Investigation of Pb–B Bonding in PbB2(BO)n− (n = 0–2): Transformation from Aromatic PbB2− to Pb[B2(BO)2]−/0 Complexes with B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) B Triple Bonds†
B Triple Bonds†
Received
15th June 2023
, Accepted 11th January 2024
First published on 13th January 2024
Abstract
Boron has been found to be able to form multiple bonds with lead. To probe Pb–B bonding, here we report an investigation of three Pb-doped boron clusters, PbB2−, PbB3O−, and PbB4O2−, which are produced by a laser ablation cluster source and characterized by photoelectron spectroscopy and ab initio calculations. The most stable structures of PbB2−, PbB3O−, and PbB4O2− are found to follow the formula, [PbB2(BO)n]− (n = 0–2), with zero, one, and two boronyl ligands coordinated to a triangular and aromatic PbB2 core, respectively. The PbB2− cluster contains a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double bond and two Pb–B single bonds. The coordination of BO is observed to weaken Pb–B bonding but strengthen the BB bond in [PbB2(BO)n]− (n = 1, 2). The anionic [PbB2(BO)2]− and its corresponding neutral closed-shell [PbB2(BO)2] contain a B
B double bond and two Pb–B single bonds. The coordination of BO is observed to weaken Pb–B bonding but strengthen the BB bond in [PbB2(BO)n]− (n = 1, 2). The anionic [PbB2(BO)2]− and its corresponding neutral closed-shell [PbB2(BO)2] contain a B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B triple bond. A low-lying Y-shaped isomer is also observed for PbB4O2−, consisting of a central sp2 hybridized B atom bonded to two boronyl ligands and a PbB unit.
B triple bond. A low-lying Y-shaped isomer is also observed for PbB4O2−, consisting of a central sp2 hybridized B atom bonded to two boronyl ligands and a PbB unit.
1 Introduction
Three-dimensional (3D) cages, especially the well-known B12-icosahedral cage, are key structural features prevalent in boranes and various boron allotropes.1–3 However, in the past two decades, size-selected boron clusters have been found through systematic experimental and theoretical studies to be planar or quasi-planar (2D) up to very large sizes.4–10 The first 3D borospherene cage (B40) was discovered in 2014,11 followed by the axially chiral borospherene B39− in 2015.12 Subsequently, seashell-like borospherenes were found as minor isomers for the B28− and B29− clusters.13,14 The largest boron cluster (B48−), characterized experimentally to date, was found to have a bilayer-type structure.15 Significant experimental efforts have also been devoted to investigate metal-doped boron clusters, which have been shown to display a wide variety of novel structural patterns,10,16–18 such as fan-shaped structures,19–22 double-chain ribbons,23,24 metal-centered boron wheels,16,25–29 half-sandwich structures,30–35 metallo-borophenes,8,36,37 metal-centered boron drums,37–42 metallo-borospherenes,43 and metal-borozene complexes.24,44,45 The most important chemical bonding features of boron and metal-doped boron clusters are found to be dominated by both delocalized σ and π bonds as a result of boron's electron deficiency.6–8,18,46–49
In addition to forming delocalized bonds, boron is also capable of engaging in localized bonds. For instance, the boronyl group (BO), which is isoelectronic with the cyano group (CN), has been shown to have a strong BO triple bond50 and has been synthesized.51 In fact, even BB triple bonds have been observed in isolated molecules52–54 and synthetic compounds.55 Earlier studies on boron oxide clusters showed that they all contained boronyl ligands53,56–58 and the analogy between boronyl and H/Au was established, i.e. they are all single electron σ donors. Using this analogy, we have designed and observed experimentally many boron-boronyl clusters.59–64 Recently, several interesting findings have also been made in metal-doped boron oxide clusters that feature unusual metal-boron (M–B) multiple bonds and boronyl ligands. The MB triple bonds were observed in linear [MB–BO]− (M = Bi, Re) and bent (−OB)IrB,65,66 while the bent (−OB)RhB was confirmed to contain a RhB quadruple bond.67 The umbrella-like [(η7-B7)-B-BO]− cluster was even found to embody metallomimetic properties of boron.68
Lead is one of the earliest metals acquainted and used by human beings. Despite their known toxicity, lead and its compounds are still important today and have wide applications in industries, including pigments, lead-acid batteries, and lead glasses, etc.69 In fact, lead is the most common gamma-ray shielding material due to its high atomic number, high density, and wide availability.70–72 Boron, on the other hand, has good shielding properties against neutron radiation because of its high thermal neutron absorption cross-section.73,74 Recently, a series of lead borate crystals, Pb3B10O16[OH]4, have been synthesized and shown to have better co-shielding properties for gamma and neutron radiations than the simple PbO/B2O3 mixture with the same molar ratio of Pb and B.75 Despite the progress achieved for synthesizing lead borate compounds with various morphologies, few lead-boron molecules are known and our knowledge about the lead-boron bonding is still limited. We have studied recently two lead-doped boron oxide clusters, PbB2O−/2− and PbB3O2− and found a PbB triple bond in the linear PbB2O2− species, a B Pb multiple bond with a bond order of 2.5 in the linear PbB2O−, and a BPb double bond in the Y-shaped PbB3O2−.76
Pb multiple bond with a bond order of 2.5 in the linear PbB2O−, and a BPb double bond in the Y-shaped PbB3O2−.76
In this article, we report the observation of three lead-doped boron and boron oxide clusters, PbB2−, PbB3O−, and PbB4O2−, which are studied via photoelectron spectroscopy (PES) and ab initio calculations. The global minimum (GM) of PbB2− is found to be an isosceles triangle, while the GM structures of PbB3O− and PbB4O2− can be viewed as successively coordinating a boronyl ligand colinearly to the B atoms of the triangular PbB2−. The triangular PbB2− is aromatic and contains a BB double bond and two Pb–B single bonds. The coordination of BO is observed to strengthen the BB double bond in PbB2− but weaken Pb–B bonding. Thus, the GM structures of the three lead-doped boron clusters can be formulated as [PbB2(BO)n]− (n = 0–2), with zero, one, and two boronyl units bonded to an aromatic PbB2 triangle. It is found that Pb bonding with the B2 part of the B2(BO)2 motif in [PbB2(BO)2]−/0 is very weak and these entities can be characterized as the Pb+/2+[B2(BO)22−] charge-transfer complexes containing a BB triple bond. A low-lying Y-shaped isomer is also observed for PbB4O2−, which consists of a central B atom in sp2 hybridization and coordinated by two boronyl ligands and a BB double bond with the PbB unit [PbB = B(BO)2−]. These findings not only broaden our understanding of Pb–B bonding but also help design novel lead-boron molecules and lead boride materials with excellent co-shielding properties for high-energy gamma and neutron radiations.
2 Method
2.1 Photoelectron spectroscopy
The lead-boron clusters were produced using a laser vaporization cluster source and were investigated using a magnetic-bottle PES apparatus, as described previously.7,77 Briefly, the PbB2−, PbB3O−, and PbB4O2− clusters were formed by laser vaporization of a cold-pressed Pb/10B mixed target with a helium carrier seeded with 5% Ar, and the 532 nm vaporization laser beam was directed collinearly with the cluster beam. The trace amount of oxide impurities on the target surface was sufficient to produce the O-containing species, PbB3O− and PbB4O2−. Seeding the helium carrier gas with O2 would produce highly oxidized clusters (PbBxOy−). The clusters formed in the source were entrained in the carrier gas pulse delivered by two symmetrically mounted molecular beam valves and underwent a supersonic expansion. We used a large waiting room nozzle and a strong supersonic expansion to remove the substantial internal energies to maximize cluster cooling. After passing a skimmer, negatively charged clusters were extracted from the collimated cluster beam and analyzed using a time-of-flight mass spectrometer. The clusters of current interest were mass-selected and decelerated before photodetached by a pulsed laser beam at 266 nm (4.661 eV) produced from the fourth harmonics of a Nd:YAG laser. Photoelectron kinetic energies were calibrated using the known spectrum of the Bi− atomic anion. The resolution of the magnetic-bottle PES apparatus was around 2.5%, that is, ∼25 meV for 1 eV electrons.
