
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodynamics of azaindoles in polar media: the influence of the environment†
Iker
Lamas
 a,
Raúl
Montero
*b,
Virginia
Martínez-Martínez
a and
Asier
Longarte
*a
a,
Raúl
Montero
*b,
Virginia
Martínez-Martínez
a and
Asier
Longarte
*a
aDepartamento de Química-Física Facultad de Ciencia y Tecnología. Universidad del País Vasco (UPV/EHU) Apart. 644, 48080 Bilbao, Spain. E-mail: asier.longarte@ehu.es; Tel: +34 946018086
bSGIKER Laser Facility Facultad de Ciencia y Tecnología. Universidad del País Vasco (UPV/EHU) 48940, Leioa, Spain. E-mail: raul.montero@ehu.es; Tel: +346013497
First published on 3rd January 2024
Abstract
We have studied the relaxation dynamics of a family of azaindole (AI) structural isomers, 4-, 5-, 6- and 7-AI, by steady-state and time-resolved methods (fs-transient absorption and fluorescence up-conversion), in solvents of different polarity. The measurements in aprotic solvents show distinctive fluorescence yields and excited state lifetimes among the isomers, which are tuned by the polarity of the medium. Guided by simple TD-DFT calculations and based on the behavior observed in the isolated species, it has been possible to address the influence of the environment polarity on the relaxation route. According to the obtained picture, the energy of the nπ* state, which is strongly dependent on the position of the pyridinic nitrogen, controls the rate of the internal conversion channel that accounts for the distinctive photophysical behavior of the isomers. On the other hand, preliminary measurements in protic media (methanol) show a very different photodynamical behavior, in which the anomalous measured fluorescent patterns are very likely the result of reactive channels (proton transfer) triggered by the electronic excitation.
Introduction
Azaindoles (AIs) can be considered prototype molecules to understand the deactivation channels operative, after near UV excitation, in the more complex purine DNA bases. More precisely, as part of a bottom-up approach, the information collected in these simpler models can guide the interpretation of the relaxation mechanisms observed in their biologically active relatives, which in principle would involve a more extensive set of electronic and vibrational coordinates.1–4 With this perspective, the photodynamics of azaindoles has attracted considerable attention in recent times.5–7 In particular, our group has investigated the relaxation of a set of neutral structural isomers, 4-, 5-, 6- and 7-azaindole (Fig. 1), in the gas phase.5 The theoretical and experimental results collected there showed that a common electronic structure of three excited states, two ππ* and one nπ* states, is responsible for the different photophysical properties exhibited by the four isomers. While the ππ* states provide the ability to be efficiently excited in the near-UV, the forbidden nπ* state, is non-adiabatically coupled to them, functioning as a gate state to access a conical intersection (CI) with the ground state. The relative energy between the ππ* and the nπ* states, determines the access to the latter, and ultimately, the lifetime of the particular isomer. The calculations also illustrated how the position of the pyridinic nitrogen modulates the energy of this nπ* state, permitting to link the structure of the isomer with its photophysical behavior. Thereby, for 7-AI, the nπ* surface lies at higher energy than those of the ππ* surfaces, hampering the access to the former after photoexcitation at 267 nm, which results in a long ns lifetime and a high fluorescence quantum yield. On the contrary, in the other three isomers, the internal conversion (IC) channel mediated by the accessible nπ* surface shortens the lifetime to the fs time-scale. | ||
| Fig. 1 Steady-state normalized absorption (a), (c) and (e) and fluorescent emission (b), (d) and (f) spectra of 4-, 5-, 6- and 7-AI in CH (a) and (b), DCM (c) and (d) and ACN (e) and (f). | ||
The detailed knowledge obtained in isolation conditions can be employed to address the role of the environment on the photodynamics observed in the more complex solution media, closer to the “natural” environment of these molecules. In this respect, 7-AI is the isomer that has received more attention in literature, mainly due to its ability to establish H-bond interactions, either with other monomer units to form homodimers in the gas phase8–14 and non-polar solvents,15–19 or with molecules of protic solvents in clusters20–29 and solutions.30–32 These systems are seen as excellent models to study excited state proton transfer (ESPT) processes33–36 that can lead to tautomerization. Studies targeting the photophysics of the other isomers are more scarce. The ESPT reaction in 6-AI trimers formed in non-polar medium has also been investigated,37 while the photophysics of 6-, 5- and 4-AI have been explored in the context of their fluorescence properties.38–40
The present work, grounding on the pathway established for the isolated species, aims to address the influence of the environment, in particular the solvent polarity, on the mechanisms that drive the relaxation and determine the observed lifetimes for this family of structural isomers. With this purpose, we have collected steady-state and femtoseconds (fs) time-resolved absorption and emission measurements in aprotic solvents of different polarity. Protic solvents have not been used to avoid proton transfer processes that can result in the appearance of new tautomers. Nevertheless, for comparison purposes, we have included some preliminary steady state and time-resolved measurements in methanol (MeOH), which exhibit a distinctive behavior that very likely involves proton transfer channels. The relaxation dynamics observed in the increasingly polar media dichloromethane (DCM) and acetonitrile (ACN) can be explained in terms of the same excited states identified in the excitation/relaxation of the isolated molecules. The collected experimental data, guided by conventional TD-DFT theoretical calculations, evidence how the polarity of the medium tunes the energy of the more polar nπ* state that controls the access to the IC channel, which is the mechanism behind the observed photophysical behavior. The study also permits to rationalize the different fluorescence properties of the isomers in terms of their chemical structure and the influence of the medium polarity.
Experimental
4-, 5- and 6-AI (98%) were acquired from Alfa Aesar, while 7-AI (98%) was obtained from Sigma-Aldrich. All the samples were employed without further purification. MeOH (99.9%, Fischer Chemical), DCM (99.9%, Merk) and ACN (99.9%, Sigma-Aldrich) were used to prepare the solutions. The concentrations reached for the time-resolved experiments were in the 6 to 33 mM interval. (See below for an extended explanation on concentration effects).Steady-state measurements
Steady-state absorption spectra of the AIs in cyclohexane (CH, 99.9%, Merk), ACN, DCM and MeOH were taken using a commercial UV/Vis spectrometer (Cintra 303 GBC Scientific Equipment Ltd). The fluorescence measurements were recorded with an Edinburgh Instruments Spectrofluorimeter (FLSP920 model, Livingston, UK) equipped with a xenon flash lamp (450 W) as the excitation source. The samples, at concentrations ∼10−5 M, were excited at 270 nm. The fluorescence spectra were corrected from the wavelength dependence on the detector sensibility.Fs-time resolved measurements
Ultrashort laser pulses were generated in an oscillator-regenerative amplifier laser system (Coherent, Mantis-Legend) that provides a 1 KHz train of 35 fs pulses at 800 nm. Pump pulses at ∼267 nm for fluorescence up-conversion (FuC) and transient absorption (TA), were generated by a third harmonic generation.The FuC set-up is based on a commercial kit (CDP FOG 100), which has been modified to allow the introduction of external pump pulses. The pump beam is focused by a lens (f = 60 mm) on a rotatory cuvette of 0.3 mm path that contains the solution under study. The sample emission is collected by an f = 60 mm lens and focused by a second one with f = 150 mm, on a 0.2 mm thick BBO crystal, where it interacts with the 800 nm gate beam to generate the up-converted signal by type-I phase matching. The latter is detected by a photomultiplier (PMT) coupled to a monochromator (CDP 220D), whose signal is integrated by a boxcar (CDP 2021A). The temporal overlap between the excitation and gate beams is controlled by a retroreflector mounted on a motorized delay stage allowing a maximum delay of ∼2 ns with a precision of 1.5 fs. The angle of the nonlinear crystal is optimized for the emission wavelength of choice, which is selected by the monochromator. For a typical measurement, 5 to 10 delay scans containing 1500 laser shots at each delay position are accumulated. Temporal resolutions around 300 fs are reached at the studied excitation and emission wavelengths.
