
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A model study of ceria–Pt electrocatalysts: stability, redox properties and hydrogen intercalation†
Lukáš
Fusek
 ab,
Pankaj Kumar
Samal
a,
Jiří
Keresteš
a,
Ivan
Khalakhan
a,
Viktor
Johánek
a,
Yaroslava
Lykhach
b,
Jörg
Libuda
b,
Olaf
Brummel
*b and
Josef
Mysliveček
*a
ab,
Pankaj Kumar
Samal
a,
Jiří
Keresteš
a,
Ivan
Khalakhan
a,
Viktor
Johánek
a,
Yaroslava
Lykhach
b,
Jörg
Libuda
b,
Olaf
Brummel
*b and
Josef
Mysliveček
*a
aCharles University, Faculty of Mathematics and Physics, Department of Surface and Plasma Science, V Holešovičkách 2, 180 00 Praha 8, Czech Republic. E-mail: josef.myslivecek@mff.cuni.cz
bInterface Research and Catalysis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany. E-mail: olaf.brummel@fau.de
First published on 18th October 2023
Abstract
The electrocatalytic properties of advanced metal-oxide catalysts are often related to a synergistic interplay between multiple active catalyst phases. The structure and chemical nature of these active phases are typically established under reaction conditions, i.e. upon interaction of the catalyst with the electrolyte. Here, we present a fundamental surface science (scanning tunneling microscopy, X-ray photoelectron spectroscopy, and low-energy electron diffraction) and electrochemical (cyclic voltammetry) study of CeO2(111) nanoislands on Pt(111) in blank alkaline electrolyte (0.1 M KOH) in a potential window between −0.05 and 0.9 VRHE. We observe a size- and preparation-dependent behavior. Large ceria nanoislands prepared at high temperatures exhibit stable redox behavior with Ce3+/Ce4+ electrooxidation/reduction limited to the surface only. In contrast, ceria nanoislands, smaller than ∼5 nm prepared at a lower temperature, undergo conversion into a fully hydrated phase with Ce3+/Ce4+ redox transitions, which are extended to the subsurface region. While the formation of adsorbed OH species on Pt depends strongly on the ceria coverage, the formation of adsorbed Hads on Pt is independent of the ceria coverage. We assign this observation to intercalation of Hads at the Pt/ceria interface. The intercalated Hads cannot participate in the hydrogen evolution reaction, resulting in the moderation of this reaction by ceria nanoparticles on Pt.
Introduction
Understanding the catalytic properties of advanced electrocatalysts for energy conversion and storage requires detailed characterization of the catalyst–electrolyte interface under operating conditions. Upon contact with the electrolyte at operating potential, the as-prepared electrocatalysts undergo structural and chemical changes yielding active catalyst phases.1,2 These changes may include the hydration and hydroxylation of the catalyst surfaces,3–5 leaching of electrochemically unstable species,6,7 or structural transformations.8,9 In specific cases, electrochemical activities of a broad spectrum of the as-prepared catalysts can be traced back to a considerably narrower range of electrochemically active phases. A representative example is the formation of transition metal oxyhydroxides on the surface of various transition metal compounds during water electrolysis.10 Combining multiple electrochemically active phases can yield new reaction pathways and increase significantly the activity of the catalyst, as shown for noble and transition metal-based electrocatalysts activated with transition metal compounds.11–13The complex nature of real high-area electrocatalysts1,2 and their interaction with electrolytes underlines the importance of model approaches which utilize well-defined model systems. In such model studies, quantitative information on the stability and catalytic activity can be obtained with atomic resolution on single crystalline, thin film or nanoparticulate samples combining experimental approaches of surface science and electrochemistry.14–18 The importance of transition metal-based catalysts for alkaline water electrolysis is fueling current interest in model electrocatalytic studies predominantly on Co, Fe, and Ni oxides and oxyhydroxides. Model studies reveal the formation of active hydroxylated phases,19–21 the stability of electrocatalyst surfaces under operating conditions,22–25 and activation of electrocatalysts via doping26–28 or bifunctional interactions in the presence of noble metals.19,29
Here, we present a model electrocatalytic study on a specific class of catalytically relevant oxides – rare earth oxides, as represented by cerium oxide (ceria). Apart from its wide application in heterogeneous catalysis,30,31 ceria finds promising applications in electrocatalysis mainly as a co-catalyst for activation of ammonia, alcohol, and CO electrooxidation,32–34 and for reactions related to electrolysis and fuel cells – the hydrogen evolution reaction (HER),35 oxygen reduction reaction (ORR),36–38 and oxygen evolution reaction (OER).39 Ceria-based electrocatalysts are generally synthesized in the form of cerium oxide32–38 or cerium hydroxide39 nanoparticles, interfacing nanoparticles of a second catalytically active phase – metal32–38 or metal oxyhydroxide.39 The heterogeneous interface between the catalytically active phases is, correspondingly, identified as the source of improvement of ceria-based electrocatalyst properties via charge transfer between a metal and ceria,32,36,37,39 enhanced water dissociation and OH supply,33,35 or via formation of specific defects in ceria.37,38 The enhancement of the ceria-based electrocatalyst properties can be co-determined by the size and structure of the ceria nanoparticles.32,38 All the above-mentioned synergistic effects of ceria in ceria-based electrocatalysts must critically depend on the nature of the cerium phases established under electrochemical conditions. These, however, have remained largely unknown so far.
To obtain elementary information on the nature of the cerium phases under electrochemical conditions, we investigate the stability and electrochemical reduction/oxidation properties of CeO2(111) nanoislands prepared on Pt(111) in alkaline electrolyte (0.1 M KOH). The properties of the as-prepared and electrochemically treated ceria/Pt model catalysts are quantified using experimental methods of surface science – scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS), low-energy electron diffraction (LEED), and atomic force microscopy (AFM), in combination with cyclic voltammetry (CV). We use different preparation methods to obtain samples with varying lateral sizes of ceria nanoislands. We collect evidence for hydration of ceria nanoislands smaller than 5 nm, and the corresponding surface or subsurface Ce3+/Ce4+ electrooxidation/reduction of large CeO2(111) and small CeO2·2H2O nanoislands, respectively. An evaluation of ceria coverage, in comparison with the charges assigned to Ce3+/Ce4+ transition and to H and OH adsorption/desorption on Pt(111), indicates reversible intercalation of adsorbed Hads at the ceria/Pt interface. The intercalated Hads is unable to participate in the hydrogen evolution reaction (HER).
