively long-lived species that do not decay rapidly in the effluent, such as HO2 and H2O2, as well as short-lived species such as H and O whose densities decay quickly in the effluent (see Fig. 6). With increasing distance from the active plasma region the reactions involving short-lived species decrease rapidly such that reactions involving longer-lived species dominate the OH consumption at larger distances. It is also visible that the main production mechanism (reaction (1) → c4) and the main loss mechanism (reaction (2) → c2) for H2O2 are still present in the effluent, while the main production reaction of OH in the plasma region (reaction (3)) is not present in the effluent as the electron density decreases rapidly when transitioning between the active plasma and the effluent most common reactions in the effluent.
 | ||
| Fig. 10 Reaction rates of the main (a) production and (b) consumption reactions of OH in the effluent. Simulations were performed for a humidity of 6400 ppm and a plasma power of 6.2 W using the pseudo-1D plug flow model. | ||
The distribution of OH visualised by the chemiluminescence with the plasma source at various distances from the liquid surfaces is shown in Fig. 11. The highest intensity is visible at the lowest distance of 14 mm. With increasing distance, the signal becomes weaker, which is consistent with the measurements of the OH concentration. Furthermore, the widening of the gas flow by diffusion can be observed. The bright spot in the center of the liquid surface can again be observed at a distance of 14 mm. With increasing distance, the bright spot disappears and at a distance of 34 mm, a uniformly distributed signal appears at the liquid surface. The depth of the chemiluminescence signal also decreases with increasing treatment distance.
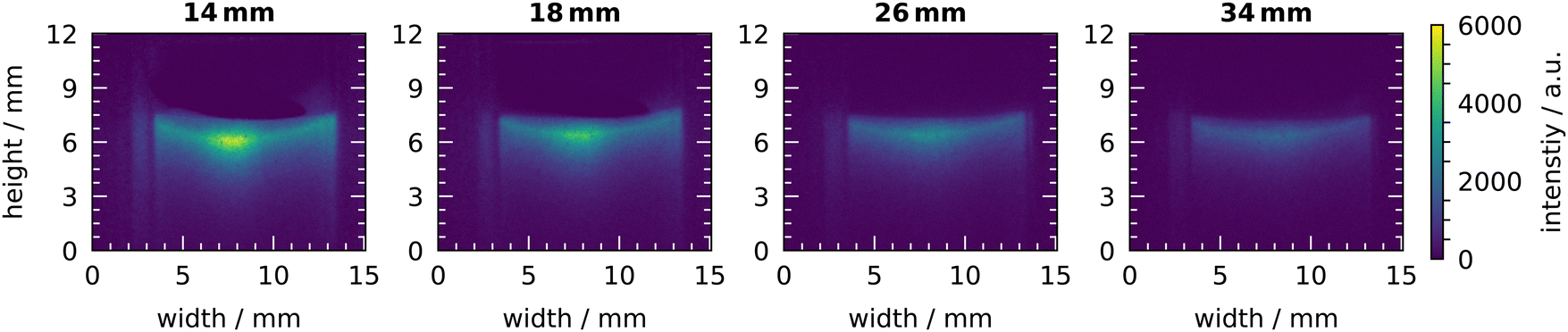 | ||
| Fig. 11 Chemiluminescence of luminol under plasma treatment for various treatment distances. Measurements were taken at a humidity of 640 ppm and a plasma power of 6 W. | ||
All in all, the treatment distance can be used to enhance the selectivity towards H2O2. At the shortest distance of 15 mm, the H2O2 concentration is fifty times higher compared to OH. At 25 mm this factor increases to 450 and at higher distances than 35 mm only H2O2 can be detected in the liquid.
 | ||
| Fig. 12 Effect of gas flow rate on species concentration in the liquid for (a) H2O2 and (b) OH. Measurements were taken at a humidity of 640 ppm and a plasma power of 6 W. Dashed lines correspond to particle fluxes obtained from model results in the gas-phase. It should be emphasised that the measured time-accumulated corrected HTA concentration in the liquid is proportional to the OH concentration, but does not reflect an instantaneous OH concentration in the liquid. | ||
Thus, the two species behave differently when the gas flow rate is changed. The correspondence between the H2O2 particle flux and the measured H2O2 concentration implies that all H2O2 molecules arriving at the liquid surface enter the liquid. This is consistent with the very high Henry's law solubility constant of H2O2, corresponding to a high solubility. On the other hand, OH has a Henry's law solubility constant that is three orders of magnitude lower.14,52 Thus, not all OH radicals that are in front of the surface might enter the liquid. This can be seen from the measurements, as the increase flattens out at high gas flow rates, while the simulation shows an increase in OH particle flux. It can be concluded that the concentration of H2O2 in the liquid follows the H2O2 flux from the plasma to the liquid, while the concentration of OH in the liquid is more dependent on the density of OH in front of the liquid.