The TA measurements were conducted in a homemade set-up, where solutions of the problem samples were excited at 267 nm. In order to refresh the sample for every pump pulse, the solutions were recirculated through a 0.2 mm optical path flow cell (Starna Scientific). The evolution of the prepared species was probed by a broadband white-light continuum (WLC), covering from 340 to 750 nm. This WLC was produced by focusing (f = 100 mm fused silica lens) an ∼1 μJ of the 800 nm fundamental beam on a 2 mm CaF2 plate, which is mounted on a linear translation stage to periodically refresh the exposed area The relative polarization of the pump–probe beams was set at magic angle configuration (54.7°) by a Berek's waveplate. The excitation and probe beams are focused into the sample, after collimation of the latter, by two spherical mirrors with focal lengths of 500 and 250 mm, respectively. Typically, pump energies of 0.5 μJ and spot radii of 200 and 75 μm at the interaction region were employed for the excitation and probe beams, in that order. The pump–probe delay was controlled by a linear translation stage (Thorlabs ODL220-FS) that permits a maximum delay of ∼2.5 ns, after a second pass through the delay line. The probe continuum transmitted through the sample is focused by an f = 100 mm lens onto an optical fiber coupled to a spectrometer (Avaspec ULS2048XL). A small fraction (40%) of the WLC beam, which is used as reference beam to significantly improve signal-to-noise ratio, is directly coupled to the second channel of the spectrometer by means of a f = 120 mm lens. The data collection and processing is carried out by a Labview written software. Roughly, the average of 3 × 104 laser shots are required to reach sensitivities in the order of 0.1 mΔOD. The instrumental response function (IRF) was provided from the coherent artifact signal CAS41 derived from pure solvent measurements. For MeOH, values between 120 and 190 fs were obtained in the 340–750 nm probe range, while for ACN the IRF was slightly longer.
In order to analyze the TA data, first the spectra were aligned to correct from the broadband pulse chirp by using the CAS as a zero delay time reference. Then, the scattering and spontaneous emission contributions were eliminated from the baseline by subtracting a spectrum collected at negative time delays.
The FuC and TA transients at the different excitation/probe wavelengths where modelled by the convolution function:

is a multi-exponential molecular response and R(λ,t) the cross-correlation (CC) function. At all measurements, a four lifetimes (τi = 1–4) exponential function was employed for the fittings. The number of lifetimes employed is dictated by the minimum set of constants required to fit the isomer with the most complex decays, 4-AI. This number of lifetimes is conserved to model the other isomers, although in some cases certain redundancy among the lifetimes is found. For the TA data, a global analysis procedure permits to extract a single set of τi constants for a collection of ai pre-exponential factors to build the decay associated spectra (DAS).

Computational methods
In order to explore the electronic structure of the AI isomers, TD-DFT(CAM-B3LYP)/6-31++G(d,p) calculations were carried out with the Gaussian 16 package.42 Solvent effects were introduced by the polarizable continuum model (PCM). The default PCM parameters, including the dielectric constants of 2.02 for CH, 35.89 for ACN, 8.93 for DCM and 32.62 for MeOH, were employed. After optimizing the ground state geometries, the vertical excitation energies (VEE) for the lowest ππ* and nπ* states were calculated by the corrected linear-response approach.Results
Calculations
Table 1 summarizes the computed VEEs of the relevant excited states, in the gas phase and solvents of different polarity. The molecular orbitals corresponding to the gas phase ππ* and nπ* vertical excitations (S1, S2 and S3) can be found in ESI1.† A more visual representation of the relative energies of these VEEs in the gas-phase and in ACN is shown in Scheme 1.| 4-AI | 5-AI | 6-AI | 7-AI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| VEE | f | VEE | f | VEE | f | VEE | f | ||
| a S4 for the 5-AI. | |||||||||
| Gas phase | S1 | 4.92 (ππ*) | 0.1622 | 5.14 (ππ*) | 0.1063 | 4.90 (ππ*) | 0.0855 | 4.99 (ππ*) | 0.1353 |
| S2 | 5.04 (nπ*) | 0.0035 | 5.16 (nπ*) | 0.0020 | 5.11 (nπ*) | 0.0032 | 5.03 (ππ*) | 0.0685 | |
| S3a | 5.05 (ππ*) | 0.0059 | 5.34 (ππ*) | 0.0092 | 5.34 (ππ*) | 0.0632 | 5.35 (nπ*) | 0.0042 | |
| CH | S1 | 4.92 (ππ*) | 0.2259 | 5.10 (ππ*) | 0.1452 | 4.96 (ππ*) | 0.1228 | 4.96 (ππ*) | 0.1961 |
| S2 | 5.01 (ππ*) | 0.0620 | 5.26 (nπ*) | 0.0026 | 5.21 (nπ*) | 0.0040 | 5.02 (ππ*) | 0.0975 | |
| S3 | 5.05 (nπ*) | 0.0043 | 5.33 (ππ*) | 0.0087 | 5.32 (ππ*) | 0.0901 | 5.38 (nπ*) | 0.0050 | |
| DCM | S1 | 4.89 (ππ*) | 0.2056 | 5.04 (ππ*) | 0.1309 | 4.92 (ππ*) | 0.1221 | 4.92 (ππ*) | 0.1656 |
| S2 | 5.03 (ππ*) | 0.0772 | 5.31 (ππ*) | 0.0080 | 5.29 (ππ*) | 0.0782 | 5.01 (ππ*) | 0.1202 | |