2.2 Computational methods
The GM structures were searched for PbB2−, PbB3O−, and PbB4O2− using the Coalescence-Kick method78 at the PBE0/lanl2dz79–82 level of theory. Low-lying candidates were fully optimized at the PBE0 level with the augmented Dunning's all-electron basis set (aug-cc-pVTZ) for B and O atoms and the aug-cc-pVTZ-pp basis set with the relativistic pseudopotentials (ECP60MDF) for the Pb atom (abbreviated as AVTZ).83,84 Vibrational frequencies were calculated at the same level to make sure that the obtained structures exhibited true minima of the systems. The relative energies of the top isomers within 1.0 eV were further refined at the CCSD(T)85–87 /AVTZ//PBE0/AVTZ level [abbreviated as CCSD(T)/AVTZ]. To obtain more reliable structural parameters, we re-optimized the lowest-lying isomer for each species and the second lowest-lying isomer of PbB4O2− at the CCSD/AVTZ level.88,89 The first vertical detachment energies (VDE1) and the adiabatic detachment energies (ADE) for the GM structures of PbB2−, PbB3O−, and PbB4O2− were calculated at the CCSD(T)/AVTZ level. The VDE1 was computed as the energy difference between the anionic ground state and the corresponding neutral ground state at the optimized anion geometry, whereas the ADE was calculated as the difference in energy between the optimized anion and the corresponding neutral at its optimized structure.
The higher VDEs were obtained by adding the neutral excitation energies to the VDE1. To obtain the energies of the neutral excited states, the state-averaged (SA) complete active space self-consistent field (CASSCF) calculations90,91 were performed using the AVTZ basis set, followed by multi-reference configuration interaction (MRCI) calculations,92 in which the spin–orbit (SO) coupling effect was considered.93 The active space employed consisted of 8 electrons in 10 orbitals for PbB2, 5 electrons in 6 orbitals for PbB3O, and 4 electrons in 6 orbitals for PbB4O2. For the second isomer of PbB4O2−, the first two VDEs were calculated at the CCSD(T)/AVTZ level as the transition from the ground state of the anionic species to the lowest singlet and triplet states of the corresponding neutral species at the optimized anion geometry. The higher VDEs were calculated using the time-dependent (TD)-PBE0/AVTZ method. Vibrational frequencies for the ground state of neutral PbB3O were computed at the PBE0/AVTZ level. Chemical bonding analyses were conducted using the adaptive natural density partitioning (AdNDP) method94 and were then visualized using the VMD software.95 The out-of-plane tensor components of the nucleus-independent chemical shift at ζ distance above the molecular plane [NICSzz(ζ)]96 were computed at the PBE0/TZP97 level to examine the aromaticity of the experimentally confirmed species. Considering the influence of heavy metal doping on the aromaticity indices,98 the centres of the ring currents were used instead of the geometry centres to calculate the NICSzz(ζ) values, and the spin–orbit coupling effects were included in the NICS calculations.99,100 Natural bond orbital (NBO) and natural resonance theory (NRT) analyses101 were performed at the PBE0/AVTZ level using the NBO 7.0 program.102 All geometry optimizations and frequency calculations at the PBE0 and CCSD levels and TD-PBE0 calculations were performed using the Gaussian 09 program.103 The CCSD(T), CASSCF, and MRCI calculations were carried out using the Molpro package.104 The NICS calculations were implemented using the ADF code,105 while the centres of the ring current were searched using the GIMIC program.106
3 Experimental results
The photoelectron spectra of PbB2−, PbB3O−, and PbB4O2− at 266 nm are shown in Fig. 1–3, respectively. The well-resolved PES bands are labeled with letters, X, A, B, …, where band X represents the transition from the anionic ground state to the electronic ground state of the corresponding neutral cluster and bands A, B, … indicate detachment transitions to the excited electronic states of the neutral cluster. The electron affinity (EA) of the corresponding neutral cluster or the ADE was determined by drawing a straight line at the leading edge of band X and then adding the instrumental resolution. The VDEs were measured using the band maxima, as given in Tables 1–3, where they are compared with the theoretical results. A larger difference between the VDE1 and ADE usually indicates a more significant geometry change from the anion to neutral cluster upon electron detachment.
 |
| | Fig. 1 Photoelectron spectrum of PbB2− at 266 nm (4.661 eV). | |
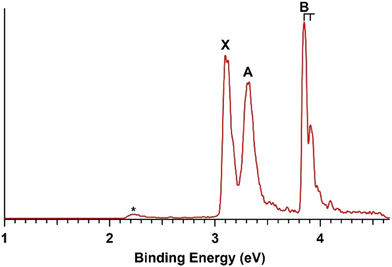 |
| | Fig. 2 Photoelectron spectrum of PbB3O− at 266 nm. | |
 |
| | Fig. 3 Photoelectron spectrum of PbB4O2− at 266 nm. | |
Table 1 The experimental VDEs for PbB2−, and their assignments and comparison with the theoretical results. All energies are given in eV. The first VDE was calculated using the CCSD(T) method. MRCI and SO coupling calculations were conducted to calculate the higher VDEs
| VDE (Exp)a |
Configurations |
Terms |
VDE (MRCI) |
Levels |
VDE (SO) |
Composition of SO coupled states |
|
The experimental uncertainty was estimated to be ±0.02 eV.
|
| X |
2.08 |
1a122a121b223a121b124a10 |
1A1 |
1.94 |
1A1(0) |
1.94 |
96.4%1A1 + 3.3%3B2 + 0.3%3B1 |
| A |
2.48 |
1a122a121b223a111b124a11 |
3A1 |
2.54 |
3A1(0) |
2.52 |
65.2%3A1 + 34.8%3B1 |
|
3A1(−1) |
2.52 |
63.8%3A1 + 36.2%3B1 |
| B |
2.65 |
1a122a121b223a121b114a11 |
3B1 |
2.65 |
3A1(+1) |
2.61 |
94.8%3A1 + 3.6%1B1 + 1.6%3B2 |
|
3B1(+1) |
2.69 |
87.0%3B1 + 12.5%1A1 + 0.4%3B2 |
| C |
2.91 |
|
|
|
3B1(−1) |
2.86 |
62.2%3B1 + 31.7%3A1 + 6.1%1B2 |
|
3B1(0) |
2.90 |
63.6%3B1 + 36.0%3A1 + 0.4%1B2 |
| D |
3.03 |
1a122a121b223a111b124a11 |
1A1 |
3.00 |
1A1(0) |
3.11 |
68.6%1A1 + 23.0%3B2 + 8.4%3B1 |
| E |
3.15 |
1a122a121b223a121b114a11 |
1B1 |
3.09 |
1B1(0) |
3.25 |
81.0%1B1 + 17.0%3B2 + 2.0%3A1 |
| F |
∼3.6 |
1a122a121b213a121b124a11 |
3B2 |
3.85 |
3B2(0) |
4.06 |
96.4%3B2 + 1.9%3A1 + 1.7%1B1 |
|
3B2(+1) |
4.06 |
97.0%3B2 + 1.9%3A1 + 1.1%1B1 |
|
3B2(−1) |
4.08 |
96.7%3B2 + 2.4%1A1 + 0.9%3B1 |
Table 2 The experimental VDEs for PbB3O−, their assignments and comparison with the theoretical results. All energies are given in eV. The first VDE was calculated using the CCSD(T) method. MRCI and SO coupling calculations were conducted to calculate higher VDEs
| VDE (exp.)a |
Configurations |
Terms |
VDE (MRCI) |
Levels |
VDE (SO) |
Composition of SO coupled states |
|
The experimental uncertainty was estimated to be ±0.02 eV.