The distribution of OH on the liquid surface at different gas flow rates is shown in Fig. 13. At 1 slm, the known shape of the centred bright spot is visible with a lower signal at the sides. This is also the shape for the gas flow rates of 0.5 slm and 1.5 slm, however, the intensity is slightly higher at 0.5 slm and slightly lower at 1.5 slm. In case of the 1.5 slm gas flow rate, the beginning of a change in the shape of the liquid surface can be recognized. In the center, where the gas flow hits the surface, a small dip appears. This dip is strongly pronounced at the high gas flow rate of 2 slm. There, the shape of the liquid surface has changed completely and is no longer flat. In the center, the dip reaches up to 4 mm deep. The intensity has also changed and is uniformly distributed on the liquid surface. The change in intensity might be due to a change of the area of the liquid surface. At 0.5 slm the liquid surface is nearly undisturbed. Thus, the surface has an area of 100 mm2 and the dominant signal originates from the center with an area of only 1 mm2. At 2 slm, the liquid surface has changed and shows a more cone-like shape. Thus, the volume from which the chemiluminescence is emitted has increased significantly. This lowers the total volume-averaged intensity and might explain the lower intensity.
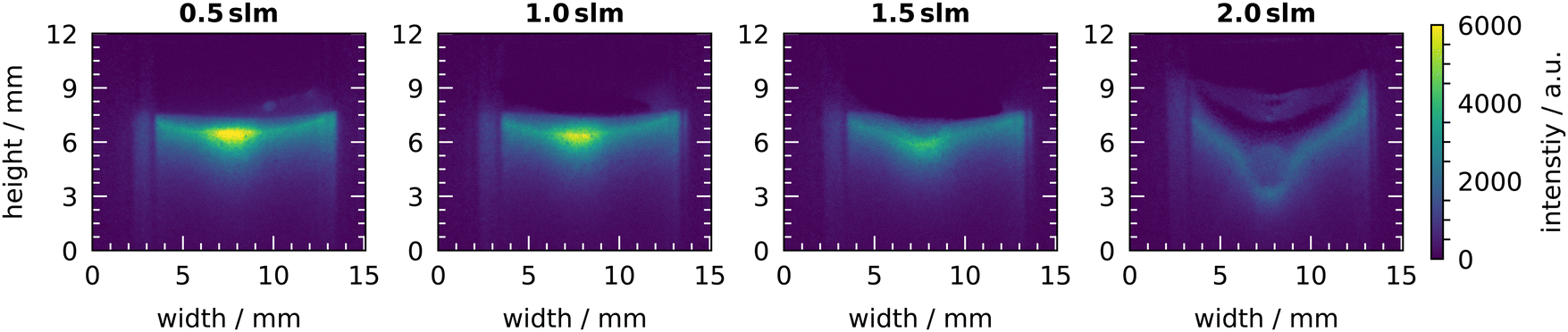 | ||
| Fig. 13 Chemiluminescence of luminol under plasma treatment at different gas flow rates. Measurements were taken at a humidity of 640 ppm and a plasma power of 6 W. | ||
As a result of these investigations, the gas flow rate can be used to increase the flux of H2O2, resulting in a higher concentration in the liquid after the same treatment time. At low gas flow rates, the selectivity towards H2O2 is increased, which is consistent with the results of a longer transport distance discussed earlier. Additionally, at lower gas flow rates, the time required for the reactive species to reach the liquid after leaving the plasma is increased, which is equivalent to an increase in the distance from plasma to liquid. However, at high gas flow rates, the increased flow comes into play with a strong interaction between effluent and liquid. This might not be advantageous for applications where stable and reproducible conditions are required.
