
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT+U and quantum Monte Carlo study of electronic and optical properties of AgNiO2 and AgNi1−xCoxO2 delafossite†
Hyeondeok
Shin
 *a,
Panchapakesan
Ganesh
b,
Paul R. C.
Kent
c,
Anouar
Benali
a,
Anand
Bhattacharya
d,
Ho Nyung
Lee
e,
Olle
Heinonen‡
d and
Jaron T.
Krogel
*f
*a,
Panchapakesan
Ganesh
b,
Paul R. C.
Kent
c,
Anouar
Benali
a,
Anand
Bhattacharya
d,
Ho Nyung
Lee
e,
Olle
Heinonen‡
d and
Jaron T.
Krogel
*f
aComputational Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA. E-mail: hshin@anl.gov; Tel: +1-630-252-7933
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
cComputational Sciences and Engineering Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
dMaterials Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
eMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
fMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA. E-mail: krogeljt@ornl.gov; Tel: +1-865-576-6204
First published on 6th February 2024
Abstract
As the only semimetallic d10-based delafossite, AgNiO2 has received a great deal of attention due to both its unique semimetallicity and its antiferromagnetism in the NiO2 layer that is coupled with a lattice distortion. In contrast, other delafossites such as AgCoO2 are insulating. Here we study how the electronic structure of AgNi1−xCoxO2 alloys vary with Ni/Co concentration, in order to investigate the electronic properties and phase stability of the intermetallics. While the electronic and magnetic structure of delafossites have been studied using density functional theory (DFT), earlier studies have not included corrections for strong on-site Coulomb interactions. In order to treat these interactions accurately, in this study we use Quantum Monte Carlo (QMC) simulations to obtain accurate estimates for the electronic and magnetic properties of AgNiO2. By comparison to DFT results we show that these electron correlations are critical to account for. We show that Co doping on the magnetic Ni sites results in a metal–insulator transition near x ∼0.33, and reentrant behavior near x ∼ 0.66.
1 Introduction
Delafossites are minerals with the generic formula ABO2, where A is a monovalent and B is a trivalent metal, and the structure consists of layers of the metal A cations interspersed between layers of BO2 that are arranged in edge-sharing BO6 octahedra, as seen in Fig. 1. A particular feature of delafossites is that the cation A is bonded vertically (along the c axis) to oxygen atoms in planes above and below. Recently, delafossites have attracted a great deal of attention because of their interesting properties that arise as a consequence of interplay between the metal A and the BO2 layers. Since the discovery of the naturally occurring delafossite form of CuFeO2,1 various types of delafossites have been synthesized and studied extensively in order to understand what gives rise to the wide ranges of electronic properties for different combinations of A and B elements.2–10 The monovalent A site is usually occupied by d9 or d10 noble metals or transition metal atoms. Most of the d9 and d10 delafossites, such as PdCoO2 and PtCoO2, exhibit a large electrical conductivity, but a much wider range of electronic properties, including metallic, semiconducting, and insulating, has been observed in d10 A-site compounds, and it appears that the B-site component is the dominating factor in the resulting electronic and optical properties.11–13 | ||
| Fig. 1 (a) Side and (b) top view of 2H-AgNiO2 structure. There are two different Ni sites, Ni1 with very small magnetic moments as Ni3.5+/d6.5 state, and Ni2 with large magnetic moments as Ni2+/d8 state; the Ni2 sites form a triangular planar antiferromagnet. | ||
Delafossites with d10 cations (A = Ag and Cu) have reported to possess wide direct electronic band gaps and p-type behavior, which makes them interesting for potential future applications of p-type transparent materials.7,14–18 Among d10-based delafossites AgBO2 and CuBO2, the Ni B-site compound AgNiO2 is known to possess rather unique electronic and magnetic properties: while most of the d10-based delafossites exhibit insulating or semiconducting behavior, only AgNiO2 exhibits metallic features in 2H polytype of hexagonal space group of P63/mmc.11,19,20 According to previous studies on 2H-AgNiO2, its ideal P63/mmc crystal structure is transformed into the P6322 structure because of lattice distortions induced by strong antiferromagnetic (AFM) interactions in the NiO2 layers of AgNiO2.11,21 Interestingly, the lattice distortion in AFM 2H-AgNiO2 is not the well-known Jahn–Teller distortion, but a charge-ordering distortion induced by charge transfer on eg states on the Ni sites. This leads to two different Ni sites, Ni1 with small magnetic moments (Ni3.5+), and Ni2 sites with large magnetic moments (Ni2+); the Ni2 sites form a triangular antiferromagnet within the Ni ab-plane. This leads to charge disproportionation on the Ni sites and AFM 2H-AgNiO2 is consequently interpreted as a strongly charge-ordered system. Moreover, because of the insulating properties of AgCoO2 while AgNiO2 exhibits a semimetallic phase, the existence of a metal–insulator transition has been predicted on AgNi1−xCoxO2 structures wherein the NiO2 layers in AgNiO2 are mixed with CoO2 layers of the insulating AgCoO2.22
In addition to experimental investigations, there have been a few reports from studies using density functional theory (DFT) to study AFM 2H-AgNiO2, in particular to address the magnetic order that has been observed experimentally.11,23 A fundamental question that can be raised in this context is to what extent electronic correlations play a role in the magnetic ordering in delafossites in general, and in AgNiO2 in particular; linked to this is the well-known broader issue of how to accurately account for electronic correlations within DFT. This is an important question for the delafossites as they contain 3d, 4d, 4f, and 5f metals with highly localized electrons bound to oxygen. It is therefore important to accurately assess the effects of electronic correlations on delafossites, and also to devise computational schemes that allow for including correlations at a known level of accuracy. One such scheme is DFT+U, in which a Hubbard U term is added to selected localized orbitals to approximately account for on-site Coulomb correlations.24,25 While the actual value of U can be used as a fitting variable, there are nowadays methods to self-consistently calculate U, reducing empiricism. Nevertheless, an on-site Coulomb interaction Hubbard U has not been considered at all in previous DFT studies of 2H-AgNiO2, mainly because it has been predicted that the effect of U is small in metallic 2H-AgNiO2.11 Furthermore, a previous DFT study for 3R-AgNiO2 concluded that projected density of state from local spin density approximation (LSDA) is in better agreement with corresponding experimental partial spectral weight (PSW) distributions than LSDA+U.26 Therefore, appropriate values of U for 2H-AgNiO2 have not been studied systematically. Previous studies have utilized the local density approximation (LDA) or the generalized gradient approximation (GGA) without any attempt to correct for on-site correlations with a Hubbard U have been used.11,12,21,26
