
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of temperature-dependent non-PAN peroxynitrate formation, RO2NO2, on nighttime atmospheric chemistry†‡
Michelle
Färber
 a,
Luc
Vereecken
a,
Hendrik
Fuchs
ab,
Georgios I.
Gkatzelis
a,
Franz
Rohrer
a,
Sergej
Wedel
a,
Andreas
Wahner
a and
Anna
Novelli
*a
a,
Luc
Vereecken
a,
Hendrik
Fuchs
ab,
Georgios I.
Gkatzelis
a,
Franz
Rohrer
a,
Sergej
Wedel
a,
Andreas
Wahner
a and
Anna
Novelli
*a
aInstitute for Energy and Climate Research, IEK-8: Troposphere, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: m.faerber@fz-juelich.de; a.novelli@fz-juelich.de
bDepartment of Physics, University of Cologne, 50932 Cologne, Germany
First published on 17th January 2024
Abstract
The formation of peroxynitrates (RO2NO2) from the reaction of peroxy radicals (RO2) and nitrogen dioxide (NO2) and their subsequent redissociation are typically not included in chemical mechanisms. This is often done to save computational time as the assumption is that the equilibrium is strongly towards the RO2 + NO2 reaction for most conditions. Exceptions are the reactions of the methyl peroxy radical due to its abundance in the atmosphere and of acyl-RO2 radicals due to the long lifetime of peroxyacyl nitrates RO2NO2 (PANs). In this study, the nighttime oxidation of cis-2-butene and trans-2-hexene in the presence of NO2 is investigated in the atmospheric simulation chamber SAPHIR, Forschungszentrum Jülich, Germany, at atmospherically-relevant conditions at different temperatures (≈276 K, ≈293 K, ≈305 K). Measured concentrations of peroxy and hydroperoxy radicals as well as other trace gases (ozone, NO2, volatile organic compounds) are compared to state-of-the-art zero-dimensional box model calculations. Good model-measurement agreement can only be achieved when reversible RO2 + NO2 reactions are included for all RO2 species using literature values available from the latest SAR by [Jenkin et al., Atmos. Chem. Phys., 2019, 19, 7691]. The good agreement observed gives confidence that the SAR, derived originally for aliphatic RO2, can be applied to a large range of substituted RO2 radicals, simplifying generalised implementation in chemical models. RO2NO2 concentrations from non-acyl RO2 radicals of up to 2 × 10 cm−3 are predicted at 276 K, impacting effectively the kinetics of RO2 radicals. Under these conditions, peroxy radicals are slowly regenerated downwind of the pollution source and may be lost in the atmosphere through deposition of RO2NO2. Based on this study, 60% of RO2 radicals would be stored as RO2NO2 at a temperature of 10 °C and in the presence of a few ppbv of NO2. The fraction increases further at colder temperatures and/or higher NO2 mixing ratios. This does not only affect the expected concentrations of RO2 radicals but, as the peroxynitrates can react with OH radicals or photolyse, they could comprise a net sink for RO2 radicals as well as increase the production of NOx (= NO + NO2) in different locations depending on their lifetime. Omitting this chemistry from the kinetic model can lead to misinterpreted product formation and may prevent reconciling observations and model predictions.
1 Introduction
In the atmosphere, volatile organic compounds (VOCs) and volatile inorganic compounds are removed via oxidation which drives their chemical degradation. In the lower troposphere, the daytime oxidation processes are driven by ozone (O3), and the hydroxyl radical (OH), which is often found to be the predominant oxidant. Another tropospheric oxidant is the nitrate radical (NO3) which plays a major role during the night.1 NO3 is formed by the reaction of nitrogen dioxide (NO2) with O3:| NO2 + O3 → NO3+ O2, | (1) |
In recent field studies utilising the laser-induced-fluorescence (LIF) technique for radical measurements, discrepancies between measured and modelled RO2 radical concentrations were found at night at moderate O3 (∼(1–70) ppbv), NO2 (∼(3–50) ppbv), and low NO (∼(0–0.3) ppbv) mixing ratios in the vicinity of Beijing,5,6 China, Wangdu,7 China, and London,8 UK. Just recently, two studies focussing on investigating the oxidation of isoprene9 and anthropogenic alkenes10 by NO3 resulted in new detailed chemical mechanisms for these compounds as intricate as their oxidation by OH. These studies highlighted the inability of detecting a large fraction of NO3 -containing isoprene-RO29 as well as NO3–RO2 from short-chain alkenes.10 Both the isomerisation (isoprene)9 and the decomposition (short-chain alkenes)10 of the NO3 -alkoxy radical do not lead to the formation of HO2 or OH, which is required for the RO2 measurement by the LIF technique:
 | (2) |
However, for long-chain alkenes such as for example trans-2-hexene, all RO2 are detectable.10 The discrepancies found at nighttime in the field5–8 may be partly explainable by the missing detection of the LIF instrument of single short-chain alkenes, contained in the measured air mixture. Still large discrepancies remain between measured and modelled RO2 radicals for high NO (up to ∼100 ppbv) and NO2 (up to ∼70 ppbv).5–8,11
Despite improvements in the agreement between measured and modelled RO2 radicals observed in the experiments in the SAPHIR chamber,10 discrepancies are still observed in particular right after the injection of the VOC when the RO2 radical production is the highest, as well as in the time dependence of the RO2 radical concentrations. The conditions in the chamber were chosen to facilitate the formation of NO3 radicals, and were characterised by zero NO and medium NO2 (17–40 ppbv) and ozone (7–30 ppbv). For these conditions, RO2 radicals formed in the oxidation by either O3 or NO3 are assumed to be mainly lost by their reaction with HO2 or RO2. NO2 will be the dominant reaction partner for these RO2 radicals, producing a short-lived peroxynitrate (RO2NO2) which is in a very fast equilibrium (τRO2NO2 ≈ 0.2 s at 298 K) with its decomposition in the lower troposphere:
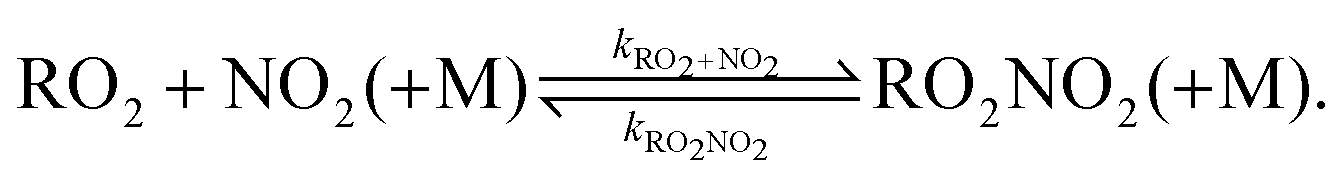 | (3) |
The impact of eqn (3) on the RO2 radical and the formed RO2NO2 is thought to be negligible due to the instability of RO2NO2 in lower tropospheric oxidation processes, therefore, the reaction of RO2 and NO2 forming RO2NO2 is omitted in most atmospheric models for non-acyl RO2 radicals except for CH3O2.12 For the latter, the formation of alkyl peroxynitrates has been thought to be mostly relevant at cold temperatures in polar regions,13,14 in the upper troposphere, where Browne et al.14 found that including the methyl peroxynitrate (CH3O2NO2) chemistry is relevant at temperatures below 240 K, or in biomass burning plumes that are lofted to high altitudes.14 In contrast, the reactions of acyl-RO2 radicals with NO2 are typically implemented in all chemical models due to the long lifetime (∼40–45 min at 298 K) of the corresponding peroxyacyl nitrate RO2NO2 (PANs) and, consequently, their relevance on regional and even global scales.13
A recent study by Khan et al.15 investigated the global effect of the reversible formation of RO2NO2 on NOx, i.e. NO + NO2, OH, and O3, using the generic rate coefficients from Jenkin et al.12 for more than 40 non-acyl RO2 radicals. Loss rates for the formed RO2NO2 species via reaction of OH and photolysis were also included. In their study, it was shown that even on the ground, up to 25% more NOx would be expected at the equator as photolysis and reaction with OH accelerate the consumption of RO2NO2. This indicates that their role and importance might need to be reevaluated.