|
| X |
3.10 |
…5a′26a′21a′′27a′28a′22a′′1 |
2A′′ |
3.21 |
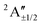
|
3.21 |
70.1%2A′′ + 26.1%2A′ + 2.1%4A′ + 1.7%4A′′ |
| A |
3.32 |
…5a′26a′21a′′27a′18a′22a′′2 |
2A′ |
3.23 |
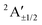
|
3.48 |
80.4%2A′ + 15.9%2A′′ + 1.6%4A′ + 2.1%4A′′ |
| B |
3.85 |
…5a′26a′21a′′27a′28a′12a′′2 |
2A′ |
3.66 |
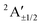
|
3.96 |
84.2%2A′ + 9.6%2A′′ + 3.3%4A′ + 2.9%4A′′ |
Table 3 The experimental VDEs for PbB4O2−, their assignments and comparison with the theoretical results. All energies are given in eV. The first VDE was calculated using the CCSD(T) method. MRCI and SO coupling calculations were conducted to calculate higher VDEs
| VDE (exp)a |
Configurations |
Terms |
VDE (MRCI) |
Levels |
VDE (SO) |
Composition of SO coupled states |
|
The experimental uncertainty was estimated to be ±0.02 eV.
|
| X |
2.88 |
…1b121a224b226a122b127a125b20 |
1A1 |
2.88 |
1A1(0) |
2.88 |
85.0%1A1 + 9.6%3B2 + 2.8%3A2 + 2.5%3B1 |
| A |
3.82 |
…1b121a224b226a122b127a115b21 |
3B2 |
3.85 |
3B2(0) |
3.86 |
59.0%3B2 + 36.3%3A2 + 2.3%3A1 + 2.4%1B1 |
|
3B2(−1) |
3.86 |
51.8%3B2 + 41.3%3A2 + 4.6%1A1 + 2.2%3B1 |
|
3B2(+1) |
3.91 |
82.8%3B2 +12.9%1A2 + 3.2%3B1 + 1.1%3A1 |
| B |
4.01 |
…1b121a224b226a122b117a115b21 |
3A2 |
3.96 |
3A2(+1) |
4.03 |
89.6%3A2 + 6.9%3A1 + 3.4%1B2 + 0.1%3B1 |
| C |
4.15 |
|
|
|
3A2(−1) |
4.15 |
56.1%3A2 + 38.0%3B2 + 4.7%3A1 + 1.3%1B1 |
| D |
4.33 |
…1b121a224b226a122b117a115b21 |
1A2 |
4.19 |
3A2(0) |
4.38 |
54.4%3A2 + 34.0%3B2 + 11.6%1A1 |
|
1A2(0) |
4.34 |
76.2%1A2 + 12.9%3B2 + 9.7%3A1 + 1.3%3B1 |
| E |
4.53 |
…1b121a224b226a122b127a115b21 |
1B2 |
4.56 |
1B2(0) |
4.70 |
81.9%1B2 + 14.6%3B1 + 3.4%3A2 |
3.1 PbB2−
The 266 nm spectrum of PbB2− shown in Fig. 1 displays six sharp and intense peaks on the low binding energy side and weak and broader PES features above 3.5 eV. The first VDE of PbB2− is obtained from band X as 2.08 eV and the ADE is estimated to be 2.03 eV, which also represents the EA of PbB2. An intense band A is observed at 2.48 eV, followed by a similarly intense band B at 2.65 eV. Three closely spaced bands C, D, E are observed at 2.91, 3.03, and 3.15 eV, respectively. Features beyond 3.5 eV are weak and congested, and they are labeled as F for the sake of discussion. All the VDEs for PbB2− are given in Table 1 and compared with the theoretical data.
3.2 PbB3O−
The 266 nm spectrum of PbB3O− (Fig. 2) is relatively simple with three sharp and well-resolved bands. Band X gives rise to the first VDE at 3.10 eV, with its ADE estimated as 3.08 eV, followed by band A at 3.32 eV. PbB3O− has the highest VDE among all three clusters due to its closed-shell electronic structure. Band B at 3.85 eV was vibrationally resolved with a vibrational spacing of ∼480 cm−1. A very weak feature marked by an asterisk was observed at the low binding energy side, which might come from a contaminant due to the congested mass spectrum caused by the isotopic distribution of lead. The measured VDEs for PbB3O− are summarized in Table 2, where they are compared with the theoretical results.
3.3 PbB4O2−
The photoelectron spectrum of PbB4O2− (Fig. 3) seems much more complicated than those for PbB2− and PbB3O− with more congested features. The PES bands labeled with X, A–E are intense and relatively sharp, which are likely from one species. On the other hand, the two bands labeled with X′ (3.13 eV) and A′ (3.40 eV) are weak and broad and they appear to come from a minor isomer of PbB4O2−. Band X yields a VDE of 2.88 eV with an estimated ADE of 2.84 eV. Following a relatively large energy gap of 1 eV, a sharp and intense band A is observed at 3.82 eV. The spectral features beyond band A become more complex, and a weaker peak B is observed at 4.01 eV, while a relatively broad band C is resolved at 4.15 eV. Two sharp bands, D and E, are observed at 4.33 and 4.53 eV, respectively. The bands C, D, and E appear to contain partially resolved vibrational features. The VDEs for band X, A–E are given in Table 3 and compared with the theoretical results to be presented below.
4 Theoretical results
Fig. 4 displays the optimized GM structures and their bond lengths of PbB2− and PbB3O− and the top two lowest-lying isomers of PbB4O2− at the CCSD/AVTZ level. All low-lying structures within about 2.0 eV of the GM structure at the PBE0/AVTZ level are presented in Fig. S1–S3 (ESI†) for PbB2−, PbB3O−, and PbB4O2−, respectively, along with their symmetries, electronic states, and relative energies. The top isomers within 1.0 eV were further calculated at the single-point CCSD(T)/AVTZ level.
 |
| | Fig. 4 The optimized structures of the GM of PbB2− (1), PbB3O− (2), PbB4O2− (3) and its closed-shell PbB4O2 (3′), and a low-lying isomer of PbB4O2− (4) and its closed-shell PbB4O22− (4′). The bond lengths are given in Å at the CCSD/AVTZ level. | |
4.1 PbB2−
Only six structures with different spin multiplicities (doublet, quartet, and sextet) are located within ∼2.0 eV of the GM for the PbB2− cluster (Fig. S1, ESI†). These structures can be divided into two categories, i.e., C2v isosceles triangle and linear Pb–B–B. It is found that the doublet isosceles triangle (C2v, 2A1) is the GM for PbB2−, consisting of a B2 dimer bridged by a Pb atom (Fig. 4, 1). The second lowest-lying isomer, a doublet linear Pb–B–B (C∞v, 2Σ) structure, is more than 0.45 eV above the GM at both the PBE0 and CCSD(T) levels. The remaining isomers are at least 1.11 eV higher in energy at the PBE0 level.