Results and discussion
Preparation and characterization of the samples by surface science methods
Samples of ceria–Pt electrocatalysts were prepared by means of physical vapor deposition in the form of inverse model catalysts consisting of CeO2(111) nanoislands supported on Pt(111). The samples were prepared using two methods which yielded distinctly different CeO2(111) nanoisland morphologies. The methods differed in thermal treatments during deposition and post-deposition, and are denoted as high-temperature (HT, final annealing temperature 1050 K) and low-temperature (LT, final annealing temperature 850 K) methods. Detailed description of the experimental procedures is given in the Materials and methods section of the ESI.† Samples with the amounts of deposited CeO2 of approximately 1, 2, and 3 monolayers (ML) were prepared using both methods. STM images of the as-prepared samples are shown in Fig. 1. In the following discussion, 1 ML represents a basic O–Ce–O repeat unit of CeO2(111) containing 7.9 × 1014 cm−2 Ce atoms, twice that of O atoms, and featuring a thickness of 3.1 Å.30,31 | ||
| Fig. 1 STM images (150 × 150 nm2) of CeO2/Pt(111) samples prepared using two different methods. High temperature preparation (HT, final annealing at 1050 K) yields compact CeO2(111) islands with straight edges (a)–(c). Low temperature preparation (LT, final annealing at 850 K) yields open CeO2(111) islands with rounded edges, and a characteristic size <5 nm (d)–(f). Samples were prepared using the amounts of deposited CeO2 of approximately 1 (a), (d), 2 (b), (e), and 3 monolayers (ML, (c) and (f)). | ||
HT samples (Fig. 1a–c) exhibit a flat geometry and compact island shapes with straight edges aligned along high-symmetry substrate directions. With increasing amount of deposited CeO2(111), the nanoislands extend laterally and coalesce to an almost continuous CeO2(111) thin film for 3 ML of deposited ceria (Fig. 1c). The growth of the ceria nanoislands resembles a layer-by-layer growth and is consistent with previous observations of CeO2(111) growth on Pt(111).40,41 The LEED diffraction pattern shows diffraction spots of the clean Pt(111) substrate and unreconstructed CeO2(111) (Fig. S1a, ESI†). The LT samples (Fig. 1d–f) exhibit a similarly flat geometry but with distinctly smaller ceria nanoisland sizes compared to HT samples (Fig. 1d). With increasing amount of deposited ceria, coalescence yields open island shapes with rounded edges (Fig. 1e), and coalescence proceeds at a slower rate as compared to the HT samples (Fig. 1f). The round edges of the LT samples are expected to contain an increased amount of kink sites compared to the straight step edges of HT samples. Similar to HT samples, LT samples show a LEED pattern corresponding to CeO2(111) on Pt(111) (Fig. S1b, ESI†). The effect of lowering the final annealing temperature on the morphology of LT samples is similar to previous observations of CeO2(111) growth on Pt(111).40,41
Based on the STM images, we can quantify the evolution of morphological features of the HT and the LT samples as a function of the amount of deposited ceria. The results are summarized in Fig. S2 and Table S1 (ESI†). For both the HT and the LT samples, the coverage of the CeO2(111) layer as determined by STM (θCeSTM) scales linearly with the amount of deposited ceria, yielding a thickness of the CeO2(111) nanoislands (4 ± 1) ML for all samples shown in Fig. 1. A distinct difference between the HT and the LT samples is the different lateral size of the CeO2(111) structures, as identified by autocorrelation length obtained from the STM images (Fig. S2a, ESI†). The autocorrelation length of the LT samples is almost constant, reflecting the characteristic width of the CeO2(111) structures (∼5 nm), much smaller than the characteristic lateral size of HT CeO2(111) nanoislands (∼20 nm). The HT and the LT samples differ in the length of the metal-oxide boundary. For HT samples, the metal-oxide boundary amounts to 1–3% of the Pt(111) surface sites, while for the LT samples, the value is 5–7% (Fig. S2b, ESI†).
In addition to morphological differences, XPS reveals differences in the chemical state of the as-prepared HT and LT samples. This, particularly the concentration of Ce3+ species, is determined from the Ce 3d spectrum (Fig. S2c, ESI†). HT and LT samples show Ce3+ concentrations in the range of 4–8% and 12–16%, respectively. This corresponds, formally, to 1–2% and 3–4% O vacancy concentration relative to stoichiometric CeO2 for HT and LT samples, respectively. The increased concentration of Ce3+ in the LT samples can be attributed to an increased concentration of structural defects (step edges, kinks) and oxygen vacancies as compared to the HT samples.
Electrochemical characterization of the samples
After preparation and characterization by surface science methods in an ultra-high vacuum (UHV), the ceria/Pt model catalysts were transferred through air into an electrochemical cell and characterized by CV in alkaline electrolyte (0.1 M KOH electrolyte, purged with Ar). All measurements were performed at potentials ranging from −0.05 to 0.9 VRHE [potential versus the reversible hydrogen electrode (RHE)], and at a scan rate of 400 mV s−1. An overview of the CV results for the HT and LT samples is presented in Fig. 2 and 3.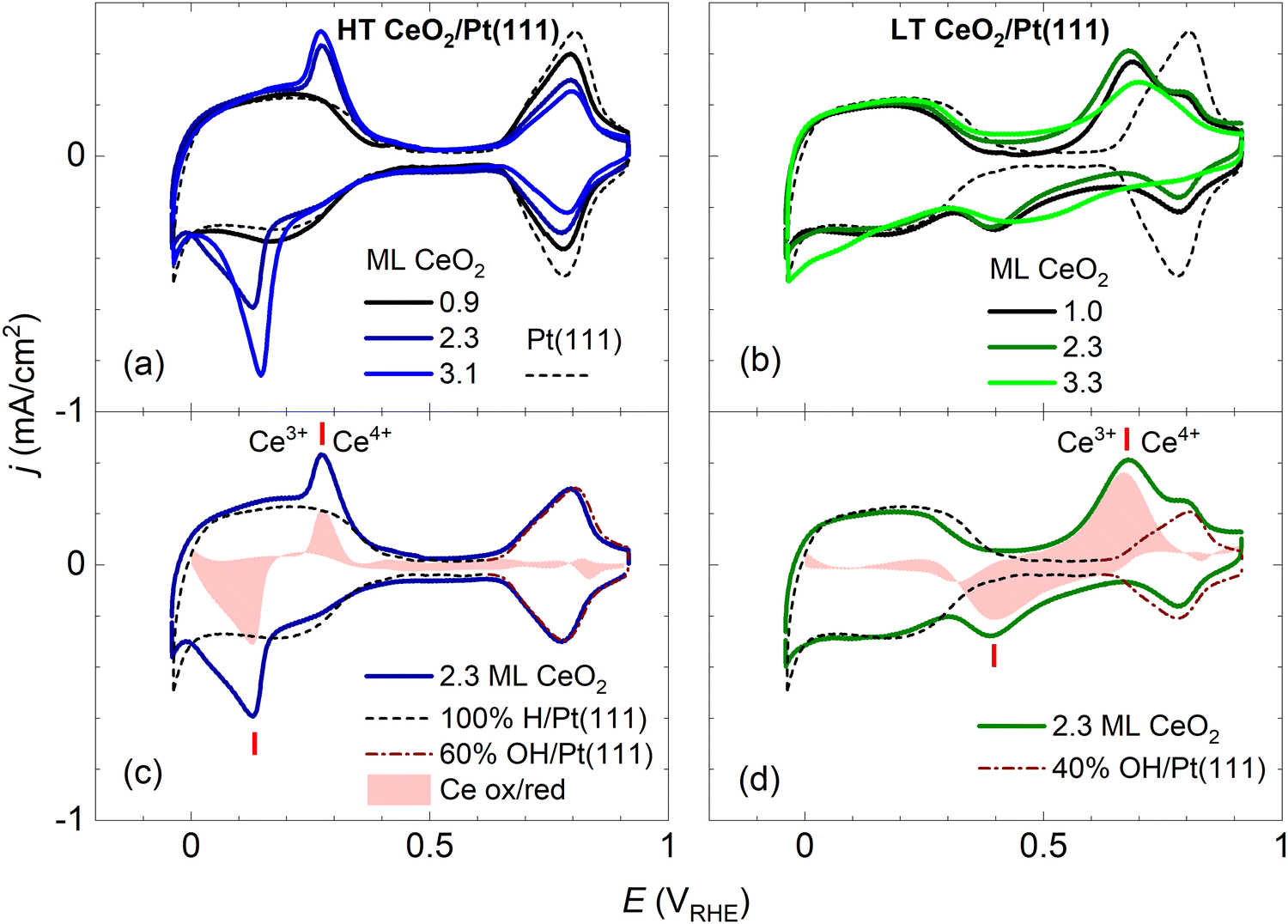 | ||