A measure for the ratio of RO2 and RO2NO2 concentrations at equilibrium is given by the equilibrium constant K, the ratio of the forward and backward reaction rate constants kRO2+NO2 and kRO2NO2, respectively:
| K = kRO2+NO2/kRO2NO2. | (4) |
For the alkyl peroxynitrates CH3O2NO2 and ethyl peroxynitrate (C2H5O2NO2), the equilibrium constant is 7 × 10−11 cm3 at 276 K, respectively (Table 1). In contrast, peroxyacetyl nitrate (CH3C(O)O2) has an equilibrium constant of 1 × 10−8 cm3 due to its longer lifetime. While at high temperatures formation of alkyl peroxynitrates is not important, the equilibrium shifts more towards the RO2NO2 products at lower temperatures, making it worthwhile to evaluate the impact of RO2NO2 formation during winter conditions.
| RO2 or RO2NO2 | k 0 | k ∞ | F c |
k
276![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K K
|
k
292K,1atm |
k
305K,1atm
|
|---|---|---|---|---|---|---|
| a Recommended by IUPAC.16 b Determined from isomeric mixtures formed from the reaction of chlorine with butane17,18 and recommended by Jenkin et al.12 c Determined from isomeric mixtures formed from the reaction of chlorine with hexane18 and recommended by Jenkin et al.12 d Pressure-independent generic rate coefficient recommended by Jenkin et al.12 | ||||||
| Forward reaction, kRO2+NO2 [cm3 s−1] | ||||||
| CH3O2a | 1.2 × 10−30 (T/300)−6.9[M] | 1.8 × 10−11 | 0.36 | 7.1 × 10−12 | 6.2 × 10−12 | 5.5 × 10−12 |
| C2H5O2a | 1.3 × 10−29 (T/300)−6.2 [M] | 8.8 × 10−12 | 0.31 | 5.6 × 10−12 | 5.3 × 10−12 | 5.0 × 10−12 |
| n- and sec- C4H9O2b | 9.6 × 10−12 | 9.6 × 10−12 | 9.6 × 10−12 | 9.6 × 10−12 | ||
| RO2d | 9.0 × 10−12 (=kfPN) | 9.0 × 10−12 | 9.0 × 10−12 | 9.0 × 10−12 | ||
| Reverse reaction, kRO2NO2 [s−1] | ||||||
| CH3O2NO2a | 9.0 × 10−5exp (−9690/T)[M] | 1.1 × 1016 exp (−10560/T) |
0.36 | 0.1 | 0.7 | 3.2 |
| C2H5O2NO2a | 4.8 × 10−4exp (−9285/T)[M] | 8.8 × 1015 exp (−10440/T) |
0.31 | 0.2 | 1.7 | 7.5 |
| n- and sec- C4H9O2NO2b | 8.3 × 1015 exp (−10368/T) |
0.4 | 3.2 | 14.3 | ||
| C 6H13O2NO2 isomersc | 7.5 × 1015 exp (−10368/T) |
0.4 | 2.8 | 12.9 | ||
| RO2NO2d | 7.6 × 1015 exp (−10400/T)(=kbPN) |
0.3 | 2.6 | 11.8 | ||
In this work, the impact of the RO2 + NO2 reaction on modelled RO2 radical concentrations is studied for RO2 radicals from the oxidation of cis-2-butene and trans-2-hexene by O3 and NO3 at different temperatures (≈276 K, ≈293 K, ≈305 K). Experiments were performed in the atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany. Nighttime conditions with high NO2 (>20 ppbv) and moderate O3 (∼10 ppbv) mixing ratios were tested which facilitate the formation of NO3 and mimic conditions often found at night. Concentrations of HO2 and RO2 radicals, as well as of O3, NO2, and VOCs were measured and are compared to zero-dimensional box model calculations, incorporating state-of-the-art oxidation schemes. The reaction of non-acyl RO2 radicals with NO2 forming RO2NO2 and its backward reaction are implemented in the chemical mechanisms of cis-2-butene and trans-2-hexene by using literature values,12,16–18 and the implications of the findings on the nighttime atmospheric RO2 chemistry are discussed. The newly introduced ozonolysis scheme and RO2 isomerisation reactions, suggested by Novelli et al.10 for cis-2-butene and trans-2-hexene, as well as their temperature dependence are tested and model-measurement comparisons are used to improve the chemical mechanisms for these species.
2 Methodology
2.1 Experiments in the atmospheric simulation chamber SAPHIR
The experiments were conducted in the outdoor atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany. The 270 m3 chamber (5 m diameter, 18 m length) allows to study atmospheric processes in a well-characterised system and is confined by an inert double-wall Teflon (FEP) film, enabling high transmittance of impinging solar radiation. A shutter system allows the shielding of the chamber from solar radiation to also mimic nighttime conditions. The temperature inside the chamber is not controlled and thus dependent on ambient conditions. The chamber utilises ultra-pure synthetic air, mixed from ultra-pure nitrogen and oxygen (79.1% N2, 20.9% O2, Linde, purity > 99.9999%). Contaminations are prevented from entering the chamber by an over-pressure of ≈33 Pa above ambient pressure. A replenishment flow is applied to account for small leakages and the air sampled by the instruments, causing a dilution for all trace gases with an average first order loss rate of 9.7 × 10−6 s−1 in the experiments in this study. Two fans are installed to mix the air in the chamber, so that all instruments sample the same air. A detailed description of SAPHIR can be found in previous works.19–22Nighttime experiments for cis-2-butene and trans-2-hexene were performed at different temperatures. The nighttime oxidation of cis-2-butene was studied at two different temperatures (T ≈ 276 K (cold), 295 K (medium)). The nighttime chemistry of trans-2-hexene was investigated at three temperatures (T ≈ 276 K (cold), 292 K (medium), 305 K (hot)). At low temperatures (T ≈ 280 K), a pure ozonolysis experiment in the absence of NO2 was also performed for trans-2-hexene.
The experimental procedure of the experiments is shown in Fig. 1–3. Before each experiment, the chamber was flushed to remove trace gases from the previous experiment. No detectable OH reactivity, equivalent to the inverse lifetime of OH, was observed in the dark, clean, and dry chamber. Cis-2-Butene (Air Liquide, 1% in N2, purity 99.4%) or trans-2-hexene (Sigma Aldrich, purity 97%) were first injected in the clean, dry, and dark chamber, reaching mixing ratios close to atmospheric conditions (<6 ppbv). Afterwards, 170–210 ppmv of carbon monoxide (CO, Air Liquide, purity 99.997%) was added to the chamber acting as OH scavenger by converting OH into HO2 to avoid its reaction with cis-2-butene or trans-2-hexene. In one oxidation experiment of trans-2-hexene, 142 ppmv of methane (CH4, Air Liquide, purity 99.5%) were injected instead to aim for lower HO2 radical concentrations. Since more reactions are involved to form HO2, a slower production of HO2 can be achieved, and higher RO2 radical concentrations are expected due to the formation of CH3O2. Nitrogen dioxide (Linde, 500 ppmv in N2, purity 99.991%) of up to 35 ppbv was then injected in all experiments with the exception of the ozonolysis study. Afterwards, ozone generated by a silent discharge ozoniser (O3onia) was injected to reach mixing ratios between 12 ppbv and 33 ppbv (96 ppbv in the ozonolysis experiment), initiating the production of NO3 in the reaction with NO2 (eqn (1)). Due to the fast reaction of cis-2-butene and trans-2-hexene with O3 and NO3, they are consumed on a timescale of ∼2–5 hours, and the alkene was injected a second time and third time (experiments with cis-2-butene at 295 K and trans-2-hexene at 305 K). To boost the oxidation by NO3, NO2 was injected a second time as well. In all experiments, no NO was present in the chamber and OH radical concentrations were below the detection limit, leading to a negligible contribution of OH chemistry. All experiments were conducted in dry conditions (H2O < 0.07 ppmv) and in complete darkness.