The main motivation for our work is to accurately assess the effect of electronic correlations on the electronic and magnetic properties of 2H-AgNiO2, and also on intermetallic phases AgNi1−xCoxO2 as well as their stability. In our work, we use quantum Monte Carlo (QMC) methods, specifically real-space variational Monte Carlo (VMC) and diffusion Monte Carlo (DMC). QMC methods are computationally expensive but highly-accurate stochastic wavefunction methods that fully incorporate electronic many-body effects.27,28 Weak through strong electronic correlations are well described. The total energy obeys a variational principle allowing the effect of different choices for the input trail wavefunctions to be assessed. QMC methods have provided accurate ground state properties for strongly-correlated transition metal oxides, including VO2, AFM NiO, and various alloys.29–34 In this study, we use QMC to obtain accurate ground state properties of 2H-AgNiO2. In addition, we study various structures of phases of the mixtures AgNi1−xCoxO2 to assess their phase stability and electronic properties. Our results show that large concentrations of substitutional Co in AgNi1−xCoxO2, x ≥ 0.33, lead to an opening of an electronic band gap and stable formation energies. This suggests an interesting way to generate a metal-insulating transition concomitant with a magnetic transition, different from, e.g., metal-insulating transitions in more classical correlated oxides, such as VO2.35–37
2 Methods
We used DMC within the fixed-node approximation as implemented in the QMCPACK code.38 Single Slater-determinant wavefunctions were used as trial wavefunctions in the QMC algorithm, with up to three-body Jastrow correlation coefficients in order to incorporate electron–ion, electron–electron, and electron–electron–ion correlations. Cut-offs for the one- and two-body Jastrows were set as the Wigner–Seitz radius of the given supercell while a maximum of 5.0 bohr was used as the cut-off for the three-body term. Single-particle orbitals in the QMC trial wavefunctions were generated by solving the Kohn–Sham equations using DFT. All DFT calculations in this study were performed with a plane-wave basis set with a 700 Ry kinetic-energy cut-off and 8 × 8 × 8 k-point grids using the QUANTUM ESPRESSO code.39 Kohn–Sham orbitals in the Slater determinant were generated using Perdew–Burke–Ernzerhof (PBE) parametrization40 of the generalized gradient approximation (GGA) exchange–correlation (XC) functional. First-order Methfessel–Paxton scheme was applied as smearing method with 0.001 Ry smearing parameter for Brillouin zone integration.41 In order to account for on-site Coulomb interactions of strongly localized d orbital in Ni, we used a Hubbard “U” for the Hubbard correction within the DFT+U formalism.24,25 Norm-conserving pseudopotentials for Ni and O in this study were the same as used in a previous QMC study of AFM NiO.32 The Ag and Co pseudopotentials were correlation-consistent effective-core potentials (ccECPs) wherein fully-correlated all-electron calculations – primarily coupled-cluster calculations – were used as references for the parameterization of the ECPs.42–45 Because ccECP pseudopotentials are hard-core and therefore require large kinetic-energy cut-off, 700 Ry kinetic-energy, for Ag, we applied the hybrid orbital representation that combines a local atomic basis set and B-splines in order to reduce memory requirements of the QMC.46 DMC calculations were done using 0.005 Ha−1 time steps within the non-local T-move approximation47 In order to reduce one-body finite-size effects from the periodic boundary conditions applied in the DMC calculations, we employed twist-averaged boundary conditions48 with up to a maximum of 64 twists for the AgNiO2 supercells.Two-body finite-size effects were reduced using the modified periodic Coulomb interaction49 and Chiesa's kinetic energy correction.50 In addition to those finite-size corrections, we estimated twist-averaged DMC energies at different sizes of supercells, 48, 96, and 144 atoms cells, and extrapolated the energies to the bulk limit in order to further reduce two-body finite size effects.
3 Results
3.1 Properties of pure 2H-AgNiO2
Previous DFT studies of AgNiO2 delafossites assumed that on-site Coulomb interactions were not important and so did not use DFT+U.11,21 One of the aims of our work is to examine the role of on-site Coulomb interactions in detail in order to ascertain their importance. Because there are no previously reported values for an optimal value of U, Uopt, we first estimated Uopt. We used a procedure established in previous works32,51–54 that has proven to be a reliable and unbiased way to estimate Uopt for transition-metal oxides. In this procedure, we minimize the DMC total energy of the PBE+U trial wavefunction as a function of U. Because the DMC total energy obeys a variational principle, this energy will exhibit a minimum. Specifically, in DMC, the minimization of the energy with respect to U is a one-parameter optimization of the many-body wavefunction nodal surface. For simplicity we assume the same U value for all Ni atoms, regardless of their local charge states. Fig. 2 shows the DMC total energy for the AFM AgNiO2 unit cell as function of the value of U in the PBE+U trial wavefunction. Using a quartic fit, we estimated an optimal U value of Uopt = 4.4(1) eV for Ni, which is close to the DMC Uopt = 4.7(2) eV found in AFM NiO.32 We use the value of U = 4.4 eV obtained from DMC for all subsequent PBE+U calculations in this study. | ||
| Fig. 2 DMC total energy of AgNiO2 as function of Hubbard U in the PBE+U trial wavefunction. | ||
To investigate how varied p−d hybridization within the DFT+U scheme may change the electronic properties of AgNiO2, we first compare the electron density-of-states (DOS) obtained using PBE and PBE+U. As expected, the DOS for 2H-AgNiO2 clearly exhibits metallic features with filled states at the Fermi level both for PBE and PBE+U (Fig. 3). Under hole doping, PBE predicts preservation of the metallic state for all doping levels. In contrast, PBE+U predicts a gap opening about 0.5 eV below the Fermi level, raising the possibility of a metal–insulator transition in 2H-AgNiO2 under hole doping. In addition, we confirmed that the Hubbard U leads to more semimetallic electronic properties of AgNiO2 as the conduction band minimum in PBE+U is closer to the Fermi level with lower DOS than in PBE. This suggests that localized Ni 3d orbitals induce a semimetallic nature in AgNiO2, and that AgNiO2 possesses an intriguing potential of tuning the band gap to a semiconductor or insulator.