3.5 Power modulation
As discussed above, the treatment distance and the gas flow rate influence the concentrations in the liquid. Increasing the distance lengthens the transport time from the plasma to the liquid. This time variable is also changed by a change of the gas flow rate. However, the change in the gas flow rate also changes the residence time. One way to separate the effects of residence time and transport time on the reactive species concentrations in the liquid is to modulate the coupled RF power by a low frequency signal. Power modulation can be used to control the on and off time of the plasma and thus the time in which the particles are located in the active plasma. At a gas flow rate of 1 slm, the particles need a time of 2.4 ms to pass the 40 mm long plasma channel. For example, pulsing the RF capillary plasma jet with a square wave signal with a frequency of 1000 Hz and a duty cycle of 50%, the plasma-on time is 1 ms, so that the particles experience more than two plasma pulses on their travel through the discharge gap. All investigated pulse frequencies and their corresponding plasma-on and plasma-off time with the number of pulses per residence time are listed in Table 2.| Duty cycle % | Frequency Hz | Plasma-on time ms | Plasma-off time ms | Number of pulses per residence time |
|---|---|---|---|---|
| 10 | 42 | 2.4 | 21.4 | 0.1 |
| 67 | 1.5 | 13.4 | 0.2 | |
| 100 | 1.0 | 9.0 | 0.2 | |
| 200 | 0.5 | 4.5 | 0.5 | |
| 400 | 0.3 | 2.3 | 1 | |
| 833 | 0.1 | 1.1 | 2 | |
| 1250 | 0.1 | 0.7 | 3 | |
| 50 | 208 | 2.4 | 2.4 | 0.5 |
| 333 | 1.5 | 1.5 | 0.8 | |
| 500 | 1.0 | 1.0 | 1.2 | |
| 1000 | 0.5 | 0.5 | 2.4 | |
| 2000 | 0.3 | 0.3 | 4.8 | |
Fig. 14 shows the H2O2 and OH concentration for the power modulation of the RF capillary plasma jet. The H2O2 concentration is not affected by the number of pulses per residence time at a duty cycle of 50% and reaches a value of about 0.7 mM. At the lower duty cycle of 10%, the H2O2 concentration rises with increasing number of pulses. With three pulses per residence time, the concentration is about 0.4 mM. In case of OH, the concentration is low and fluctuates when changing the number of pulses for both duty cycles. It appears that the OH concentration is slightly higher for the 50% duty cycle, but in both cases the concentration reaches a maximum of only 0.01 mM. The OH radical might be strongly influenced by pulsing the RF jet, as it can react in the plasma-off time, similar to the mechanism in the effluent described above, and the number of OH radicals reaching the liquid surface is lower. Thus, trends can not be detected by the diagnostic due to the low concentration and low sensitivity. Comparing both concentrations with the unmodulated cases, indicated with bars in the figure, a clear difference between OH and H2O2 can be recognized. The H2O2 concentration for a duty cycle of 50% reaches close to the value for the unmodulated case, while the OH concentration is only half of the concentration achieved in the unmodulated case. Considering the lifetime of the two species, this behaviour is quite reasonable. In the plasma-on time, the species are created and transported through the discharge gap. During the plasma-off time, the production disappears and the destruction of the species takes place. The highly reactive OH interacts more strongly in the plasma-off time than the long-lived H2O2. Thus, the concentration of OH is lower due to the pulsing of the RF jet, while H2O2 is less affected. In case of the duty cycle of 10%, another effect can be observed. As the number of pulses increases, the H2O2 concentration increases. This indicates that at these parameters a production of H2O2 with an accumulation takes place. Thus, by pulsing the RF jet with low frequencies, the production of reactive species within the plasma jet can be controlled.
 | ||
| Fig. 14 Effect of low frequency power modulation on species concentration in the liquid for (a) H2O2 and (b) OH. Measurements were taken at a humidity of 6400 and an unmodulated plasma power of 6 W. It should be emphasised that the measured OH concentration shown in the figure is formally the time-accumulated HTA concentration in the liquid and does not reflect an instantaneous OH concentration in the liquid. It should be emphasised that the measured time-accumulated corrected HTA concentration in the liquid is proportional to the OH concentration, but does not reflect an instantaneous OH concentration in the liquid. | ||
Pulsing the RF jet with low frequencies significantly lowers the gas temperature of the plasma.53,54 There, all investigated frequencies between 50 Hz and 10 kHz showed lower gas temperatures than in the unmodulated case. It can therefore be assumed that the gas temperature for the capillary jet (this work) is also lower for the pulsed cases, which strongly influences the chemistry of the plasma.