 | ||
| Fig. 1 Comparison of modelled and measured (5 minutes average) trace gases and HO2 and RO2 radicals in the experiment with cis-2-butene for cold (276 K) and medium (295 K) temperatures. Model results displayed as FZJ (blue) and FZJ + RO2NO2 (brown) models refer to the FZJ mechanism without and with including additional formation of non-acyl RO2NO2, respectively. Dashed lines indicate the total RO2 radical concentration predicted by the model, while solid lines are the RO2 radical concentration predicted to be detectable by the ROx LIF system, as discussed in Section 2.2. Injections of chemical species into the chamber are marked by vertical lines. | ||
 | ||
| Fig. 2 Comparison between modelled and measured (5 minutes average) trace gases and HO2 and RO2 radical concentrations in the trans-2-hexene experiment performed at ≈276 K with CO as OH scavenger. Model results displayed as FZJ (blue) and FZJ + RO2NO2 (brown) models refer to the FZJ mechanism with and without including additional formation of non-acyl RO2NO2, respectively. In addition, FZJ + RO2NO2 disc.-corr. (red) denotes the model run considering the overestimation of ozonolysis-generated RO2 radical concentrations by a factor of 1.7, observed in the ozonolysis of trans-2-hexene (see text). Injections of chemical species into the chamber are marked by vertical lines. | ||
 | ||
| Fig. 3 Comparison between modelled and measured (5 minutes average) trace gases and HO2 and RO2 radical concentrations in the nighttime oxidation experiment of trans-2-hexene in presence of CH4. Model results displayed as FZJ (blue) and FZJ + RO2NO2 (brown) models refer to the FZJ mechanism with and without including additional formation of non-acyl RO2NO2, respectively. Solid lines indicate the total RO2 radical concentration predicted by the model without the RO2NO2 interference, while dashed lines are the RO2 radical concentration predicted by the model considering a 2% interference by RO2NO2. Injections of chemical species into the chamber are marked by vertical lines. | ||
2.2 Instrumentation
Measurements of the radicals OH, HO2, and RO2 were conducted with a laser-induced fluorescence (LIF) instrument (ROx LIF).23,24 Chamber air is sucked into a low-pressure detection cell (≈ 4 hPa) where OH radicals are excited by a laser pulse (repetition frequency = 8.5 kHz) at a wavelength of 308 nm. The subsequently emitted fluorescence light, which is directly proportional to the sampled OH radical concentration, is detected by gated photon counting.25–27 By adding NO in a second detection cell, a fraction of HO2 radicals is chemically converted to OH and, thus, HOx (= OH + f HO2, f < 1) can be detected.24–26,28 Possible interferences in the HOx measurement may appear in the presence of specific RO2 radicals, when HO2 is rapidly produced by the RO2 + NO reaction.28 By working at lower NO concentrations, RO2 interferences are minimised and do not play a role in the presented experiments. RO2 radicals are indirectly measured after they are converted into HO2 or OH in a converter (≈25 hPa) by adding NO. OH, formed from the reaction of HO2 with NO, is converted back to HO2 in the converter by addition of excess CO. The HO2 radicals are then sampled by a detection cell and converted to OH by a continuous addition of pure NO, which enables a high conversion efficiency of HO2,23 so that the sum of OH, HO2, and RO2 (=ROx) is measured, from which [RO2] can be derived. An interference signal in the presence of NO3 that is equivalent to a RO2 radical concentration of 3.6 × 106 cm−3 per pptv of NO3 was observed in the ROx system. A previous study29 also reported a NO3 interference signal equivalent to HO2 and RO2 radical concentrations of 1.0 × 106 cm−3 and 1.7 × 106 cm−3 per pptv of NO3, respectively. More details can be found in the ESI‡ Section B. Modelled RO2 radical concentrations were corrected for this interference which only impacted the experiment at highest temperature when very high concentrations of NO3 were reached. Modelled HO2 radical concentrations were corrected using the parametrisation determined by Fuchs et al.29 Recently, it was found that -nitrate-alkoxy radicals decompose in the converter of the RO2 detection system forming NO2 and therefore these radicals are not detectable with the LIF technique that requires the formation of either OH or HO2 in the detection system.9,10 Examples include alkoxy radicals (RO) produced in the oxidation of anthropogenic alkenes by NO3 such as CH3 CH(NO3)CH(CH3)O (MCM notation: C42NO33O) formed in the oxidation of cis-2-butene by NO3. The rate coefficient of the decomposition of CH3 CH(NO3)CH(CH3)O (MCM notation: C42NO33O), forming NO2, was optimised based on the observed RO2 in the experiments of the nighttime oxidation of cis-2-butene, resulting in a decomposition rate of 3.0 × 103 s−1 (this work) compared to 9.5 × 103 s−1 predicted theoretically10 at 276 K. Further details can be found in the ESI‡ Section C.
The OH reactivity (kOH) was measured by a pump-and-probe technique utilising LIF to detect the amount of OH reacting with the sampled air in a flow tube.30,31 Time series of cis-2-butene and trans-2-hexene and calibrated time series of acetaldehyde were measured by proton-transfer-reaction time-of-flight mass spectrometry (PTR-ToF-MS, Ionicon).32,33 Normalised counts of cis-2-butene and trans-2-hexene are converted to parts per billion (ppb) by using the observed OH reactivity at the point in time of the VOC injection. Ozone was detected by UV absorption (Ansyco), and CO, CH4, formaldehyde, and water vapour were monitored utilising cavity ring-down spectroscopy (CRDS, Picarro). Furthermore, NO and NO2 were measured by a chemilumiscence instrument (EcoPhysics). More detailed information about the performance of the instruments can be found in the ESI‡ Section D. NO3 and N2O5 measurements were not available for any experiment, therefore corresponding wall loss rates were introduced and adjusted to match the observed VOC decay.
2.3 Model calculations
To compare measurements with model results, a zero-dimensional box model is used, starting from the chemical mechanistic information in the FZJ mechanisms for cis-2-butene and trans-2-hexene published by Novelli et al.10; these are themselves built upon the Master Chemical Mechanism (MCM v3.3.1, http://mcm.leeds.ac.uk).34,35 Dilution is taken into account by considering a first-order loss for all implemented species as described in Section 2.1. Temperature and pressure are constrained to measured data. The injection of O3, NO2, cis-2-butene, trans-2-hexene, CO, and CH4 into the chamber is reproduced by an active source during the injection period, with a source strength that is matched to the measured increase of the observables. The temperature-dependent reaction rate of trans-2-hexene with O3 is taken from Atkinson and Arey.1 At medium and high temperatures, the reaction rate of trans-2-hexene and NO3 is taken to be twice as large as the temperature-independent reaction rate used in the MCM; the latter is estimated from the SAR in Jenkin et al.34 The increased rate is consistent with the study by Novelli et al.10 The FZJ mechanisms for cis-2-butene and trans-2-hexene used here as the base kinetic models differ from the MCM by the following points:• Ozonolysis scheme for cis-2-butene and trans-2-hexene including the formation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OO˙ (ethanal-2-peroxy, MCM notation: HCOCH2O2) and OCHCHO(OO˙)CH2CH3 (butanal-2-peroxy, MCM notation: BUTALAO2). The subsequent bimolecular chemistry for these radicals follows the MCM.
CHCH2OO˙ (ethanal-2-peroxy, MCM notation: HCOCH2O2) and OCHCHO(OO˙)CH2CH3 (butanal-2-peroxy, MCM notation: BUTALAO2). The subsequent bimolecular chemistry for these radicals follows the MCM.
• Isomerisation reactions for ethanal-2-peroxy and butanal-2-peroxy based on quantum chemical calculations, leading to the formation of OC(OOH)CH2OO˙ (ethyl-1-peracid-2-peroxy) and OC(OOH)CH(OO˙)C2H5 (butyl-1-peracid-2-peroxy), respectively (Fig. S1, ESI‡).
• Updated alkoxy decomposition rates for RO radicals formed in the oxidation of cis-2-butene and trans-2-hexene by NO3 based on quantum chemical calculations.
• Optimised yield of the RO and OH radical from the reaction of RO2 with HO2 for the first generation NO3–RO2 (MCM notation: C62NO33O2, C63NO32O2) formed in the oxidation of trans-2-hexene by NO3.