4.2 PbB3O−
Candidate structures for PbB3O− are composed of five isomers containing a terminal BO group bonded to a PbB2 moiety and another five isomers based on a B3 triangle (Fig. S2, ESI†). The GM of PbB3O− is 2 (Cs, 1A′), as shown in Fig. 4 with all bond lengths labeled. The GM can be viewed as attaching a BO group to a B atom of the PbB2 triangle along the B–B bond, weakening the Pb–B bond while strengthening the terminal Pb–B bond. The closest low-lying isomer consists of a B3 triangle with a bridging Pb atom and a bridging O atom, which is 0.84 eV higher in energy than the GM at the PBE0 level and 1.00 eV higher at the CCSD(T) level. The large energy gap between the first two isomers indicates the high stability of GM 2.
4.3 PbB4O2−
For the PbB4O2− cluster, we found fourteen isomers within 2 eV of the GM, most of which possess at least one terminal BO group (Fig. S3, ESI†). The GM of PbB4O2− (3C2v, 2B2) is a planar structure, consisting of two terminal BO groups and a PbB2 isosceles triangle (Fig. 4). It can be viewed as attaching a BO unit to each of the two B atoms of the GM of PbB2− (1) colinearly, significantly weakening the B–Pb bonds while strengthening the B–B bonds of the PbB2 unit. The second lowest-lying isomer (4C2v, 2B2) is less stable than the GM by ∼0.95 eV at both the PBE0 and CCSD(T) levels (Fig. S3, ESI†). It has a Y-shaped structure (Fig. 4), similar to the recently reported C2v PbB3O2−,76 and can be viewed as two boronyl groups and one Pb–B unit coordinated to a central B atom. Overall, the GM structures of PbB2− (1), PbB3O− (2), and PbB4O2− (3) can be expressed as [PbB2(BO)n]− (n = 0–2), i.e., zero, one, and two boronyl ligands coordinated to a triangular PbB2 core.
5 Discussion
5.1 Comparison between experiment and theory
The predicted VDEs are summarized in Tables 1–3 for the GM structures of PbB2−, PbB3O−, and PbB4O2−, respectively, in comparison with the experimentally measured values, as well as in Fig. 5–7. The computed ADE/VDE1 values for the GM structures of PbB2(BO)n− (n = 0–2) and the VDE1 for the low-lying isomer of PbB4O2− are compared with the experimental results in Table 4. The theoretical VDEs were calculated using the CCSD(T) method for VDE1 and using the SA-CASSCF method for higher VDEs. The valence molecular orbitals (MOs) of the GM structures of PbB2−, PbB3O−, and PbB4O2− are depicted in Fig. S4 (ESI†).
 |
| | Fig. 5 Comparison of the photoelectron spectrum of PbB2− with the computed VDEs. The vertical bars correspond to the computed VDEs using the CCSD(T) + SA-CASSCF method. The shortest bars correspond to singlet final states and the slightly longer bars correspond to triplet final states, and the longest bars indicate degenerate states at the same energy. | |
 |
| | Fig. 6 Comparison of the photoelectron spectrum with the computed VDEs for the GM of PbB3O−. The vertical bars correspond to the computed VDEs using the CCSD(T) + SA-CASSCF method. | |
 |
| | Fig. 7 Comparison of the photoelectron spectrum with the computed VDEs for the C2v GM of PbB4O2−. The vertical bars correspond to the computed VDEs using the CCSD(T) + SA-CASSCF method. The shortest bars correspond to singlet final states and the slightly longer bars correspond to triplet final states, and the longest bars indicate degenerate states at the same energy. Note that the detachment channel corresponding to band E has a computed VDE of 4.70 eV (Table 3), slightly out of the scale of the figure. The PES bands X′ and A′ are due to isomer 4 (see Fig. S5, ESI†). | |
Table 4 Comparison of the experimental ADE and VDE1 with calculated values for the GM of PbB2(BO)n− (n = 0–2) and a low-lying isomer for n = 2 at the PBE0/AVTZ and CCSD(T)/AVTZ levels. All energies are given in eV
| Species |
final state |
ADE (theo.) |
VDE1 (theo.) |
ADE (exp)a |
VDE1 (exp)a |
| PBE0 |
CCSD(T) |
PBE0 |
CCSD(T) |
|
The experimental uncertainty was estimated to be ±0.02 eV.
Measured from the observed weak band X′ in Fig. 3.
|
| PbB2−C2v (1) |
1A1 |
2.26 |
1.91 |
2.29 |
1.94 |
2.03 |
2.08 |
| PbB3O−, Cs (2) |
2A′′ |
3.10 |
3.16 |
3.18 |
3.21 |
3.08 |
3.10 |
| PbB4O2−, C2v (3) |
1A1 |
2.86 |
2.75 |
3.01 |
2.88 |
2.84 |
2.88 |
|
C
2v (4) |
1A1 |
3.27 |
3.16 |
3.29 |
3.16 |
|
3.13b |
5.1.1. PbB2−.
For the C2v GM of PbB2− (1), detachment of the electron from the 4a1 SOMO (Fig. S4a, ESI†) leads to the singlet 1A1 ground state of neutral PbB2O (Table 1). The calculated ADE of 1.91 eV and VDE1 of 1.94 eV at the CCST(T) level agree well with the experimental values at 2.03 eV and 2.08 eV, respectively (Table 4). The next two detachment channels are derived from the removal of the β electron from the 3a1 HOMO-2 and the 1b1 HOMO-1, resulting in the triplet 3A1 and 3B1 final states, respectively (Table 1). The 3A1 state splits into three closely-spaced SO states 3A1(0), 3A1(−1), and 3A1(+1) with predicted VDEs of 2.52, 2.52, and 2.61 eV, while the 3B1 state also gives rise to three SO states 3B1(+1), 3B1(−1), and 3B1(0) with computed VDEs of 2.69, 2.86, and 2.90 eV, respectively (Table 1). These SO states should account for the observed PES bands A (2.48 eV), B (2.65 eV), and C (2.91 eV). The next two singlet final states 1A1 and 1B1 with the calculated VDEs of 3.11 and 3.25 eV are derived by removing the α electron from the HOMO-2 (3a1) and the HOMO-1 (1b1), in good agreement with the observed bands D and E at 3.03 and 3.15 eV, respectively. Detachment of the β electron from the deeper HOMO-3 (1b2) results in the triplet 3B2 final state, which gives rise to three SO-split final states, 3B2(0), 3B2(+1), and 3B2(−1). These final states along with detachment from even deeper MOs should account for the congested PES signals beyond 3.5 eV. Overall, the theoretical VDEs for PbB2− with the SO effects are in good agreement with the experimental data (Fig. 5), confirming triangular structure 1 as the GM of PbB2−.
5.1.2. PbB3O−.
The Cs GM of PbB3O− (2) possesses a closed-shell electronic configuration with a 1A′ ground state and thus only doublet final states are expected upon one-electron detachment. The first detachment channel corresponds to the removal of an electron from the HOMO (2a′′) (Fig. S4b, ESI†), giving rise to a 2A′′ neutral ground state (Table 2). The calculated ADE/VDE1 values are 3.16/3.21 eV at the CCSD(T) level (Table 4), in reasonable agreement with experimental values of 3.08/3.10 eV. Following a small energy gap of 0.27 eV, the second and third VDEs are predicted to be 3.48 eV and 3.96 eV (Table 2), resulting from electron detachment from the HOMO-2 (7a′) and the HOMO-1 (8a′), respectively. These two VDEs, separated by 0.48 eV, are consistent with observed bands A (3.32 eV) and B (3.85 eV). The energy intervals of these three detachment channels can well reproduce the observed energy gaps between bands X and A (0.22 eV) and between bands A and B (0.53 eV), as shown in Table 2 and Fig. 6. The observed vibrational structures with a spacing of ∼480 cm−1 in band B are consistent with the calculated frequency for the terminal Pb–B stretching mode at 533 cm−1. The good agreement between the theoretical and experimental results lends considerable credence to the predicted GM of PbB3O− (2).