 | ||
| Fig. 3 Projected density of states of AFM 2H-AgNiO2 from (a) PBE and (b) PBE+U, with U = 4.4 eV. | ||
For further analyses of the effects of U on semimetallic AgNiO2, we compared total charge and spin densities obtained from PBE and PBE+U. Fig. 4(a) and (b) show significant differences in both charge and spin densities between PBE+U and PBE near the Ni sites – accumulation and depletion can be found near the Ni sites in both the charge and spin density differences. The charge density differences between PBE+U and PBE induced by the Hubbard U are mainly located on the octahedral NiO6 structures, with no significant changes near the Ag sites. Within the NiO2 layers, there is a rather pronounced charge density redistribution induced by Hubbard U on the Ni–O bond. This shows that the Hubbard U strongly affects the p−d hybridization of the Ni–O bonds, even though AgNiO2 is in a semimetallic phase. Among the Ni sites, there is a large charge accumulation on the Ni2+ sites (Ni2) that also possess large magnetic moments. This indicates that there is discrepancy between the magnetic moments obtained by PBE and PBE+U, as the Hubbard U affects the magnetic moment on Ni. In addition to the charge density difference, we can also see that PBE underestimates the spin density on Ni sites relative to PBE+U (see Fig. 4), which is analogous to results obtained in an earlier DMC study of AFM NiO,32 although the spin density difference is smaller for AgNiO2 than for insulating NiO. As assumed, it is clear that influence of the Hubbard U is not as large in semimetallic AgNiO2 compared to its effect in insulating NiO; however, we conclude that the existence of localized 3d orbitals is still leads to moderate effects in AgNiO2 because of the large density differences between PBE and PBE+U.
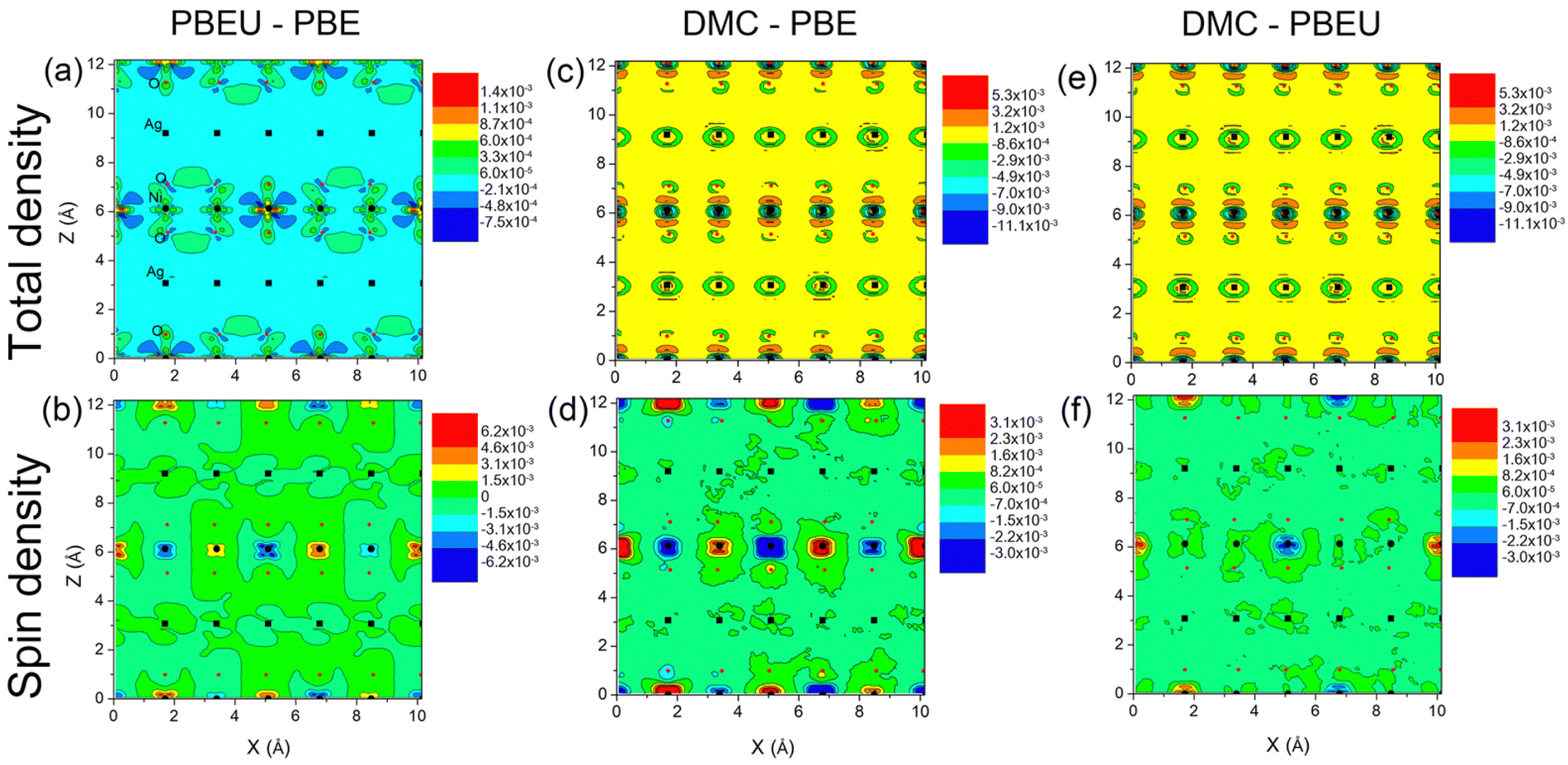 | ||
| Fig. 4 (a) Charge and (b) spin density difference between PBE+U and PBE of a NiO2 layer of AgNiO2, those between DMC and PBE ((c) and (d)), and between DMC and PBE+U ((e) and (f)). The density differences are projected onto the (010) plane and in units of Å−3. | ||
In order to further accurately assess the electronic properties of 2H-AgNiO2, we performed DMC calculations of AgNiO2 using a PBE+U trial wavefunction with the optimal value of U. We estimated the cohesive energy of AgNiO2 by computing E(AgNiO2) − E(Ag) − E(Ni) − 2E(O), where E(AgNiO2), E(Ag), E(Ni), E(O) are the DMC total energy of AgNiO2 and that of atomic Ag, Ni, and O, respectively. The computed DMC AgNiO2 cohesive energy with full incorporation of the finite-size analysis is 14.23(3) eV per f.u., which is significantly smaller than the PBE result of 15.21 eV per f.u. but consistent with PBE+U one of 14.21 eV per f.u. Significantly larger PBE cohesive energy than DMC seems to be related with overestimation of NiO cohesive energy compared to corresponding experimental result within PBE functionals.32 Although experimental values of the AgNiO2 cohesive energy are not available to the best of our knowledge, the large differences in cohesive energy clearly shows a large discrepancy between the DMC, DFT, and DFT+U schemes in dealing with the electronic structure of AgNiO2. The charge density difference between DMC and PBE+U, ρ(DMC) − ρ(PBE+U), shows a charge density accumulation on Ni–O complexes in DMC relative to PBE+U, somewhat similar to the charge density difference ρ(PBE+U) − ρ(PBE), but the charge accumulation in ρ(DMC) − ρ(PBE+U) is concentrated on specific Ni–O pairs in the yz plane, while density difference ρ(PBE+U) − ρ(PBE) is more spread out over the entire NiO6 layer. From this anisotropic density accumulation in DMC relative to PBE+U, we suspect there is a similar symmetry-breaking in the Ni–O bonds to that already seen in DMC studies of NiO and HfO2.32,55 In Fig. 4(d) and (f), we see strong spin accumulation and depletion only on the AFM Ni sites. This tells us that magnetic moment on the Ni sites is significantly underestimated in both PBE and PBE+U compared to DMC. In order to compare the DMC and DFT magnetic moments, we computed the magnetic moments on Ni sites as function of U. Fig. 5 shows that the DFT magnetic moment increases monotonically on the AFM Ni sites Ni2 as U increases. However, even at large values of U, up to 6 eV where PBE+U magnetic moment shows the largest value, the PBE+U moment is still smaller than DMC magnetic moment. The estimated DMC magnetic moment on the AFM Ni sites is 1.71(1) μB, which is slightly larger but in the good agreement with the reported local magnetization of Ni, 1.552(7) μB.56 We see that PBE+U magnetic moment shows empirically closest result with the experimental one in U ∼ 2 eV with 1.58 μB while PBE without U exhibits smaller value of 1.46 μB. From this analysis, we conclude that the Hubbard U significantly affects the band gap and magnetic moment of 2H-AgNiO2, and the addition of a Hubbard U is necessary in order to achieve reasonably accurate magnetic moment and charge density within DFT.