| Fig. 2 Dependence of the stable CVs on the amount of deposited CeO2 for HT samples shown in Fig. 1a–c (a), and for LT samples shown in Fig. 1d–f (b). For reference, the CV of clean Pt(111) is shown. Determination of the faradaic contribution to Ce3+/Ce4+ electrooxidation/reduction for HT (c) and LT (d) samples. The red shaded area represents a difference between the experimental CV of a ceria/Pt sample (solid line) and scaled reference data for clean Pt(111). The red ticks indicate the positions of Ce3+/Ce4+ redox peaks. CVs in 0.1 M KOH, at a scan rate of 400 mV s−1. | ||
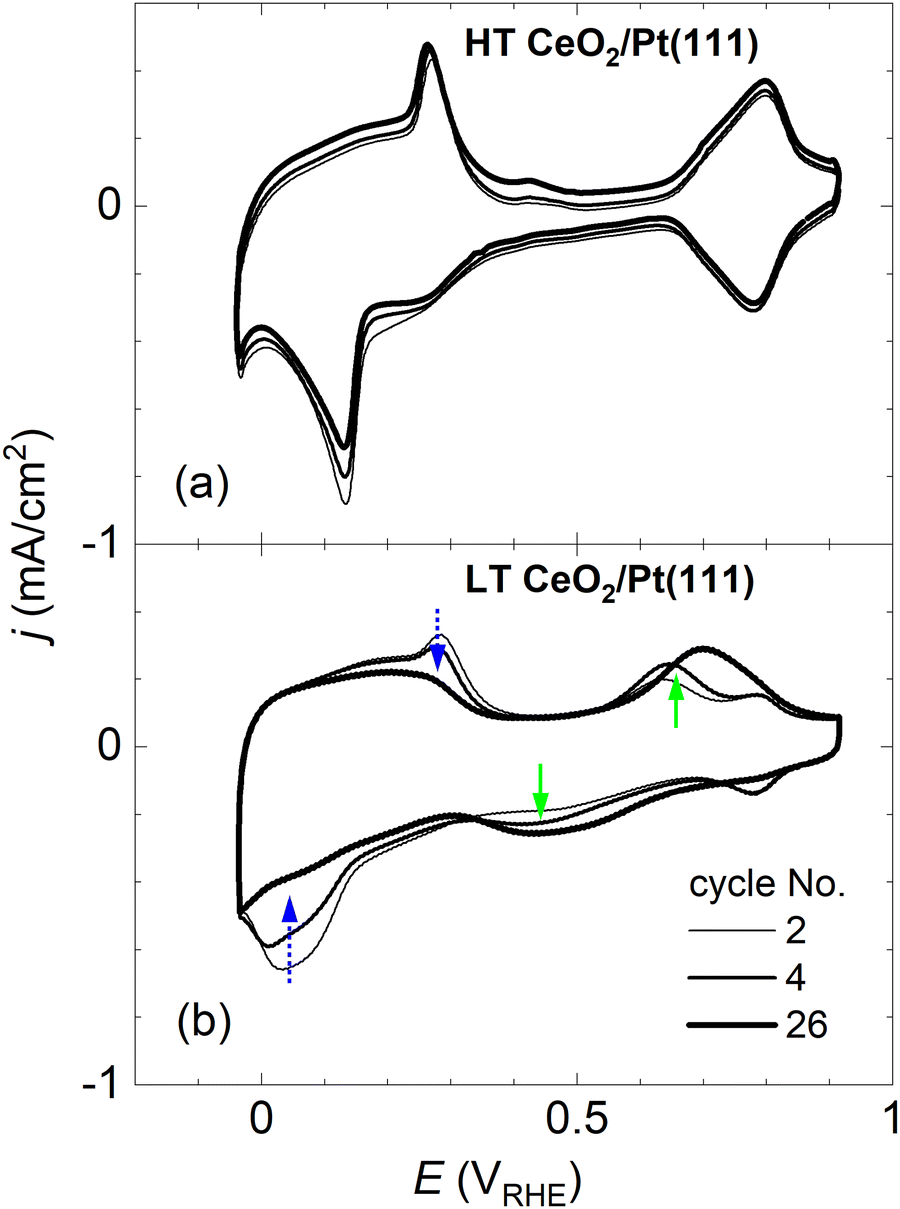 | ||
| Fig. 3 Stability of the CVs upon immersion of the samples into the electrolyte. The representative HT sample in (a), and the LT sample in (b). In (b), the dotted arrows indicate disappearing peaks, and the full arrows indicate growing peaks. CVs in 0.1 M KOH, at a scan rate of 400 mV s−1. | ||
In Fig. 2, the CV of clean Pt(111) is presented for comparison. A detailed view of the clean Pt(111) reference as obtained in our experiments is presented in Fig. S3 (ESI†). The CV consists of three distinct regions: The hydrogen region (H desorption/adsorption) between −0.05 and 0.35 VRHE, the double layer region between 0.35 and 0.65 VRHE, and the oxygen region (OH adsorption/desorption) between 0.65 and 0.90 VRHE. The total integrated charges associated with these regions qrefH/Pt = 350 μC cm−2, qrefDL =125 μC cm−2, and qrefOH/Pt = 305 μC cm−2 for the hydrogen, double layer, and oxygen region, respectively, are in good agreement with values reported in the literature.15
Fig. 2a and b show CVs of HT samples shown in Fig. 1a–c, and CVs of LT samples shown in Fig. 1 d–f, respectively. Both figures illustrate the dependence of the CV shape on the amount of deposited CeO2. Compared to the CV of clean Pt(111), CVs of HT samples include a new pair of irreversible peaks positioned in the hydrogen region. CVs of LT samples include a new pair of irreversible peaks as well, however, it is positioned at higher potential (by approx. 0.3 V) in the double layer region of clean Pt(111). The features observable on Pt(111), corresponding to H desorption/adsorption, the double layer, and the OH adsorption/desorption, are clearly resolved for all samples. Qualitatively, for both the HT and the LT samples, the areas of the irreversible peaks increase with increasing amount of deposited ceria, while areas of the OH adsorption/desorption on Pt(111) decrease. The area of H desorption/adsorption on Pt(111) does not show apparent changes.
CVs in Fig. 2 represent stabilized CVs obtained upon immersion of the sample in the electrolyte and application of approx. 25 cycles. CVs obtained immediately after the immersion of the samples in the electrolyte are shown in Fig. 3. Fig. 3a illustrates that the CVs of the HT samples are stable from the first complete cycle, and remain stable on the time scale of the present experiments (∼10 min contact of the samples with the electrolyte, ∼50 CV cycles). In contrast, distinct changes are observable in the shape of the CVs of the LT samples. Initially, irreversible peaks similar to the peaks of HT samples appear in the H adsorption/desorption region of Pt(111). These peaks, however, are quickly replaced by a pair of irreversible peaks localized at higher potentials. CVs with irreversible peaks in the double layer region stabilize within ∼20 cycles.
Assignment of the CV peaks
The irreversible peaks in the CVs of both HT and LT samples can be assigned to particular Ce phases as follows: At pH = 13, corresponding to the 0.1 M KOH electrolyte, the solubility of Ce3+ ions is negligible42 and electrochemical phase transitions are expected to occur between hydroxylated phases, and, in the presence of oxygen (or traces of oxygen, such as in the Ar-saturated electrolytes), oxidized solid ceria phases.43 In chemical compounds, Ce adopts exclusively Ce3+ and Ce4+ oxidation states,30 and Ce3+ and Ce4+ oxidation states are also the only states considered in aqueous electrolytes under potential.43 The observed irreversible peaks in both HT and LT CVs thus correspond to Ce3+/Ce4+ redox transitions. At pH =13, and potentials >0.7 VRHE (highest-potential irreversible peak in our observations, Fig. 2b and d), Ce4+ containing phases must dominate.43 The observed shift of the irreversible peaks by ∼0.3 V between HT and LT samples however indicates that the Ce4+ containing phases in the HT and LT samples are distinctly different.The Ce phase present in a vacuum, under ambient conditions, and in water-based electrolytes is predominantly the cubic phase of CeO2 which is observed across the whole range of ceria oxygen deficiency between CeO2 and Ce2O3.44–46 In the presence of water, the reduced cubic phase CeO2−x can become partly hydroxylated.31,45,47 At strong oxygen deficiency and in the presence of water, CeO2−x thin films or nanoparticles are observed to convert to hexagonal Ce(OH)3.47–50 Finally, for highly dispersed CeO2 powders, the amorphous hydrated phase with a stoichiometry of CeO2·2H2O, also denoted as Ce(OH)4, has been identified.45,51,52 Given the properties of the known Ce phases, the HT and LT samples at oxidizing potentials >0.7 VRHE are expected to consist of cubic CeO2 or amorphous CeO2·2H2O phases, respectively.