| S3 | 5.15 (nπ*) | 0.0044 | 5.39 (nπ*) | 0.0029 | 5.31 (nπ*) | 0.0042 | 5.42 (nπ*) | 0.0050 | |
| ACN | S1 | 4.88 (ππ*) | 0.1888 | 5.02 (ππ*) | 0.1203 | 4.91 (ππ*) | 0.1156 | 4.91 (ππ*) | 0.1463 |
| S2 | 5.03 (ππ*) | 0.078 | 5.31 (ππ*) | 0.0076 | 5.28 (ππ*) | 0.0698 | 5.01 (ππ*) | 0.1225 | |
| S3 | 5.18 (nπ*) | 0.0042 | 5.43 (nπ*) | 0.0030 | 5.35 (nπ*) | 0.0041 | 5.44 (nπ*) | 0.0049 | |
| MeOH | S1 | 4.88 (ππ*) | 0.1867 | 5.02 (ππ*) | 0.1191 | 4.91 (ππ*) | 0.1147 | 4.91 (ππ*) | 0.1441 |
| S2 | 5.03 (ππ*) | 0.0771 | 5.31 (ππ*) | 0.0075 | 5.26 (ππ*) | 0.0687 | 5.01 (ππ*) | 0.1217 | |
| S3 | 5.18 (nπ*) | 0.0042 | 5.43 (nπ*) | 0.0029 | 5.34 (nπ*) | 0.0041 | 5.44 (nπ*) | 0.0049 | |
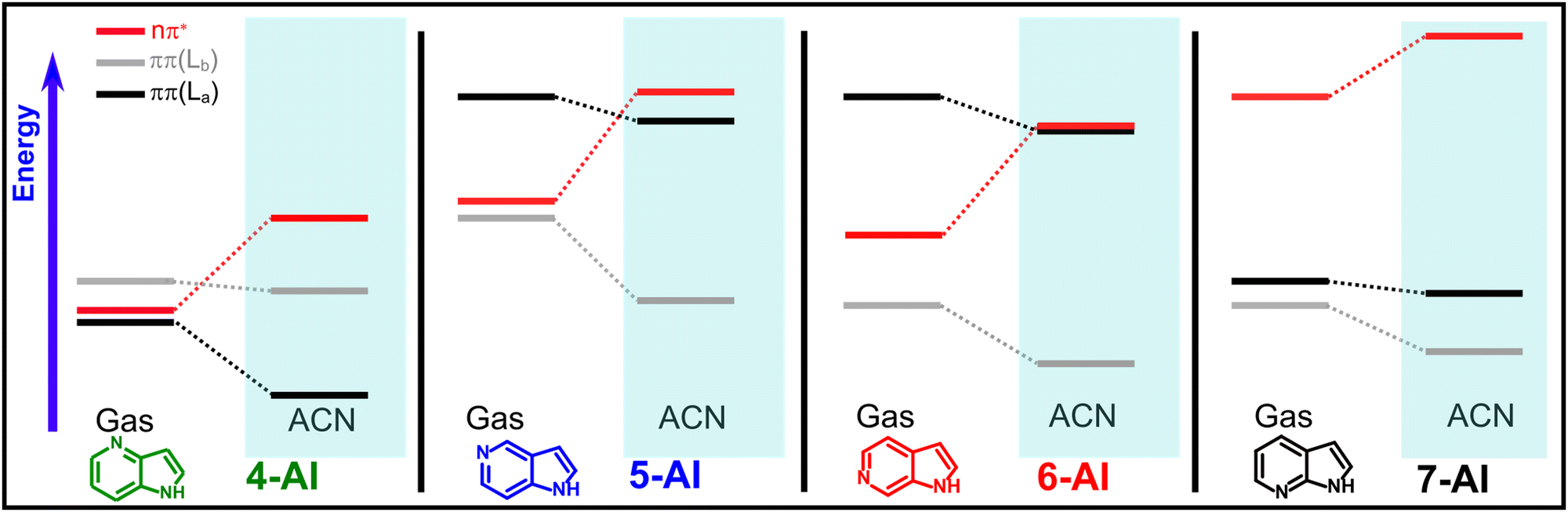 | ||
| Scheme 1 Schematic representation of the effect of solvation by ACN on the VEEs of the AI isomers, according to the calculated values shown in Table 1. | ||
Regarding the isolated compounds, despite of some differences, due to the limitations of TD-DFT calculations to reproduce absolute energies and oscillator strengths, the VEEs are in agreement with the values obtained at the MS-CASPT2 level.5 Three close-lying singlet states are computed in the excitation region, two with ππ* and one with nπ* character. According to the oscillator strengths obtained, and depending on the isomer, one of the ππ* states, or alternatively, a mixture of both, will be prepared by photoexcitation in the studied energy region. For 7-AI, the nπ* dark surface is calculated at energies considerably higher than the ππ* states, which lie at very close energies. Contrarily, in 4-, 5- and 6-AI, the nπ* state is located between the ππ* states. For 4-AI the three excitations are in a narrow 0.13 eV interval of energy. Characteristically, in 5-AI, the absorption onset is predicted at higher energies than in the other isomers. On the other hand, the energy gap between the two bright ππ* states in 6-AI is computed to be particularly large. A comparison between these predictions and the absorption spectra collected in the non-polar CH, which in principle should reflect the bright ππ* excitations, is offered in the Discussion section.
Concerning the data in different solvents, according to the calculated VEEs, the main effect of the polarity is the destabilization of the dark nπ* state, which is reflected by the small changes exhibited in the absorption spectra recorded as a function of the solvent polarity (Fig. 1a, c and e and Table 2). Conversely, it will be shown below how the emission properties (Fig. 1b, d and f and Table 2) are highly affected by the solvent polarity, as the nπ* is involved in the relaxation process.
| 4-AI | 5-AI | 6-AI | 7-AI | ||
|---|---|---|---|---|---|
| CH | Abs. λmax | 286(4.34) | 259(4.79) | 286(4.34)/255(4.86) | 286(4.34) |
| Emis. λmax | 317(3.91) | 311(3.99) | 312(3.97) | 311(3.99) | |
| Φ fl | 0.022 | 0.043 | 0.015 | 1.0 | |
| DCM | Abs. λmax | 289(4.29) | 263(4.71) | 288(4.31)/260(4.77) | 288(4.31) |
| Emis. λmax | 339(3.66) | 335(3.70) | 354(3.50) | 339(3.66) | |
| Φ fl | 0.592 | 0.001 | 0.003 | 1.0 | |
| ACN | Abs. λmax | 288(4.31) | 264(4.70) | 288(4.31)/260(4.77) | 287(4.32) |
| Emis. λmax | 350(3.54) | 379(3.27) | 344(3.60) | 350(3.54) | |
| Φ fl | 0.507 | 0.022 | 0.009 | 1.0 | |
| MeOH | Abs. λmax | 291(4.26) | 266(4.66) | 293(4.23)/260(4.77) | 290(4.28) |
| Emis. λmax | 400(3.10) | 403(3.08) | 421(2.94) | 364(3.41) | |
| Φ fl | 4.53 | 19.68 | 34.78 | 1.0 |
The predictions for MeOH are analogous to those calculated for ACN, as the model only considers the polarity of the solvents, which are very similar.
Steady-state absorption and emission
Fig. 1 shows normalized absorption and fluorescence (λexc = 270 nm) spectra of 4-, 5-, 6- and 7-AI dissolved in CH, DCM and ACN, while the absorption/emission maxima and relative fluorescence quantum yields are summarized in Table 2.In the polar solvents, the absorption spectra of the AIs are slightly redshifted and exhibit less resolved vibrational structure, but they do not show substantial differences with respect to those collected in CH. As it will be discussed below, the position of the bands observed for the different compounds can be correlated with the energies that the calculations predict for the two lowest ππ* excitations.
However, respect to the non-polar CH, the fluorescence spectra in polar media (Table 2 and Fig. 1d and f) exhibit more profound changes. The gas phase long-living 7-AI5 exhibits strong fluorescence in all the studied solvents. Contrarily, the compounds showing ultrafast relaxation in the isolated phase, 5- and 6-AI,5 have very weak fluorescence in all the aprotic solvents. Accordingly, in the following we will refer to them as non-fluorescent isomers. On the other hand, 4-AI shows an intermediate behavior, switching on its fluorescence when placed in polar environments. These observations will be correlated with the time-resolved measurements shown below.