 | ||
| Fig. 5 Magnetic moments of (a) Ni1 and (b) Ni2 sites as function of U obtained using PBE+U (black squares) and DMC (red circle). | ||
3.2 Moderate Co doping: metal–insulator transition for x = 0.33
The PBE+U and DMC results for AgNiO2 described in the previous section indicate the possibility of a metal–insulator transition based on the observations both in PBE+U and DMC of a small of electron density from states in the conduction band just below the Fermi level. This suggests that the introduction of dopants or other defects into stoichiometric AgNiO2 may provide a path to move the conduction band minimum above the Fermi level. Various transition-metal doped delafossites have in fact been studied previously as transition-metal doping has been known to enhance p-type semiconductor properties. Among various transition metal candidates, we consider here Co as a dopant and investigate how Co doping influences the electronic properties and band gap opening in 2H-AgNiO2. When studying these intermetallics, it is crucial first to obtain an accurate structure as the equilibrium structure and geometry vary with the concentration of dopants and with their locations, and the electronic structure in turn depends strongly on the geometry of the structure. We attempted to obtain a good quality lattice structure for AgNi1−xCoxO2 by considering both the pure AgNiO2 geometry but with dopants on Ni1 sites, and a fully relaxed structure within DFT+U framework. To compare these two geometries and to choose an energetically stable geometry for the intermetallic, we estimated the DMC total energy for these structures. The result is that the pure 2H-AgNiO2 structure with Co on the Ni1 sites exhibits a lower FN-DMC energy than the fully relaxed structure. Therefore, we used the pure 2H-AgNiO2 as a structure for the intermetallic AgNi1−xCoxO2. Details in FN-energy comparison between different geometries are in ESI.† In order to optimize the trial wavefunction for AgNi1−xCoxO2, we determined an optimal U-value of 4.0(1) eV for the Co dopants by minimizing the DMC total energy for the 2H-AgCoO2 structure (see ESI†). Among potentially available Co concentrations of AgNi1−xCoxO2, we first considered AgNi0.66Co0.33O2 structure. Although the existence of MIT can be expected on AgNi0.66Co0.33O2 since it is experimentally reported on x ∼ 0.3,22 the energetic stability of phases that result from random Co substitutions at various Co concentration is unclear. In order to investigate the relative stability of various random phases and the dependencies of their electronic and optical properties on substitutional sites, we considered additional phases of AgNi0.66Co0.33O2 wherein four Ni2+ Ni1 sites out of a total of eight are replaced by Co dopants. We did not consider substitution of Co on the AFM Ni2 sites because calculations showed that this leads to a collapse of the magnetic order and an energetically unstable structure of the mixture. Because there are too many AgNi0.66Co0.33O2 configurations with four Co atoms on eight possible sites to make comprehensive DMC calculations practical, we selected only four different phases, shown in Fig. 6. Other various phases with larger sizes of cell can be found on ESI.† | ||
| Fig. 6 Upper and lower Co-doped NiO2 layers for four different phases of AgNi0.66Co0.33O2. The blue spheres indicate Co dopants. | ||
We first compute the PBE+U density-of-states of these four phases in order to compare their optical properties. As can be seen in Fig. 7, the optical properties of the AgNi0.66Co0.33O2 mixture depends strongly on which of the Ni1 sites are substituted with Co. Phases 1 and 4 show completely closed band gaps and metallic densities-of-states; however, phases 2 and 3 exhibit open band gaps. Because of the completely different electronic properties of the four phases, with phases 1 and 4 metallic and phase 2 and 3 semiconductor-like, and the very large differences in densities-of-states near the Fermi level, we conclude that the electronic and optical properties vary strongly with the specific sites used for Co-substitution, and the detailed properties of AgNi0.66Co0.33O2 can potentially be controlled by selectively choosing the sites for substitution.