The follow-up chemistry for additionally included RO2 and RO radicals was implemented following the SARs in Jenkin et al.,12 Vereecken et al.,10,36–38 and Novelli et al.10 For the model-measurement comparison when NO3 chemistry is contributing, the fraction of RO2 which is not measurable by the radical instrument10 (Section 2.2) must be taken into account, i.e. distinguishing between “total RO2” and “detectable RO2”.
The effect of the isomerisation reactions of the RO2 radicals from the ozonolysis reaction, ethanal-2-peroxy and butanal-2-peroxy, on the predicted RO2 speciation is investigated by comparing model runs using the FZJ mechanisms with and without including RO2 isomerisation reactions. In another sensitivity model run, not only reactions of methyl peroxy (CH3O2) and PAN-like RO2 (RC(O)OO˙) with NO2 are included, as implemented in the FZJ mechanism, but also the reaction of NO2 with all formed non-acyl RO2 species. In this sensitivity run, RO2 + NO2 reactions forming RO2NO2 as well as the corresponding backward reactions (eqn (3)) are introduced for all formed RO2 species. Rate constants for the forward and backward reactions for CH3O2, ethyl peroxy (C2H5O2), and acetonyl peroxy (CH3 C(O)CH2O2) are taken from recommendations by IUPAC.16 Recommendations in Jenkin et al.,12 partially based on Zabel et al.,18 are used for the forward and backward reaction rate constants for C4 peroxy radicals, and for the backward reaction rate constant for C6–RO2NO2 in this work. For all RO2 for which rate constants cannot be taken from literature, such as for n-C3H7O2, a pressure-independent generic forward reaction rate constant, kfPN, and a rounded average of reported backward reaction rate constants for C2 to C8 alkyl RO2, kbPN, recommended by Jenkin et al.,12 are used. However, the contribution of n-C3H7O2 to the total RO2 amounts to 2% and is thus negligible. It is important to mention that all the rate coefficients used from literature and SAR are for non-NO3 substituted RO2 radicals but are used here due to the lack of specific values for nitrate RO2 radicals. An overview of temperature-dependent rate coefficients for the formation and decomposition of non-acyl RO2NO2 is shown in Table 1, together with the corresponding rate coefficients at T = 276 K, 292 K, and 305 K. We also determined the kbPN and kfPN values by a fitting procedure against the experimental data, retrieving values that are within 30% of the literature data above. This supports applying literature data for alkyl-RO2 to our NO3-RO2 radicals, but the fitted values carry a large uncertainty and are not used in the results and discussion below
3 Results
3.1 Nighttime chemistry of cis-2-butene
Measured and modelled time series of HO2 and RO2 radical concentrations in the two nighttime experiments investigating the oxidation of cis-2-butene are shown in Fig. 1. For the two experiments in this work, injected O3, NO2, and cis-2-butene concentrations are below 15 ppbv, 30 ppbv and 15 ppbv, respectively, and expected NO3 concentrations do not exceed 8 pptv. For these conditions, on average, NO3 contributed about 40% at 276 K and 50% at 295 K to the oxidation of cis-2-butene (Table S2, ESI‡). As described in Section 2.2, only 16% and 5% of the most dominant RO2 that is formed, CH3 CH(OO˙)CH(ONO2)CH3 (MCM notation: C42NO33O2), are detected by the instrument at cold (T ≈ 276 K) and medium (T ≈ 295 K) temperatures, respectively. The higher the temperature becomes, the faster the decomposition of the alkoxy radical is. Therefore, the modelled total RO2 and the modelled detectable RO2 are discussed separately in the following.For cold conditions, there is a large difference in the total modelled RO2 between the FZJ mechanisms with and without including the additional formation of RO2NO2, with the latter predicting a much lower concentration (by a factor of 7 on average) in the first two hours of oxidation and a much slower increase. When the detectable modelled RO2 are compared to the measured RO2 radical concentrations at low temperatures, the mechanism including non-acyl RO2NO2 formation (FZJ + RO2NO2 model) reproduces the data very well with a model-to-measurement ratio of 0.84, and excellent reproduction of the time dependence. The mechanism without including RO2 + NO2 reactions (FZJ model) can reasonably reproduce the detectable RO2 after one hour after the cis-2-butene injections (average model-to-measurement ratio = 0.8). However, their concentration is largely overestimated by up to a factor of 4.5 right after the injection, with a distinctly earlier concentration peak. The smaller difference between the two model results observed for the second injection of cis-2-butene is due to a larger contribution of CH3O2 (from close to 0% after the first cis-2-butene injection to ∼ 20% after the second). As the reaction of CH3O2 with NO2 is included in both the FZJ and MCM mechanisms, the effect of the correction is smaller.
Differences of total RO2 radical concentrations between the two FZJ mechanisms are also observed in the experiment at medium temperatures. However, the difference is much less pronounced and the average ratio of concentrations obtained between the two FZJ mechanisms is 1.1. A small underestimation of the measured RO2 (by a factor of 1.2) by both mechanisms can be observed by comparing it to the detectable RO2. The FZJ mechanism without alkyl-RO2 + NO2 reactions would predict a small peak of the detectable RO2, while concentrations are better described if these reactions are considered (Fig. 1). From the second injection on, RO2 radical concentrations predicted by both mechanisms converge to the same value.
The measured HO2 radical concentrations are well described by both FZJ mechanisms and agree within 36% (FZJ model) and 21% (FZJ + RO2NO2 model) after the first VOC injection and within 5% (both FZJ models) after the second VOC injection in the experiment at cold conditions and within 7% (both FZJ mechanisms) in the experiment at medium temperatures. A comparison of the concentration of acetaldehyde, a major product of the ozonolysis of cis-2-butene, obtained by the different models is shown in Fig. S11 (ESI‡) for the experiment at cold conditions. The different mechanisms predict similar product concentrations with a model-to-measurement ratio of 0.7.
3.2 Nighttime chemistry of trans-2-hexene
Fig. 2 shows measured HO2 and RO2 radical concentrations for the experiment with trans-2-hexene performed at ≈276 K. In contrast to cis-2-butene, the amount of RO2 that cannot be detected is negligible,10 so that the total RO2 can be directly compared to the measurements.The addition of RO2 + NO2 reactions leads to a less steep increase of the RO2 radical concentrations which is in much better agreement with the measurements after the first injection of trans-2-hexene (within 2% compared to 20% for the FZJ model). Similar to cis-2-butene, at 276 K the FZJ mechanism expects a fast increase of RO2 radicals, which is not observed in the measurements.
As ozone contributes up to 40% to the oxidation of trans-2-hexene, and discrepancies were observed earlier when ozone dominated the oxidation at 292 K in the study by Novelli et al.,10 an ozonolysis experiment was performed for trans-2-hexene. Fig. S2 (ESI‡) shows the comparison between measured and modelled HO2 and RO2 radicals for the MCM and the FZJ mechanisms. An agreement within 20% and 30% between measured and modelled HO2 is found for the MCM and the FZJ mechanism, respectively. Although the FZJ mechanism improves the agreement between measured and modelled RO2 radicals, still a ratio of modelled to measured of 1.7 remains at the point of injection of trans-2-hexene. The impact of this discrepancy, in the presence of NO2, was estimated by assuming an overall overestimation of a factor of 1.7 for RO2 formed from ozonolysis and correcting their concentrations modelled by the FZJ mechanism including RO2NO2 formation by this factor. As a consequence, the total modelled RO2 radical concentrations would be 25% lower (cold temperatures) at the peak RO2 after the first injection (Fig. 2) with an average ratio of modelled to measured RO2 of ≈0.7. Although the agreement between measured and modelled RO2 is decreased when an overestimation of RO2 of 1.7 is assumed (from 12% to 37% for the first 2.5 hours of oxidation), the modelled RO2 shows the same behaviour in reaching the peak RO2 radical concentrations and a worse agreement is found for the mechanism without additional NO2 reactions (model-to-measurement ratio of 1.9 for the first 2.5 hours of oxidation). More details about the interpretation of the ozonolysis experiment can be found in the ESI‡ Section A.
As compared to cis-2-butene (Fig. 1) and to the ozonolysis experiment (Fig. S2, ESI‡), the HO2 radicals are largely underestimated by both models. HO2 radicals are mainly formed from the reaction of CO with OH (≈61% of the total production rate) which is formed directly from the ozonolysis of trans-2-hexene, among other production pathways such as isomerisation reactions of second-generation RO2 (30% of the total production rate). A similar discrepancy between measured and modelled HO2 radicals was observed when performing the same experiment but using CH4 instead of CO as an OH scavenger (Fig. 3).
