
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobustness and stability of the L-carnosine copper(II) dimer: changes in the copper coordination sphere upon heating/solvent substitution†
Draginja
Mrvoš-Sermek
 a,
Marina
Tašner
a,
Darko
Vušak
a,
Nenad
Judaš
a,
Kinga
Wzgarda-Raj
*ab,
Ivica
Đilović
*a and
Dubravka
Matković-Čalogović
a
a,
Marina
Tašner
a,
Darko
Vušak
a,
Nenad
Judaš
a,
Kinga
Wzgarda-Raj
*ab,
Ivica
Đilović
*a and
Dubravka
Matković-Čalogović
a
aDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, Zagreb, Croatia. E-mail: idilovic@chem.pmf.hr
bDepartment of Physical Chemistry, Faculty of Chemistry, University of Lodz, Pomorska 163/165, Lodz, Poland
First published on 26th November 2024
Abstract
Coordination compounds of L-carnosine (H2car) have been the subject of much research, as they have potential therapeutic applications due to their antioxidant and anti-inflammatory properties. This paper analyzes the coordination possibilities of L-carnosine in reactions with copper(II) under different conditions. Three solvates (1–3) of copper(II) coordination compounds with car ligands were prepared, their molecular and crystal structures determined, and their interconversion conditions described. The Cu(II) ion coordination polyhedron is square-pyramidal, with water/methanol molecules in apical positions [Cu2(car)2(solv)2] (solv = water or methanol). All three compounds consist of discrete dimers in which two car ligands coordinate two Cu(II) ions. The methanol solvates transform to the most stable compound, [Cu2(car)2(H2O)2]·2H2O, upon exposure to moist air. Upon heating, this compound loses both coordinated and solvent water molecules, and the copper coordination changes to square-planar, yet the dimer is stable up to 200 °C. When cooled in moist air, it readily converts to the original compound [Cu2(car)2(H2O)2]·2H2O. An exhaustive temperature-dependent structural and spectroscopic solid-state analysis indicates the robustness and reactivity of the activated dimer without coordinated molecules (coordinatively unsaturated sites, CUS). Preliminary results indicate that the generation of CUS requires heating after which the pyridine-based linkers can be introduced by prolonged milling using polar aprotic dimethylformamide as a liquid. These findings present potential routes for utilizing the activated dimer in creating novel compounds.
Introduction
Carnosine (β-alanyl-L-histidine) participates in numerous physiological processes involving the coordination of metal ions.1–6 Undoubtedly, the molecules of this dipeptide have high potential to achieve a large number of bonds because they possess a versatile set of interacting groups (imidazole moiety, amide, amino, and carboxyl groups) that can be used in numerous ways – from the simple discrete binding of metal ions, more complex coordination compounds, and supramolecular systems to MOFs (metal–organic frameworks). Carnosine readily forms coordination compounds with copper, zinc, cobalt, vanadium, nickel, and manganese ions,7–12 as confirmed by different experimental techniques that are dominantly related to solutions.2 A branch of studies has led to numerous examples where carnosine compounds are used as active substances (for example, a chelate complex with Zn(II), polaprezinc is a commercially available drug). Along the line of structural characteristics, much attention has been devoted to studying the binding preferences of carnosine toward metal ions, especially with Cu(II) ions. In 1955, Dobbie and Kermack proposed the first model of a monomer (Scheme 1, I), which other authors further updated.13 A wealth of provided data corroborated the existence of an equilibrium between monomeric (Scheme 1, II and III) and dimeric species (Scheme 1, IV) that is affected by the pH value and ionic strength of the medium (higher pH values favor dimers).14 Later, Schröder et al. showed that dimers in vivo are improbable due to low Cu(II) concentration.15 In 1965, Freeman and Szymanski published the structure of the dimeric complex in which two deprotonated carnosine molecules bridge two Cu(II) ions (by N atoms in imidazole rings, Scheme 1, IV). Their dimer model had four water molecules, two coordinating the Cu(II) ions and two being crystallization molecules.16 They collected X-ray data using the Weissenberg method and measured data photometrically, resulting in 1319 reflections. Although they recognized a disordered dimeric structure, the lack of high-angle diffraction data prevented modelling of the disorder and placement of H atoms, which resulted in a less desirable R-factor of 0.10.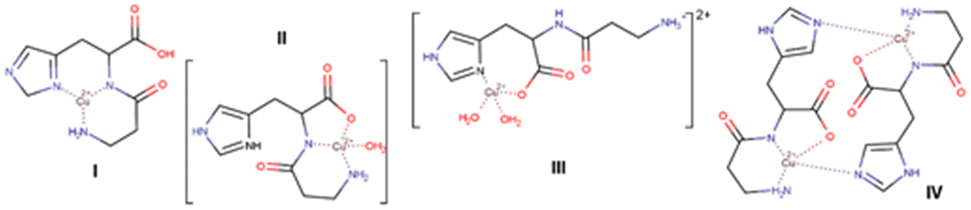 | ||
| Scheme 1 Proposed molecular structures of copper(II) carnosine coordination compounds. | ||
We are interested in studying the interactions of amino acids and their derivatives with essential metal ions, especially copper(II). In recent years, we have described numerous systems with exciting properties (biological and magnetic) and applicable concepts (supramolecular control of the synthesis outcome or separation in the solid state).17–24 Here, we present the synthesis and thorough solid-state characterization of three dimeric Cu(II) coordination compounds with carnosine. We determined the structures, by single-crystal X-ray diffraction, of the three compounds: [Cu2(car)2(H2O)2]·2H2O (1) (at 170, 270, and 370 K), [Cu2(car)2(MeOH)2x(H2O)2(1−x)]·mH2O·nMeOH (2) (solvent could not be defined), and [Cu2(car)2(MeOH)2]·5.5 MeOH (3) (car = doubly deprotonated L-carnosine; MeOH = methanol). Interestingly, the occurrence of distinct products depends highly on the presence of water, and the investigated coordination compounds can mutually interconvert, some even in the solid state. By heating compound 1, water loss may lead to an activated precursor usable for synthesizing more complex systems of these biocompatible materials. An excellent example of a similar approach is the Zn(II)–carnosine porous material used for guest adsorption.25 The activation of common materials such as MOFs or supramolecular porous coordination compounds, which involves the generation of CUS, is usually too long and can lead to the breakdown of the system. Recently, systems that can be activated under milder conditions and further used in catalysis and similar chemical processes have been sought.26
Results and discussion
Liquid-assisted mechanochemical synthesis resulted (LAG, Scheme 2) in the same dimeric coordination compounds as the one obtained by the solution-based synthesis [Cu2(car)2(H2O)2]·2H2O (1).27 Copper(II) hydroxide was ground with L-carnosine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (a small amount of water was added to accelerate the reaction, Scheme 2). The second compound, [Cu2(car)2(MeOH)2x(H2O)2(1−x)]·mH2O·nMeOH (2), was synthesized in methanol by using copper(II) acetate. Methanol solvate [Cu2(car)2(MeOH)2]·5.5MeOH (3) was prepared exclusively by using dry methanol in the presence of sodium methoxide. In order to successfully obtain compound 3, it is essential to maintain a moisture-free environment. When compound 1 was recrystallized from methanol, compound 2 was obtained. Transformation 2 → 1 is reversible because when compound 2 is left in moist air, 1 is obtained.
