
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystal structures of potassium and cesium salts of adenine: the role of alkali cations†
Sarabjeet
Kaur
 a,
Jeremy
Harvey
b,
Luc
Van Meervelt
c and
Christine E. A.
Kirschhock
*a
a,
Jeremy
Harvey
b,
Luc
Van Meervelt
c and
Christine E. A.
Kirschhock
*a
aCentre for Surface Chemistry and Catalysis Characterisation and Application Team, Department of Microbial and Molecular Systems (M2S), Katholieke Universiteit Leuven, Celestijnenlaan 200f – box 2461, 3001 Leuven, Belgium. E-mail: christine.kirschhock@kuleuven.be
bQuantum Chemistry and Physicochemistry, Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200f – box 2404, 3001 Leuven, Belgium
cBiochemistry, Molecular and Structural Biology, Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200f – box 2404, 3001 Leuven, Belgium
First published on 4th November 2024
Abstract
This study reports the crystal structures of potassium and cesium salts of adenine (K-adenine and Cs-adenine) from the perspective of the interaction of alkali cations with purine nucleobases. Unlike previously-known guanine salts, both K-adenine and Cs-adenine are anhydrous, with the counter ions (K+ and Cs+) directly coordinating to the ring nitrogens of adenine anions. In both structures, the crystal packing is predominantly determined by cation–anion interactions, with additional stabilization through hydrogen-bonding of neighbouring adenines. Attempts to crystallise either the cesium salt of guanine or the sodium salt of adenine were unsuccessful. To explain this trend, quantum-chemical calculations were performed to rationalise the preferences of sodium, potassium, and cesium cations to coordinate either with water or adenylate/guanylate anions. The exchange energies of cation–anion complexes reveal that sodium cations exhibit a preference for water or guanylate coordination via oxygen, while cesium cations prefer adenylate coordination via nitrogen functions, avoiding water interaction. Potassium exhibits an intermediate trend. Overall, this research offers insights into interactions between alkali-cations and organic anions, aiding the development of new crystalline compounds and co-crystals.
Introduction
Nucleobases are nitrogenous heterocycles, which combined with a phosphate group and a ribose or deoxyribose unit form nucleotides, the structural units of RNA or DNA, respectively. While cytosine, thymine and uracil are derived from pyrimidine, guanine and adenine are based on the bicyclic purine skeleton.1,2 Containing hydrogen-donor, as well as -acceptor functionality, nucleobases readily engage in hydrogen-bonding between and among themselves, potentially also involving water molecules.3,4 Furthermore, the aromatic pi-systems of neighboring molecules can interact, representing an additional factor governing self-organisation into a macromolecule or a crystal. For instance, guanine forms stacked hydrogen-bonded layers resulting in three different crystalline phases: guanine monohydrate and two anhydrous polymorphs, α and β.5–7 Such ordered multilayers of guanine have interesting properties, for example, imparting a metallic luster to fish scales by constructive interference8 or a silver hue to spiders by reflection along guanine plates.9Owing to the amphiprotic nature of nucleobases, they occur as neutrals, cations or anions, depending on the pH. In the gas phase or in the presence of 1 or 2 water molecules, N9H and N7H are the dominant keto-amino tautomer forms for purines.10–13 Purines are protonated at N1 (N9H, major tautomer of adenine) or N3 (N7H, minor tautomer of adenine),14,15 and N7 (N9H, major tautomer of guanine) or N9 (N7H, minor tautomer of guanine) (Fig. 1a–d).15,16 The pKa values for the protonation of guanine and adenine are 3.2 (ref. 17 and 18) and 4.1, respectively.18,19 The most acidic proton of adenine is associated with one of the nitrogen atoms in the imidazole ring, position N7 or N9 depending on the tautomer and it dissociates with a pKa value of 9.6.17,20 Owing to the presence of the carbonyl group in guanine on position 6, the guanine initially deprotonates at N1 or N3 with pKa of 9.8 (ref. 17 and 20) and a second time at N7 or N9 at high pH (pKa = 12.4).21 These observations can be explained on the basis of quantitative molecular electrostatic potentials (MEP) maps of N9H tautomers revealing the most positive region of adenine along the N9–H bond (53.81 kcal mol−1) and the most negative region at the N1 atom (−32.39 kcal mol−1). On the other hand, the most positive value for the guanine is along the N7–H bond (54.11 kcal mol−1) and the most negative value is observed close to the carbonyl oxygen and N1 (−51.63 kcal mol−1).22Fig. 1e and f illustrates the protonation–de-protonation equilibria of guanine and adenine, as well as their tautomerism.