The assignment of the redox transitions on the CeO2 phase of the HT samples to Ce3+/Ce4+can be verified by recording a CV of a continuous thin film of CeO2(111) on Pt(111) (see Fig. S4, ESI†). The CV of the continuous film does not show the peaks characteristic of clean Pt(111) and is dominated by a pair of irreversible peaks at potentials comparable to those observed on the HT samples (cf.Fig. 2). The CV of the continuous film corresponds well to previous observations on polycrystalline CeO2 films.53 Assignment of redox transitions observed on the LT samples to the Ce3+/Ce4+ couple of hydrated CeO2·2H2O is supported by following indirect evidence: The initial instability of the CV on LT samples can be assigned to a progressive hydration of the as-prepared CeO2(111) nanoislands during cycling. Similar as for the case of unsupported ceria nanoparticles,45,51,52 hydration is observed only for ceria nanoislands with sizes smaller than ∼5 nm, i.e. for the LT samples. Finally, the mutual shift of the irreversible peaks in the CVs of HT and LT samples corresponds well to phase transitions predicted for ceria phases in water-based electrolytes, where the redox potential of hydrated ceria CeO2·xH2O ⇌ Ce(OH)3 at pH = 13 is found to be ∼0.3 V higher than the one of anhydrous ceria CeO2 ⇌ Ce(OH)3.43
Characterization of samples after electrochemical treatment
After finishing the CV experiments, the samples were taken out of the electrolyte under potential control at ≥0.75 VRHE, ensuring that the oxidized character of both the LT and the HT samples is preserved. Samples were further examined via AFM in air and via XPS. AFM images of the samples indicate that the coverage, the flat character, and the compact shape of ceria nanoislands in HT samples are conserved during CV experiments (Fig. S5a–d, ESI†). For the LT samples (Fig. S5e–h, ESI†), the morphology of ceria and the distribution of ceria on the substrate are altered. These observations are in agreement with the electrochemical stability observed for the as-prepared HT samples (Fig. 3a), and with the transformation to hydrated CeO2·2H2O observed for the LT samples (Fig. 3b). A more detailed analysis of the AFM images is hampered by high noise level and sample contamination during the manipulation of samples in air. XPS analysis of Ce 3d spectra on the HT samples after the CV experiments shows no changes with respect to the as-prepared state (Fig. S6a and b, ESI†), indicating a low concentration of structural defects and oxygen vacancies, supporting our assumption that the HT samples are stable under electrochemical conditions. In contrast, we observed reduction of ceria after the CV experiments for the LT samples (increase of Ce3+ fraction from 15 to 23%, Fig. S6c and d, ESI†). Reduction is observed in spite of several hour exposure of LT samples to air under ambient conditions prior to the XPS measurement. This may reflect an increased disorder or a partly hydroxylated nature of the LT samples after finishing the CV experiments. Both in air, and in the water-based solutions, amorphous nanoparticles of hydrated CeO2·2 H2O were previously reported to convert spontaneously to a defect-rich, highly reduced cubic CeO2−x phase.45,51 Poor control over water exposure and carbon contamination during the manipulation of model catalyst samples in air did not allow a systematic investigation of the O 1s and C 1s XPS signals after the CV experiments.Faradaic charges in the CV experiments of the HT and LT samples
We attempted to determine the charges related to the faradaic processes in the CV of the HT and LT samples, in particular the charge corresponding to Ce3+/Ce4+ transition qCe, the charge corresponding to H adsorption/desorption on Pt(111) qH/Pt, and the charge corresponding to OH adsorption/desorption on Pt(111) qOH/Pt. Our estimate of the charges is based on the following CV features characteristic for both HT and LT samples:(i) The presence of ceria nanoislands blocks the OH adsorption on the Pt(111) surface which results in decreasing qOH/Pt. This effect is clearly visible in HT samples (Fig. 2a), where the OH adsorption/desorption peaks do not overlap with the Ce3+/Ce4+ redox peaks. This effect is apparent in LT samples as well (Fig. 2b), where OH adsorption/desorption and Ce3+/Ce4+ redox peaks are still clearly separated in the cathodic scan (Fig. 2b). In the following discussion, we determine the electrochemically active area of the ceria nanoislands θCeEC [and the complementary electrochemically active area of clean Pt(111), θPtEC = 1 − θCeEC] from the attenuation of the OH adsorption/desorption peak on the HT and LT samples with respect to the reference CV of clean Pt(111), and the corresponding charge qrefOH/Pt (Fig. S3, ESI†). θCeEC and θPtEC are evaluated as fractions of the clean Pt(111) surface.
(ii) The presence of ceria nanoislands does not have a pronounced effect on the H adsorption/desorption and qH/Pt in the CV of HT and LT samples, relative to the reference CV of clean Pt. This effect is clearly visible on LT samples (Fig. 2b), where the H adsorption/desorption peaks do not overlap with Ce3+/Ce4+ redox peaks. This effect is apparent in HT samples as well (Fig. 2a), where the Ce3+/Ce4+ redox peaks seem superimposed on the H adsorption/desorption region of clean Pt(111). In the following discussion, we assume qH/Pt to be constant for all HT and LT samples, and equal to qrefH/Pt determined on clean Pt(111) (Fig. S3, ESI†).
(iii) The presence of ceria nanoislands does not influence significantly the double layer charge qDL. In the CVs of HT samples, the double layer regions of HT samples show no or small broadening relative to clean Pt(111) (Fig. 2a, Fig. S7a–c, ESI†). For LT samples, the double layer region is occupied by broad Ce3+/Ce4+ redox peaks (Fig. 2b, Fig. S7d–f, ESI†). For the 1.0 ML LT sample, it is however apparent that the double layer region is not significantly broadened compared to clean Pt(111) either (Fig. S7d, ESI†). Since qDL is the smallest charge contribution, we assume qDL to be constant for all HT and LT samples, and equal to qrefDL determined on clean Pt(111) (Fig. S3, ESI†).
Considering points (i)–(iii), the charge corresponding to Ce3+/Ce4+ redox peaks in the HT and LT samples can be determined as qCe = qexptot − qrefDL − qrefH/Pt − (1 − θCeEC)·qrefOH/Pt, where qexptot is the total charge experimentally determined in the CV of HT or LT samples, θCeEC is the electrochemically active area of the ceria nanoislands determined from the attenuation of the OH peak in the CV of the HT and LT samples, and qrefDL, qrefH/Pt, and qrefOH/Pt are the reference charges determined for a clean Pt(111) surface. Determining qCe for HT and LT samples is illustrated graphically in Fig. 2c, d, and Fig. S7 (ESI†). A reference CV of clean Pt(111) and the corresponding scaling procedure to determine the attenuation of the OH peak are illustrated in Fig. S3 (ESI†).
The electrochemically active CeO2 area θCeEC for all HT and LT samples is shown in Fig. 4, and compared to the geometrical coverage of CeO2 nanoislands θCeSTM determined from STM images of the as-prepared samples (Fig. 1, Table S1, ESI†). θCeEC and θCeSTM are plotted as a function of the amount of deposited CeO2. We observe that θCeSTM of both the HT and the LT samples, and θCeEC of HT samples scale linearly with the amount of CeO2. θCeEC of LT samples is nonlinear with respect to the amount of CeO2. θCeEC of the LT samples is also systematically higher than θCeEC of HT samples. These observations can be again assigned to the instability and redistribution of ceria nanoislands in LT samples during electrochemical cycling (Fig. 3b and Fig. S5e–h, ESI†), and, potentially, to the increase in volume of the nanoislands due to hydration of CeO2 to CeO2·2H2O. Another systematic trend can be observed in the θCeEC of HT samples. In spite of the electrochemical and morphological stability of the HT samples (Fig. 3a and Fig. S5a–d, ESI†), θCeEC is significantly lower than θCeSTM. In this case, inhomogeneity of the samples represents the most likely explanation.