The observed model-measurement discrepancy could be due to an artefact in the LIF instrument detecting HO2, if there was a mechanism artificially producing HO2 in the HOx detection cell. An instrumental interference by NO2 and peroxynitric acid (HNO4, formed in the reaction of HO2 with NO2) can be excluded because this would also impact the HO2 radical concentration measured in the experiment with cis-2-butene, for which no model-measurement discrepancy for HO2 was observed (Fig. 1). Furthermore, an interference from the contemporary decomposition of RO2 radicals is minimised by running the HOx detection cell at reduced NO (Section 2.2) and no indication of an interference was observed when changing the NO concentrations in the HOx cell. A clear correlation of the model-measurement difference in HO2 radical concentrations is only found with the modelled non-acyl RO2NO2 concentrations (Fig. 4), indicating that the model-measurement discrepancy of HO2 is likely due to an instrumental interference in the HO2 measurements from RO2NO2. This would be consistent with the good agreement observed for the ozonolysis experiment (Fig. S2, ESI‡). An interference of ≈2% of the non-acyl RO2NO2 in the HOx cell would be enough to explain the measured HO2 radical concentration (Fig. 2 and 3).
 | ||
| Fig. 4 Correlation between observed model-measurement deviations of HO2 with modelled non-acyl RO2NO2 concentrations. Displayed are NO3 oxidation experiments of trans-2-hexene. | ||
It is currently not clear how the non-acyl RO2NO2 would be detected as HO2 in the HOx cell of the LIF instrument, especially as no discrepancy was observed for cis-2-butene. RO2 radicals are obtained by subtracting the measured HO2 and OH radicals from the sum of all three radical species (OH + HO2 + RO2), as detected in the ROx LIF system. Therefore, an interference in the HOx measurement would result in a lower measured RO2 radical concentration as compared to the “real” value. Using the modelled HO2 radical instead of the measured value would increase the RO2 radical concentrations by 20% and 30% in the experiments at low temperatures with CO (Fig. S15, ESI‡) and CH4 (Fig. 3) as OH scavenger, respectively. As the HO2 radical concentrations for the conditions of the experiments in this study are low, their impact is limited and no change in the time profile of the RO2 radical is observed. More details can be found in Section G of the ESI.‡
Two more experiments at 292 K and 305 K were performed with trans-2-hexene (Fig. S12, ESI‡). Although a worse agreement between measured and modelled RO2 was observed (average model-measurement ratio of 1.5 and 1.2 for 292 K and 305 K, respectively) as compared with the experiment at 276 K, a shift in the peak for measured RO2 radical concentration could be observed which does not show any “delay” as found in the experiment at cold temperatures (Fig. 2). This is consistent with the faster decomposition rate of the formed RO2NO2 at those temperatures and is also reproduced by both models where negligible differences are seen already maximum 30 minutes after the VOC injection.
As compared to the experiment at 276 K (cold), a better agreement between modelled and measured HO2 radical is obtained. Model calculations tend to overestimate the HO2 radicals at 276 K. Just recently, a study by McKee et al.39 showed that at high temperatures production of nitryl hydride (HNO2) from the reaction of HO2 and NO2 can be expected. By including this reaction in the model calculations of the experiment at hot conditions (T = 305 K), the comparison of the model with the measurement improves leading to a model-to-measurement ratio of 1.3 on average (compared to 1.6 if the HNO2 formation is not considered, Fig. S12, ESI‡). More discussion about possible reasons for the observed discrepancies found at 292 K and 305 K for trans-2-hexene can be found in the ESI‡ Section G.
The comparison of acetaldehyde concentrations predicted by the mechanisms is shown in Fig. S14 (ESI‡), showing similar concentrations for the different temperatures. The mechanisms overall underestimate the measured acetaldehyde concentrations by a factor of 1.7 and 3 in the experiments at medium and hot temperatures, respectively, but in the experiment at cold conditions a good agreement is achieved (within 15%). The reason for this temperature-dependent discrepancy cannot be easily explained and needs further investigations.
4 Discussion
An overview of the concentrations of non-acyl peroxynitrates, formed in the nighttime oxidation, is shown in Fig. 5 for cis-2-butene and in Fig. S13 (ESI‡) for trans-2-hexene. Acyl peroxynitrates are excluded here because their formation is implemented identically in both mechanisms, and in the following, RO2 and RO2NO2 refer only to non-acyl peroxy radicals and their respective peroxynitrates, unless noted otherwise. | ||
| Fig. 5 Formed non-acyl RO2NO2 concentrations and the fraction of non-acyl RO2 stored as RO2NO2 (RO2NO2/(RO2 +RO2NO2)) for the two cis-2-butene oxidation experiments in the presence of NO2 applying the FZJ mechanism either without (FZJ model, blue) or with (FZJ + RO2NO2 model, brown) the additional formation of alkyl-RO2NO2. Model results of the RO2NO2 concentrations, based on the FZJ mechanism without including additional formation of RO2NO2 (FZJ model), refer to CH3O2NO2 concentrations. Vertical lines refer to the injection of chemical species. | ||
The RO2NO2 concentrations expected from the FZJ mechanism are systematically lower than the RO2NO2 concentrations expected from the FZJ + RO2NO2 mechanism, including non-acyl RO2NO2 formation, as only the formation of CH3O2NO2 is implemented (Section 2.3). A strong increase of peak RO2NO2 concentrations from 140 pptv to up to 480 pptv is observed over the temperature range of 295–276 K. While the main loss path for RO2 radicals in the FZJ model is the loss in the reaction with HO2 (k∼ (2 − 10) × 10−3 s−1), the reaction with NO2 (k∼ (5 − 9) s−1, Table 1) is the predominant, temporary loss of RO2 in the FZJ + RO2NO2 mechanism, including formation of non-acyl RO2NO2, due to the large NO2 mixing ratios (up to 40 ppbv).
The fraction of RO2 stored in the reservoir RO2NO2 species can be derived from the ratio of corresponding concentrations: RO2NO2/(RO2 + RO2NO2) (Table 2, Fig. 5 and Fig. S13, ESI‡). In the experiment at cold conditions, the conversion of RO2via reaction with NO2 leads to more than 90% of non-acyl RO2 that is present in form of the reservoir species RO2NO2 throughout the experiment. In comparison, 80% to 40% of the non-acyl RO2 are stored as RO2NO2 throughout the experiment at medium temperatures (Fig. 5), in which the temperature increased from 290 K to 300 K, leading to a shift of equilibrium towards RO2. When one of the VOCs is injected a large fraction of the RO2 radicals formed is converted into the reservoir species RO2NO2, leading to the slower increase of the free, measurable RO2 radical concentrations in the FZJ + RO2NO2 mechanism, including additional RO2NO2 formation, compared to the FZJ mechanism (Fig. 1 and 2). This emphasises the importance of including the formation of peroxynitrates in chemical mechanisms, especially at lower temperatures where the RO2NO2/RO2 ratio can reach a factor of 20.
| Temperature [K] | K|eqn (3) [cm3] |
|---|---|
| 305 | 7.0 × 10−13 |
| 292 | 3.2 × 10−12 |
| 276 | 2.5 × 10−11 |
At temperatures typically reached in mid latitudes, RO2NO2 concentrations are expected to be on the order of 1 × 1010 cm−3 at the conditions tested in this work and can therefore impact the fate of ROx species. In cities as Beijing,5 China, temperatures around 255 K can be reached in winter, leading to a significant fraction of non-acyl RO2 stored as reservoir RO2NO2 species, and thus to different RO2 radical concentrations. Low temperatures are also present at high altitudes, where biomass burning plumes can introduce polluted conditions, allowing alkyl-RO2NO2 other than CH3O2NO2 to be formed and impacting the effective RO2 radical concentration.
One important finding of this study is that the rate coefficients used for forward and backward reactions (Table 1 and 2) do an excellent job in bringing measurements and model results in agreement despite not being derived for the NO3-substituted RO2 radicals as investigated here. This indicates that the substituent on the RO2 radical does not seem to have an impact on the reaction rate. This allows to generalise the finding beyond the molecules and conditions explored in the SAPHIR chamber.