As anticipated in the introduction, the studied AIs exhibit in the protic MeOH medium (Table 2 and Fig. 6) distinctive fluorescence patterns probably due to the occurrence of ESPT processes, which would be only briefly discussed below.
Fs time-resolved fluorescence and absorption
On the other hand, in the used polar solvents the AI derivatives do not show any significant dependence of their absorption or emission spectra on the concentration (Fig. SI2, ESI†). Actually, polar solvents preclude the formation of self-aggregates because the solvent/solute interactions overpower those found in the self-aggregates,37,40 making viable the study of monomers photodynamics at the high concentration required for the time-resolved techniques.
 | ||
| Fig. 2 FuC transients of 4-(a1;a2), 5-(b1;b2), 6-(c1;c2) and 7-AI(d1;d2) recorded in ACN, after excitation at 267 nm, at the 338 (a1–d1) and 381 (a2–d2) nm emission wavelengths. The dots are the experimental data, the red line the global exponential fitting and the color lines the individual exponential components employed. The lifetimes and weights corresponding to each component are indicated in the graphs and summarized in Table 3. | ||
| Compound | λ emis. (nm) | Lifetimes (ps) | Coefficients |
|---|---|---|---|
| 4-AI | 338 | τ 1 = 0.7 (0.9) | 16.8 (8.0) |
| τ 2 = 13.4 (9.2) | 4.6 (−3.2) | ||
| τ 3 = 162 (24.8) | 6.4 (2.3) | ||
| τ 4 = 3200 (1300) | 23.8 (34.0) | ||
| 381 (375) | τ 1 = 0.5 (0.3) | 3.2 (−16.7) | |
| τ 2 = 16.5 (3.2) | 1.6 (−3.5) | ||
| τ 3 = 176 (45.0) | 2.3 (5.8) | ||
| τ 4 = 4900 (1500) | 44.0 (70.9) | ||
| 5-AI | 338 | τ 1 = 1.0 (1.7) | 8.0 (18.1) |
| τ 2 = 6.0 (7.8) | 13.6 (7.2) | ||
| τ 3 = 34.5 (45.3) | 16.0 (1.3) | ||
| τ 4 = 220 (150) | 1.5 (1.9) | ||
| 381 (375) | τ 1 = 0.1 (0.2) | −15.0 (8.3) | |
| τ 2 = 4.1 (2.2) | 22.4 (19.0) | ||
| τ 3 = 36.0 (13.0) | 16.5 (4.6) | ||
| τ 4 = 220 (206) | 1.7 (2.9) | ||
| 6-AI | 338 | τ 1 = 1.0 (1.5) | 21.2 (31.3) |
| τ 2 = 4.8 (7.0) | 15.2 (5.2) | ||
| τ 3 = 38.6 (32.4) | 8.1 (1.7) | ||
| τ 4 = 205 (235) | 37.2 (0.3) | ||
| 381 (375) | τ 1 = 0.3 (1.6) | −11.2 (44.5) | |
| τ 2 = 3.2 (8.7) | 33.0 (5.3) | ||
| τ 3 = 17.5 (56.0) | 8.2 (2.4) | ||
| τ 4 = 206 (913) | 42.2 (2.2) | ||
| 7-AI | 338 | τ 1 = 0.7 (1.6) | 25.0 (7.4) |
| τ 2 = 40.0 (13.6) | −21.3 (−3.5) | ||
| τ 3 = 55.0 (25.0) | 22.7 (1.5) | ||
| τ 4 = 1400 (2450) | 30.3 (19.7) | ||
| 381 (375) | τ 1 = 0.3 (0.8) | −19.5 (−57.7) | |
| τ 2 = 23.0 (3.0) | −13.4 (3.4) | ||
| τ 3 = 31.0 (72.0) | 13.5 (16.2) | ||
| τ 4 = 1600 (2400) | 91.9 (371) |
In order to confirm the above observations, fluorescence measurements were conducted in the less polar DCM (PDCM = 3.1 < PACN = 5.8),44,45Fig. 3. As in the former solvent, in general, a four component multi-exponential function was employed to fit the signals. As in ACN, the 4- and 7-AI decays are differentiated from the 5- and 6-AI signals. However, interestingly, the latter compounds show notable differences with respect to the data collected in ACN. Although the multi-exponential lifetimes are similar in both solvents, the amplitude of shortest τ1 constant is remarkably larger in DCM, yielding a faster average decay-time.
 | ||
| Fig. 3 FuC transients of 4-(a1;a2), 5-(b1;b2), 6-(c1;c2) and 7-AI(d1;d2) recorded in DCM, after excitation at 267 nm, at the 338 (a1–d1) and 375 (a2–d2) nm emission wavelengths. The dots are the experimental data, the red line the global exponential fitting and the color lines the individual exponential components employed. The lifetimes and weights corresponding to each component are indicated in the graphs and summarized in Table 3 (in parentheses). | ||
 | ||
| Fig. 4 TA spectra recorded at selected delays for 4- (a), 5- (b), 6- (c), and 7-AI (d) dissolved in ACN after exciting at 267 nm. | ||
 | ||
| Fig. 5 Decay associated spectra (DAS) for the four studied AIs in ACN. The graphs show the weights along the spectra of the lifetimes derived from the modelling of the TA data obtained after excitation at 267 nm. See Experimental section for details. | ||
Discussion
It is important to remark that due to solubility/aggregation issues it was not possible to obtain time-resolved data of the AIs monomers in non-polar medium. Then, the picture obtained for the isolated molecules5 is the starting point to interpret the relaxation dynamics observed in the different solvents. As it was sketched in the introduction, the three excited states whose vertical VEEs are summarized in Table 1, are involved in the photodynamics of the AI isomers. Their common relaxation pathway that was calculated in the gas phase5 leads from the Franck–Condon (FC) ππ* structures, to the nπ* surface, and from there to a CI with the ground state. The relative position of the nπ* state respect to the ππ* states controls the access to this route. While for 5- and 6-AI a low barrier is calculated along the path, 7-AI presents a higher barrier to reach the CI, which results in the dramatically different lifetimes measured. Although the 4-AI relaxation could not be tracked in the gas-phase, the absence of fluorescent emission seem to point, in agreement with calculations, to a mechanism analogous to 5- and 6-AI.In order to address how this picture is altered by the environment, we can start by comparing the results of the conducted PCM calculations that are summarized in Table 1, with the recorded steady-state absorption and emission spectra (Fig. 1 and Table 2). In general, the relative VEEs obtained for the two ππ* excitations of the isolated molecules validate the interpretation of the absorption spectra in CH. For 4- and 7-AI the two ππ* excitations are calculated at very close energies, which results in the single band present in the spectrum. For 5-AI, these excitations are also predicted to be similar, but shifted to higher energies, in good agreement with the recorded spectrum. On the other hand, in 6-AI the energy gap between the two ππ* states, which carry comparable oscillator strengths, is computed to be particularly large, as reflected by the two well defined bands present in the spectrum.