5.1.3. PbB4O2−.
The calculated VDE1 for the C2v GM of PbB4O2− (3) at the CCSD(T) level is 2.88 eV due to the electron detachment of the 5b2 SOMO (Fig. S4c, ESI†), in excellent agreement with the observed band X at 2.88 eV. The ground state ADE is predicted as 2.75 eV at the CCSD(T) level, compared to the experimental value of 2.84 eV (Table 4). Detachment of the β electron from the HOMO-1 (7a1) gives rise to the triplet 3B2 final state, which results in three SO states 3B2(0), 3B2(−1), and 3B2(+1) with calculated VDEs of 3.86, 3.86, and 3.91 eV, respectively, in agreement with the observed intense band A at 3.82 eV (Table 3). The removal of one electron from the 2b1 HOMO-2 can produce a triplet 3A2 state and a singlet 1A2 final state. The 3A2 state splits into three SO states 3A2(1), 3A2(−1), and 3A2(0) with calculated VDEs of 4.03, 4.15, and 4.38 eV, respectively. The singlet 1A2 final state yields a calculated VDE of 4.34 eV. The SO states 3A2(1) and 3A2(−1) agree well with the PES bands B and C at 4.01 and 4.15 eV, respectively, whereas the SO state 3A2(0) and the singlet 1A2 state have similar VDEs, in agreement with band D at 4.33 eV. The next detachment channel with a calculated VDE of 4.70 eV corresponds to the 1B2 singlet final state due to the detachment of the α electron from the 7a1 HOMO-1, in good agreement with the observed band E at 4.53 eV.
It should be noted that the calculated VDEs for the C2v GM of PbB4O2− (3) cannot interpret the observed minor bands X′ at 3.13 eV and A′ at 3.40 eV. However, the first two detachment channels of the second isomer, the Y-shaped C2v PbB4O2− (4), with the calculated VDEs of 3.16 and 3.45 eV (Table S1 and Fig. S5, ESI†), respectively, reproduce well the experimental VDEs of bands X′ and A′. Moreover, the next two detachment channels (3.71 and 4.52 eV) of 4 may contribute weakly to bands A and E at 3.82 and 4.53 eV, respectively (Table S1 and Fig. S5, ESI†). The Y-shaped isomer 4 is 0.95 eV higher in energy than the GM structure 3 at the CCSD(T) level (Fig. S3, ESI†). Thus, it was surprising that Y-shaped isomer 4 was present in the experiment at all. There is likely a large kinetic barrier between the two structures, providing dynamic stability for 4 relative to GM 3. Overall, the excellent agreement between the theoretical and experimental data provides considerable credence to the identified GM of PbB4O2− (3) and the existence of the low-lying isomer 4.
5.2 Aromaticity in [PbB2(BO)n]− (n = 0–2)
The GM structures of the three Pb/B clusters can be viewed as starting from a triangular PbB2− unit with successive addition of a boronyl unit in PbB3O− [PbB2(BO)−] and PbB4O2− [PbB2(BO)2−]. These structural features again confirm the previous finding that the boronyl unit, isoelectronic to CN,50 is a strong ligand.51,53,56–68 To understand the intrinsic nature of the high stability of the experimentally observed [PbB2(BO)n]− (n = 0–2) clusters, we analyzed the chemical bonding for the GM structures of PbB2− (1) and PbB3O− (2), as well as that of PbB4O2− (3) and its corresponding neutral PbB4O2 (3′, Fig. 4) using the AdNDP method, as shown in Fig. 8. The AdNDP bonding analyses for the low-lying Y-shaped C2v PbB4O2− (4) is presented in Fig. 9.
 |
| | Fig. 8 AdNDP bonding analyses for the GM of (a) C2v PbB2− (1), (b) Cs PbB3O− (2), (c) C2v PbB4O2− (3), and (d) C2v PbB4O2 (3′) at the PBE0/B,O/def2-TZVP/Pb/def2-TZVPP level. Isovalues for σ orbitals and π orbitals are 0.06 and 0.05, respectively. ON indicates the occupation numbers. | |
 |
| | Fig. 9 The AdNDP bonding analysis for the Y-shaped C2v PbB4O2− (4) at the PBE0/B,O/def2-TZVP/Pb/def2-TZVPP level. Isovalues for σ orbitals and π orbitals are 0.06 and 0.05, respectively. ON indicates the occupation numbers. | |
The AdNDP bonding analysis revealed that the C2v GM of PbB2− (1) possesses one Pb 6s lone pair, one 2c-1e B–B σ bond, two 2c-2e Pb–B σ bonds, one delocalized 3c-2e Pb–B–B σ bond, and one delocalized 3c-2e Pb–B–B π bond (Fig. 8a). Due to the strong relativistic effects,107 the 6s orbital is significantly stabilized and acts like a lone pair with little 6s-6p hybridization, similar to that found in Bi–B clusters.66,108 The Pb–B bonding is basically obtained through the 6p orbitals of Pb and the sp orbitals of B. The PbB2− cluster exhibits σ + π double aromaticity with two σ and two π delocalized electrons fulfilling the 4N + 2 Hückel rule for each system. When boronyl units are successively added to PbB2− to produce PbB3O− [PbB2(BO)−] and PbB4O2− [PbB2(BO)2−], the B–B σ bond in PbB2− is consumed to form new B–BO σ bonds, while the two localized Pb–B σ bonds are transformed to two delocalized Pb–B2 σ bonds. As shown in Fig. 8b, the Cs GM structure of PbB3O− (2) displays one Pb 6s lone pair, one 2c-2e B–BO σ bond, and a terminal BO group (one 1c-2e O lone pair, the BO triple bond: one 2c-2e B–O σ bond, and two 2c-2e B–O π bonds). The bonding between Pb and B2 in 2 consists of three delocalized σ bonds and one delocalized π bond. Thus, the closed-shell Cs PbB3O− (2) is doubly aromatic with six σ and two π delocalized electrons.
The AdNDP analysis for the C2v GM structure of PbB4O2− (3) readily identified the Pb 6s lone pair, two 2c-2e B–BO σ bonds, and two terminal BO triple bonds (Fig. 8c). The remaining valence electrons in the open-shell 3 consist of one 3c-1e σ bond, two 3c-2e σ bonds, and one 3c-2e delocalized π bond over the triangular PbB2 core, giving rise to five delocalized σ electrons and two delocalized π electrons. When the unpaired electron of 3 is detached to form the closed-shell neutral C2v PbB4O2 (3′), the singly occupied 3c-1e σ bond is removed, and one of the delocalized 3c-2e σ bonds is transformed to the localized 2c-2e B–B bond. The closed-shell 3′, thus, possesses one delocalized 3c-2e σ bond and one delocalized 3c-2e π bond on its PbB2 unit (Fig. 8d), making it σ + π doubly aromatic with two σ and two π delocalized electrons.
To quantitatively assess the double aromatic character of C2v PbB2− (1), Cs PbB3O− (2), and C2v PbB4O2−/0 (3 and 3′), NICSzz(ζ) values of these clusters were calculated and compared with those of B3− clusters shown in Table 5. NICS values are usually calculated using the ring critical points,109,110 but the PbB2 triangle does not have one. The centres of the ring currents (Fig. S6 and Table S2, ESI†) are used instead of the ring critical points. For these Pb/B species, all examined points located within or above the molecular planes possess large negative NICSzz values, and the NICSzz values decrease significantly as the ζ values increase from 0 to 1 Å (Table 5). These phenomena appear to be similar to the computed NICSzz values for the prototypical σ + π doubly aromatic B3−. We conclude that C2v PbB2− (1), Cs PbB3O− (2), and C2v PbB4O2−/0 (3 and 3′) are σ + π doubly aromatic in nature, in agreement with the AdNDP results discussed above.