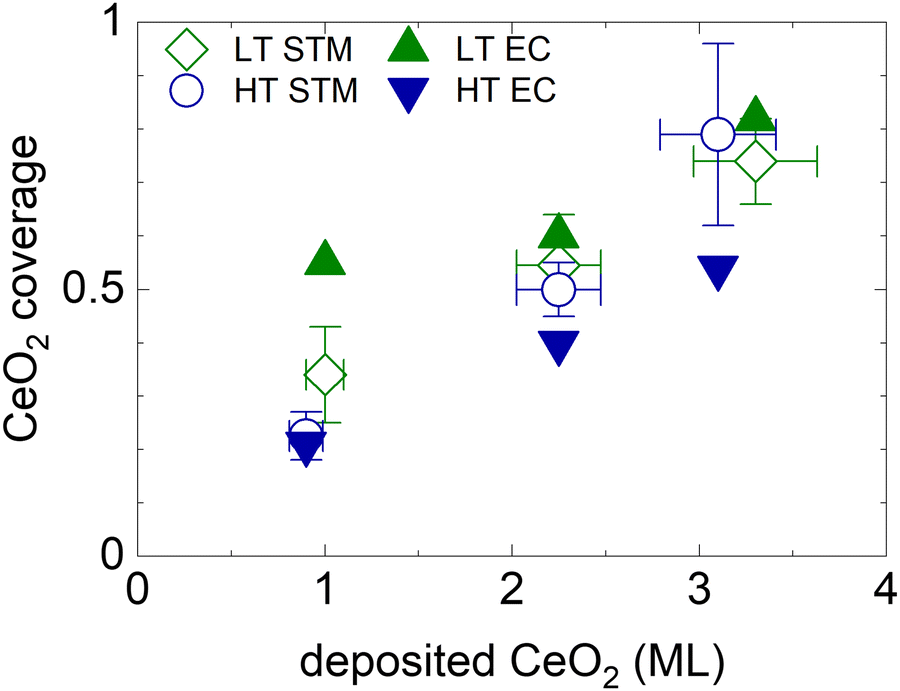 | ||
| Fig. 4 Electrochemically active area of ceria nanoislands θCeEC in HT and LT samples (full symbols) in comparison with the coverage of CeO2(111) nanoislands θCeSTM estimated from STM images of the as-prepared samples shown in Fig. 1 and Table S1 (ESI†) (open symbols). The error bars represent the ±10% uncertainty in the amount of deposited CeO2, and the upper and the lower estimates of CeO2(111) STM coverage. | ||
An evaluation of qCe and other charge contributions as a function of the amount of the deposited CeO2 is shown in Fig. 5a for HT samples, and 5b for LT samples. qexptot in all samples is, within ±10%, equal to qtot on the clean Pt(111). The charge contribution qCe increases in all samples monotonically with the amount of deposited ceria. The HT samples show a smaller qCe value as compared to the LT samples. To estimate a bulk or a surface character of the Ce3+/Ce4+ redox transition, we compare the evaluated qCe for HT and LT samples to the total charge density available for the Ce3+/Ce4+ redox reaction in the bulk and on the surface of the as-prepared samples. The total charge density available for the Ce3+/Ce4+ redox reaction in the bulk of the as-prepared samples is obtained as a product of Ce atom density in 1 ML of CeO2(111) (7.9 × 1014 cm−2), and the amount of deposited Ce, and it is indicated by the dashed line shown in Fig. 5a and b. Given the average thickness of ceria island (4 ± 1) ML for all as-prepared HT and LT samples (Table S1, ESI†), the total charge density available on the surface of the as-prepared samples is estimated as 1/4 of the charge density available in the bulk, and it is indicated by a dotted line shown in Fig. 5a and b. For HT samples, qCe corresponds well to the charge available at the surface of CeO2(111), indicating the surface character of the Ce3+/Ce4+ redox transition. For LT samples, the Ce3+/Ce4+ redox transition involves more Ce atoms than originally available on the surface of the as-prepared LT samples. This indicates that the Ce3+/Ce4+ redox transition in LT samples involves subsurface Ce atoms as well. These subsurface Ce atoms might have been exposed during redistribution of the as-prepared CeO2 nanoislands in LT samples.
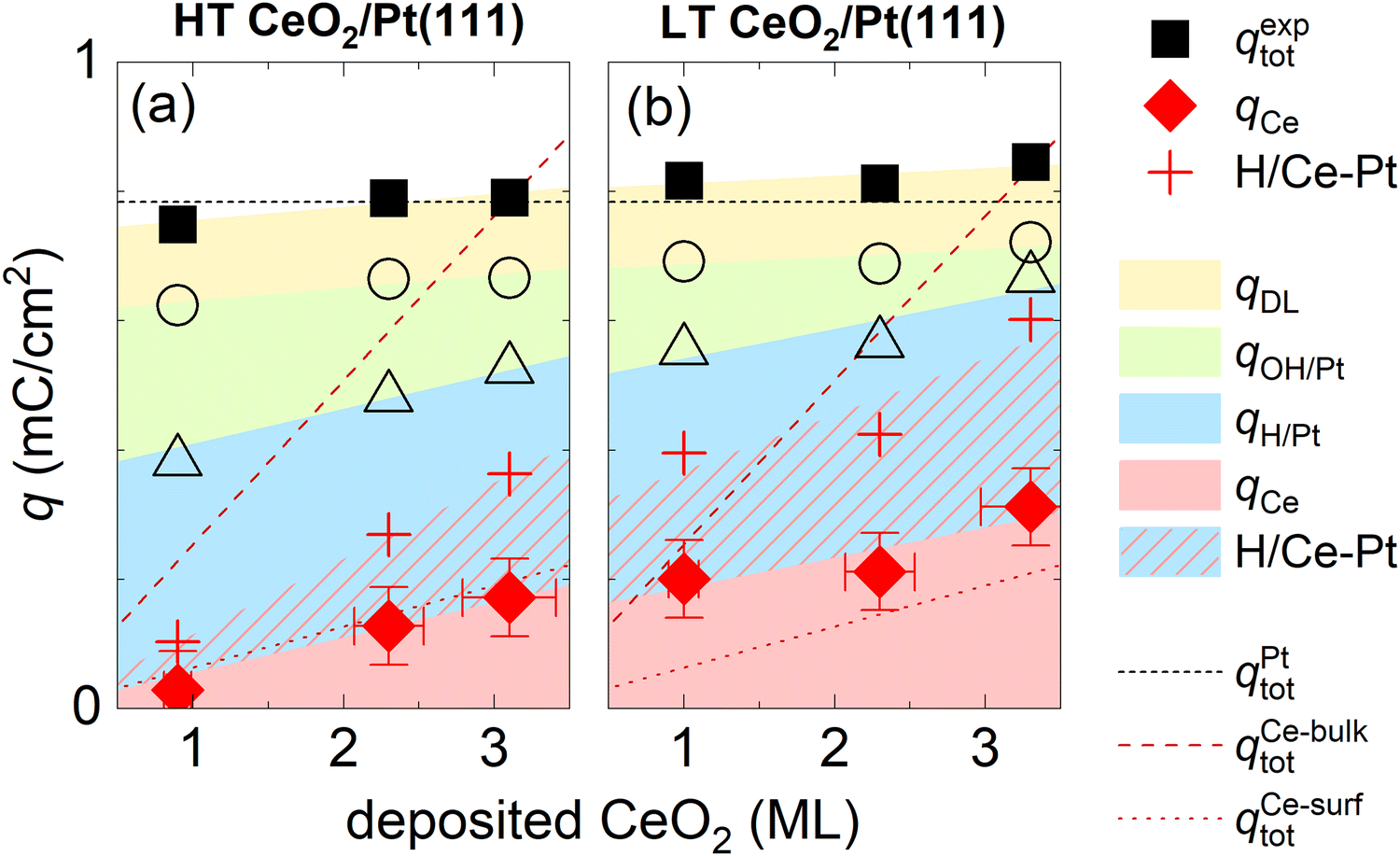 | ||
| Fig. 5 Assignment of charge contributions in the CVs of HT and LT samples shown in Fig. 1 and 2 as a function of the amount of deposited CeO2. Total charge in a CV (qexptot), and the faradaic contribution to Ce3+/Ce4+ electrooxidation/reduction (qCe) are marked with full symbols, incremental contributions to the total charge from H adsorption/desorption on Pt(111) (qH/Pt), OH adsorption/desorption on Pt(111) (qOH/Pt), and the double layer (qDL) are marked with open symbols. The red crosses indicate the amount of H adsorbed/desorbed on Pt(111), which intercalates at the ceria/Pt interface (H/Ce-Pt). The color shading/hatching of the charge contributions represents a guide to the eyes. The dotted lines represent the total charge in the CV of clean Pt(111) (qPttot), and charge required for a complete oxidation/reduction of all deposited Ce atoms (qCe-bulktot) and surface Ce atoms (qCe-surftot). | ||
Hydrogen intercalation at the ceria–Pt interface
CVs of ceria nanoislands in both the HT and the LT samples presented in this study differ qualitatively from CVs reported previously for transition metal inverse model catalysts, i.e. nanoislands of transition metal oxyhydroxides on Pt(111).54,55 Particularly, in contrast to ceria nanoislands, transition metal oxyhydroxide nanoislands on Pt(111) block both the OH adsorption/desorption and H adsorption/desorption on Pt(111).54,55 This different behavior is also reproduced in our experimental setup. Transition metal ions represent a trace contaminant in commercial ultrapure KOH, and the deposition of transition metal oxyhydroxide nanoparticles can be observed on Pt(111) upon extended electrochemical cycling.56 The CV of Pt(111) upon such extended cycling is displayed in Fig. S8 (ESI†). The CV shows peaks assigned to transition metal oxyhydroxides, and the corresponding attenuation of both OH adsorption/desorption and H adsorption/desorption on Pt(111).56To explain this somewhat counterintuitive behavior of ceria nanoislands, i.e. our observation that H adsorption/desorption on Pt(111) remains apparently unchanged even in cases when OH adsorption/desorption is significantly attenuated for the HT and LT samples (Fig. 2a, b and Fig. S7, ESI†), we must invoke a fast and reversible reaction removing the adsorbed H atoms from the free Pt(111) surface in the cathodic scan, and replenishing them back on Pt(111) in the anodic scan. We may estimate the amount of adsorbed H atoms that must take part in this reaction as θCeEC·qrefH/Pt. This amount is indicated in Fig. 5a and b using red crosses. We propose that the adsorbed H atoms are removed from Pt(111) by surface diffusion and intercalated at the Pt/ceria interface. Such non-faradaic process involves no charge transfer between H atoms and the working electrode, and, consequently, no additional features in the CV. Intercalated H atoms still interact with the Pt(111) substrate, which limits the charge of H adsorbed on Pt(111) in HT and LT samples to 2/3 of the density of Pt(111) surface atoms,15 as is the case for clean Pt(111).