:1 ratio (a small amount of water was added to accelerate the reaction, Scheme 2). The second compound, [Cu2(car)2(MeOH)2x(H2O)2(1−x)]·mH2O·nMeOH (2), was synthesized in methanol by using copper(II) acetate. Methanol solvate [Cu2(car)2(MeOH)2]·5.5MeOH (3) was prepared exclusively by using dry methanol in the presence of sodium methoxide. In order to successfully obtain compound 3, it is essential to maintain a moisture-free environment. When compound 1 was recrystallized from methanol, compound 2 was obtained. Transformation 2 → 1 is reversible because when compound 2 is left in moist air, 1 is obtained.
 | ||
| Scheme 2 Reaction pathways to compounds 1, 2, and 3. Decomposition of 2 and 3 and their transformation to 1 proceed upon exposure to moist air. Compound 1 can be transformed to 2 by recrystallization from methanol. | ||
In our study of solvated compounds of copper(II) with serine and phenanthroline, we found a solid-state-to-solid-state transformation of five different solvates (H2O/MeOH) into one stable compound with a water molecule coordinated to the copper ion and water molecules of crystallization, similar to that found in this case.17
All three compounds, 1–3, consist of discrete dimers in which two car ligands coordinate two Cu(II) ions. Solvent molecules are present in voids and channels of the crystal structures. The Cu(II) coordination polyhedron is a distorted square pyramid at low and room temperature. In the equatorial plane, the Cu(II) ion is coordinated by a terminal β-alanyl amino N atom, a peptide N atom, a carboxyl O atom of one car, and an N atom from the L-histidine imidazole ring of the other car molecule (Table 1). A water molecule occupies the apical position in 1 [at 170 (1170) and 270 K (1270)], a disordered water/methanol molecule in 2 [at 100 K (2100)], a water molecule in 2 [at 170 K (2170)], and a methanol molecule in 3 (data collected only at 150 K). The shortest bond lengths in the low (and RT) temperature structures are those of Cu–N2, while the longest equatorial bonds are Cu–N1. Due to the Jahn–Teller effect, the longest bonds are to the apical O atoms. For 5-coordinated compounds, the τ5 parameter can provide insight into the geometry of the coordination sphere and deviation from an ideal square pyramidal or trigonal bipyramidal geometry, with tau values of 0 and 1, respectively.271170 and 1270 display τ5 parameters of 0.29 and 0.28, respectively, indicating a moderate deviation from the ideal square pyramidal geometry. The inclusion of methanol in the solvent environment is postulated to exert an influence on the geometry of the complex, resulting in minimised distortions. 2100 and 2170 both exhibit τ5 parameters of 0.14, signifying a lesser deviation from the ideal geometry, similar to 3 with a τ5 parameter of 0.19. In the high-temperature structure 1370, there is no apical ligand, the coordination is distorted square-planar, and all distances to the Cu(II) ions are shortened. Devoid of apical solvent this is a new compound – its properties will be discussed later.