Table 5 NICSzz(ζ) values (ppm) of C2v PbB2− (1), Cs PbB3O− (2), C2v PbB4O2−/0 (3 and 3′), and D3h B3− computed at the centres of the ring current using the ADF code with the PBE0/TZP all-electron basis, including the noncollinear spin–orbit coupling effect
|
|
PbB2− |
PbB3O− |
PbB4O2− |
PbB4O2 |
B3− |
|
ζ = 0.0 |
−183.9 |
−136.4 |
−193.5 |
−209.1 |
−76.5 |
|
ζ = 0.2 |
−173.4 |
−128.7 |
−185.3 |
−188.7 |
−74.2 |
|
ζ = 0.4 |
−149.8 |
−111.5 |
−165.3 |
−144.3 |
−67.7 |
|
ζ = 0.6 |
−126.0 |
−94.9 |
−142.0 |
−103.2 |
−58.6 |
|
ζ = 0.8 |
−106.8 |
−82.1 |
−119.8 |
−75.4 |
−49.0 |
|
ζ = 1.0 |
−90.4 |
−71.2 |
−99.4 |
−56.9 |
−40.0 |
5.3 Multiple B–B bonding in [PbB2(BO)n]− (n = 0–2)
According to the chemical bonding analyses shown in Fig. 8, [PbB2(BO)n]− (n = 0–2) exhibit obvious multiple B–B bonding characteristics. With two 2c-2e Pb–B σ bonds, one 2c-1e B–B σ bond, and two delocalized 3c-2e σ/π bonds (Fig. 8a), C2v PbB2− (1) possesses two Pb–B single bonds and one B–B double bond with the NRT bond orders of 1.04 and 1.68 (Table S3, ESI†), respectively. The Pb–B bond length and B–B bond length in 1 are 2.283 Å and 1.553 Å (Fig. 4), respectively, in good accord with the Pb–B single-bond length (2.29 Å) and BB double-bond length (1.56 Å) predicted from Pyykkö's additive atomic covalent radii,111 respectively. Compared with the C2v PbB2− (1) which possesses three localized σ bonds (Fig. 8a), Cs [PbB2(BO)]− (2) and C2v [PbB2(BO)2]− (3) both have four delocalized σ/π bonds covered on their PbB2 cores (Fig. 8b and c), causing the Pb–B bonding to be weakened, while the B–B bonds to be strengthened. The less symmetric 2 contains one weakened Pb–B bond with a NRT bond order of 0.63, one strengthened terminal Pb–B bond with a bond order of 1.55, and one strengthened B–B bond with a bond order of 1.78 (Table S3, ESI†). The terminal Pb–B bond length of 2.147 Å in 2 (Fig. 4) is consistent with the PbB double-bond length of 2.13 Å, based on Pyykkö's atomic covalent radii.111 The central B–B bond length of 1.515 Å lies between the BB double-bond length (1.56 Å) and the BB triple-bond length (1.46 Å) predicted by Pyykkö's atomic covalent radii.111
The two delocalized 3c-2e σ bonds and one delocalized 3c-2e π bond in the open-shell C2v PbB4O2− (3) are mainly concentrated on the B–B moiety of the PbB2 core (Fig. 8c). The central B–B bond with a NRT bond order of 1.76 is shortened to 1.498 Å (Fig. 4) which is close to the B≡B triple bond (1.46 Å) based on Pyykkö's atomic covalent radii. It is worth noting that the delocalized 3c-1e σ bond in 3 exhibits clearly partial anti-bonding nature, mainly derived from a Pb 6p orbital (Fig. 8c). With the unpaired electron detached, the length of the central B–B bond with the NRT bond order of 1.86 in neutral C2v PbB4O2− (3′) is further shortened to 1.478 Å which can be approximately treated as a B≡B triple bond. In fact, the B≡B triple bond length in 3′ is slightly shorter than the corresponding value of 1.481 Å previously reported in the perfect linear D∞h B2(BO)22− and slightly longer than that (1.468 Å) observed in D∞h B2(CO)2.53,112 Natural bonding orbital (NBO) analyses indicated that there exist substantial charge transfers from the Pb atoms to the B4O2 units in C2v PbB4O2−/0 (3 and 3′) (0.45 |e| for 3, 1.09 |e| for 3′). Thus, these two charge-transfer complexes can be approximately viewed as Pb+/Pb2+ units weakly interacting with the linear B2(BO)22− motifs, i.e., Pb+[B2(BO)22−]/Pb2+[B2(BO)22−], with Pb–B bond lengths (2.383 Å for 3, 2.399 Å for 3′; Fig. 4) obviously longer than the Pb–B single bond (2.29 Å).111
5.4 Multiple Pb–B and B–B bonding in Y-shaped PbB4O2−/2−
The structure of the low-lying Y-shaped isomer C2v PbB4O2− (4) is interesting. Our AdNDP analysis for 4 (Fig. 9) showed the Pb 6s lone pair, two O lone pair lone pairs, two BO triple bonds, and two B–BO single bonds. The remaining valence electrons form one 2c-2e B–B σ bond, one 3c-2e B–B–Pb π bond, one 2c-2e Pb–B σ bond, and one 2c-1e Pb–B π bond, resulting in the BB bond and the Pb–B bond along the two-fold main molecular axis which have the formal bond orders of 2.0 and 1.5, respectively. The BB double bond length of 1.565 Å in 4 (Fig. 4) is comparable to the corresponding value of 1.56 Å obtained from Pyykkö's double-bond covalent atomic radii.111 The Pb–B length of 2.195 Å lies between the Pb–B single bond length (2.29 Å) and the PbB double-bond length (2.13 Å) computed from Pyykkö's covalent atomic radii, consistent with its formal bond order of 1.5. Adding an electron to 4 yields the closed-shell C2v PbB4O22− (4′, Fig. 4) with an even shortened Pb–B bond length of 2.188 Å, closer to a PbB double bond (2.13 Å).111 Thus, the Y-shaped C2v PbB4O22− (4′) can be formulated as [PbBB(BO)2]2−. NRT analyses predicted a B–B bond order of 1.59 for 4 and 1.69 for 4′, which are slightly lower than 2.0 due to the existence of the delocalized B–B–Pb 3c-2e π bond. The calculated NRT bond orders for Pb–B in 4 and 4′ are 1.72 and 2.08, respectively, consistent with the 1.5 bond order for the Pb–B multiple bond in 4 and the PbB double bond in 4′, respectively. The central B atom in 4′ undergoes sp2 hybridization and forms two single B–B bonds with the two boronyl ligands and a double bond with the B atom bonded to Pb. The B atom that is bonded to Pb undergoes sp hybridization, forming a PbB double bond and a BB double bond, similar to the central C atom in the allene molecule (H2CCCH2). In other words, the closed-shell C2v PbB4O22− (4′) is an electron-precise molecule with distinct hybridizations for the two central B atom and it is isovalent with PbCCH2 and should be a highly stable species. However, the open-shell C2v PbB4O2− (4) misses one electron, which may explain why it is less stable than the C2v GM of PbB4O2− (3). Nevertheless, it is conceivable that a large energy barrier may exist from isomer 4 to GM 3, allowing 4 to be present in the experiment despite its relatively high energy.