 | ||
| Fig. 7 Projected density-of-states for four different phases of AgNi0.66Co0.33O2. | ||
Because there are many possible metallic and semiconducting phases of AgNi0.66Co0.33O2, it is important to find the most stable one. We estimated the DMC total energy of four candidates based on symmetry. Fig. 8 shows the PBE+U and the DMC total energy differences between the four phases with the energy (PBE+U and DMC, respectively) of phase 1 as reference at zero total energy. As can be seen in the figure, the semiconducting phases 2 and 3 have lower DMC total energy than the metallic phases 1 and 4, indicating that the semiconducting phases are more energetically favored and stable than metallic ones for the AgNi0.66Co0.33O2 mixture. There is a large DMC energy difference between the metallic phases 1 and 4 in DMC and a smaller PBE+U energy difference, but a relatively small energy difference between phases 2 and 3 both for PBE+U and DMC. The much smaller PBE+U energy difference between the metallic and semiconducting phases than the DMC energy difference, about 0.05 eV per f.u. and 0.21(1) eV per f.u., respectively, strongly suggests that the semiconducting phases driven by Co-substitution are due to electron correlations between the Co and Ni sites, effects that are well accounted for in DMC but not as accurately in DFT or DFT+U. From lower PBE+U and DMC total energies on semiconducting phases than semimetallic ones confirmed the existence of MIT, transiting favored phase from semimetallic on pristine AgNiO2 to semiconducting phase on AgNi0.66Co0.33O2. In addition, since coexistence of semimetallic and semiconducting phase is observed at the concentration of x = 0.33, we assume that the critical Co concentration of MIT is located nearby x = 0.33, which is consistent with the experimental measurement of MIT on x = 0.3.22 In further calculations at x = 0.33 in larger cells (96 and 144 atom cells, see ESI†), we see largely the same behavior, with a coexistence of locally gapped and ungapped phases. However, the expanded results also show that the ungapped phases are characterized by low density of states at the Fermi level. These findings are interesting because the experimental thermal behavior of the thermoelectric power22 also suggested the presence of a residual finite density of states at the Fermi level for this concentration.
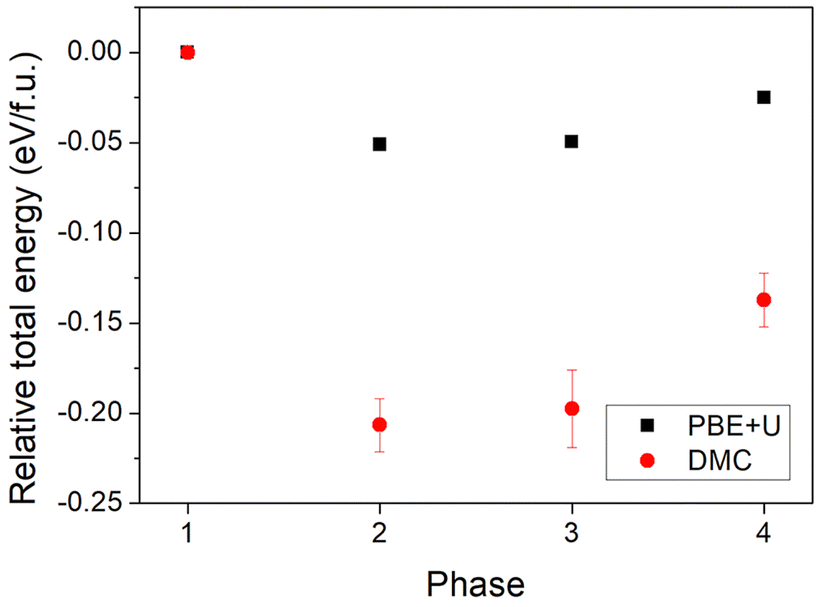 | ||
| Fig. 8 PBE+U and DMC relative energy between four different phases of AgNi0.66Co0.33O2. | ||
On the other hand, we see the metallic phase in higher Co concentration on the single NiO6 layer than x = 0.33 as seen in both phases 1 and 4. Since the varied structure are in-layer density fluctuations and each of these contain a layer at higher Co concentration, these results leads us to suspect the existence of reentrant phase to the metallic phase on high Co concentration over x = 0.33.
3.3 High Co doping: AgNi0.33Co0.66O2
In order to investigate optical properties on high Co concentration for AgNi1−xCoxO2, we additionally considered high Co concentration of x = 0.66, substituting all non-magnetic Ni1 sites in a hexagonal pattern (see Fig. 9). This x = 0.66 seems a hypothetical structure at a concentration where no experimental result for the electronic and optical properties has been reported. | ||
| Fig. 9 (a) Side and (b) top view of AgNi0.33Co0.66O2 cell. Blue atoms represent Co. | ||
The PBE+U density-of-states of AgNi0.33Co0.66O2 (see Fig. 10(a)) shows that Co-doping moves the valence band edge very close to the Fermi level, and at the conduction band edge, 3d-Co states have fully replaced d-Ni ones. Although the valence band edge still lies above Fermi level, the closeness of the band edge to the Fermi levels suggests that Co-substitution on the Ni3.5+ Ni1 sites results in the electronic properties of AgNi0.33Co0.66O2 moving from those of a semimetal closer to those of an insulator.
 | ||
| Fig. 10 (a) PBE+U projected density-of-states of AgNi0.33Co0.66O2, and (b) DMC spin density difference between AgNiO2 and AgNi0.33Co0.66O2. | ||
Fig. 3(b) shows the DMC spin density difference between AgNi0.33Co0.66O2 and AgNiO2. The figure shows a density changes on the AFM Ni sites Ni2, but density change is opposite in sign to the induced AFM magnetic moments. This tells us that the magnetic moments on Ni sites on AgNi0.33Co0.66O2 are smaller than in pristine AgNiO2, and this is confirmed by a DMC estimate of the magnetic moment of 1.52(1) μB for AgNi0.33Co0.66O2, which is smaller than the moment of 1.71(1) μB for the same Ni2 sites in AgNiO2.
These results suggest that reentrance to the metallic phase can be possible at high Co doping. This behavior is also consistent with phase 1 and 4 in Fig. 6 for AgNi0.66Co0.33O2, and confirms that reentrance to the metallic phase from insulator can be possible in high Co concentration.