Due to the duty cycle of the modulation pulses, the dissipated plasma power is only half or even only one tenth of the plasma power compared to the unmodulated case. Thus, the energy efficiency of the species production can be enhanced by pulsing the RF jet, as shown in Fig. 15. The energy efficiency  is calculated via
is calculated via
 | (5) |
 | ||
| Fig. 15 Energy efficiency for the low frequency modulated plasma calculated from measured species concentration in the liquid and averaged plasma power for (a) H2O2 and (b) OH. Measurements were taken at a humidity of 6400 and an unmodulated plasma power of 6 W. | ||
In case of OH, the energy efficiency is lower, except for three number of pulses at the low duty cycle. Under these conditions, strong fluctuations in the concentration measurements occur due to the low sensitivity of the OH measurement for the modulated plasma treatments. These fluctuations become even more pronounced in the post-processing for calculating the energy efficiency and no trend is discernible here either. However, the energy efficiency of OH appears to be lower when the RF jet is pulsed compared to plasma treatments without pulsing.
All in all, the power modulation of the RF jet can be used to control the production of reactive species in the plasma with a higher selectivity towards stable molecule such as H2O2, and a higher energy efficiency can be achieved in the production of these stable molecules.
4 Conclusion
An atmospheric pressure plasma jet operated with humid helium was used to treat a liquid and the concentrations of the hydroxyl radical and hydrogen peroxide were measured in the liquid. The concentration of H2O2 reached values up to 1 mM while the OH concentration was a factor of 50 lower. The production of the reactive species and thus their concentration in the liquid could be controlled by the humidity of the feed gas and the dissipated plasma power. By increasing both parameters, the H2O2 concentration increases and reaches saturation for higher plasma powers between 6 W and 12 W. The OH concentration shows a different behaviour as it increases linearly with the plasma power and has a maximum at low humidity concentrations.Simulation results demonstrate generally good agreement to the measurements, with certain limitations, and have been used to outline the main production and consumption reactions for OH and H2O2 in the plasma and the effluent. The density distribution of the species from the simulations, along the direction of the gas flow, showed a steep increase for OH followed by an increase of H2O2 in the discharge channel, and both densities remained constant for more than half of the channel to the outlet. In the effluent, the density of OH decreased strongly, while the H2O2 density remained relatively constant.
Experimentally, the OH distribution in the liquid was visualised by the chemiluminescence of luminol. Chemiluminescence is present at the liquid surface and only spans over a few millimeter into the liquid. The main signal originated from the position where the gas flow hit the liquid surface.
Transport phenomena were investigated by varying the treatment distance and the gas flow rate. With increasing treatment distance the H2O2 concentration decreased only slightly while the OH concentration decreased exponentially. The highly reactive OH radical is strongly affected by a longer reaction time in the gas phase at larger distances leading to increased consumption in effluent region while the long-lived H2O2 is less affected. By varying the gas flow rate, the flux of reactive species reaching the liquid surface is varied. Our results indicate that a large fraction of H2O2 leaving the jet enters the liquid and a higher gas flow rate results in a higher concentration of H2O2 in the liquid. In contrast, a higher gas flow rate did not drastically enhance the concentration of OH in the liquid as modelled by the simulation, showing that it is not completely dissolved in the liquid.
Low frequency pulsing of the RF jet was shown to be a promising tool to control the plasma chemistry and to enhance the selectivity towards long-lived plasma species. During the plasma-off time, reactive species such as OH are consumed, while long-species can survive. Thus, an accumulation of the long-lived species is obtained, resulting in a higher selectivity towards these species and the energy efficiency of long-lived species production can be enhanced.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the DFG within CRC 1316 (Sub-project B11, project number 327886311). The authors would like to thank the research group Applied Microbiology of Prof. J. Bandow at Ruhr University Bochum for providing the buffer solution. Furthermore, the authors thank Prof. Kushner for providing the GlobalKin code.References
- T. Haruyama, T. Namise, N. Shimoshimizu, S. Uemura, Y. Takatsuji, M. Hino, R. Yamasaki, T. Kamachi and M. Kohno, Non-catalyzed one-step synthesis of ammonia from atmospheric air and water, Green Chem., 2016, 18(16), 4536–4541, 10.1039/C6GC01560C.
- Y. Gorbanev, E. Vervloessem, A. Nikiforov and A. Bogaerts, Nitrogen Fixation with Water Vapor by Nonequilibrium Plasma: toward Sustainable Ammonia Production, ACS Sustainable Chem. Eng., 2020, 8(7), 2996–3004, DOI:10.1021/acssuschemeng.9b07849.