6. Conclusion
In conclusion, we report an investigation on the structures and chemical bonding of three lead-boron clusters, PbB2−, PbB3O−, and PbB4O2−, using photoelectron spectroscopy and ab initio calculations. The global minima of these three species are found to be planar and can be formulated as [PbB2(BO)n]− (n = 0–2), consisting of a triangular PbB2 core coordinated by zero, one, and two boronyl ligands, respectively. In addition, a Y-shaped low-lying isomer with a PbBB unit coordinated by two BO ligands to the terminal B atom was also observed to be present in the cluster beam of PbB4O2−. Chemical bonding analyses indicate that the global minima of PbB2− and PbB3O− have 2σ/2π and 6σ/2π delocalized electrons, respectively, rendering them doubly aromatic. The global minimum of the C2v PbB4O2− contains 5σ/2π delocalized electrons and is also shown to display double aromaticity. The Pb and B2 bonding is weak in the global minimum of PbB4O2− and its corresponding neutral species of PbB4O2, which can be alternatively viewed as Pb+/2+ units coordinated to the linear B4O22− ligand with the B≡B triple bond. The open-shell Y-shaped C2v PbB4O2− isomer contains a Pb–B multiple bond with a bond order of 1.5, while the closed-shell Y-shaped PbB4O22− possesses a PbB double bond with the bond order of 2.0, which is an electron-precise molecule with distinct hybridization for the two B atoms, [PbBB(BO)2]2−. Investigation of the lead-boron cluster enriches the understanding of Pb–B bonding and provides new insights for the rational design of novel Pb–B molecules and potential lead boride materials.
Author contributions
W. J. C. and T. T. C. performed the experiments. Q. C., X. Y. W., R. N. Y., and H. G. L. performed the computational calculations. S. D. L. and L. S. W. guided the work. Q. C., W. J. C., D. F. Y., S. D. L., and L. S. W. wrote and revised the manuscript. All authors analyzed the experimental and theoretical results.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We thank Dr H. S. Hu for help with the NICSzz calculations. This work was supported by the US National Science Foundation (CHE-2053541 to L.S.W.) and the National Natural Science Foundation of China (22003034 to Q. C., 21973057 and 21720102006 to S.D.L.). Q.C. gratefully acknowledges the start-up fund from Shanxi University for the support.
References
- W. N. Lipscomb, Science, 1977, 196, 1047 CrossRef CAS PubMed.
- E. D. Jemmis and D. L. V. K. Prasad, J. Solid State Chem., 2006, 179, 2768 CrossRef CAS.
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640 CrossRef CAS PubMed.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811 CrossRef CAS.
- E. Oger, N. R. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503 CrossRef CAS PubMed.
- A. P. Sergeeva, I. A. Popov, Z. A. Piazza, W. L. Li, C. Romanescu, L. S. Wang and A. I. Boldyrev, Acc. Chem. Res., 2014, 47, 1349 CrossRef CAS PubMed.
- L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69 Search PubMed.
- W. L. Li, X. Chen, T. Jian, T. T. Chen, J. Li and L. S. Wang, Nat. Rev. Chem., 2017, 1, 0071 Search PubMed.
- M. R. Fagiani, X. Song, P. Petkov, S. Debnath, S. Gewinner, W. Schöllkopf, T. Heine, A. Fielicke and K. R. Asmis, Angew. Chem., Int. Ed., 2017, 56, 501 CrossRef CAS PubMed.
- T. Jian, X. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550 RSC.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727 CrossRef CAS PubMed.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754 CrossRef CAS PubMed.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed.
- H. R. Li, T. Jian, W. L. Li, C. Q. Miao, Y. J. Wang, Q. Chen, X. M. Luo, K. Wang, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 29147 RSC.
- W. J. Chen, Y. Y. Ma, T. T. Chen, M. Z. Ao, D. F. Yuan, Q. Chen, X. X. Tian, Y. W. Mu, S. D. Li and L. S. Wang, Nanoscale, 2021, 13, 3868 RSC.
- T. T. Chen, L. F. Cheung and L. S. Wang, Annu. Rev. Phys. Chem., 2022, 73, 233 CrossRef CAS PubMed.
- J. Barroso, S. Pan and G. Merino, Chem. Soc. Rev., 2022, 51, 1098 RSC.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem.
Res., 2013, 46, 350 CrossRef CAS PubMed.
- W. L. Li, A. S. Ivanov, J. Federic, C. Romanescu, I. Cernusak, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2013, 139, 104312 CrossRef PubMed.
- L. F. Cheung, G. S. Kocheril, J. Czekner and L. S. Wang, J. Phys. Chem. A, 2020, 124, 2820 CrossRef CAS PubMed.
- L. F. Cheung, G. S. Kocheril, J. Czekner and L. S. Wang, J. Am. Chem. Soc., 2020, 142, 3356 CrossRef CAS PubMed.
- L. F. Cheung, J. Czekner, G. S. Kocheril and L. S. Wang, J. Am. Chem. Soc., 2019, 141, 17854 CrossRef CAS PubMed.
- Q. Chen, H. J. Zhai, S. D. Li and L. S. Wang, J. Chem. Phys., 2013, 138, 084306 CrossRef PubMed.
- W. J. Chen, M. Kulichenko, H. W. Choi, J. Cavanagh, D. F. Yuan, A. I. Boldyrev and L. S. Wang, J. Phys. Chem. A, 2021, 125, 6751 CrossRef CAS PubMed.
- T. T. Chen, W. L. Li, H. Bai, W. J. Chen, X. R. Dong, J. Li and L. S. Wang, J. Phys. Chem. A, 2019, 123, 5317 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, A. P. Sergeeva, W. L. Li, L. S. Wang and A. I. Boldyrev, J. Organomet. Chem., 2012, 721, 148 CrossRef.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101 CrossRef CAS PubMed.
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334 CrossRef CAS PubMed.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, J. Chem. Phys., 2011, 135, 104301 CrossRef PubMed.
- I. A. Popov, W. L. Li, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Phys. Chem. A, 2014, 118, 8098 CrossRef CAS PubMed.
- B. Le Chen, W. G. Sun, X. Y. Kuang, C. Lu, X. X. Xia, H. X. Shi and G. Maroulis, Inorg. Chem., 2018, 57, 343 CrossRef PubMed.
- M. Ren, S. Jin, D. Wei, Y. Y. Jin, Y. Tian, C. Lu and G. L. Gutsev, Phys. Chem. Chem. Phys., 2019, 21, 21746 RSC.
- W. L. Li, T. T. Chen, W. J. Chen, J. Li and L. S. Wang, Nat. Commun., 2021, 12, 6467 CrossRef CAS PubMed.
- T. T. Chen, W. L. Li, T. Jian, X. Chen, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2017, 56, 6916 CrossRef CAS PubMed.
- W. L. Li, T. Jian, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2016, 55, 7358 CrossRef CAS PubMed.
- T. Jian, W. L. Li, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Chem. Sci., 2016, 7, 7020 RSC.
- W. L. Li, T. T. Chen, Z. Y. Jiang, W. J. Chen, H. S. Hu, L. S. Wang and J. Li, Chin. J. Chem. Phys., 2019, 32, 241 CrossRef CAS.
- W. L. Li, T. Jian, X. Chen, H. R. Li, T. T. Chen, X. M. Luo, S. D. Li, J. Li and L. S. Wang, Chem. Commun., 2017, 53, 1587 RSC.
- T. Jian, W. L. Li, I. A. Popov, G. V. Lopez, X. Chen, A. I. Boldyrev, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 154310 CrossRef PubMed.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L. S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS PubMed.
- C. Xu, L. Cheng and J. Yang, J. Chem. Phys., 2014, 141, 124301 CrossRef PubMed.
- T. T. Chen, W. L. Li, W. J. Chen, X. H. Yu, X. R. Dong, J. Li and L. S. Wang, Nat. Commun., 2020, 11, 2766 CrossRef CAS PubMed.