3.4 Stability of AgNi1−xCoxO2
The thermodynamic stability of the AgNiO2 delafossite and its doped variants depends strongly on their formation energies. In order to estimate optimal conditions for formation of AgNi1−xCoxO2, we calculated the enthalpy of formation for AgNiO2 and its doped variants under different growth conditions. The enthalpy of formation of AgNiO2 can be estimated by computing where μX indicates the chemical potential for given atom X. In order to prevent formation of competing phases and phase separation or decomposition of AgNiO2, these chemical potentials should be constrained as follows:
where μX indicates the chemical potential for given atom X. In order to prevent formation of competing phases and phase separation or decomposition of AgNiO2, these chemical potentials should be constrained as follows: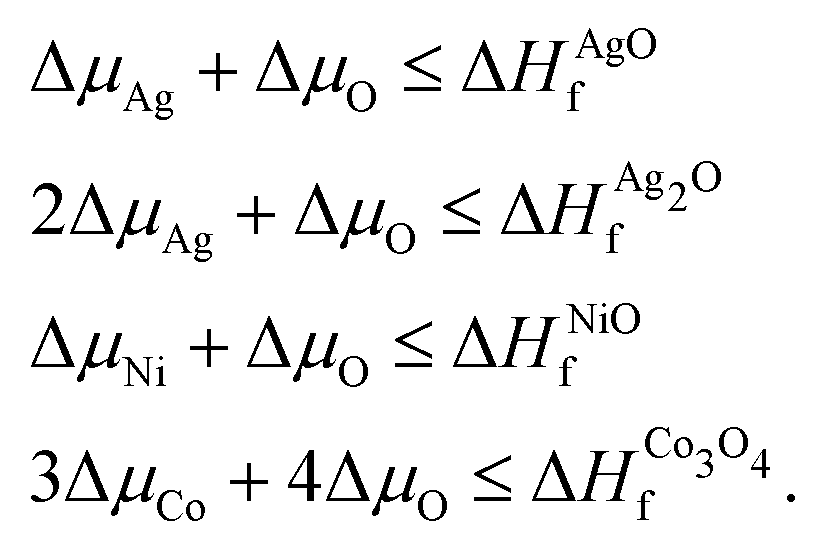 | (1) |
Based on the computed ΔHf using PBE+U, the phase diagram of AgNiO2 can be illustrated as a function of the allowed ranges of the chemical potentials of Ag, Ni, and O, given the constraints on them, as shown in Fig. 11.57,58 Within the boundaries given by the constraints on the enthalpy of formation of AgO, Ag2O, and NiO, we obtained the following chemical potentials of (ΔμAg, ΔμNi, ΔμO) under different growth conditions: Ag-poor:Ni-poor:O-rich A(−0.10, −2.85, −0.05), Ag-rich:Ni-poor:O-poor B(0.00, −2.75, −0.15), Ag-poor:Ni-rich:O-rich C(−0.08, −2.78, −0.10), and Ag-rich:Ni-rich:O-poor D(0.00, −2.71, −0.17). Using these chemical potentials, the formation energy of a Co defect is given by
 | (2) |
 | ||
| Fig. 11 Illustration of the accessible chemical potential range for AgNiO2 from PBE+U. | ||
Table 1 summarizes the computed formation energies of Co dopants in AgNi0.66Co0.33O2 and AgNi0.33Co0.66O2. As is seen in the table, PBE+U predicts spontaneous formation of Co-defects for all phases and growth conditions for which the formation energy of the defect is negative. It has previous been reported that PBE+U tends to underestimate the formation energy of defects in transition metal oxide systems,32,55,59,60 so spontaneous defect formation may not occur. The PBE+U results lead us to confirm that the Ag-rich:Ni-poor:O-poor growth condition is the most favorable one for pure AgNiO2 and AgNi1−xCoxO2 with the lowest formation energy within the given constraints. Although PBE+U does not provide quantitatively accurate formation energies for Co doping, a qualitative comparison between various growth conditions does give guidelines for the best growth conditions for synthesizing AgNiO2 and AgNi1−xCoxO2.
| AgNi0.33Co0.66O2 | AgNi0.66Co0.33O2 | ||||
|---|---|---|---|---|---|
| Phase 1 | Phase 2 | Phase 3 | Phase 4 | ||
| Ag-poor:Ni-poor:O-rich | −0.13 | −0.97 | −1.02 | −1.02 | −0.99 |
| Ag-rich:Ni-poor:O-poor | −0.16 | −0.98 | −1.03 | −1.03 | −1.00 |
| Ag-poor:Ni-rich:O-rich | −0.13 | −0.97 | −1.02 | −1.02 | −0.99 |
| Ag-rich:Ni-rich-O-poor | −0.15 | −0.98 | −1.02 | −1.03 | −1.00 |
In order to compare stability of AgNi1−xCoxO2 with the binary oxides, we compute PBE+U formation energy against elemental solids and binary oxides under stoichiometric conditions. In Table 2, we see large formation energy gap of ∼8 eV between one relative with elemental solids and the binaries. With comparison of formation energies Tables 1 and 2, we see that formation energies of AgNi1−xCoxO2 under the chemical potential constraints are significantly closer to formation energies from the binary oxides than those from the elemental solids in Table 2, which tells us formation of AgNi1−xCoxO2 is almost energetically consistent with the ideal formation against binary oxides. In addition, smaller formation energies in x = 0.66 than x = 0.33 in all growth conditions lead us to conclude relative difficulty of AgNi0.33Co0.66O2 synthesis.