- T. von Woedtke, H.-R. Metelmann and K.-D. Weltmann, Clinical Plasma Medicine: State and Perspectives of in Vivo Application of Cold Atmospheric Plasma: Clinical Plasma Medicine: State and Perspectives of in Vivo Application of Cold Atmospheric Plasma, Contrib. Plasma Phys., 2014, 54(2), 104–117, DOI:10.1002/ctpp.201310068.
- T. Von Woedtke, A. Schmidt, S. Bekeschus, K. Wende and K.-D. Weltmann, Plasma Medicine: A Field of Applied Redox Biology, In Vivo, 2019, 33(4), 1011–1026, DOI:10.21873/invivo.11570.
- J.-W. Lackmann and J. E. Bandow, Inactivation of microbes and macromolecules by atmospheric-pressure plasma jets, Appl. Microbiol. Biotechnol., 2014, 98(14), 6205–6213, DOI:10.1007/s00253-014-5781-9.
- V. S. K. Kondeti, C. Q. Phan, K. Wende, H. Jablonowski, U. Gangal, J. L. Granick, R. C. Hunter and P. J. Bruggeman, Long-lived and short-lived reactive species produced by a cold atmospheric pressure plasma jet for the inactivation of Pseudomonas aeruginosa and Staphylococcus aureus, Free Radical Biol. Med., 2018, 124, 275–287, DOI:10.1016/j.freeradbiomed.2018.05.083.
- A. Yayci, T. Dirks, F. Kogelheide, M. Alcalde, F. Hollmann, P. Awakowicz and J. E. Bandow, Microscale Atmospheric Pressure Plasma Jet as a Source for Plasma-Driven Biocatalysis, ChemCatChem, 2020, 12(23), 5893–5897, DOI:10.1002/cctc.202001225.
- A. Yayci, A. G. Baraibar, M. Krewing, E. F. Fueyo, F. Hollmann, M. Alcalde, R. Kourist and J. E. Bandow, Plasma-Driven in Situ Production of Hydrogen Peroxide for Biocatalysis, ChemSusChem, 2020, 13(8), 2072–2079, DOI:10.1002/cssc.201903438.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. Fernandez Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. Machala, I. Marinov, D. Mariotti, S. Mededovic Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schroter, M. Shiraiwa, B. Tarabova, P. A. Tsai, J. R. R. Verlet, T. von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma–liquid interactions: a review and roadmap, Plasma Sources Sci. Technol., 2016, 25(5), 053002, DOI:10.1088/0963-0252/25/5/053002.
- Y. Morabit, M. I. Hasan, R. D. Whalley, E. Robert, M. Modic and J. L. Walsh, A review of the gas and liquid phase interactions in low-temperature plasma jets used for biomedical applications, Eur. Phys. J. D, 2021, 75(1), 32, DOI:10.1140/epjd/s10053-020-00004-4.
- I. Adamovich, S. Agarwal, E. Ahedo, L. L. Alves, S. Baalrud, N. Babaeva, A. Bogaerts, A. Bourdon, P. J. Bruggeman, C. Canal, E. H. Choi, S. Coulombe, Z. Donko, D. B. Graves, S. Hamaguchi, D. Hegemann, M. Hori, H.-H. Kim, G. M. W. Kroesen, M. J. Kushner, A. Laricchiuta, X. Li, T. E. Magin, S. Mededovic Thagard, V. Miller, A. B. Murphy, G. S. Oehrlein, N. Puac, R. M. Sankaran, S. Samukawa, M. Shiratani, M. Simek, N. Tarasenko, K. Terashima, E. Thomas Jr, J. Trieschmann, S. Tsikata, M. M. Turner, I. J. van der Walt, M. C. M. van de Sanden and T. von Woedtke, The 2022 Plasma Roadmap: low temperature plasma science and technology, J. Phys. D: Appl. Phys., 2022, 55(37), 373001, DOI:10.1088/1361-6463/ac5e1c.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges?, Curr. Opin. Chem. Biol., 2017, 37, 1–9, DOI:10.1016/j.cbpa.2016.10.007.
- J. Winter, K. Wende, K. Masur, S. Iseni, M. Dunnbier, M. U. Hammer, H. Tresp, K.-D. Weltmann and S. Reuter, Feed gas humidity: a vital parameter affecting a cold atmospheric-pressure plasma jet and plasma-treated human skin cells, J. Phys. D: Appl. Phys., 2013, 46(29), 295401, DOI:10.1088/0022-3727/46/29/295401.