Fig. 6 shows the fraction of RO2NO2/(RO2 +RO2NO2) for different temperatures and NO2 mixing ratios. Conditions observed in different cities5,6,8,40 are highlighted and are found to favour the formation of non-acyl RO2NO2. Especially at wintertime in Beijing, a significant amount of RO2 is expected to be stored as RO2NO2. In contrast, NO2 and temperature observed in September in a boreal forest2 do not facilitate the production of RO2NO2. Though, for relatively low values of NO2 of ∼1 ppbv and ∼10 °C (280 K) that can also be found in forested environments, more than 20% of RO2 radical is stored as RO2NO2. For a mixing ratio of NO2 up to 10 ppbv, the fraction can increase up to 80%. Although a large part of the RO2NO2 will decompose back to RO2 and NO2, depending on temperature and lifetime, RO2NO2 can be transported and contribute to the NOx levels further away from their emission sources. In addition, nothing is known about the additional loss rate of RO2NO2 by reaction with OH and photolysis. Khan et al.15 introduced these loss reactions based on similarity with other molecules and show they could have an impact in particular at the equator.
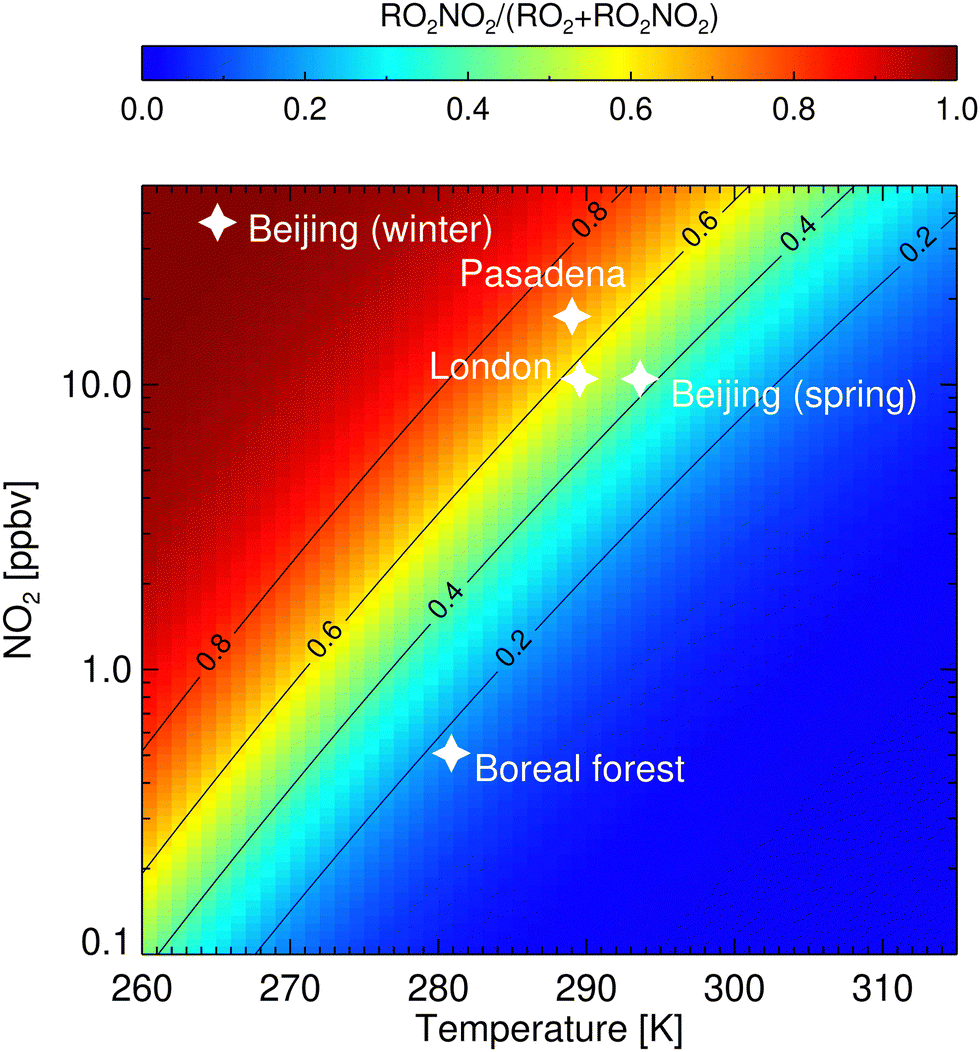 | ||
| Fig. 6 Fraction of peroxy radicals stored as RO2NO2 as a function of temperature and NO2 mixing ratios. Contour lines mark certain RO2NO2/(RO2 + RO2NO2) ratios. Values were determined from a steady-state calculation, assuming the generic forward and backward reaction rate coefficients kfPN and kbPN (Table 1) for reaction (3), respectively. White stars mark regimes that were observed in a boreal forest,2 in London,8 Pasadena,40 and Beijing at cold5 and medium temperatures.6 | ||
5 Summary & conclusions
In this study, the nighttime chemistry of cis-2-butene and trans-2-hexene was investigated under nighttime conditions. Experiments were performed in an outdoor simulation chamber under controlled conditions. The measurements show a clear dependence of the RO2 radical concentration time profiles on temperature, with the lowest temperatures having a significantly delayed RO2 peak concentration compared to what would be expected for direct formation of short-lived RO2 radicals. Measured trace gases and radicals were compared with different chemical mechanisms, implemented in zero-dimensional box model calculations based on the FZJ mechanism.10 This model improves upon the Master Chemical Mechanism by including a state-of-the-art ozonolysis scheme for cis-2-butene and trans-2-hexene, updated chemistry for nitrate peroxy radicals (NO3–RO2) from NO3 radical chemistry as well as for oxygenated peroxy radicals from ozonolysis. An extended model, FZJ + RO2NO2, additionally includes the reversible reactions of all non-acyl RO2 radicals with NO2 forming RO2NO2. In addition, the decomposition rate of –NO3–RO radicals, first introduced by a study by Novelli et al.,10 is adjusted to best match observed radical concentrations.The measured time profiles of the RO2 radical concentrations are reproduced best when accounting for the reaction of RO2 with NO2 for all RO2, not solely those for CH3O2 and acyl-RO2 radicals as typically included in atmospheric models. Especially for cold conditions, including the formation of reservoir RO2NO2 species significantly improves the modelled RO2 and an average agreement with the measured RO2 within 16% is achieved for cis-2-butene. This reaction allows reversible formation of alkyl peroxynitrate reservoir species, leading to a reduction of the effective concentration of free RO2 radicals that is especially pronounced at lower temperatures. In addition, the RO2 radical concentrations observed are equally (within 20% on average) described by both FZJ mechanisms (FZJ model with and without including additional RO2NO2 formation) above 290 K. With decreasing temperatures, including the formation and subsequent decomposition of RO2NO2 in the model reduces the predicted total free RO2 radical concentration and delays peak RO2 radical concentrations as observed in the experiments, thereby significantly improving the shape of modelled RO2 time profiles.
A discrepancy between modelled and measured HO2 radical concentration, which for the low-temperature conditions reached a factor of 3 on average, was observed for trans-2-hexene. Sensitivity analyses indicate that this discrepancy correlates best with a measurement interference by non-acyl RO2NO2, where an interference of ≈2% would be sufficient to reproduce the measured HO2. However, as this discrepancy does not influence the time-dependence of the RO2 radical concentration profiles in the chamber experiments, it has no impact on the conclusions.