| Cu | 1 170 | 1 270 | 1 370 | 2 100 | 2 170 |
|---|---|---|---|---|---|
| 1aSymmetry operators: 1a−x, −x + y, 4/3 − z; 1by, x, 1 − z; 1c1 − x, +y, 1 − z; 1d1 − x, 1 − y, z. | |||||
| N1A/N1 | 2.019(5) | 2.017(6) | 1.960(15) | 1.993(2) | 1.990(3) |
| N1B | 2.009(7) | 2.012(6) | 1.960(16) | — | — |
| N2 | 1.953(3) | 1.948(2) | 1.912(6) | 1.939(2) | 1.944(2) |
| O1 | 1.982(3) | 1.978(3) | 1.899(12) | 1.985(2) | 1.983(2) |
| N31 | 1.998(3)1a | 1.998(2)1a | 1.991(7)1b | 1.966(2)1c | 1.972(2)1d |
| OW1/O4 | 2.405(5) | 2.453(6) | — | 2.518(2) | 2.545(2) |
General and crystal data, a summary of intensity data collection, and structure refinement for the compounds are given in Table 2. A list of bond lengths, angles, and hydrogen bonds for the multiple temperature structures of 1, 2, and 3 are in ESI Tables S1–S27.†
| Identification code | 1 170 | 2 100 |
|---|---|---|
| Formula | [Cu2(car)2(H2O)2]·2H2O | [Cu2(car)2(H2O)0.70(MeOH)0.30]·mH2O·nMeOH |
| Empirical formula | C18H32Cu2N8O10 | — |
| Formula weight | 647.60 | — |
| Temperature/K | 170(1) | 100(1) |
| Crystal system | Trigonal | Orthorhombic |
| Space group | P3121 | I222 |
| a/Å | 8.56884(6) | 12.08960(8) |
| b/Å | 8.56884(6) | 20.35323(15) |
| c/Å | 30.61946(18) | 13.73806(9) |
| α/° | 90 | 90.0 |
| β/° | 90 | 90.0 |
| γ/° | 120 | 90.0 |
| Volume/Å3 | 1947.03(3) | 3380.42(4) |
| Z | 3 | 4 |
| ρ calc/g cm−3 | 1.657 | — |
| μ/mm−1 | 2.620 | — |
| F(000) | 1002.0 | — |
| Crystal size/mm3 | 0.11 × 0.11 × 0.03 | 0.06 × 0.05 × 0.04 |
| Radiation/Å | CuKα (λ = 1.54184) | Synchrotron, (λ = 0.700) |
| 2θ range for data collection/° | 8.66 to 150.15 | 3.86 to 60 |
| Index ranges | −10 ≤ h ≤ 10, −10 ≤ k ≤ 9, −38 ≤ l ≤ 38 | −17 ≤ h ≤ 17, −27 ≤ k ≤ 27, −19 ≤ l ≤ 19 |
| Reflections collected | 48652 |
29910 |
| Independent reflections | 2655 [Rint = 0.0346] | 4990 [Rint = 0.0370] |
| Data/restraints/param. | 2655/38/224 | 4990/10/185 |
| Goodness-of-fit on F2 | 1.055 | 1.047 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0318, wR2 = 0.0746 | R 1 = 0.0296, wR2 = 0.0805 |
| Final R indexes [all data] | R 1 = 0.0336, wR2 = 0.0757 | R 1 = 0.0296, wR2 = 0.0805 |
| Max. diff. peak/hole/e Å−3 | 0.42/−0.44 | 0.43/−0.37 |
| Flack parameter | −0.019(6) | 0.024(5) |
Compound 1, [Cu2(car)2(H2O)2]·2H2O, (Fig. 1) crystallizes in the trigonal P3121 space group. A two-fold axis relates the two dimeric parts, so the asymmetric unit comprises half of the dimer. The water molecule is in the general position. The structure is disordered in the N1–C1–C2–C3–O3 part of the car ligand. It was modeled in two positions, with the major part having an occupancy of 0.601(11) in 1170.
 | ||
| Fig. 1 Structure of 1170 with the atom numbering scheme. The major disordered part is colored according to the atom coloring scheme (A part), while the minor part (B) is colored in orange and shown as a ball and sticks only in one half of the molecule for clarity. | ||
Almost all donor and acceptor atoms are included in the hydrogen bonding, linking the molecules into a complex 3D network. The shortest hydrogen bonds N4–H4⋯O3A/B (−1 + y, x, 1 − z), OW1–HW1A⋯OW2 (y, 1 + x, 1 − z), OW2–HW2A⋯O2 (−1 + y, 1 + x, 1 − z) and OW2–HW2B⋯O3A are in the range 2.661(13)–2.742(5) Å (ESI† Fig. S1, Table S3). There are no solvent-accessible voids in this crystal structure.
In the high-temperature structure 1370, no apical water molecule is bonded to the Cu(II) ion, so the coordination polyhedron is distorted square-planar (Fig. 2). The same part of the molecule is disordered as in the low-temperature structure, but there is only a tiny difference in the two occupancies, which are almost 0.5. In the 1370 structure, part A is present in 52% of the coordination moieties, and the configuration corresponds to part B in 1170. There is only one solvent O atom (from a water molecule) with an occupancy of 50%. Even if this water molecule is not considered, the N4–H4⋯O3A/B hydrogen bonds connect the dimers into a 3D network, and the solvent accessible volume is 116 Å3.
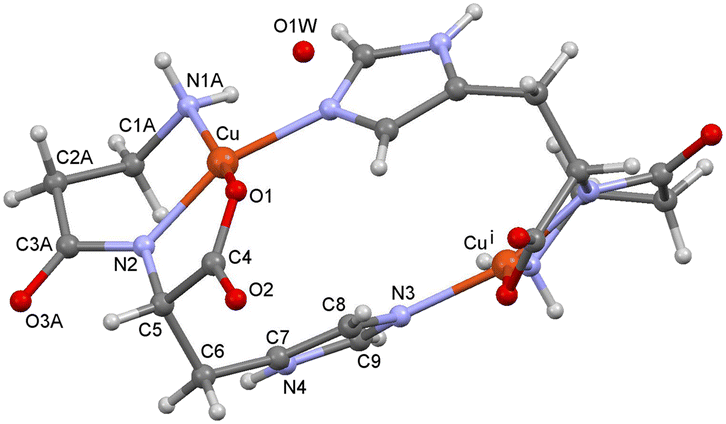 | ||
| Fig. 2 Structure of 1 at 370 K (1370) with the atom numbering scheme. Only the major disordered part is shown (part A). O1W is a water molecule, but the H atoms could not be located. | ||
Fig. 3 presents the molecular surfaces and 2D fingerprint plots, i.e., histograms presenting the number of surface points as a function of the internal (di) and external (de) distances to the closest atoms, calculated for the crystal structures of 1170 and 1370. Analysis of the percentages of the different atom–atom contacts in 1170 and (1370) shows that the most abundant contacts are H⋯H with 46.7% and 34.8%, respectively. The second most significant interaction is O⋯H/H⋯O, contributing 33.8% for 1170 and 45.4% for 1370, being attributed to O–H⋯O, N–H⋯O, O–H⋯N and C–H⋯O hydrogen bonding. Sharp and symmetrical spikes indicate strong hydrogen bonding in the 2D fingerprint plot. Apart from the mentioned, N⋯H/H⋯N (3.8% for 1170 and 4.9% for 1370) and C⋯H/H⋯C (7.5% for 1170 and 7.3% for 1370) interactions are also evident. The interaction schemes are similar in both structures, thus confirming that the area around the O and N atoms is essential in the formation of hydrogen bonds and in stabilizing the crystal architectures.