- J. Winter, H. Tresp, M. U. Hammer, S. Iseni, S. Kupsch, A. Schmidt-Bleker, K. Wende, M. Dunnbier, K. Masur, K.-D. Weltmann and S. Reuter, Tracking plasma generated H2O2 from gas into liquid phase and revealing its dominant impact on human skin cells, J. Phys. D: Appl. Phys., 2014, 47(28), 285401, DOI:10.1088/0022-3727/47/28/285401.
- M. M. Hefny, C. Pattyn, P. Lukes and J. Benedikt, Atmospheric plasma generates oxygen atoms as oxidizing species in aqueous solutions, J. Phys. D: Appl. Phys., 2016, 49(40), 404002, DOI:10.1088/0022-3727/49/40/404002.
- Z. Chen, D. Liu, C. Chen, D. Xu, Z. Liu, W. Xia, M. Rong and M. G. Kong, Analysis of the production mechanism of H2O2 in water treated by helium DC plasma jets, J. Phys. D: Appl. Phys., 2018, 51(32), 325201, DOI:10.1088/1361-6463/aad0eb.
- D. X. Liu, P. Bruggeman, F. Iza, M. Z. Rong and M. G. Kong, Global model of low-temperature atmospheric-pressure He + H2O plasmas, Plasma Sources Sci. Technol., 2010, 19(2), 025018, DOI:10.1088/0963-0252/19/2/025018.
- S. Schroter, A. R. Gibson, M. J. Kushner, T. Gans and D. O’Connell, Numerical study of the influence of surface reaction probabilities on reactive species in an rf atmospheric pressure plasma containing humidity, Plasma Phys. Controlled Fusion, 2018, 60(1), 014035, DOI:10.1088/1361-6587/aa8fe9.
- C. A. Vasko, D. X. Liu, E. M. van Veldhuizen, F. Iza and P. J. Bruggeman, Hydrogen Peroxide Production in an Atmospheric Pressure RF Glow Discharge: Comparison of Models and Experiments, Plasma Chem. Plasma Process., 2014, 34(5), 1081–1099, DOI:10.1007/s11090-014-9559-8.
- G. Willems, J. Benedikt and A. von Keudell, Absolutely calibrated mass spectrometry measurement of reactive and stable plasma chemistry products in the effluent of a He/H2O atmospheric plasma, J. Phys. D: Appl. Phys., 2017, 50(33), 335204, DOI:10.1088/1361-6463/aa77ca.
- Y. Gorbanev, C. C. W. Verlackt, S. Tinck, E. Tuenter, K. Foubert, P. Cos and A. Bogaerts, Combining experimental and modelling approaches to study the sources of reactive species induced in water by the COST RF plasma jet, Phys. Chem. Chem. Phys., 2018, 20(4), 2797–2808, 10.1039/C7CP07616A.
- S. Schroter, A. Wijaikhum, A. R. Gibson, A. West, H. L. Davies, N. Minesi, J. Dedrick, E. Wagenaars, N. de Oliveira, L. Nahon, M. J. Kushner, J.-P. Booth, K. Niemi, T. Gans and D. O’Connell, Chemical kinetics in an atmospheric pressure helium plasma containing humidity, Phys. Chem. Chem. Phys., 2018, 20(37), 24263–24286, 10.1039/C8CP02473A.
- S. Schroter, J. Bredin, A. R. Gibson, A. West, J. P. Dedrick, E. Wagenaars, K. Niemi, T. Gans and D. O’Connell, The formation of atomic oxygen and hydrogen in atmospheric pressure plasmas containing humidity: picosecond twophoton absorption laser induced fluorescence and numerical simulations, Plasma Sources Sci. Technol., 2020, 29(10), 105001, DOI:10.1088/1361-6595/abab55.
- J. Jiang, V. S. S. K. Kondeti, G. Nayak and P. J. Bruggeman, Experimental and modeling studies of the plasma chemistry in a humid Ar radiofrequency atmospheric pressure plasma jet, J. Phys. D: Appl. Phys., 2022, 55(22), 225206, DOI:10.1088/1361-6463/ac570a.
- J. Golda, J. Held, B. Redeker, M. Konkowski, P. Beijer, A. Sobota, G. Kroesen, N. S. J. Braithwaite, S. Reuter, M. M. Turner, T. Gans, D. O’Connell and V. Schulz-von der Gathen, Concepts and characteristics of the ‘COST Reference Microplasma Jet’, J. Phys. D: Appl. Phys., 2016, 49(8), 084003, DOI:10.1088/0022-3727/49/8/084003.