Under the conditions of the experiments in this work, up to 2 × 10 cm−3 of non-acyl RO2NO2 are expected at 276 K, comprising about 95% of RO2. Under winter conditions in polluted cities such as Beijing, China, where temperatures of 255 K are reached,5 even higher fractions of the RO2 present as RO2NO2 reservoir species are expected. This will have consequences for radical chemistry, the spatial distribution of RO2 and NO2, and the loss of radicals through deposition of RO2NO2.13
The general forward and backward reaction rates from SAR12 which refer to non-substituted RO2 radicals do an excellent job in bringing measured and modelled RO2 radicals in agreement for the conditions of this study (NO3–RO2). This suggests that substituents on the RO2 radical have a small effect on the reaction rates making the implementation in global models easier. Although the largest impact of these reactions can be expected for cold temperatures (higher altitudes), already for NO2 mixing ratios as low as few ppbv and a temperature of 280 K, a fraction of RO2NO2/(RO2 + RO2NO2) of 20% can be expected (Fig. 6). Given the large uncertainties of the follow-up chemistry of RO2NO2, it is not easy to assess, a priori, their effect in different environments and more studies are needed to shed light on their chemistry. Generally, the impact of the formation of RO2NO2 manifests likely as a time delay in the response of the chemical mixture to changes in VOC emission and oxidation, NOx levels, temperature, and other environmental factors.
Discrepancies remain between modelled and measured RO2 radicals in the ozonolysis experiment of trans-2-hexene and for NO2 + O3 experiments of trans-2-hexene at higher temperatures (292 K and 305 K). Uncertainties in the ozonolysis may be related to the chemistry of peracid-substituted RO2 radicals formed in RO2H-migration reactions. Insufficient data are available to resolve this issue at this time, but it is shown that the discrepancy is too small to significantly affect the O3 + NO3 nighttime experiments in this work.
Data availability
Data from the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page (https://data.eurochamp.org/). Experiments with cis-2-butene performed at T ≈ 276 K and T ≈ 295 K are available at https://doi.org/10.25326/MBQ2-QY9541 and https://doi.org/10.25326/6BV7-MR14,42 respectively. Experimental data from the nighttime oxidation experiments of trans-2-hexene in the presence of NO2 and CO conducted at T ≈ 276 K, T ≈ 292 K, and T ≈ 305 K are accessible at https://doi.org/10.25326/DV74-3P36,43 https://doi.org/10.25326/E63B-9J58,44 and https://doi.org/10.25326/5CDB-Q698,45 respectively. Data from the nighttime experiment of trans-2-hexene in the presence of NO2 and CH4 as well as from the ozonolysis experiment of trans-2-hexene are available at https://doi.org/10.25326/DSQH-4 × 7146 and https://doi.org/10.25326/89B1-GR69,47 respectively.Conflicts of interest
The authors declare that they have no conflict of interest.References
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- J. Liebmann, E. Karu, N. Sobanski, J. Schuladen, M. Ehn, S. Schallhart, L. Quéléver, H. Hellen, H. Hakola, T. Hoffmann, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Atmos. Chem. Phys., 2018, 18, 3799–3815 CrossRef CAS.
- J. M. Liebmann, J. B. A. Muller, D. Kubistin, A. Claude, R. Holla, C. Plass-Dülmer, J. Lelieveld and J. N. Crowley, Atmos. Chem. Phys., 2018, 18, 12045–12059 CrossRef CAS.
- P. M. Edwards, K. C. Aikin, W. P. Dube, J. L. Fry, J. B. Gilman, J. A. De Gouw, M. G. Graus, T. F. Hanisco, J. Holloway, G. Hübler, J. Kaiser, F. N. Keutsch, B. M. Lerner, J. A. Neuman, D. D. Parrish, J. Peischl, I. B. Pollack, A. R. Ravishankara, J. M. Roberts, T. B. Ryerson, M. Trainer, P. R. Veres, G. M. Wolfe, C. Warneke and S. S. Brown, Nat. Geosci., 2017, 10, 490–495 CrossRef CAS.
- Z. Tan, F. Rohrer, K. Lu, X. Ma, B. Bohn, S. Broch, H. Dong, H. Fuchs, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, X. Li, Y. Liu, Y. Liu, A. Novelli, M. Shao, H. Wang, Y. Wu, L. Zeng, M. Hu, A. Kiendler-Scharr, A. Wahner and Y. Zhang, Atmos. Chem. Phys., 2018, 18, 12391–12411 CrossRef CAS.
- L. K. Whalley, E. J. Slater, R. Woodward-Massey, C. Ye, J. D. Lee, F. Squires, J. R. Hopkins, R. E. Dunmore, M. Shaw, J. F. Hamilton, A. C. Lewis, A. Mehra, S. D. Worrall, A. Bacak, T. J. Bannan, H. Coe, C. J. Percival, B. Ouyang, R. L. Jones, L. R. Crilley, L. J. Kramer, W. J. Bloss, T. Vu, S. Kotthaus, S. Grimmond, Y. Sun, W. Xu, S. Yue, L. Ren, W. J. F. Acton, C. N. Hewitt, X. Wang, P. Fu and D. E. Heard, Atmos. Chem. Phys., 2021, 21, 2125–2147 CrossRef CAS.
- Z. Tan, H. Fuchs, K. Lu, A. Hofzumahaus, B. Bohn, S. Broch, H. Dong, S. Gomm, R. Häseler, L. He, F. Holland, X. Li, Y. Liu, S. Lu, F. Rohrer, M. Shao, B. Wang, M. Wang, Y. Wu, L. Zeng, Y. Zhang, A. Wahner and Y. Zhang, Atmos. Chem. Phys., 2017, 17, 663–690 CrossRef CAS.
- L. K. Whalley, D. Stone, R. Dunmore, J. Hamilton, J. R. Hopkins, J. D. Lee, A. C. Lewis, P. Williams, J. Kleffmann, S. Laufs, R. Woodward-Massey and D. E. Heard, Atmos. Chem. Phys., 2018, 18, 2547–2571 CrossRef CAS.
- L. Vereecken, P. T. M. Carlsson, A. Novelli, F. Bernard, S. S. Brown, C. Cho, J. N. Crowley, H. Fuchs, W. Mellouki, D. Reimer, J. Shenolikar, R. Tillmann, L. Zhou, A. Kiendler-Scharr and A. Wahner, Phys. Chem. Chem. Phys., 2021, 23, 5496–5515 RSC.
- A. Novelli, C. Cho, H. Fuchs, A. Hofzumahaus, F. Rohrer, R. Tillmann, A. Kiendler-Scharr, A. Wahner and L. Vereecken, Phys. Chem. Chem. Phys., 2021, 23, 5474–5495 RSC.
- E. J. Slater, L. K. Whalley, R. Woodward-Massey, C. Ye, J. D. Lee, F. Squires, J. R. Hopkins, R. E. Dunmore, M. Shaw, J. F. Hamilton, A. C. Lewis, L. R. Crilley, L. Kramer, W. Bloss, T. Vu, Y. Sun, W. Xu, S. Yue, L. Ren, W. J. F. Acton, C. N. Hewitt, X. Wang, P. Fu and D. E. Heard, Atmos. Chem. Phys., 2020, 20, 14847–14871 CrossRef CAS.
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- E. C. Browne, A. E. Perring, P. J. Wooldridge, E. Apel, S. R. Hall, L. G. Huey, J. Mao, K. M. Spencer, J. M. S. Clair, A. J. Weinheimer, A. Wisthaler and R. C. Cohen, Atmos. Chem. Phys., 2011, 11, 4209–4219 CrossRef CAS.
- M. A. H. Khan, B. Miles, M. E. Jenkin, R. G. Derwent, C. J. Percival and D. E. Shallcross, ACS Earth Space Chem., 2020, 4, 1201–1212 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and I. Subcommittee, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- K. McKee, M. A. Blitz and M. J. Pilling, J. Phys. Chem. A, 2016, 120, 1408–1420 CrossRef CAS PubMed.
- F. Zabel, A. Reimer, K. H. Becker and E. H. Fink, J. Phys. Chem., 1989, 93, 5500–5507 CrossRef CAS.
- F. Rohrer, B. Bohn, T. Brauers, D. Brüning, F.-J. Johnen, A. Wahner and J. Kleffmann, Atmos. Chem. Phys., 2005, 5, 2189–2201 CrossRef CAS.
- E. Schlosser, B. Bohn, T. Brauers, H.-P. Dorn, H. Fuchs, R. Hässler, A. Hofzumahaus, F. Holland, F. Rohrer, L.-O. Rupp, M. Siese, R. Tillmann and A. Wahner, J. Atmos. Chem., 2007, 56, 187–205 CrossRef CAS.