We note that the intercalated atomic hydrogen may eventually further react with ceria. Some reaction pathways were previously identified in hydrogen reactions with ceria, particularly the creation of O–H bonds followed by reduction of Ce4+ to Ce3+ as observed for nearly stoichiometric samples of cubic ceria,31 and creation of Ce–H bonds followed by oxidation of Ce3+ to Ce4+ as observed for reduced ceria containing high concentration of oxygen vacancies.57–59 Such non-faradaic reactions may be the cause for the irreversible nature and asymmetry of Ce3+/Ce4+ redox peaks in the CV curves of the HT and LT samples (Fig. 2). Identification and analysis of the involved ceria phases are however beyond the scope of this manuscript.
Suppressing the onset of the HER reaction
In spite of the fact that due to H intercalation, ceria/Pt model electrocatalysts can accommodate an amount of adsorbed H which is comparable to that of a clean Pt(111) surface, the presence of ceria nanoislands suppresses the onset of the hydrogen evolution reaction for the HT and the LT samples. The onset of the HER in the CVs shown in Fig. 2 is summarized in Fig. S9 (ESI†). For all HT and LT samples, the onset of the HER is suppressed in comparison with the clean Pt(111) surface. In LT samples, suppression of the HER onset gets stronger with increasing ceria coverage. In the first approximation, the suppressed onset of the HER observed on our model ceria/Pt samples can be interpreted as a geometrical effect with the HER proceeding only on the fraction of Pt(111) exposed to the electrolyte, and with no participation of H intercalated at the ceria surface. In the case of a continuous ceria layer on Pt(111), and at potentials investigated in this study (Fig. S4, ESI†), no HER is observed at all.Conclusions
We identified structure–property relationships and bifunctional electrocatalytic mechanisms in a rare-earth oxide-based model electrocatalyst, i.e. CeO2(111)/Pt(111). We used a model electrocatalytic approach with the catalysts prepared and characterized by experimental techniques of surface science (physical vapor deposition, STM, AFM, XPS, LEED). The structural and chemical properties of the model catalysts were then correlated to electrochemical properties investigated via cyclic voltammetry in alkaline electrolyte (CV, 0.1 M KOH).Stability and redox properties
Flat and compact CeO2(111) nanoislands with lateral sizes of approx. 20 nm or larger, and straight edges are stable during cycling at potentials between −0.05 and 0.9 VRHE. The CV peaks assigned to the electrooxidation/electroreduction of surface Ce atoms are irreversible and appear at approx. 0.15/0.30 VRHE (cathodic/anodic scan). Similar peaks are identified for a continuous CeO2(111) layer on Pt(111). The peaks are assigned to CeO2 ⇌ Ce(OH)3 transition.Flat and open CeO2(111) nanoislands with lateral sizes below approx. 5 nm, and with rounded edges are initially unstable. The CV peaks assigned to the electrooxidation/electroreduction of surface and subsurface Ce atoms are irreversible and stabilize at higher potentials of approx. 0.45/0.70 VRHE (cathodic/anodic scan). This observation is interpreted as a phase change of ceria nanoislands from cubic CeO2 to amorphous ceria hydrate CeO2·2H2O, and the shifted irreversible peaks assigned to the CeO2·2H2O ⇌ Ce(OH)3 transition.
Large vs. small CeO2(111) nanoislands were prepared using different preparation methods, but their stability/instability in CV is interpreted as a predominantly structural effect. The small CeO2(111) nanoislands include a substantial fraction of undercoordinated surface sites (oxygen vacancies, kink sites at rounded edges) as compared to the larger and compact nanoislands. Upon potential cycling, these undercoordinated sites react with H2O to yield a hydrated CeO2·2H2O phase. Both CeO2 and CeO2·2H2O nanoislands, which appear at oxidizing potentials (above 0.3 and 0.7 VRHE, respectively), are reduced at lower potentials (0.15 and 0.45 VRHE, respectively), resulting in the creation of Ce3+ containing inhomogeneous and partly hydroxylated phases that can be denoted as cerium oxyhydroxide.
Bifunctionality and hydrogen intercalation
Quantitative analysis of the charge in the CVs for the transformation of CeO2(111) and CeO2·2H2O nanoislands on Pt(111) reveals a bifunctional mechanism in which a fraction of H atoms adsorbed on Pt(111) undergoes surface diffusion and intercalation at the Pt/ceria interface. This non-faradaic mechanism is reversible and allows the accommodation of H atoms on the whole Pt(111) surface in spite of the fact that part of the Pt is occupied by CeO2(111) or CeO2·2H2O. This behavior is contrary to what is observed in the case of nanoislands of transition metal oxyhydroxides on Pt(111), which tend to block the adsorption/desorption of H. H accommodated at the Pt/ceria interface likely contributes to establishing cerium oxyhydroxide phases at reducing potentials, but it cannot participate in the HER on ceria/Pt model catalysts.Outlook
Our investigations illustrate the power of model electrocatalyst studies in identifying the conditions and consequences of physicochemical and electrochemical processes relevant to metal-oxide electrocatalysis. The redox properties of the ceria/Pt(111) model system in the electrochemical environment are complex and involve the formation of different cerium phases which can be partly controlled via preparation procedures and the applied potential. The present identification of the cerium phases established under electrochemical conditions may contribute to understanding and knowledge-based improvement of technologically favorable properties of ceria-based electrocatalysts.Data availability
Original experimental data are available via Zenodo, DOI: https://doi.org/10.5281/zenodo.8231965.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Czech Science Foundation (project GAČR 20-11688J) and the Deutsche Forschungsgemeinschaft (DFG) (project 431733372). Support from the DFG via Collaborative Research Centre SFB 1452 – Catalysis at Liquid Interfaces (project 431791331), and project 453560721 is acknowledged. P. K. S. thanks the Grant Agency of the Charles University for the support (project GAUK 326122).References
- L. Liu and A. Corma, Structural Transformations of Solid Electrocatalysts and Photocatalysts, Nat. Rev. Chem., 2021, 5, 256 CrossRef CAS.
- S. Zhao, Y. Yang and Z. Tang, Insight into Structural Evolution, Active Sites, and Stability of Heterogeneous Electrocatalysts, Angew. Chem., Int. Ed., 2022, 61, e202110186 CrossRef CAS.
- K. Fan, H. Zou, Y. Lu, H. Chen, F. Li, J. Liu, L. Sun, L. Tong, M. F. Toney and M. Sui, et al., Direct Observation of Structural Evolution of Metal Chalcogenide in Electrocatalytic Water Oxidation, ACS Nano, 2018, 12, 12369 CrossRef CAS.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, In Operando Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4, J. Am. Chem. Soc., 2016, 138, 36 CrossRef CAS.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, Understanding the Oxygen Evolution Reaction Mechanism on CoOx Using Operando Ambient-Pressure X-Ray Photoelectron Spectroscopy, J. Am. Chem. Soc., 2017, 139, 8960 CrossRef CAS PubMed.