 | ||
| Fig. 1 (a) N7H and (b) N9H tautomeric states of guanine; (c) N7H and (d) N9H tautomeric states of adenine; protonation and deprotonation equilibria of (e) guanine and, (f) adenine. The numbering scheme of atoms corresponds to those traditionally used for purines.23 | ||
The ionic state of the molecules critically affects their crystallization. Under acidic conditions, guanine tends to crystallise as a mono-hydrate, but in protonated form (also known as guanidinium, guaninium ion or H2G+) it forms salts with various anions, including Cl−,24,25 NO3−,26 CO32−,27 and SO42−.28 These salts have been studied for their potential applications in organic electronics and in the development of sensors for detecting metal ions.29,30 Neutral conditions result in the two anhydrous guanine polymorphs,31 while in the form at high pH crystalline guanine salts with alkali metal cations such as Na+,32,33 K+,34 or Li+ are known.35 Singly deprotonated guanine also forms coordination complexes with transition metal cations such as Mn2+, Fe2+, Ni2+, Co2+, Cu2+or Zn2+.36,37
In crystalline polymorphs of guanine, the hydrogens are either attached to N1 and N7 (anhydrous guanine) or to N1 and N9 (guanine monohydrate). The molecule can be deprotonated twice at N1, N7 or N9 to yield a singly or doubly deprotonated state.5–7,34 In its sodium salt (2Na+·C5H3N5O2−·7H2O) guanine is doubly deprotonated at N1 and N7, crystallising in the monoclinic system (P21/c).33 The crystals consist of strictly alternating organic and inorganic zones alternating in a-direction (Fig. 2). Polyhedral edge- and corner-sharing chains of sodium in octahedral and trigonal bipyramidal coordination by water alternates with guanidinium anions, hydrogen-bonded to water-molecules of the inorganic chains (Fig. 2).33 No direct interaction between cation and guanine exists.
 | ||
| Fig. 2 The Na-guanine salt (2Na+·C5H3N5O2−·7H2O) consists of alternating organic and inorganic layers in a-direction. Sodium solely coordinates to water molecules and forms chains of edge- and corner-sharing sodium polyhedra in c-direction (progressing in view direction).33 Guanine interacts via its carbonyl-group with water molecules. | ||
The mono-potassium salt of guanine (K+·C5H3N5O−·H2O) crystallizes in the monoclinic system (C2/c) (Fig. 3).34 Reminiscent of the di-sodium salt, this structure shows polyhedral chains of edge-linked cations in the b-direction (Fig. 3). But here, chains are linked by guanine anions, which engage in direct cation coordination through their oxygen atom, as well as through their various nitrogen centres (O6, N3, N9). The structure contains two types of inorganic chains, whereof one (K1) is entirely based on coordination by oxygen atoms of water or guanine, while the other involves coordination by oxygen of water and heterocyclic nitrogen. This arrangement allows formation of hydrogen bonds between the amine functions of guanine molecules into helical channels with roughly 4-fold symmetry.33,34
 | ||
| Fig. 3 K-guanine (K+·C5H3N5O−·H2O) displays 2 types of edge-sharing polyhedral chains of the cations in b-direction. K1 (violet) entirely coordinates to oxygen, originating from water and guanine, while K2 (green) interacts with water and organic nitrogen (O6, N3, N9).34 | ||
Similar to guanine, adenine is known to form salts in protonated form with various anions, such as H2PO4−, Cl−, ClO4−, NO3− and SO42− ions.24,38–43 These salts are often used in biochemical and analytical applications.44–46 In the pH range between 4.1 and 9.6, adenine crystallizes as two anhydrous polymorphs (polymorph I and II), consisting of stacked H-bonded layers. In these polymorphs, adenine adopts its most stable N9H tautomeric form (Fig. 1c and d). The two polymorphs often occur together and can be obtained in pure form by sublimation (polymorph I)45 or recrystallisation from aqueous ethanol (polymorph II)44,45 Besides the anhydrous forms, adenine also crystallizes as adenine trihydrate wherein adenine chains are linked with two water layers via N–H⋯O and O–H⋯O hydrogen bonds.47 Unlike guanine, adenine only exists in singly de-protonated state at high pH (Fig. 1f). Adenine anions can have varying coordination (di, tri- or tetradentate) through purine ring nitrogens (N1, N3, N7 and N9) with Zn2+, Cu2+ or Co2+ forming metal–organic frameworks.48–51 Recently, the crystal structure of the hydrated salt of tetrabutylammonium and adenine has been reported.52 Therein, no direct cation–anion interaction is observed. Adenine anions are connected via hydrogen-bonding by water, forming corrugated sheets in the bc-plane. The interaction of adenine anions with Na+ and K+ has been studied in dimethylsulfoxide (DMSO) in anhydrous conditions.53 XRD combined with UV and NMR revealed the formation of ion pairs, and a crystal structure of the adenine sodium salt containing DMSO solvating molecules was described.54 In this crystal the oxygen atom of the solvent actively participates in cation coordination next to nitrogen atoms of the heterocycle.54
Interestingly, even though alkali-hydroxide (NaOH and KOH) solutions of adenine have extensively been used for heterogeneous alkylation53,55 and formation of anti-inflammatory56 and antiviral drugs,57,58 the crystal structure of alkali-adenine salts from water have not been reported in literature. The fact that crystalline alkali salts are known only for guanine but not for adenine prompted the here reported research.