- G. Gorbanev and B. Gathen, Applications of the COST Plasma Jet: More than a Reference Standard, Plasma, 2019, 2(3), 316–327, DOI:10.3390/plasma2030023.
- T. Winzer, D. Steuer, S. Schuttler, N. Blosczyk, J. Benedikt and J. Golda, RFdriven atmospheric-pressure capillary plasma jet in a He/O2 gas mixture: multidiagnostic approach to energy transport, J. Appl. Phys., 2022, 132(18), 183301, DOI:10.1063/5.0110252.
- J. Golda, F. Kogelheide, P. Awakowicz and V. S.-V. D. Gathen, Dissipated electrical power and electron density in an RF atmospheric pressure helium plasma jet, Plasma Sources Sci. Technol., 2019, 28(9), 095023, DOI:10.1088/1361-6595/ab393d.
- J. Benedikt, D. Schroder, S. Schneider, G. Willems, A. Pajdarova, J. Vlcek and V. Schulz-von der Gathen, Absolute OH and O radical densities in effluent of a He/H2O micro-scaled atmospheric pressure plasma jet, Plasma Sources Sci. Technol., 2016, 25(4), 045013, DOI:10.1088/0963-0252/25/4/045013.
- R. Nogueira, M. Oliveira and W. Paterlini, Simple and fast spectrophotometric determination of H2O2 in photo-Fenton reactions using metavanadate, Talanta, 2005, 66(1), 86–91, DOI:10.1016/j.talanta.2004.10.001.
- S. Schuttler, L. Jolmes, E. Jeß, K. Tschulik and J. Golda, Validation of in situ diagnostics for the detection of OH and H2O2 in liquids treated by a humid atmospheric pressure plasma jet, Plasma Processes Polym., 2023, e2300079, DOI:10.1002/ppap.202300079.
- J. C. Barreto, G. S. Smith, N. H. Strobel, P. A. McQuillin and T. A. Miller, Terephthalic acid: a dosimeter for the detection of hydroxyl radicals in vitro, Life Sci., 1994, 56(4), PL89–PL96, DOI:10.1016/0024-3205(94)00925-2.
- T. Mason, J. Lorimer, D. Bates and Y. Zhao, Dosimetry in sonochemistry: the use of aqueous terephthalate ion as a fluorescence monitor, Ultrasonics Sonochem., 1994, 1(2), S91–S95, DOI:10.1016/1350-4177(94)90004-3.
- S. Kanazawa, H. Kawano, S. Watanabe, T. Furuki, S. Akamine, R. Ichiki, T. Ohkubo, M. Kocik and J. Mizeraczyk, Observation of OH radicals produced by pulsed discharges on the surface of a liquid, Plasma Sources Sci. Technol., 2011, 20(3), 034010, DOI:10.1088/0963-0252/20/3/034010.
- S. Kanazawa, T. Furuki, T. Nakaji, S. Akamine and R. Ichiki, Application of chemical dosimetry to hydroxyl radical measurement during underwater discharge, J. Phys.: Conf. Ser., 2013, 418, 012102, DOI:10.1088/1742-6596/418/1/012102.
- B. Myers, P. Ranieri, T. Smirnova, P. Hewitt, D. Peterson, M. Herrera Quesada, E. Lenker and K. Stapelmann, Measuring plasma-generated ˙OH and O atoms in liquid using EPR spectroscopy and the non-selectivity of the HTA assay, J. Phys. D: Appl. Phys., 2021, 54(14), 145202, DOI:10.1088/1361-6463/abd9a6.
- T.-W. Chen, C.-T. Liu, C.-Y. Chen, M.-C. Wu, P.-C. Chien, Y.-C. Cheng and J.-S. Wu, Analysis of Hydroxyl Radical and Hydrogen Peroxide Generated in Helium-Based Atmospheric-Pressure Plasma Jet and in Different Solutions Treated by Plasma for Bioapplications, ECS J. Solid State Sci. Technol., 2020, 9(11), 115002, DOI:10.1149/2162-8777/ab9c78.
- X. Fang, G. Mark and C. von Sonntag, OH radical formation by ultrasound in aqueous solutions Part I: the chemistry underlying the terephthalate dosimeter, Ultrasonics Sonochem., 1996, 3(1), 57–63, DOI:10.1016/1350-4177(95)00032-1.
- C. Lu, G. Song and J.-M. Lin, Reactive oxygen species and their chemiluminescence-detection methods, TrAC, Trends Anal. Chem., 2006, 25(10), 985–995, DOI:10.1016/j.trac.2006.07.007.