However, the most meaningful information extracted from the calculated VEEs comes from the comparison of the values for the different solvents. They show that the main effect of the solvent polarity on the excitations involved in the photodynamics of the AIs is the destabilization of the nπ* state. This is caused by the drastic reduction of the dipole moment of the molecule associated to this transition, as the electron more localized on the n orbital of the nitrogen atom is promoted to a π* orbital that is delocalized over the molecule. On the other side, the bright ππ* states responsible of the excitation are much less affected by the solvent polarity. An overall impression about the predictions on the relevant excitations and the effect of the solvent on them can be obtained from Scheme 1. These calculations match the steady-state spectra in Fig. 1. In general, as already discussed above, the relative energies of the two lowest ππ* excitations of the different isomers calculated in Table 1, reproduce the absorption spectra of the AIs, which reflect these bright S0-ππ* excitations. In addition, as predicted by calculations, the absorption spectra are very little affected by the solvent polarity, as it is observed in the comparison of the spectra in CH and the more polar media. Contrarily, the fluorescent emission exhibits a heavy dependence on the solvent. In CH, 7-AI shows an intense fluorescence band centered at ∼310 nm, while the emission of 4-, 5- and 6-AI appears in the same region, but it is about 50 times weaker. In the more polar DCM and ACN (Table 1 and Fig. 1d and f), the fluorescence of all the isomers is progressively shifted to lower energies. Nevertheless, the more striking effect is the increment of the 4-AI fluorescence, which acquires a behavior very similar to 7-AI. On the other hand, although the fluorescence bands of 5- and 6-AI experiment a red shift with the solvent polarity, they remain weakly fluorescent in all media. Therefore, we can conclude from the steady-state data that the nπ* surface, which is mainly affected by the solvent polarity, plays an important role in controlling the fluorescence of the isomers, in correlation with the relaxation mechanism found in the gas phase.5
The FuC measurements (Fig. 2 and 3) confirm and provide further details on the effect of the solvent on the operative mechanism. These FuC transients were collected after excitation at 267 nm, which in view of the theoretical predictions and absorption measurements (Fig. 1), will produce a mixture of the two close-lying ππ* excitations, according to their respective transition probabilities (Table 1). See below for an extended explanation about eventual conversion between these states. Four lifetimes (τ1, τ2, τ3 and τ4) were used to model all the fluorescence signals in ACN and DCM. According to their evolution and wavelength dependence, the decays of the four AI isomers can be separated in two groups that correlate with the fluorescent quantum yields observed in the steady-state measurements: the fluorescent 4- and 7-AI and the non-fluorescent 5- and 6-AI.
Fluorescent samples 4- and 7-AI
The fluorescent 4- and 7-AI show a very similar behavior characterized by three initial components, from hundreds of fs to tens of picoseconds, followed by a τ4 of few nanoseconds that turns the emission off (Fig. 2 and 3). Giving the gas-phase results for 7-AI5 and the calculations on the relative energies of ππ* and nπ* states, the latter surface is too high to be accessed. It is worth to remark here that 4-AI becomes fluorescent when solvated by ACN and DCM, mimicking the behavior of 7-AI in these polar media. In this scenario, the observed dynamics can be restricted to the ππ* states. The shortest lifetime τ1 is likely related to solvation dynamics. In fact, as the detection is shifted to the red, it becomes a rising signal (Fig. 2a2;d2 and 3a2;d2). Such behavior is indicative of dynamic Stokes shift, which is a spectral redshift of the emission because of solvent reorganization during the excited state lifetime.46–48 Aiming to obtain the characteristic lifetimes related with the ACN solvation dynamics and to confirm the origin of τ1 time component, FuC measurements were conducted for Coumarin 153 (C153) in ACN. C153 has been extensively used because it is considered an ideal probe for the solvation dynamics.49–52 This study, whose details are included in the ESI3† permit to relate the τ1 component with the ACN initial response to the excited state dipole moment. The origins of τ2 and τ3 are more unclear. Very remarkably τ2 appears as a formation component for all the fluorescent compounds. We tentatively attribute these two components to non-adiabatic relaxation between the two ππ* states. Although calculations predict that the lowest S1 ππ* dominates the absorption (Table 1), according to the spectra (Fig. 1), the excitation at 267 nm could prepare a mixture of the two ππ* sates. As it has been shown for 7-AI in the gas phase,5 and for other similar systems,53,54 the IC between these two states occurs at ultrafast rates. However, after this initial decay, a population re-equilibration scenario between them, followed by vibrational cooling, is possible. In order to test this possibility, and aiming to populate exclusively the lowest ππ* state, FuC experiments in ACN were also conducted at the onset of the absorption spectrum, 300 nm, for 4- and 7-AI (Fig. SI5, ESI†). These measurements did not reveal substantial differences, other than some minor variation of the extracted lifetimes, respect to those obtained after excitation at 267 nm.Finally, the long τ4 component indicates that the prepared excited states relax in the ns time-scale toward a non-fluorescent location. For 4-AI the TA data (see below) show the formation of a non-fluorescent location that does not decay in the time-scale of the experiments. In the case of 7-AI no longer lifetimes are found. Experiments in longer time-scales would be required to know the channels that determine the measured fluorescence nanoseconds-lifetime.
Non-fluorescent samples 5- and 6-AI
The non-fluorescent 5- and 6-AI exhibit a very different time evolution (Fig. 2 and 3). The results in the gas phase showed ultrafast relaxation in the fs time-scale that was explained by the existence of a barrierless pathway from the FC ππ* structures, to the nπ* surface, and from there to a CI with the ground state. Although the calculations (Table 1 and Scheme 1) predict that the nπ* state is shifted to higher energies by the polarity of the solvent, the lack of fluorescence (Fig. 1d and f) and the FuC decays (Fig. 2bi;ci and 3 bi;ci) indicate that, in contrast with 4-AI, this non-radiative channel is still operative in ACN and DCM. In both solvents, the FuC signals at the tested emission wavelengths are modelled by a multi-exponential function with constants from some tenths up to two hundreds picoseconds. Except the τ1 component in ACN (Fig. 2bi;ci), which perhaps could be attributed to the solvent response, in general, it is not possible to assign the exponentials to specific processes. Therefore, attending to the dynamics described in the gas phase,5 the full multi-exponential decay has to be attributed to the relaxation pathway that takes the molecule from the ππ* locally excited state to a CI between the nπ* and the ground state. This path must involve the solvent response, the intramolecular vibrational relaxation and the coupling between the ππ* and nπ* states. In addition, as it was already mentioned for the fluorescent 4- and 7-AI, in view of the 5- and 6-AI absorption spectra, the 267 nm excitation would prepare a mixture of the two ππ* states, since non-adiabatic relaxation between the two surfaces should also be present during the earliest times. Very presumably, as we already mentioned above, IC between the two ππ* states should occur at very short times, even shorter than the experimental time-resolution. Then, the relaxation would proceed from this relaxed ππ* surfaces toward the nπ* state, giving raise to the measured decays. Now, it is also worth to note that multi-exponential decays can be intrinsic to relaxation processes involving barriers.55Nevertheless, to test an eventual dependence of the relaxation dynamics on the excitation wavelength, FuC measurements in ACN were conducted after exciting 5- and 6-AI at the onset of their absorption spectra, 280 nm. The decays (Fig. SI5, ESI†) show only minor changes in the extracted lifetimes, pointing to a relaxation pathway analogous to that observed at 267 nm.