 | ||
| Fig. 3 The molecular Hirshfeld surface and 2D fingerprint plots of a) crystal structure (1170) and b) crystal structure (1370), mapped with the dnorm parameter. | ||
Compound 2, [Cu2(car)2(MeOH)2x(H2O)2(1−x)]·mH2O·nMeOH, crystallizes in the orthorhombic I222 space group. As in 1, the two dimeric parts are related by a two-fold axis, so half of the dimer is in the asymmetric unit. There is no disorder (Fig. 4, top). Compound 2 transforms very fast to 1 when exposed to moist air. Compound 2 was not synthesized using dry methanol, which means that both water and methanol molecules may be present in the solvent-accessible voids. Despite being rapidly frozen, the crystals may have already lost some of their solvent methanol molecules or have some disordered molecules remaining. The solvent-accessible area in 2100 is 1344 Å3 (two voids of 672 of Å3). The solvent occupies 40% of the unit cell volume, and the voids are continuous (Fig. 4, bottom). The formula could not be defined, but such volume can be occupied by four methanol or 16 water molecules or a nonstoichiometric combination of the two solvents.
 | ||
| Fig. 4 Structure of compound 2 at 100 K with the atom numbering scheme (up). The Cu(II) coordination spheres include either a disordered methanol molecule (65%) or a coordinated water molecule and a solvent water molecule (35%). Here, the two possibilities are shown on the two Cu(II) ions (top). View down the crystallographic a-axis showing voids in the structure when the solvent molecules are omitted (bottom). | ||
It was attempted to refine some solvent molecules/atoms for the 100 K structure (ESI† Tables S13–S16 and Fig. S2), but solvent masking was found to be more reasonable since it was not clear if they were water or disordered methanol (ESI† Tables S10–S12). In the structure, at 170 K, no solvent atoms could be placed in rational positions, so it was refined only by using solvent masking. The difference between the structures at 100 and 170 K might not be due to the data collection temperature but to placing the crystals into a stream of cold air (2170) or liquid nitrogen (2100) or the cooling speed.
The apical position in 2100 is occupied by either a methanol molecule [occupancy of 0.654(14) for O4 and C10] or two water molecules [occupancy of 0.346(14) for O4 and OW2]. Therefore, when O4 is a water molecule bonded to a Cu(II) ion, it forms a hydrogen bond with a solvent water molecule OW2. The Cu–O4 bond is longer than in 1 and amounts to 2.518(2) Å. In the structure at 170 K, only a water molecule is in the apical position of the distorted square pyramid at a distance of 2.545(2) Å (Table 2). The N4–H4⋯O2 (x, 1 − y, 1 − z) hydrogen bonds connect the dimers into a 3D network, even when the solvent molecules are not present (ESI† Table S12). The crystal structure resilience is why high-quality X-ray data could be obtained, as the crystals did not decompose further when frozen.
Compound 3, [Cu2(car)2(MeOH)2]·5.5MeOH, also crystallizes in the trigonal space group P3121 as compound 1 but has a larger unit cell. Data were collected only at 150 K. The asymmetric unit comprises the whole dimer, which is not disordered. The structural model of 3 is of inferior quality (R-factor is 0.1068) due to fast transformation to 1, as can be seen by the cracking of the crystals under the microscope which begins immediately after removal from the flask onto a microscope glass slide, even under Paratone® N oil. Methanol molecules occupy the apical positions of the two Cu(II) ions (ESI† Fig. S5). The solvent molecules occupy a volume of 2048 Å3. When the solvent and two coordinated MeOH molecules are also omitted, the total solvent accessible area is 2773 Å3 (49% of the unit cell volume). The voids are continuous, and the MeOH molecules quickly exit the structure, causing it to collapse and transform into polycrystalline 1.
Transformation 2 → 1 was monitored using PXRD (Fig. 5). A freshly prepared sample of 2 was placed wet (methanol solution) on a zero-background silicon holder and covered with a Kapton foil. After data collection, the foil was removed, and a new powder diffraction pattern was recorded. The decomposition of 2 to 1 occurred shortly after removing the Kapton foil. In the green powder diffraction pattern, it can be seen that there are maxima that do not belong to any of the described structures, so along with a mixture of compounds 1 and 2 there is an unknown compound X. The subsequent measurement was performed after 90 min, showing all maxima that belong to 1, the disappearance of those related to compound 2, and a few small intensity maxima corresponding to only a small amount of X that could not be separated nor characterized. Due to the fast decomposition of 3, the powder diffraction pattern could not be recorded. The crystals of compounds 2 and 3 crack fast when taken out of the crystallization solution due to loss of methanol and uptake of water molecules (they both transform to compound 1). Such a fast decomposition prevented other analyses like elemental, IR, and TG.