- W. J. Chen, Y. Y. Zhang, W. L. Li, H. W. Choi, J. Li and L. S. Wang, Chem. Commun., 2022, 58, 3134 RSC.
- M. Kulichenko, W. J. Chen, H. W. Choi, D. F. Yuan, A. I. Boldyrev and L. S. Wang, J. Vac. Sci. Technol., A, 2022, 40, 042201 CrossRef CAS.
- A. I. Boldyrev and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589 RSC.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004 CrossRef CAS PubMed.
- A. P. Sergeeva, D. Y. Zubarev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 7244 CrossRef CAS PubMed.
- D. Y. Zubarev and A. I. Boldyrev, J. Comput. Chem., 2007, 28, 251 CrossRef CAS PubMed.
- H. J. Zhai, L. M. Wang, S. D. Li and L. S. Wang, J. Phys. Chem. A, 2007, 111, 1030 CrossRef CAS PubMed.
- H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345 CrossRef CAS PubMed.
- M. Zhou, N. Tsumori, Z. Li, K. Fan, L. Andrews and Q. Xu, J. Am. Chem. Soc., 2002, 124, 12936 CrossRef CAS PubMed.
- S. D. Li, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 2573 CrossRef CAS PubMed.
- L. C. Ducati, N. Takagi and G. Frenking, J. Phys. Chem. A, 2009, 113, 11693 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed.
- H. J. Zhai, S. D. Li and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 9254 CrossRef CAS PubMed.
- W. Z. Yao, J. C. Guo, H. G. Lu and S. D. Li, J. Phys. Chem. A, 2009, 113, 2561 CrossRef CAS PubMed.
- D. Y. Zubarev, A. I. Boldyrev, J. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2007, 111, 1648 CrossRef CAS PubMed.
- H. J. Zhai, J. C. Guo, S. D. Li and L. S. Wang, Chem. Phys. Chem., 2011, 12, 2549 CrossRef CAS PubMed.
- Q. Chen, H. Bai, H. J. Zhai, S. D. Li and L. S. Wang, J. Chem. Phys., 2013, 139, 044308 CrossRef PubMed.
- Q. Chen, H. J. Zhai, S.-D. Li and L. S. Wang, J. Chem. Phys., 2012, 137, 044307 CrossRef PubMed.
- H. Bai, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2013, 15, 9646 RSC.
- H. J. Zhai, C. Q. Miao, S. D. Li and L. S. Wang, J. Phys. Chem. A, 2010, 114, 12155 CrossRef CAS PubMed.
- H. J. Zhai, Q. Chen, H. Bai, H. G. Lu, W. L. Li, S. D. Li and L. S. Wang, J. Chem. Phys., 2013, 139, 174301 CrossRef PubMed.
- T. T. Chen, L. F. Cheung, W. J. Chen, J. Cavanagh and L. S. Wang, Angew. Chem., Int. Ed., 2020, 59, 15260 CrossRef CAS PubMed.
- T. Jian, L. F. Cheung, T. T. Chen and L. S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9551 CrossRef CAS PubMed.
- L. F. Cheung, T. T. Chen, G. S. Kocheril, W. J. Chen, J. Czekner and L. S. Wang, J. Phys. Chem. Lett., 2020, 11, 659 CrossRef CAS PubMed.
- W. J. Tian, W. J. Chen, M. Yan, R. Li, Z. H. Wei, T. T. Chen, Q. Chen, H. J. Zhai, S. D. Li and L. S. Wang, Chem. Sci., 2021, 12, 8157 RSC.
- H. Liu, S. Wang, G. Zhou, J. Wu and W. Duan, J. Chem. Phys., 2007, 126, 134705 CrossRef PubMed.
- A. Kumar, Radiat. Phys. Chem., 2017, 136, 50 CrossRef CAS.
- D. K. Gaikwad, S. S. Obaid, M. I. Sayyed, R. R. Bhosale, V. V. Awasarmol, A. Kumar, M. D. Shirsat and P. P. Pawar, Mater. Chem. Phys., 2018, 213, 508 CrossRef CAS.
- M. I. Sayyed, Y. S. Rammah, A. S. Abouhaswa, H. O. Tekin and B. O. Elbashir, Phys. B, 2018, 548, 20 CrossRef CAS.
- S. Woosley, N. Abuali Galehdari, A. Kelkar and S. Aravamudhan, J. Mater. Res., 2018, 33, 3657 CrossRef CAS.
- X. Li, J. Wu, C. Tang, Z. He, P. Yuan, Y. Sun, W.-M. Lau, K. Zhang, J. Mei and Y. Huang, Composites, Part B, 2019, 159, 355 CrossRef CAS.
- Y. Wu, Q. P. Zhang, D. Zhou, Y. L. Zhou and J. Zheng, J. Alloys Compd., 2017, 727, 1027 CrossRef CAS.
- W. J. Chen, T. T. Chen, Q. Chen, H. G. Lu, X. Y. Zhao, Y. Y. Ma, Q. Q. Yan, R. N. Yuan, S. D. Li and L. S. Wang, Commun. Chem., 2022, 5, 25 CrossRef CAS PubMed.
- L. S. Wang, H. S. Cheng and J. Fan, J. Chem. Phys., 1995, 102, 9480 CrossRef CAS.
- A. P. Sergeeva, B. B. Averkiev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2011, 134, 224304 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- J. Čížek, Adv. Chem. Phys., 1969, 14, 35 CrossRef.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef CAS.
- P. Celani and H. J. Werner, J. Chem. Phys., 2000, 112, 5546 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803 CrossRef CAS.
- A. Berning, M. Schweizer, H. J. Werner, P. J. Knowles and P. Palmieri, Mol. Phys., 2000, 98, 1823 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- P. v R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. V. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS PubMed.
- C. Foroutan-Nejad, J. Vicha and A. Ghosh, Phys. Chem. Chem. Phys., 2020, 22, 10863 RSC.
- L. Alvarado-Soto, R. Ramírez-Tagle and R. Arratia-Pérez, Chem. Phys. Lett., 2008, 467, 94 CrossRef CAS.
- E. Schott, X. Zárate and R. Arratia-Pérez, Polyhedron, 2011, 30, 846 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks and H. B. Schlegelet al., GAUSSIAN 09, Revision A.2, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhutet al., MOLPRO, version 2012.1.
- ADF and SCM, Theoretical Chemistry, Vrijie Universiteit, Amsterdam, The Netherlands, 2014, https://www.scm.com.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500 RSC.
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef.
- W. J. Chen, M. Kulichenko, H. W. Choi, J. Cavanagh, D. F. Yuan, A. I. Boldyrev and L. S. Wang, J. Phys. Chem. A, 2021, 125, 6751 CrossRef CAS PubMed.
- C. Foroutan-Nejad, S. Shahbazian and P. Rashidi-Ranjbar, Phys. Chem. Chem. Phys., 2010, 12, 12630 RSC.
- C. Foroutan-Nejad, Z. Badri, S. Shahbazian and P. Rashidi-Ranjbar, J. Phys. Chem. A, 2011, 115, 12708 CrossRef CAS PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326 CrossRef PubMed.
- M. Zhou, N. Tsumori, Z. Li, K. Fan, L. Andrews and Q. Xu, J. Am. Chem. Soc., 2002, 124, 12936 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02800c |
| ‡ Q. Chen, W.-J. Chen and X-Y. Wu contributed equally to this work. |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ac,
Wei-Jia
Chen‡
ac,
Wei-Jia
Chen‡
 Pb multiple bond with a bond order of 2.5 in the linear PbB2O−, and a B
Pb multiple bond with a bond order of 2.5 in the linear PbB2O−, and a B