As already described, although the FuC measurements collected in the less polar DCM (Fig. 3) show in general a time evolution close to that observed in ACN (Fig. 2), the transients of 5- and 6-AI exhibit interesting particularities that confirm the trends predicted by the calculations. Although the values of the components are very similar in both solvents, the faster lifetimes τ1 and τ2 show a much larger amplitude in DCM, resulting in transients with average lifetimes notoriously shorter that are closer to those found in the gas phase.se This observation can be related with the fact that the nπ* surface is less affected by the lower polarity of DCM and therefore more easily accessed by coupling to the prepared ππ* states.
TA measurements
In general, the TA data recorded in ACN are in good agreements with the fluorescence observations, supporting the photophysical behavior sketched by the latter. Nevertheless, they exhibit some differences that can provide additional information on the relaxation pathway. The most perceptible changes are found for 4-AI. The DAS (Fig. 5a) and the decays (Fig. SI4a1, ESI†), reveal the existence of a spectral feature, mainly centered around 475 nm, that does not decay in the observation window, τ4 > 10 ns. According to the DAS, Fig. 5a, this location would be formed with the τ3 = 2.9 ns component that corresponds to the τ4 = 3.2–4.9 ns derived from the fluorescence decays. Attending to its non-fluorescent and long-living nature we assign this band to the absorption of a triplet state. Then, intersystem crossing (ISC) seems to be involved in the relaxation of the excited state. Interestingly, ISC has in fact been identified as an important relaxation channel in the case of 7-AI,56 but for this compound, our spectra do not show the absorption of the triplet state that must be located out of the probing region.In the case of 5-AI, there is also a noticeable change in the value of the longest lifetime τ4 (Fig. 5b and Fig. SI4bi, ESI†) with respect to that measured in fluorescence (Fig. 2bi). The fact that this lifetime is longer across the TA indicates that the system reaches a non-fluorescent location, before leaving the TA observation window. It is conceivable to assign it as the dark nπ* surface that mediates the coupling to the ground state. For 5-AI, this surface could present a small barrier to reach the CI with the ground state, as it was calculated for the isolated 7-AI,5 explaining the longest lifetimes measured in the TA experiments.
Protic media
We will briefly comment on the results collected in protic media, MeOH, which show a different behavior that points to the appearance of specific photochemical channels. After comparing the steady-state data already seen in DCM or ACN (Fig. 1) with those recorded in MeOH (Fig. 6), two solvents with similar polarity, it is clear that while the absorption spectra are very similar, the fluorescence emission patterns show profound changes. In MeOH, the overall emission intensity of the previously considered fluorescent isomers, 4- and 7-AI, is greatly reduced, below the values obtained for 5- and 6-AI. At the same time, in all cases the fluorescence bands are strongly extended to the red, showing in some cases new distinctive maxima. Accordingly, the FuC data included in the ESI† (Fig. SI6) show lifetimes altered respect to those measured in ACN. In particular, 4- and 7-AI lifetimes are much shorter than in ACN, while for 6-AI the opposite happens.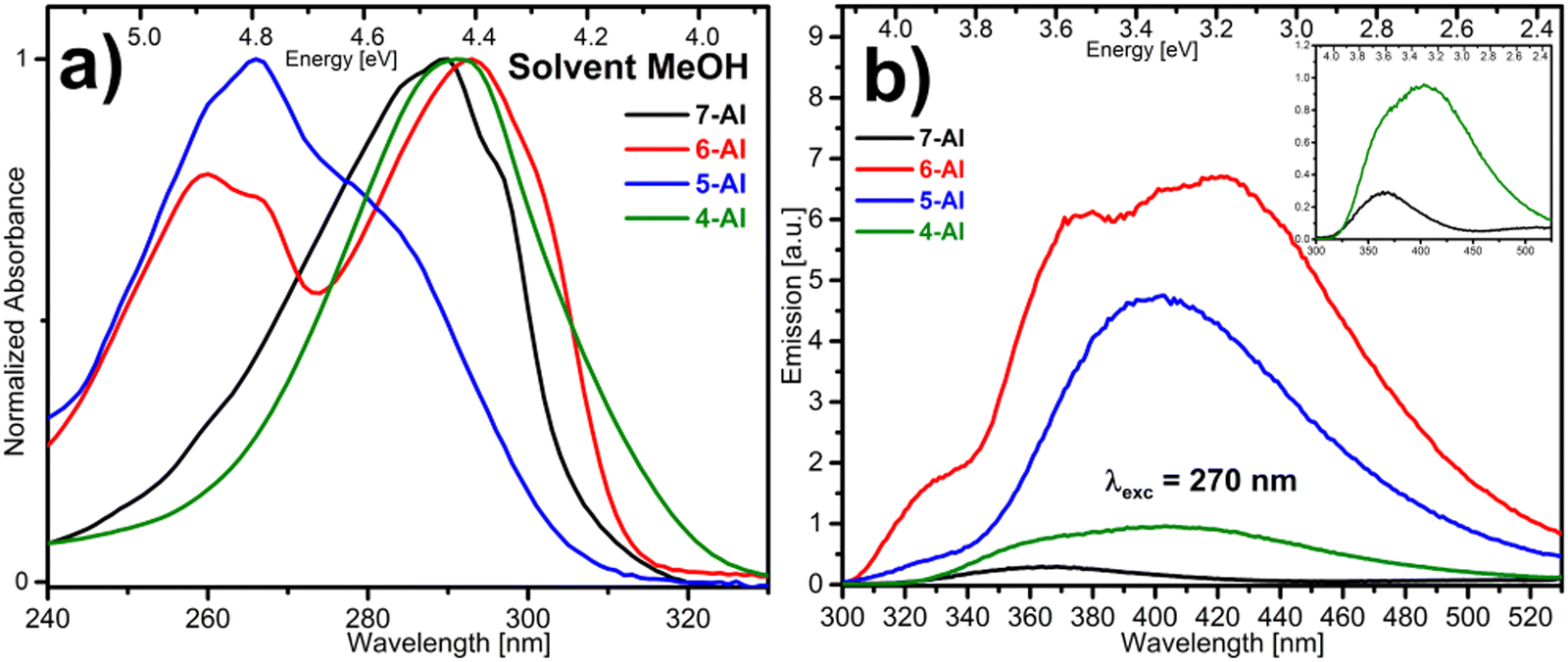 | ||
| Fig. 6 Absorption (a) and fluorescent emission (b) spectra of 4-, 5-, 6- and 7-AI in MeOH. | ||
The full interpretation of this behavior is beyond the capabilities of the present study, and we just intend to point at the main guidelines of the problem. In this sense, it can be said that the measured lifetimes, particularly the longest τ3 component, are conditioned by the occurrence of ESPT processes that may lead to the formation of tautomers. For 7-AI, apart from the extensively studied concerted ESPT in dimers,8–19 the ESPT in monomers solvated by water and MeOH molecules has been demonstrated theoretically28,29,31 and experimentally.25–27,30 In the case of 6-AI in methanol, the tautomerization has also been demonstrated and the appearance in the emission spectrum of intermediate protonated species proposed.38 In the close analogue 2,6-diazaindole in aqueous solutions, excited-state triple proton transfer reaction resulting in tautomerization has also been shown.57 To the best of our knowledge, the possibility of the ESPT process occurring in 4- and 5-AI, which would require the transfer of the proton through longer solvent chains, has not been explored to date.