 | ||
| Fig. 5 The transformation of 2 → 1 was monitored using PXRD. Bottom to up: calculated powder pattern of 2 (red), 2 recorded with a Kapton foil (blue), 2 + 1 + X after removal of Kapton foil (green), 1 + X after 90 min (pink), 1 as-synthesized (orange), and 1 calculated from the 1270 structure (brown). RT – room temperature. | ||
Upon heating compound 1 from 170 to 270 K, the unit cell expands as expected (the volume is larger by 20 Å3 with larger parameters a, b, and a slightly shorter c, at the higher temperature). On the contrary, the temperature-dependent powder X-ray pattern indicated contraction of the unit cell upon further heating (Fig. 6). Changes in unit-cell parameters are associated with the loss of water molecules. The shift of the majority of diffraction maxima (12.2°, 13.2°, 14.7°, and others) towards higher 2θ angles is consistent with changes in the unit-cell volume. The single crystal data shows that at 370 K (1370), the volume is smaller by 123.7 Å3 compared to 1270. As evidenced by a series of unit-cell parameter measurements performed on single crystals, heating has a more significant effect on the length of the crystallographic a-axis than the length of the c-axis. In the low-temperature structure 1170, the c-axis is 0.56% longer than in the high-temperature structure 1370 (the a-axis is shortened even more, 2.48%). Positions of the maxima at 2θ of 8.7° and 17.5°, which are related to the planes (003) and (006), change almost unnoticeably during heating. These planes are coplanar with the direction of dimer packing. Interestingly, when cooling to the initial temperature of 30 °C, the sample binds moisture from the environment (TTK600 camera), returning to its initial state (powder patterns collected at 30 °C at the beginning and end of the experiment are the same). Therefore, this process is reversible.
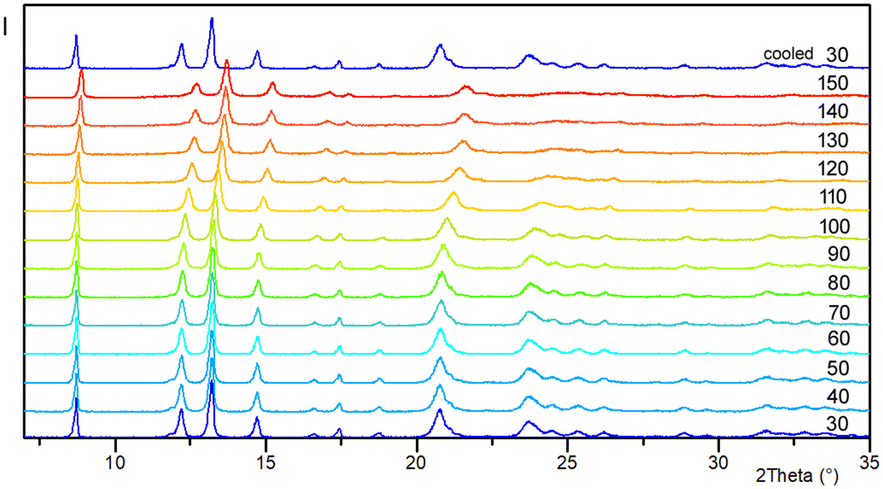 | ||
| Fig. 6 Variable-temperature PXRD study of the as-synthesized 1. The diffractograms were collected from 30 to 150 °C in intervals of 10 degrees, and the last one (top) was collected after cooling to 30 °C. The temperatures are in °C. | ||
The TGA curve of compound 1 shows a two-step degradation process (Fig. 7). The first step is associated with water loss in which the sample retains crystallinity (will be discussed later). The weight loss of 10.8% from 40 to almost 200 °C corresponds to the complete dehydration (calculated from the single-crystal structure, w(H2O) is 11.1%). Further heating ends with the total degradation (mass loss is 65.5%) of compound 1, and only CuO remains in the crucible (64.3%, calculated from the formula).
 | ||
| Fig. 7 TGA curve (black) and DSC thermogram (blue) of compound 1. | ||
In many ways, solid-state IR spectroscopy is complementary to the PXRD method. The first method addresses the changes at the molecular level, while the latter detects the changes in the crystal structure. The apparent advantage of the first method is the data acquisition time and its independence from the crystallinity of the solid-state phase (in the case of an amorphous sample, PXRD is not always informative, although some solid-state transformations are going via amorphous phases). In this case, the IR spectra are comparable to the spectra of Cu(II) coordination compounds with amino acids and peptides and confirm data from other performed experiments (solid-state transformations, DSC, PXRD).17,18,23 It also shows significant changes in regions 3750–3000 cm−1 and 750–500 cm−1 associated with water molecule interactions with other groups (Fig. 8). As expected, as the temperature increases, the proportion of water molecules in the sample decreases constantly, so the bands from the first region become lower in intensity. In the second region, noticeable changes are associated with Cu(II) ion coordination sphere changes. These observations are consistent with the electron density analysis in the 1170 and 1370 structures.
 | ||
| Fig. 8 Variable temperature FTIR spectra of compound 1. | ||
During the transformation of 1 from 270 to 370 K, the distance Cu(II)⋯OH2 changes from 2.4 Å to 3.2 Å. Comparison of the time-averaged structures is not the best way to depict the dynamics of the process, but it is evident that some of the water molecules change position as their fraction in the structure decreases. Water molecules move from the Cu(II) ions to the newly emerged cavities in the crystal structure. Except for the change of the coordination polyhedron, some conformation differences can be observed by comparing 1 at these two temperatures. At the higher temperature, the Cu(II) ions are further apart by 0.23 Å (Fig. 9, bottom). If the two Cu(II) coordination spheres are overlapped so that the coordinating atoms are at fixed points, the differences become even more visible.