| x = 0.66 | x = 0.33 | ||||
|---|---|---|---|---|---|
| Phase 1 | Phase 2 | Phase 3 | Phase 4 | ||
| Elemental | −3.58 | −4.22 | −4.27 | −4.27 | −4.25 |
| Binary | −0.09 | −0.87 | −0.92 | −0.92 | −0.89 |
4 Conclusions
We have performed DMC calculations on the AFM AgNiO2 delafossite in order to obtain accurate electronic properties and magnetic moments. We found that the addition of Hubbard U to the DFT scheme dramatically changes the electronic and magnetic properties of AgNiO2. Using DFT+U with the U value optimized using DMC, we confirmed that AgNiO2 has a semimetallic nature induced by strong p−d hybridization in AFM NiO2 layer. Our PBE+U and DMC studies of AgNi1−xCoxO2 shows a metal–insulator transition at x ∼ 0.33 by Co substitution on the non-magnetic Ni1 sites, which is in good agreement with the experimental result. In addition to the semiconducting phase in AgNi0.66Co0.33O2, it is found that the coexistence of metallic phase when more than x = 0.33 of Co dopant is substituted in the single layer of NiO2 in AgNi0.66Co0.33O2, leading to possible existence of the reentrance of metallic phase in high Co concentration. This reentrant behavior in AgNi1−xCoxO2 is confirmed in high Co concentration of AgNi0.33Co0.66O2 where semimetallic nature is discovered in the PBE+U result. PBE+U formation energies revealed that AgNi0.66Co0.33O2 possesses the lowest formation energy under oxygen-rich conditions, which suggests a path for synthesizing the AgNi0.66Co0.33O2 mixture. Our works clearly shows the difficulty in using PBE+U to quantitatively estimate formation energies of Co-doping in AgNiO2 because of the poor description of the 3d orbitals in Ni and Co. More accurate electronic structure methods than existing DFT approximations are needed for accurate prediction of electronic and magnetic properties of defective delafossites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division, as part of the Computational Materials Sciences Program and Center for Predictive Simulation of Functional Materials. An award of computer time was provided by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under contract DE-AC02-06CH11357 and resources of the Oak Ridge Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC05-00OR22725.Notes and references
- A. Pabst, Am. Mineral., 1946, 31, 539–546 CAS.
- R. D. Shannon, D. B. Rogers and C. T. Prewitt, Inorg. Chem., 1971, 10, 713–718 CrossRef CAS.
- C. T. Prewitt, R. D. Shannon and D. B. Rogers, Inorg. Chem., 1971, 10, 719 CrossRef.
- D. B. Rogers, R. D. Shannon, C. T. Prewitt and J. L. Gillson, Inorg. Chem., 1971, 10, 723 CrossRef.
- F. A. Benko and F. P. Koffyberg, J. Phys. Chem. Solids, 1984, 45, 57 CrossRef CAS.
- F. A. Benko and F. P. Koffyberg, J. Phys. Chem. Solids, 1987, 48, 431 CrossRef CAS.
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939 CrossRef CAS.
- H.-J. Noh, J. Jeong, J. Jeong, E.-J. Cho, S. B. Kim, K. Kim, B. I. Min and H.-D. Kim, Phys. Rev. Lett., 2009, 102, 256404 CrossRef PubMed.
- P. Kushiwaha, V. Sunko, P. J. W. Moll, L. Bawden, J. M. Riley, N. Nandi, H. Rosner, M. P. Schmidt, F. Arnold, E. Hassinger, T. K. Kim, M. Hoesch, A. P. Mackenzie and P. D. C. King, Sci. Adv., 2015, 1, e1500692 CrossRef PubMed.
- J. M. Ok, M. Brahlek, W. S. Choi, K. M. Roccapriore, M. F. Chisholm, S. Kim, C. Sohn, E. Skoropata, S. Yoon, J. S. Kim and H. N. Lee, APL Mater., 2020, 8, 051104 CrossRef CAS.
- E. Wawrzyńska, R. Coldea, E. M. Wheeler, I. I. Mazin, M. D. Johannes, T. Sörgel, M. Jansen, R. M. Ibberson and P. G. Radaelli, Phys. Rev. Lett., 2007, 99, 157204 CrossRef PubMed.
- J.-S. Kang, S. S. Lee, G. Kim, H. J. Lee, H. K. Song, Y. J. Shin, S. W. Han, C. Hwang, M. C. Jung, H. J. Shin, B. H. Kim, S. K. Kwon and B. I. Min, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 195122 CrossRef.
- S. Seki, Y. Onose and Y. Tokura, Phys. Rev. Lett., 2008, 101, 067204 CrossRef CAS PubMed.
- K. Ueda, T. Hase, H. Yanagi, H. Kawazoe and H. Hosono, J. Appl. Phys., 2001, 88, 1790 CrossRef.
- H. Yanagi, T. Hase, S. Ibuki, K. Ueda and H. Hosono, J. Appl. Phys., 2001, 78, 1583 CAS.
- M. Snure and A. Tiwari, Appl. Phys. Lett., 2007, 91, 092123 CrossRef.
- D. O. Scanlon, A. Walsh and G. W. Watson, Chem. Mater., 2009, 21, 4568 CrossRef CAS.
- S. Santra, N. S. Das and K. K. Chattopadhyay, Mater. Lett., 2013, 92, 198 CrossRef CAS.
- A. Wichainchai, P. Dordor, J. P. Doumerc, E. Marquestaut, E. Pouchard, P. Hagenmuller and A. Ammar, J. Solid State Chem., 1988, 74, 126 CrossRef CAS.
- A. I. Coldea, L. Seabra, A. McCollam, A. Carrington, L. Malone, A. F. Bangura, D. Vignoless, P. G. van Rhee, R. D. McDonald, T. Sörgel, M. Jansen, N. Shannon and R. Coldea, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 907, 020401 CrossRef.
- E. Wawrzyńska, R. Coldea, E. M. Wheeler, T. Sörgel, M. Jansen, R. M. Ibberson, P. G. Radaelli and M. M. Koza, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 094439 CrossRef.
- Y. Shin, J. Doumerc, P. Dordor, M. Pouchard and P. Hagenmuller, J. Solid State Chem., 1993, 107, 194–200 CrossRef CAS.
- W. E. Pickett, Y. Quan and V. Pardo, J. Phys.: Condens. Matter, 2014, 26, 274203 CrossRef PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- M. D. Johannes, S. Strelvsov, I. I. Mazin and D. I. Kohmskii, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 180404 CrossRef.
- P. J. Reynolds, D. M. Ceperley, B. J. Alder and W. A. Lester, J. Chem. Phys., 1982, 77, 5593–5603 CrossRef CAS.
- W. M. C. Foulkes, L. Mitas, R. J. Needs and G. Rajagopal, Rev. Mod. Phys., 2001, 73, 33–83 CrossRef CAS.
- H. Zheng and L. K. Wagner, Phys. Rev. Lett., 2015, 114, 176401 CrossRef PubMed.