Comparison with adenine decay dynamics
It may be instructive to compare the present results with the behavior found in the more complex purine bases, in particular, the extensively studied adenine molecule. The excitation/relaxation of isolated adenine at 267 nm involves a similar set of excited states. From experimental observations58,59 and some theoretical predictions,60,61 the dark nπ* state is found to be S1 at the FC geometry. Above, the two close-lying La and Lb ππ* states are responsible for the excitation process. According to the extensive investigation carried out by computational studies, a number of CIs connecting these states and the ground state can mediate the relaxation pathway, which highly depends on the excitation energy.60–62 Experimentally, the ultrafast bi-exponential decay found after excitation at 267 nm is composed of the τ1∼100 fs and τ2∼1.0 ps lifetimes.63–65 Although many of the computational studies have been able to predict the ultrafast relaxation of the molecule, discrepancies in the exact mechanism still remain.61,66–68 In the actual view, two main channels involving two different geometry CIs are invoked to explain the observed decay: (i) A ππ* → nπ* → S0 relaxation mediated by a S1/S0 C6-puckered CI.67 (ii) A ππ* → S0 channel through a S1/S0 C2 puckered CI, where the nπ* state does not play major role.61,66 The importance of the calculated mechanism depends on the employed methodology.68The studied AI isomers can be highly relevant regarding the theoretical description of the operative channel in adenine. The measured relaxation dynamics indicates that the involvement of the nπ* state is required for an efficient IC to S0. In those cases where the nπ* state is shifted to higher energies, as in 7-AI in any media of 4-AI in polar solvents, the excited population remains in the ππ* states for long times, which results in the high fluorescence quantum yields. Consequently, differently from adenine, the access to the C2 puckered geometries that couple the ππ*/S0 states must present higher barriers in these species. In this context, the studied AIs and related molecules of increasing complexity as diazaindoles, can be excellent models to test non-adiabatic dynamical simulations and to establish the nuclear-electronics structural base of the observed dynamical behavior.
Conclusions
The reported calculations and experiments aimed to characterize the influence of the environment on the photodynamics of the studied set of AIs structural isomers. In particular, the steady-state and time-resolved measurements conducted in aprotic solvents, and their comparison with isolated phase experiments5 and theoretical predictions has allowed us to establish the effect of the medium polarity on the relaxation pathway. We postulate, in correlation with the gas-phase results, that the lifetime of the studied AIs depends on the relative energy between the dark nπ* and the bright ππ* states, which is controlled by the polarity of the environment. As the calculations predict, the polarity of the solvent shifts the nπ* surface to higher energies. This hampers the access to this surface and to the nπ*/S0 CI, which, as it was demonstrated in the gas-phase studies, controls the relaxation dynamics of the AIs. Additionally, preliminary measurements in MeOH sketch a very different picture for the time evolution in polar media. In this case, the relaxation channels involve ESPT reactions that lead to the formation of tautomers. Further studies able to identify the spectral and dynamical signature of the intermediate and final reaction products, together with calculations on the potential reaction channels, would be required to develop an understanding of the problem.Author contributions
I. Lamas: conceptualization, formal analysis, investigation, writing – review & editing; R. Montero: conceptualization, formal analysis, investigation, methodology, supervision, writing – review & editing; V. Martínez-Martínez: formal analysis, investigation; A. Longarte: conceptualization, methodology, project administration, funding acquisition, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support from the Spanish MICIN through grants PGC2018-098561-B-C21 and PID2020-114347RB-C32. The work was also partially funded by the Basque Government (IT1491-22 and IT1639-22). I. Lamas thanks the UPV/EHU for his predoctoral fellowship. Technical and human support provided by SGIker (UPV/EHU, MICINN, GV/EJ, ESF) is also gratefully acknowledged.References
- J. L. Pecourt, J. Peon and B. Kohler, J. Am. Chem. Soc., 2001, 123, 10370–10378 CAS.
- H. Kang, B. Jung and S. K. Kim, J. Chem. Phys., 2003, 118, 6717–6719 CAS.
- B. Marchetti, T. N. V. Karsili, M. N. R. Ashfold and W. Domcke, Phys. Chem. Chem. Phys., 2016, 18, 20007–20027 CAS.
- A. A. Beckstead, Y. Zhang, M. S. de Vries and B. Kohler, Phys. Chem. Chem. Phys., 2016, 18, 24228–24238 CAS.
- I. Lamas, R. Montero, V. Martínez-Martínez, A. Longarte and L. Blancafort, Phys. Chem. Chem. Phys., 2020, 22, 18639–18645 CAS.
- J. A. Noble, E. Marceca, C. Dedonder, W. Phasayavan, G. Féraud, B. Inceesungvorn and C. Jouvet, Phys. Chem. Chem. Phys., 2020, 22, 27280–27289 CAS.
- R. Mansour, S. Mukherjee, M. Pinheiro, J. A. Noble, C. Jouvet and M. Barbatti, Phys. Chem. Chem. Phys., 2022, 24, 12346–12353 CAS.
- K. Fuke, K. H. Yoshiuchi and K. Kaya, J. Phys. Chem., 1984, 88, 5840–5844 CAS.
- K. Fuke and K. Kaya, J. Phys. Chem., 1989, 93, 614–621 CAS.
- A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 378, 260 CAS.
- D. E. Folmer, E. S. Wisniewski and A. W. Castleman, Phys. Lett., 2000, 318, 637–643 CAS.
- K. Sakota and H. Sekiya, J. Phys. Chem. A, 2005, 109, 2722–2727 CAS.
- K. Sakota, C. Okabe, N. Nishi and H. Sekiya, J. Phys. Chem. A, 2005, 109, 5245–5247 CAS.
- H. Sekiya and K. Sakota, J. Photochem. Photobiol., C, 2008, 9, 81–91 CAS.
- W. M. Hetherington, R. H. Micheels and K. B. Eisenthal, Chem. Phys. Lett., 1979, 66, 230–233 CAS.
- K. Tokumura, Y. Watanabe, M. Udagawa and M. Itoh, J. Am. Chem. Soc., 1987, 109, 1346–1350 CAS.
- P. Share, M. Pereira, M. Sarisky, S. Repinec and R. M. Hochstrasser, J. Lumin., 1991, 48–49, 204–208 CAS.
- T. Fiebig, M. Chachisvilis, M. Manger, A. H. Zewail, A. Douhal, I. Garcia-Ochoa and A. de La Hoz Ayuso, J. Phys. Chem. A, 1999, 103, 7419–7431 CAS.
- O. Kwon and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8703–8708 CAS.
- S. K. Kim and E. R. Bernstein, J. Phys. Chem., 1990, 94, 3531–3539 CAS.