 | ||
| Fig. 9 Comparison of the crystal structures 1170 (top left) and 1370 (top right). In 1370, H atoms on water molecules were not modeled due to lower-quality data. The overlayed dimer molecules are shown in purple (1170) and yellow (1370) (bottom). | ||
The high-temperature structure 1370, which lacks the apical solvent molecule, has attracted our additional interest. Creating different solvates from activated species is a well-studied phenomenon, so we attempted to explore the possibilities of creating compounds like MOFs or coordination polymers using common linkers. For this purpose, we used only a small subset of linkers, such as 4,4′-bipyridine (bpy), 1,2-bis(4-pyridyl)ethene (bpe), nicotinic (na) and isonicotinic acids (ina), and imidazole (im). The first set of reactions included milling of all reactants simultaneously in a 1:1:1 ratio [Cu(OH)2, car and linker]. It proved to be unsuccessful, probably due to the presence of water (reaction stoichiometry and the choice of copper hydroxide always promoted enough water to obtain the most stable compound [Cu2(car)2(H2O)2]·2H2O). The second set of reactions included heating of 1 to 400 K, followed by cooling to room temperature in a desiccator (with P4O10 as drying agent). The mixture was then introduced in the milling jar. The milling reaction progress was monitored by ex situ PXRD (Fig. 10). Only when bpy and bpe were used did new phases appear after 20–40 minutes in the presence of polar aprotic DMF (N,N′-dimethylformamide). The emergence of diffracted intensities at lower 2θ values correlates with longer crystallographic axes of the new phases. These preliminary results indicate the promising landscape of CUS in Cu(car) to form new interactions and compounds, and will be definitively further investigated.
 | ||
| Fig. 10 PXRD patterns of mixtures obtained after milling dehydrated 1 with bpy (up) and bpe (down) compared to PXRD patterns of reactants (1 and bpe or bpy). In successful LAG milling reactions DMF was added. Labels also contain the information of the milling time. | ||
Conclusions
Reactions of L-carnosine with copper(II) salts under different conditions resulted in three compounds. Their crystal structures comprise almost the same building blocks – isoskeletal dimers that differ in the apical ligand coordinated to Cu(II) ions (water or methanol molecule). The voids in the crystal structures are occupied with solvent molecules, giving rise to very complex hydrogen bonding networks. When the solvent molecules are methanol, they quickly leave the structure, transforming into the stable structure of 1 with water solvent molecules. These can be removed by heating, altering the Cu(II) ion coordination sphere from square-pyramidal to square-planar, resulting in a compound with CUS. This high-temperature structure is a new compound reactive toward water and also to other molecules that can coordinate Cu(II) ions in the apical position. Temperature-dependent structural and spectroscopic solid-state analyses indicated the robustness and reactivity of the Cu(car) dimers, capable of providing new phases by addition of common pyridine-based linkers, such as bpy or bpe, in a relatively simple heating–milling routine. These finding are definitively worth of further structural characterization.Experimental
Materials and methods
L-Carnosine and copper(II) acetate monohydrate were of analytical reagent grade and were used without further purification. Copper(II) hydroxide was prepared by adding an aqueous solution of sodium hydroxide to an aqueous solution of copper(II) sulfate (CuSO4·5H2O). Methanol was dried and distilled over magnesium methoxide.CHN elemental analyses were performed on a Perkin Elmer 2400 Series II CHNS analyzer in the Analytical Services Laboratories of the Ruđer Bošković Institute, Zagreb, Croatia.
IR spectra were obtained in the range 4000–450 cm−1 on a Perkin-Elmer SpectrumTM FTIR-spectrometer in an ATR mode.
Because of the fast decomposition of compounds 2 and 3 in air, only compound 1 was analyzed by IR spectroscopy. In the spectrum of L-carnosine, the characteristic ν(NH2) stretching frequency is observed at 3240 cm−1 (which overlaps with the broad OH peak), while asymmetric and symmetric stretching frequencies of carboxylate are observed at 1564 cm−1 and 1403 cm−1 respectively.28 Compound 1 has a strong absorption band around 3300 cm−1, assigned to the overlap of the N–H and O–H stretching frequencies (ESI† Table S25). The amide carbonyl frequency appeared at 1627 cm−1, a shift to around 30 cm−1 compared to free L-carnosine (1655 cm−1). This lowering of the amide frequency may be due to the deprotonation of the amide carbonyl group of L-carnosine, as suggested by Lukton and Sisti,29,30 and coordination to the Cu(II) ion through the amide N atom. The absorption peaks in 1 at 1560 and 1395 cm−1 correspond to the carboxylate groups' asymmetric and symmetric stretching frequencies, indicating that the O atoms of the carboxylate groups coordinate Cu(II) ions. The IR spectrum of the 1 agrees well with its structural characteristics and the data from Freeman and Szymanski.26
Temperature-dependent IR spectra were recorded on a Bruker VECTOR 22 spectrometer (using the KBr pellet technique, 4000–400 cm−1 range, 2 cm−1 steps, 128 scans) from 30 to 180 °C. A thin, almost transparent, bluish KBr pellet was placed in a temperature-adjustable holder. After each spectra acquisition, the temperature was raised by 10 °C and kept constant for 10 minutes to ensure equal heat distribution (in the pellet). After the experiment, the pellet partially lost its transparency and color (it became brownish, which is consistent with other observations).
Thermal analysis was performed using a simultaneous TGA-DTA analyzer (Mettler-Toledo TGA/SDTA 851e). The samples were placed in 40 μL aluminium crucibles, heated in oxygen (50 mL min−1) from room temperature up to 600 °C at a heating rate of 10 °C min−1. Data collection and analysis were done using the program package STARe Software 14.00 Mettler Toledo GmbH, 2015.31
Mechanochemical reactions were performed using a Retsch MM 200 mixer mill operating at 25 Hz.