- G. Merenyi, J. Lind and T. E. Eriksen, Luminol chemiluminescence: chemistry, excitation, emitter, J. Bioluminesci. Chemiluminesci., 1990, 5(1), 53–56, DOI:10.1002/bio.1170050111.
- N. Shirai, Y. Matsuda and K. Sasaki, Visualization of short-lived reactive species in liquid in contact with atmospheric-pressure plasma by chemiluminescence of luminol, Appl. Phys. Express, 2018, 11(2), 026201, DOI:10.7567/APEX.11.026201.
- N. Shirai, G. Suga and K. Sasaki, Correlation between gas-phase OH density and intensity of luminol chemiluminescence in liquid interacting with atmosphericpressure plasma, J. Phys. D: Appl. Phys., 2019, 52(39), 39LT02, DOI:10.1088/1361-6463/ab2ff2.
- G. Merenyi and J. S. Lind, Role of a peroxide intermediate in the chemiluminescence of luminol. A mechanistic study, J. Am. Chem. Soc., 1980, 102(18), 5830–5835, DOI:10.1021/ja00538a022.
- A. M. Lietz and M. J. Kushner, Air plasma treatment of liquid covered tissue: long timescale chemistry, J. Phys. D: Appl. Phys., 2016, 49(42), 425204, DOI:10.1088/0022-3727/49/42/425204.
- A. Brisset, A. R. Gibson, S. Schroter, K. Niemi, J.-P. Booth, T. Gans, D. O’Connell and E. Wagenaars, Chemical kinetics and density measurements of OH in an atmospheric pressure He + O2 + H2O radiofrequency plasma, J. Phys. D: Appl. Phys., 2021, 54(28), 285201, DOI:10.1088/1361-6463/abefec.
- C. Lazzaroni, P. Chabert, M. A. Lieberman, A. J. Lichtenberg and A. Leblanc, Analytical–numerical global model of atmospheric-pressure radio-frequency capacitive discharges, Plasma Sources Sci. Technol., 2012, 21(3), 035013, DOI:10.1088/0963-0252/21/3/035013.
- Y. He, P. Preissing, D. Steuer, M. Klich, V. Schulz-von der Gathen, M. Boke, I. Korolov, J. Schulze, V. Guerra, R. P. Brinkmann and E. Kemaneci, Zero-dimensional and pseudo-one-dimensional models of atmospheric-pressure plasma jets in binary and ternary mixtures of oxygen and nitrogen with helium background, Plasma Sources Sci. Technol., 2021, 30(10), 105017, DOI:10.1088/1361-6595/ac278d.
- D. W. O’Sullivan, M. Lee, B. C. Noone and B. G. Heikes, Henry's Law Constant Determinations for Hydrogen Peroxide, Methyl Hydroperoxide, Hydroxymethyl Hydroperoxide, Ethyl Hydroperoxide, and Peroxyacetic Acid, J. Phys. Chem., 1996, 100(8), 3241–3247, DOI:10.1021/jp951168n.
- P. Preissing, I. Korolov, J. Schulze, V. Schulz-von der Gathen and M. Boke, Three-dimensional density distributions of NO in the effluent of the COST reference microplasma jet operated in He/N2/O2, Plasma Sources Sci. Technol., 2020, 29(12), 125001, DOI:10.1088/1361-6595/abbd86.
- B. Myers, E. Barnat and K. Stapelmann, Atomic oxygen density determination in the effluent of the COST reference source using in situ effective lifetime measurements in the presence of a liquid interface, J. Phys. D: Appl. Phys., 2021, 54(45), 455202, DOI:10.1088/1361-6463/ac1cb5.
- D. J. Jacob, Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate, J. Geophys. Res., 1986, 91(D9), 9807, DOI:10.1029/JD091iD09p09807.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15(8), 4399–4981, DOI:10.5194/acp-15-4399-2015.
- S. Kelly and M. M. Turner, Power modulation in an atmospheric pressure plasma jet, Plasma Sources Sci. Technol., 2014, 23(6), 065012, DOI:10.1088/0963-0252/23/6/065012.
- M. Mahreen, G. V. Prakash, S. Kar, D. Sahu and A. Ganguli, Influence of pulse modulation frequency on helium RF atmospheric pressure plasma jet characteristics, Contrib. Plasma Phys., 2022, 62, 7, DOI:10.1002/ctpp.202200007.
| This journal is © the Owner Societies 2024 |