- Y. Duan, J. Y. Lee, S. Xi, Y. Sun, J. Ge, S. J. H. Ong, Y. Chen, S. Dou, F. Meng and C. Diao, et al., Anodic Oxidation Enabled Cation Leaching for Promoting Surface Reconstruction in Water Oxidation, Angew. Chem., Int. Ed., 2021, 60, 7418 CrossRef CAS PubMed.
- S. Song, L. Mu, Y. Jiang, J. Sun, Y. Zhang, G. Shi and H. Sun, Turning Electrocatalytic Activity Sites for the Oxygen Evolution Reaction on Brownmillerite to Oxyhydroxide, ACS Appl. Mater. Interfaces, 2022, 14, 47560 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik and D. Fan, et al., In-Situ Structure and Catalytic Mechanism of NiFe and CoFe Layered Double Hydroxides during Oxygen Evolution, Nat. Commun., 2020, 11, 2522 CrossRef CAS.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Identification of Key Reversible Intermediates in Self-Reconstructed Nickel-Based Hybrid Electrocatalysts for Oxygen Evolution, Angew. Chem., Int. Ed., 2019, 58, 17458 CrossRef CAS PubMed.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Recent Advances in Activating Surface Reconstruction for the High-Efficiency Oxygen Evolution Reaction, Chem. Soc. Rev., 2021, 50, 8428 RSC.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Enhancing the Alkaline Hydrogen Evolution Reaction Activity through the Bifunctionality of Ni(OH)2/Metal Catalysts, Angew. Chem., Int. Ed., 2012, 51, 12495 CrossRef CAS PubMed.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao and M. Zeng, et al., Highly Active and Durable Methanol Oxidation Electrocatalyst Based on the Synergy of Platinum–Nickel Hydroxide–Graphene, Nat. Commun., 2015, 6, 10035 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. Zhu, et al., Dynamic Stability of Active Sites in Hydr(Oxy)Oxides for the Oxygen Evolution Reaction, Nat. Energy, 2020, 5, 222 CrossRef.
- D. M. Kolb, Electrochemical Surface Science, Angew. Chem., Int. Ed., 2001, 40, 1162 CrossRef CAS.
- N. M. Markovic and P. N. J. Ross, Surface Science Studies of Model Fuel Cell Electrocatalysts, Surf. Sci. Rep., 2002, 45, 117 CrossRef CAS.
- F. Faisal, C. Stumm, M. Bertram, F. Waidhas, Y. Lykhach, S. Cherevko, F. Xiang, M. Ammon, M. Vorokhta and B. Šmíd, et al., Electrifying Model Catalysts for Understanding Electrocatalytic Reactions in Liquid Electrolytes, Nat. Mater., 2018, 17, 592 CrossRef CAS PubMed.
- A. R. Akbashev, Electrocatalysis Goes Nuts, ACS Catal., 2022, 12, 4296 CrossRef CAS.
- O. Brummel, Y. Lykhach, M. Ralaiarisoa, M. Berasategui, M. Kastenmeier, L. Fusek, A. Simanenko, W. Gu, P. C. J. Clark and R. Yivlialin, et al., A Versatile Approach to Electrochemical In Situ Ambient-Pressure X-Ray Photoelectron Spectroscopy: Application to a Complex Model Catalyst, J. Phys. Chem. Lett., 2022, 13, 11015 CrossRef CAS PubMed.
- J. Fester, A. Makoveev, D. Grumelli, R. Gutzler, Z. Sun, J. Rodríguez-Fernández, K. Kern and J. V. Lauritsen, The Structure of the Cobalt Oxide/Au Catalyst Interface in Electrochemical Water Splitting, Angew. Chem., Int. Ed., 2018, 57, 11893 CrossRef CAS PubMed.
- F. Reikowski, F. Maroun, I. Pacheco, T. Wiegmann, P. Allongue, J. Stettner and O. M. Magnussen, Operando Surface X-Ray Diffraction Studies of Structurally Defined Co3O4 and CoOOH Thin Films during Oxygen Evolution, ACS Catal., 2019, 9, 3811 CrossRef CAS.
- T. Wiegmann, I. Pacheco, F. Reikowski, J. Stettner, C. Qiu, M. Bouvier, M. Bertram, F. Faisal, O. Brummel and J. Libuda, et al., Operando Identification of the Reversible Skin Layer on Co3O4 as a Three-Dimensional Reaction Zone for Oxygen Evolution, ACS Catal., 2022, 12, 3256 CrossRef CAS PubMed.
- C. Stumm, M. Bertram, M. Kastenmeier, F. D. Speck, Z. Sun, J. Rodríguez-Fernández, J. V. Lauritsen, K. J. J. Mayrhofer, S. Cherevko and O. Brummel, et al., Structural Dynamics of Ultrathin Cobalt Oxide Nanoislands under Potential Control, Adv. Funct. Mater., 2021, 31, 2009923 CrossRef CAS.
- D. Grumelli, T. Wiegmann, S. Barja, F. Reikowski, F. Maroun, P. Allongue, J. Balajka, G. S. Parkinson, U. Diebold and K. Kern, et al., Electrochemical Stability of the Reconstructed Fe3O4(001) Surface, Angew. Chem., Int. Ed., 2020, 59, 21904 CrossRef CAS PubMed.
- M. Müllner, M. Riva, F. Kraushofer, M. Schmid, G. S. Parkinson, S. F. L. Mertens and U. Diebold, Stability and Catalytic Performance of Reconstructed Fe3O4(001) and Fe3O4(110) Surfaces during Oxygen Evolution Reaction, J. Phys. Chem. C, 2019, 123, 8304 CrossRef.
- M. Bertram, C. Prössl, M. Ronovský, J. Knöppel, P. Matvija, L. Fusek, T. Skála, N. Tsud, M. Kastenmeier and V. Matolín, et al., Cobalt Oxide-Supported Pt Electrocatalysts: Intimate Correlation between Particle Size, Electronic Metal–Support Interaction and Stability, J. Phys. Chem. Lett., 2020, 11, 8365 CrossRef CAS PubMed.
- F. Buchner, M. Eckardt, T. Böhler, J. Kim, J. Gerlach, J. Schnaidt and R. J. Behm, Oxygen Reduction and Evolution on Ni-modified Co3O4(111) Cathodes for Zn–Air Batteries: A Combined Surface Science and Electrochemical Model Study, ChemSusChem, 2020, 13, 3199 CrossRef CAS.
- F. Buchner, S. Fuchs and R. J. Behm, UHV Preparation and Electrochemical/-Catalytic Properties of Well-Defined Co– and Fe-Containing Unary and Binary Oxide Model Cathodes for the Oxygen Reduction and Oxygen Evolution Reaction in Zn-Air Batteries, J. Electroanal. Chem., 2021, 896, 115497 CrossRef CAS.
- Z. Sun, A. Curto, J. Rodríguez-Fernández, Z. Wang, A. Parikh, J. Fester, M. Dong, A. Vojvodic and J. V. Lauritsen, The Effect of Fe Dopant Location in Co(Fe)OOHx Nanoparticles for the Oxygen Evolution Reaction, ACS Nano, 2021, 15, 18226 CrossRef CAS PubMed.
- D. R. Kauffman, X. Deng, D. C. Sorescu, T.-D. Nguyen-Phan, C. Wang, C. M. Marin, E. Stavitski, I. Waluyo and A. Hunt, Edge-Enhanced Oxygen Evolution Reactivity at Ultrathin, Au-Supported Fe2O3 Electrocatalysts, ACS Catal., 2019, 9, 5375 CrossRef CAS.
- A. Trovarelli and P. Fornasiero, Catalysis by Ceria and Related Materials, 2nd edn, World Scientific, Singapore, 2013 Search PubMed.
- D. R. Mullins, The Surface Chemistry of Cerium Oxide, Surf. Sci. Rep., 2015, 70, 42 CrossRef CAS.
- J. Corchado-García, L. E. Betancourt, C. A. Vélez, S. D. Senanayake, D. Stacchiola, K. Sasaki, M. J.-F. Guinel, Y. Zhou, C. L. Cheung and C. R. Cabrera, Cerium Oxide as a Promoter for the Electro-Oxidation Reaction of Ethanol: In Situ XAFS Characterization of the Pt Nanoparticles Supported on CeO2 Nanoparticles and Nanorods, Phys. Chem. Chem. Phys., 2015, 17, 32251 RSC.
- Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Enhanced Supply of Hydroxyl Species in CeO2-Modified Platinum Catalyst Studied by in Situ ATR-FTIR Spectroscopy, ACS Catal., 2016, 6, 2026 CrossRef CAS.
- O. Brummel, F. Waidhas, F. Faisal, R. Fiala, M. Vorokhta, I. Khalakhan, M. Dubau, A. Figueroba, G. Kovács and H. A. Aleksandrov, et al., Stabilization of Small Platinum Nanoparticles on Pt–CeO2 Thin Film Electrocatalysts During Methanol Oxidation, J. Phys. Chem. C, 2016, 120, 19723 CrossRef CAS.
- Z. Weng, W. Liu, L.-C. Yin, R. Fang, M. Li, E. I. Altman, Q. Fan, F. Li, H.-M. Cheng and H. Wang, Metal/Oxide Interface Nanostructures Generated by Surface Segregation for Electrocatalysis, Nano Lett., 2015, 15, 7704 CrossRef CAS.
- K. R. Yoon, J. M. Kim, K. A. Lee, C.-K. Hwang, S. G. Akpe, Y. J. Lee, J. P. Singh, K. H. Chae, S. S. Jang and H. C. Ham, et al., Activity-Stability Benefits of Pt/C Fuel Cell Electrocatalysts Prepared via Remote CeO2 Interfacial Doping, J. Power Sources, 2021, 496, 229798 CrossRef CAS.
- K. Fugane, T. Mori, D. R. Ou, P. Yan, F. Ye, H. Yoshikawa and J. Drennan, Improvement of Cathode Performance on Pt-CeOx by Optimization of Electrochemical Pretreatment Condition for PEFC Application, Langmuir, 2012, 28, 16692 CrossRef CAS.
- S. Chauhan, T. Mori, T. Masuda, S. Ueda, G. J. Richards, J. P. Hill, K. Ariga, N. Isaka, G. Auchterlonie and J. Drennan, Design of Low Pt Concentration Electrocatalyst Surfaces with High Oxygen Reduction Reaction Activity Promoted by Formation of a Heterogeneous Interface between Pt and CeOx Nanowire, ACS Appl. Mater. Interfaces, 2016, 8, 9059 CrossRef CAS.
- M. Liu, K. Min, B. Han and L. Y. S. Lee, Interfacing or Doping? Role of Ce in Highly Promoted Water Oxidation of NiFe-Layered Double Hydroxide, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS.
- P. Luches, F. Pagliuca and S. Valeri, Morphology, Stoichiometry, and Interface Structure of CeO2 Ultrathin Films on Pt(111), J. Phys. Chem. C, 2011, 115, 10718 CrossRef CAS.
- P. Luches, F. Pagliuca and S. Valeri, Structural and Morphological Modifications in Thermally Reduced Cerium Oxide Ultrathin Epitaxial Films on Pt(111), Phys. Chem. Chem. Phys., 2014, 16, 18848 RSC.
- T. V. Plakhova, A. Y. Romanchuk, S. N. Yakunin, T. Dumas, S. Demir, S. Wang, S. G. Minasian, D. K. Shuh, T. Tyliszczak and A. A. Shiryaev, et al., Solubility of Nanocrystalline Cerium Dioxide: Experimental Data and Thermodynamic Modeling, J. Phys. Chem. C, 2016, 120, 22615 CrossRef CAS.
- P. Yu, S. A. Hayes, T. J. O’Keefe, M. J. O’Keefe and J. O. Stoffer, The Phase Stability of Cerium Species in Aqueous Systems, J. Electrochem. Soc., 2006, 153, C74 CrossRef CAS.
- T. Duchoň, F. Dvořák, M. Aulická, V. Stetsovych, M. Vorokhta, D. Mazur, K. Veltruská, T. Skála, J. Mysliveček and I. Matolínová, et al., Ordered Phases of Reduced Ceria As Epitaxial Films on Cu(111), J. Phys. Chem. C, 2014, 118, 357 CrossRef.
- C. J. Neal, T. S. Sakthivel, Y. Fu and S. Seal, Aging of Nanoscale Cerium Oxide in a Peroxide Environment: Its Influence on the Redox, Surface, and Dispersion Character, J. Phys. Chem. C, 2021, 125, 27323 CrossRef CAS.
- E. A. Kümmerle and G. Heger, The Structures of C–Ce2O3+δ, Ce7O12, and Ce11O20, J. Solid State Chem., 1999, 147, 485 CrossRef.
- D. Schweke, G. Rafailov, S. Zalkind, O. Azulai, L. Rabinovitch and S. Hayun, Elucidating the Role of Hydrogen Species Originating from Water Vapor in the Oxidation Mechanism of Cerium, Corros. Sci., 2022, 196, 110030 CrossRef CAS.
- D. Schweke, A. Rubin, L. Rabinovitch, O. Kraynis and T. Livneh, Cerium Metal Oxidation Studied by IR Reflection-Absorption and Raman Scattering Spectroscopies, J. Phys.: Condens. Matter, 2022, 34, 324002 CrossRef CAS.
- F. Dvořák, L. Szabová, V. Johánek, M. Farnesi Camellone, V. Stetsovych, M. Vorokhta, A. Tovt, T. Skála, I. Matolínová and Y. Tateyama, et al., Bulk Hydroxylation and Effective Water Splitting by Highly Reduced Cerium Oxide: The Role of O Vacancy Coordination, ACS Catal., 2018, 8, 4354 CrossRef.
- P. Abellan, T. H. Moser, I. T. Lucas, J. W. Grate, J. E. Evans and N. D. Browning, The Formation of Cerium(III) Hydroxide Nanoparticles by a Radiation Mediated Increase in Local pH, RSC Adv., 2017, 7, 3831 RSC.
- B. Djuričić and S. Pickering, Nanostructured Cerium Oxide: Preparation and Properties of Weakly-Agglomerated Powders, J. Eur. Ceram. Soc., 1999, 19, 1925 CrossRef.
- S. V. N. T. Kuchibhatla, A. S. Karakoti, A. E. Vasdekis, C. F. Windisch, S. Seal, S. Thevuthasan and D. R. Baer, An Unexpected Phase Transformation of Ceria Nanoparticles in Aqueous Media, J. Mater. Res., 2019, 34, 465 CrossRef CAS.
- U. Lavrencic Stangar, B. Orel, I. Grabec and B. Ogorvec, Optical and Electrochemical Properties of CeO2 and CeO2-TiO2 Coatings, Solar Energy Materials and Solar Cells, 1993, 31, 171 CrossRef.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang and M. Uchimura, Paulikas, a P.; Stamenkovic, V.; Markovic, N. M. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces, Science, 2011, 334, 1256 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Trends in Activity for the Water Electrolyser Reactions on 3d M(Ni,Co,Fe,Mn) Hydr(Oxy)Oxide Catalysts, Nat. Mater., 2012, 11, 550 CrossRef CAS PubMed.
- R. Subbaraman, N. Danilovic, P. P. Lopes, D. Tripkovic, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, Origin of Anomalous Activities for Electrocatalysts in Alkaline Electrolytes, J. Phys. Chem. C, 2012, 116, 22231 CrossRef CAS.
- Z. Li, K. Werner, K. Qian, R. You, A. Płucienik, A. Jia, L. Wu, L. Zhang, H. Pan and H. Kuhlenbeck, et al., Oxidation of Reduced Ceria by Incorporation of Hydrogen, Angew. Chem., Int. Ed., 2019, 58, 14686 CrossRef CAS.
- Z. Li, K. Werner, L. Chen, A. Jia, K. Qian, J. Zhong, R. You, L. Wu, L. Zhang and H. Pan, et al., Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk, Chem. – Eur. J., 2021, 27, 5268 CrossRef CAS.
- T. Matsukawa, K. Iida, M. Nakamura and T. Ishigaki, Detection of Hydroxyl and Hydride Functional Groups in a Ceria Crystal under Hydrogen Reduction, CrystEngComm, 2021, 23, 2355 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03831a |
| This journal is © the Owner Societies 2024 |