Synthesis
Crystallography
Powder X-ray diffraction data (PXRD) were collected on a Malvern PANalytical Aeris diffractometer in a Bragg–Brentano geometry with CuKα radiation (λ = 1.54184 Å) at room temperature. The samples were placed on a silicon holder, and the diffractograms were measured in the 2θ range 5–40° with a step size of 0.022° and an exposition of 15.0 s per step.Temperature-dependent powder X-ray diffraction (PXRD) patterns were collected on a Malvern PANalytical Empyrean diffractometer in reflection mode over the 3–40° 2θ range (step size 0.0066°; exposition 9.5 s), using a PIXcel3D-MEdipix3 detector and Cu radiation (Cu Kα1: 1.540598 Å; Cu Kα2: 1.544426 Å). The sample was mounted in a zero-background holder and mounted inside the Anton Paar TTK 600 chamber in air. The diffraction data were collected in the range from 30 to 150 °C. After single temperature data collection, the temperature was increased by ten °C increments and kept constant for 10 min before subsequent measurement. After collecting the highest temperature diffraction data at 150 °C, the sample was cooled back to 30 °C, and the data collection was repeated using the same parameters. HighScore Plus32 and Data Viewer were used for data analysis and visualization.
Single-crystal X-ray diffraction data were collected on two diffractometers and at synchrotron Elettra in Trieste. Crystals were mounted on cryo-loops with Paratone® N oil and transferred into the stream of cold air or nitrogen. Several data collections were performed for the stable crystals of compound 1, of which the three most representative are presented here. Data for 1170, 1270, and 1370 were collected at 170 K, 270 K, and 370 K, respectively, by ω-scans on a XtaLAB Synergy S diffractometer with CuKα radiation (λ = 1.54184 Å). The Smartstream system used cold air for low-temperature measurements. The 270 K and 370 K measurements were performed on the same crystal. Crystals of compounds 2 and 3 decomposed very fast after being out of the solution, so they were transferred into Paratone® N oil immediately from the solution by a dropping pipette, quickly picked up by a plastic loop, and transferred to a cold air stream on the diffractometer. X-ray data for 2170 were collected as described above. Data for 2100 was collected from a smaller crystal at synchrotron Elettra on the XRD2 beamline at 100 K (nitrogen stream) using 0.7000 Å radiation. An Oxford Diffraction Xcalibur 3 CCD diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) was used for data collection at 150 K (nitrogen stream) for 3150. These crystals also decomposed during data collection, so a fast and unique set was collected for this data.
Data collection and reduction were performed using the CrysAlis software package33 for all in-house data collection and reduction. Synchrotron data were also reduced by the CrysAlis Red program. The structures were solved by direct methods using the SHELXT program34 and refined by the full-matrix least squares method based on F2 against all reflections using the SHELXL program.34 Using the riding model, the H atoms on C and N atoms were placed in calculated positions with Uiso(H) = 1.2Ueq(C, N). Final refinement was done using the OLEX2 program.35 Non-hydrogen atoms were refined anisotropically except in 3150, where only non-solvent heteroatoms were refined anisotropically. All solvent methanol molecules in this structure had fixed bond lengths and angles. In the structure of 2100, four disordered O atoms were found. The H atoms could not be found or modelled on two disordered O atoms in 2100. There was still 465.1 Å3 of potential solvent accessible area, probably due to disordered methanol molecules. No solvent molecules could be modelled in 2170. The solvent masking routine34 was better for both structures, 2100 and 2170.
In all data sets of 1, one part of the car ligand is disordered in two positions. Atoms N1 to C3 were refined by using restraints for distances involving these atoms and for their anisotropic displacement in parts A and B. The occupancy of part A was refined to 0.601(11), 0.540(13), and 0.52(3) in 1170, 1270 and 1370, respectively. Geometrical parameters were calculated using Olex2 and PLATON.36 The drawings of the structures were prepared in ORTEP and MERCURY.36–39 Details of data collection and structure refinement of 1 at 170 K (1170), 2 at 100 K (2100) using the solvent masking routine are given in Table 2. Details of data collection and structure refinement of 1 at 270 and 370 K, 2 at 100 K (2100) with some modelled atoms and 2 at 170 K (2170), and 3 are given in ESI† Tables S25–S27 respectively.39
CCDC numbers 2327404, 2327405, 2327412, 2327414, 2327416, and 2327429 contain the ESI† crystallographic data for 1170, 1270 and 1370, 2100, 2170 and 3, respectively.
The Hirshfeld surfaces were generated with CrystalExplorer17.0 using the automatic procedures implemented in the program.40 The surfaces are mapped with a normalized contact distance (dnorm), with values ranging from −0.7000 Å to 2.1000 Å, which indicates the different close contacts with the molecules' neighboring species. The dnorm image is transparent to visualize the molecular structure of 1170 and 1370, with the color scale reflecting intermolecular distances. The hydrogen-bonding contacts of distances shorter than the corresponding sum of the van der Waals radii are presented as large red circular areas.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC numbers 2327404, 2327405, 2327412, 2327414, 2327416, and 2327429 contain the ESI† crystallographic data for 1170, 1270 and 1370, 2100, 2170 and 3, respectively.Author contributions
DM-S, IĐ, and DM-Č, conceptualized and designed the study. DV, NJ, MT, KW-R, IĐ, and DM-Č performed the experiments and analysed the data. All authors contributed to the original draft of the manuscript. KW-R, IĐ, and DM-Č reviewed and edited the manuscript. DM-Č acquired funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Croatian Science Foundation (grant no. IP-2014-09-4274) and a grant from the University of Zagreb Synthesis and structural characterization of organic and coordination compounds, structure of proteins, for 2022. We also acknowledge the support of project CIuK, which is co-financed by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grants KK.01.1.1.02.0016). We thank Annie Heroux and Nicola Demitri for their valuable support at Synchrotron Elettra in Trieste and Prof. Adriana Kenđel for her help in setting TD-IR measurements.Notes and references
- A. A. Boldyrev, Biochemistry, 2000, 65, 751–756 CAS
.