- P. Chou, C. Wei, C. Chang and M. Kuo, J. Phys. Chem., 1995, 99, 11994–12000 CAS.
- Y. Huang, S. Arnold and M. Sulkes, J. Phys. Chem., 1996, 100, 4734–4738 CAS.
- D. E. Folmer, E. S. Wisniewski, S. M. Hurley and A. W. Castleman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12980–12986 CAS.
- T. Bao Chau Vu, I. Kalkman, W. Leo Meerts, Y. N. Svartsov, C. Jacoby and M. Schmitt, J. Chem. Phys., 2008, 128, 214311 Search PubMed.
- Y. Koizumi, C. Jouvet, T. Norihiro, S. Ishiuchi, C. Dedonder-Lardeux and M. Fujii, J. Chem. Phys., 2008, 129, 104311 Search PubMed.
- K. Sakota, C. Jouvet, C. Dedonder, M. Fujii and H. Sekiya, J. Phys. Chem. A, 2010, 114, 11161–11166 CAS.
- G. A. Pino, I. Alata, C. Dedonder, C. Jouvet, K. Sakota and H. Sekiya, Phys. Chem. Chem. Phys., 2011, 13, 6325–6331 CAS.
- R. Daengngern, N. Kungwan, P. Wolschann, A. J. A. Aquino, H. Lischka and M. Barbatti, J. Phys. Chem. A, 2011, 115, 14129–14136 CAS.
- N. Kungwan, K. Kerdpol, R. Daengngern, S. Hannongbua and M. Barbatti, Theor. Chem. Acc., 2014, 133, 1480 Search PubMed.
- R. S. Moog and M. Maroncelli, J. Phys. Chem., 1991, 95, 10359–10369 CAS.
- S. Mente and M. Maroncelli, J. Phys. Chem. A, 1998, 102, 3860–3876 CAS.
- O.-H. Kwon, Y.-S. Lee, H. J. Park, Y. Kim and D.-J. Jang, Angew. Chem., Int. Ed., 2004, 43, 5792–5796 CAS.
- J. Catalán and J. L. G. de Paz, Chem. Phys., 2005, 123, 114302 Search PubMed.
- J. Catalán, J. C. del Valle and M. Kasha, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8338–8343 Search PubMed.
- S. Takeuchi and T. Tahara, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5285–5290 CAS.
- R. Crespo-Otero, N. Kungwan and M. Barbatti, Chem. Sci., 2015, 6, 5762–5767 CAS.
- T. Tu, Y. Chen, Y. Chen, Y. Wei, Y. Chen, C. Chen, J. Shen, Y. Chen, S. Ho, K. Cheng, S. Lee, C. Chen and P. Chou, Angew. Chem., Int. Ed., 2018, 57, 5020–5024 CAS.
- S. M. Twine, L. Murphy, R. S. Phillips, P. Callis, M. T. Cash and A. G. Szabo, J. Phys. Chem. B, 2003, 107, 637–645 CAS.
- M. T. Cash, P. R. Schreiner and R. S. Phillips, Org. Biomol. Chem., 2005, 3, 3701–3706 CAS.
- L. Merkel, M. G. Hoesl, M. Albrecht, A. Schmidt and N. Budisa, ChemBioChem, 2010, 11, 305–314 CAS.
- B. Dietzek, T. Pascher, V. Sundström and A. Yartsev, Laser Phys. Lett., 2007, 4, 38 Search PubMed.
- M. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- Y. Liu, Y. He, Y. Yang and Y. Liu, Chem. Phys. Lett., 2021, 762, 138137 CAS.
- E. Katz, R. Eksteen, P. Schoenmakers and N. Miller, In Handbook of HPLC, ed. M. Dekker, New York, 1998, Appl. Organomet. Chem., 2000, 14, 130–131 Search PubMed.
- J. A. C. Broekaert and D. C. Harris, Quantitative chemical analysis, Anal. Bioanal. Chem., 9th edn, 2015, vol. 407, pp. 8943–8944 Search PubMed.
- R. Jimenez, G. R. Fleming, P. V. Kumar and M. Maroncelli, Nature, 1994, 369, 471–473 CAS.
- P. Abbyad, W. Childs, X. Shi and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20189–20194 CAS.
- A. Rosspeintner, B. Lang and E. Vauthey, Annu. Rev. Phys. Chem., 2013, 64, 247–271 CAS.
- S. J. Rosenthal, X. Xie, M. Du and G. R. Fleming, J. Chem. Phys., 1991, 95, 4715–4718 CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CAS.
- R. Karmakar and A. Samanta, J. Phys. Chem. A, 2002, 106, 4447–4452 CAS.
- I. Eom and T. Joo, J. Chem. Phys., 2009, 131, 244507 Search PubMed.
- F. Messina, M. Prémont-Schwarz, O. Braem, D. Xiao, V. S. Batista, E. T. J. Nibbering and M. Chergui, Angew. Chem., Int. Ed., 2013, 52, 6871–6875 CAS.
- T. Gustavsson, N. Sarkar, I. Vayá, M. C. Jiménez, D. Markovitsia and R. Improta, Photochem. Photobiol. Sci., 2013, 12, 1375–1386 CAS.
- M. Olivucci, A. Lami and F. A. Santoro, Angew. Chem., Int. Ed., 2005, 44, 5118–5121 CAS.
- P. Ilich, J. Mol. Struct., 1995, 354, 37 CAS.
- K.-Y. Chung, Y.-H. Chen, Y.-T. Chen, Y.-H. Hsu, J.-Y. Shen, C.-L. Chen, Y.-A. Chen and P.-T. Chou, J. Am. Chem. Soc., 2017, 139, 6396–6402 CAS.
- N. J. Kim, G. Jeong, Y. S. Kim, J. Sung, S. K. Kim and Y. D. Park, J. Chem. Phys., 2000, 113, 10051–10055 CAS.
- C. Canuel, M. Mons, F. Piuzzi, B. Tardivel, I. Dimicoli and M. Elhanine, J. Chem. Phys., 2005, 122, 074316 Search PubMed.
- L. Blancafort, J. Am. Chem. Soc., 2006, 128, 210–219 CAS.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, Chem. – Eur. J., 2006, 12, 6559–6571 Search PubMed.
- S. Matsika, J. Phys. Chem. A, 2005, 109, 7538–7545 CAS.
- S. Ullrich, T. Schultz, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2004, 126, 2262–2263 CAS.
- H.-H. Ritze, H. Lippert, E. Samoylova, V. R. Smith, I. V. Hertel, W. Radloff and T. Schultz, J. Chem. Phys., 2005, 122, 224320 Search PubMed.
- H. Satzger, D. Townsend, M. Z. Zgierski, S. Patchkovskii, S. Ullrich and A. Stolow, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 10196–10201 CAS.
- M. Barbatti and H. Lischka, J. Am. Chem. Soc., 2008, 130, 6831–6839 CAS.
- E. Fabiano and W. Thiel, J. Phys. Chem. A, 2008, 112, 6859–6863 CAS.
- M. Barbatti, Z. Lan, R. Crespo-Otero, J. J. Szymczak, H. Lischka and W. Thiel, J. Chem. Phys., 2012, 137, 22A503 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03412g |
| This journal is © the Owner Societies 2024 |