- H. Fuchs, T. Brauers, H.-P. Dorn, H. Harder, R. Häseler, A. Hofzumahaus, F. Holland, Y. Kanaya, Y. Kajii, D. Kubistin, S. Lou, M. Martinez, K. Miyamoto, S. Nishida, M. Rudolf, E. Schlosser, A. Wahner, A. Yoshino and U. Schurath, Atmos. Chem. Phys., 2010, 10, 12233–12250 CrossRef CAS.
- E. C. Apel, T. Brauers, R. Koppmann, B. Bandowe, J. Boßmeyer, C. Holzke, R. Tillmann, A. Wahner, R. Wegener, A. Brunner, M. Jocher, T. Ruuskanen, C. Spirig, D. Steigner, R. Steinbrecher, E. Gomez Alvarez, K. Müller, J. P. Burrows, G. Schade, S. J. Solomon, A. Ladstätter-Weißenmayer, P. Simmonds, D. Young, J. R. Hopkins, A. C. Lewis, G. Legreid, S. Reimann, A. Hansel, A. Wisthaler, R. S. Blake, A. M. Ellis, P. S. Monks and K. P. Wyche, J. Geophys. Res. Atmos., 2008, 113, D20307 CrossRef.
- H. Fuchs, F. Holland and A. Hofzumahaus, Rev. Sci. Instrum., 2008, 79, 084104 CrossRef PubMed.
- F. Holland, A. Hofzumahaus, J. Schäfer, A. Kraus and H.-W. Pätz, J. Geophys. Res. Atmos., 2003, 108, 2–23 CrossRef.
- F. Holland, M. Hessling and A. Hofzumahaus, J. Atmos. Sci., 1995, 52, 3393–3401 CrossRef.
- H. Fuchs, H.-P. Dorn, M. Bachner, B. Bohn, T. Brauers, S. Gomm, A. Hofzumahaus, F. Holland, S. Nehr, F. Rohrer, R. Tillmann and A. Wahner, Atmos. Meas. Tech., 2012, 5, 1611–1626 CrossRef CAS.
- C. Cho, A. Hofzumahaus, H. Fuchs, H.-P. Dorn, M. Glowania, F. Holland, F. Rohrer, V. Vardhan, A. Kiendler-Scharr, A. Wahner and A. Novelli, Atmos. Meas. Tech., 2021, 14, 1851–1877 CrossRef CAS.
- H. Fuchs, B. Bohn, A. Hofzumahaus, F. Holland, K. D. Lu, S. Nehr, F. Rohrer and A. Wahner, Atmos. Meas. Tech., 2011, 4, 1209–1225 CrossRef CAS.
- H. Fuchs, Z. Tan, A. Hofzumahaus, S. Broch, H.-P. Dorn, F. Holland, C. Künstler, S. Gomm, F. Rohrer, S. Schrade, R. Tillmann and A. Wahner, Atmos. Meas. Tech., 2016, 9, 1431–1447 CrossRef CAS.
- S. Lou, F. Holland, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, R. Häseler, K. Kita, Y. Kondo, X. Li, M. Shao, L. Zeng, A. Wahner, Y. Zhang, W. Wang and A. Hofzumahaus, Atmos. Chem. Phys., 2010, 10, 11243–11260 CrossRef CAS.
- H. Fuchs, A. Novelli, M. Rolletter, A. Hofzumahaus, E. Y. Pfannerstill, S. Kessel, A. Edtbauer, J. Williams, V. Michoud, S. Dusanter, N. Locoge, N. Zannoni, V. Gros, F. Truong, R. Sarda-Esteve, D. R. Cryer, C. A. Brumby, L. K. Whalley, D. Stone, P. W. Seakins, D. E. Heard, C. Schoemaecker, M. Blocquet, S. Coudert, S. Batut, C. Fittschen, A. B. Thames, W. H. Brune, C. Ernest, H. Harder, J. B. A. Muller, T. Elste, D. Kubistin, S. Andres, B. Bohn, T. Hohaus, F. Holland, X. Li, F. Rohrer, A. Kiendler-Scharr, R. Tillmann, R. Wegener, Z. Yu, Q. Zou and A. Wahner, Atmos. Meas. Tech., 2017, 10, 4023–4053 CrossRef CAS.
- A. Jordan, S. Haidacher, G. Hanel, E. Hartungen, L. Märk, H. Seehauser, R. Schottkowsky, P. Sulzer and T. D. Märk, Int. J. Mass Spectrom., 2009, 286, 122–128 CrossRef CAS.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191–241 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2009, 11, 9062–9074 RSC.
- L. Vereecken and B. Nozière, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC.
- K. McKee, M. A. Blitz, R. J. Shannon and M. J. Pilling, J. Phys. Chem. A, 2022, 126, 7514–7522 CrossRef CAS PubMed.
- S. M. Griffith, R. F. Hansen, S. Dusanter, V. Michoud, J. B. Gilman, W. C. Kuster, P. R. Veres, M. Graus, J. A. De Gouw, J. Roberts, C. Young, R. Washenfelder, S. S. Brown, R. Thalman, E. Waxman, R. Volkamer, C. Tsai, J. Stutz, J. H. Flynn, N. Grossberg, B. Lefer, S. L. Alvarez, B. Rappenglueck, L. H. Mielke, H. D. Osthoff and P. S. Stevens, J. Geophys. Res.: Atmos., 2016, 121, 4211–4232 CrossRef CAS.
- M. Färber, H. Fuchs, G. Gkatzelis, F. Rohrer, S. Wedel and A. Novelli, Atmospheric simulation chamber study: cis-2-butene + NO3 - Gas-phase oxidation - kinetic study - 2022-02-11, 2023, (Version 1.0) [Data set]. AERIS.
- A. Novelli, C. Cho, H. Fuchs, A. Hofzumahaus, F. Rohrer and R. Tillmann, Atmospheric simulation chamber study: cis-2-butene + NO3 - Gas-phase oxidation - product study - 2020-05-19, 2021, (Version 1.0) [Data set]. AERIS.
- M. Färber, H. Fuchs, G. Gkatzelis, F. Rohrer, S. Wedel and A. Novelli, Atmospheric simulation chamber study: trans-2-hexene + NO3 - Gas-phase oxidation - kinetic study - 2022-01-26, 2023, (Version 1.0) [Data set]. AERIS.
- A. Novelli, C. Cho, H. Fuchs, A. Hofzumahaus, F. Rohrer and R. Tillmann, Atmospheric simulation chamber study: trans-2-hexene + NO3 - Gas-phase oxidation - product study - 2020-05-23, 2021, (Version 1.0) [Data set]. AERIS.
- M. Färber, H. Fuchs, G. Gkatzelis, F. Rohrer, S. Wedel and A. Novelli, Atmospheric simulation chamber study: trans-2-hexene + NO3 - Gas-phase oxidation - kinetic study - 2022-07-20, 2023, (Version 1.0) [Data set]. AERIS.
- M. Färber, H. Fuchs, G. Gkatzelis, F. Rohrer, S. Wedel and A. Novelli, Atmospheric simulation chamber study: trans-2-hexene + NO3 - Gas-phase oxidation - kinetic study - 2022-02-21, 2023, (Version 1.0) [Data set]. AERIS.
- M. Färber, H. Fuchs, G. Gkatzelis, F. Rohrer, S. Wedel and A. Novelli, Atmospheric simulation chamber study: trans-2-hexene + O3 - Gas-phase oxidation - kinetic study - 2021-12-08, 2023, (Version 1.0) [Data set]. AERIS.
Footnotes |
| † The data of the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page (https://data.eurochamp.org/). More information can be found in the Data availability statement. |
| ‡ Electronic supplementary information (ESI) available: The supplement related to this article is available online, and contains detailed information about the ozonolysis experiment of trans-2-hexene, the NO3 interference of the radical instrument, the modified alkoxy decomposition rate used for cis-2-butene-RO, instrumentation details, contributions of NO3 and O3 to the VOC oxidation, the time series of measured acetaldehyde concentrations, and the discussion of the nighttime oxidation of trans-2-hexene at medium and hot temperatures. See DOI: https://doi.org/10.1039/d3cp04163h |
| This journal is © the Owner Societies 2024 |