- A. A. Boldyrev, G. Aldini and W. Derave, Physiol. Rev., 2013, 93, 1803–1845 CrossRef CAS
- C. Abate, D. Aiello, M. Cordaro, O. Giuffrè, A. Napoli and C. Foti, J. Mol. Liq., 2022, 368, 120772 CrossRef CAS
- F. Bellia, V. Lanza, I. Naletova, B. Tomasello, V. Ciaffaglione, V. Greco, S. Sciuto, P. Amico, R. Inturri, S. Vaccaro, T. Campagna, F. Attanasio, G. Tabbì and E. Rizzarelli, Antioxidants, 2023, 12, 1632 CrossRef CAS PubMed
- H. Oppermann, J. Dietterle, K. Purcz, M. Morawski, C. Eisenlöffel, W. Müller, J. Meixenberger and F. Gaunitz, Cancer Cell Int., 2018, 18, 111–121 CrossRef
- K. Chmielewska, K. Dzierzbicka, I. Inkielewicz-Stępniak and M. Przybylowska, Chem. Res. Toxicol., 2020, 33, 1561–1571 Search PubMed
- E. J. Baran, Biochemistry, 2000, 65, 789–797 CAS
- T. Matsukura, T. Takahashi, Y. Nishimura, T. Ohtani, M. Sawada and K. Shibata, Chem. Pharm. Bull., 1990, 38, 3140–3146 CrossRef CAS
- M. Förster and H. Vahrenkamp, Chem. Ber., 1995, 128, 541–550 CrossRef
- M. L. Branham, P. Singh, K. Bisetty, M. Sabela and T. Govender, Molecules, 2011, 16, 10269–10291 CrossRef CAS
- A. Torreggiani, P. Taddei and G. Fini, Biopolymers, 2002, 67, 70–81 CrossRef CAS
- T. Ama, F. Kawaguchi, M. Uchijima, N. Koine and T. Yasui, Bull. Chem. Soc. Jpn., 1989, 62, 3464–3468 CrossRef CAS
- H. Dobbie and W. O. Kermack, Biochemistry, 1955, 59, 246–257 CAS
- M. Tamba and A. Torreggiani, Int. J. Radiat. Biol., 1999, 75, 1177–1188 CrossRef CAS PubMed
- L. Schröder, C. H. Schmitz and P. Bachert, J. Inorg. Biochem., 2008, 102, 174–183 CrossRef
- H. C. Freeman and J. T. Szymanski, Chem. Commun., 1965, 598–599 RSC
- D. Vušak, B. Prugovečki, D. Milić, M. Marković, I. Petković, M. Kralj and D. Matković-Čalogović, Cryst. Growth Des., 2017, 17, 6049–6061 CrossRef
- J. Pejić, D. Vušak, G. Szalontai, B. Prugovečki, D. Mrvoš-Sermek, D. Matković-Čalogović and J. Sabolović, Cryst. Growth Des., 2018, 18, 5138–5154 CrossRef
- D. Vušak, J. Pejić, M. Jurković, G. Szalontai and J. Sabolović, CrystEngComm, 2020, 22, 5587–5600 RSC
- D. Vušak, K. M. Špoljarić, J. Jurec, D. Žilić and B. Prugovečki, Croat. Chem. Acta, 2022, 95(4), 157–165 CrossRef
- D. Vušak, K. Ležaić, J. Jurec, D. Žilić and B. Prugovečki, Heliyon, 2022, 8, e09556 CrossRef PubMed
- M. Tašner, D. Vušak, I. Kekez, A. Gabud, V. Pilepić, D. Mrvoš-Sermek and D. Matković-Čalogović, Heliyon, 2022, 8, e11100 CrossRef
- K. Smokrović, S. Muratović, B. Karadeniz, K. Užarević, D. Žilić and I. Đilović, Cryst. Growth Des., 2020, 20, 2415–2423 CrossRef
- K. Smokrović, I. Đilović and D. Matković-Čalogović, CrystEngComm, 2020, 22, 4963–4968 RSC
- A. P. Katsoulidis, K. S. Park, D. Antypov, C. Martí-Gastaldo, G. J. Miller, J. E. Warren, C. M. Robertson, F. Blanc, G. R. Darling, N. G. Berry, J. A. Purton, D. J. Adams and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2014, 53, 193–198 CrossRef CAS PubMed
- H. C. Freeman and J. T. Szymanski, Acta Crystallogr., 1967, 22, 406–417 CrossRef CAS
- Ü. K. Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- A. Torreggiani, M. Tomba and G. Fini, Biopolymers, 2000, 57, 149–159 CrossRef CAS
- A. Lukton and A. Sisti, J. Org. Chem., 1961, 26, 617–619 CrossRef CAS
-
STARe, Evaluation Software, vers. 14.00, Mettler-Toledo GmbH, 2015 Search PubMed
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, Powder Diffr., 2014, 29(S2), S13–S18 CrossRef CAS
-
CrysAlisPro 1.171.41.123a, Rigaku Oxford Diffraction, 2022 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A:Found. Crystallogr., 1990, 46, 194–201 CrossRef
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2327404, 2327405, 2327412, 2327414, 2327416 and 2327429. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01070a |
| This journal is © The Royal Society of Chemistry 2025 |