- C. Mitra, J. T. Krogel, J. A. Santana and F. A. Reboredo, J. Chem. Phys., 2015, 143, 164710 CrossRef PubMed.
- J. Trail, B. Monserrat, P. L. Ríos, R. Maezono and R. J. Needs, Phys. Rev. B, 2017, 95, 121108 CrossRef.
- H. Shin, Y. Luo, P. Ganesh, J. Balachandran, J. T. Krogel, P. R. C. Kent, A. Benali and O. Heinonen, Phys. Rev. Materials, 2017, 1, 173603 Search PubMed.
- I. Kylänpää, Y. Luo, O. Heinonen, P. R. C. Kent and J. T. Krogel, Phys. Rev. B, 2019, 99, 075154 CrossRef.
- D. Wines, K. Saritas and C. Ataca, J. Chem. Phys., 2021, 155, 194112 CrossRef CAS PubMed.
- F. J. Morin, Phys. Rev. Lett., 1959, 3, 34 CrossRef CAS.
- A. Tseleve, I. A. Luk'yanchuk, I. N. Ivanov, J. D. Budai, J. Z. Tischler, E. Strelcov, A. Kolmakov and S. V. Kalinin, Nano Lett., 2010, 10, 4409 CrossRef PubMed.
- M. A. Huber, M. Plankl, M. Eisele, R. E. Marvel, F. Sandner, T. Korn, C. Schüller, R. F. H. Jr., R. Huber and T. L. Cocker, Nano Lett., 2016, 16, 1421 CrossRef CAS PubMed.
- J. Kim, A. Baczewski, T. Beaudet, A. Benali, C. Bennett, M. Berrill, N. Blunt, E. J. L. Borda, M. Casula, D. Ceperley, S. Chiesa, B. K. Clark, R. Clay, K. Delaney, M. Dewing, K. Esler, H. Hao, O. Heinonen, P. R. C. Kent, J. T. Krogel, I. Kylanpaa, Y. W. Li, M. G. Lopez, Y. Luo, F. Malone, R. Martin, A. Mathuriya, J. McMinis, C. Melton, L. Mitas, M. A. Morales, E. Neuscamman, W. Parker, S. Flores, N. A. Romero, B. Rubenstein, J. Shea, H. Shin, L. Shulenburger, A. Tillack, J. Townsend, N. Tubman, B. van der Goetz, J. Vincent, D. C. Yang, Y. Yang, S. Zhang and L. Zhao, J. Phys.: Condens. Matter, 2018, 30, 195901 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. M. Samos, N. Mazari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616 CrossRef CAS PubMed.
- M. C. Bennett, C. A. Melton, A. Annaberdiyev, G. Wang, L. Shulenburger and L. Mitas, J. Chem. Phys., 2017, 147, 224106 CrossRef PubMed.
- M. C. Bennett, G. Wang, A. Annaberdiyev, C. A. Melton, L. Shulenburger and L. Mitas, J. Chem. Phys., 2018, 149, 104108 CrossRef PubMed.
- A. Annaberdiyev, G. Wang, C. A. Melton, M. C. Bennett, L. Shulenburger and L. Mitas, J. Chem. Phys., 2018, 149, 134108 CrossRef PubMed.
- G. Wang, A. Annaberdiyev, C. A. Melton, M. C. Bennett, L. Shulenburger and L. Mitas, J. Chem. Phys., 2019, 151, 144110 CrossRef PubMed.
- Y. Luo, K. P. Esler, P. R. C. Kent and L. Shulenburger, J. Chem. Phys., 2018, 149, 084107 CrossRef PubMed.
- M. Casula, S. Moroni, S. Sorella and C. Filippi, J. Chem. Phys., 2010, 132, 154113 CrossRef PubMed.
- C. Lin, F. H. Zong and D. M. Ceperley, Phys. Rev. E, 2001, 64, 016702 CrossRef CAS PubMed.
- N. D. Drummond, R. J. Needs, A. Sorouri and W. M. C. Foulkes, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 125106 CrossRef.
- S. Chiesa, D. M. Ceperley, R. M. Martin and M. Holzmann, Phys. Rev. Lett., 2006, 97, 076404 CrossRef PubMed.
- K. Foyevstova, J. T. Krogel, J. Kim, P. R. C. Kent, E. Dagotto and F. A. Reboredo, Phys. Rev. X, 2014, 4, 031003 Search PubMed.
- Y. Luo, A. Benali, L. Shulenburger, J. T. Krogel, O. Heinonen and P. R. C. Kent, New J. Phys., 2016, 18, 113049 CrossRef.
- H. Shin, A. Benali, Y. Luo, E. Crabb, A. Lopez-Bezanilla, L. E. Ratcliff, A. M. Jokisaari and O. Heinonen, Phys. Rev. Mater., 2018, 2, 075001 CrossRef CAS.
- K. Saritas, J. T. Krogel, S. Okamoto, H. N. Lee and F. A. Reboredo, Phys. Rev. Mater., 2019, 3, 124414 CrossRef CAS.
- R. Chimata, H. Shin, A. Benali and O. Heinonen, Phys. Rev. Mater., 2019, 3, 075005 CrossRef CAS.
- E. M. Wheeler, R. Coldea, E. Wawrzyńska, T. Sörgel, M. Jansen, M. M. Koza, J. Taylor, P. Adroguer and N. Shannon, Phys. Rev. B, 2009, 79, 104421 CrossRef.
- C. Persson, Y.-J. Zhao, S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035211 CrossRef.
- A. Walsh, Y. Yan, M. M. Al-Jassim and S. H. Wei, J. Phys. Chem. C, 2008, 125, 12044 CrossRef.
- J. Santana, J. T. Krogel, J. Kim, P. R. C. Kent and F. A. Reboredo, J. Chem. Phys., 2015, 142, 164705 CrossRef PubMed.
- T. Ichibha, K. Saritas, J. T. Krogel, Y. Luo, P. R. C. Kent and F. A. Reboredo, Sci. Rep., 2023, 13, 6703 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Geometry for AgNi1−xCoxO2, U scanning for AgCoO2. See DOI: https://doi.org/10.1039/d3cp03477a |
| ‡ Present address: Seagate Technology, Computer Ave. 7801, Bloomington, MN 55435, USA. |
| This journal is © the Owner Societies 2024 |