It is well known that guanine chemistry is quite different from that of adenine, owing to the presence of an amide group on the pyrimidine ring in guanine.33,34,59–61 For example, even under neutral conditions and in the presence of alkali metals, guanine quadruplexes have been observed to form spontaneously in water, involving proton-accepting properties of the carbonyl- and proton-donating properties of the amine-group.62 Adenine only forms tetrads in the nucleotide form in human telomeric DNA regions, but not as a lone nucleobase. However, despite and because of the differences in chemical functionality of these two purine nucleobases it is quite intriguing to attempt the crystallization of alkali-adeninium salts to compare to their guanine counterparts.
Experimental section
Single crystal preparation and measurement
A similar methodology at pH 14 was followed for the preparation of single crystals of Cs+C5H4N5−. The initial solution consisted of 5 CsOH and 15 H2O molecules per 1 adenine molecule. Large dark pink crystals were obtained after three weeks at ambient conditions. Using the same protocol, also the crystallization of the sodium salt of adenine and cesium salt of guanine were attempted, but no crystalline solid was obtained. Instead, the experiments led to poorly crystalline polymorphs of adenine (in case of sodium salt of adenine) or guanine (in case of cesium salt of guanine), next to amorphous solids.
Suitable crystals were selected and mounted on an Agilent SuperNova diffractometer equipped with an Eos CCD detector, using Mo Kα radiation (λ = 0.71073 Å). The crystal was kept at 293(2) K during data collection. Using Olex2,63 the structure was solved with the SHELXT64 structure solution program using intrinsic phasing and refined with the SHELXL65 refinement package using least squares minimization. Hydrogen atoms were located in difference density maps and subsequently refined freely, except for atoms H2 and H8 in K+C5H4N5− which were placed in idealized positions and refined in the ‘riding mode’. Non-hydrogen atoms were refined anisotropically and hydrogen atoms with isotropic temperature factors at 1.2 times Ueq of the parent atoms. Crystal data, data collection and structure refinement details are summarized in Tables S1–S13 and Fig. S1 and S2.†
Computational details
The structure of each of the nucleobase anions (adenylate and guanylate) was optimized at the B3LYP-D3BJ/6-311+G(d) level of theory, including the dispersion correction at the D3 level with Becke–Johnson damping, and using the Gaussian 16 programme code.66 The anions are found to be nearly but not quite planar at this level of theory, with the exocyclic nitrogen atoms found to prefer a pyramidal structure. The energy at the optimized structure was then recomputed using the DLPNO-CCSD(T) method with the def2-QZVPP basis set on all atoms, using the ORCA 5.0 program package.67 Complexes with a single sodium, potassium or cesium cation have been studied at the same levels of theory. For the case of cesium, the SDD ECP and associated basis set was used, while the version of the def2-QZVPP basis set including an ECP was used for the single point calculations.Results and discussion
Crystal structure of K+C5H4N5−
The crystal of the mono-potassium salt of adenine (K-adenine) has monoclinic space group P21 with one formula unit in the asymmetric unit (Table S1†). Unlike K+ and Na+ salts of guanine, the structure contains no water molecules. In this absence of water, the hepta-coordinated potassium cation is entirely coordinated by nitrogen from adenine (Fig. 4a).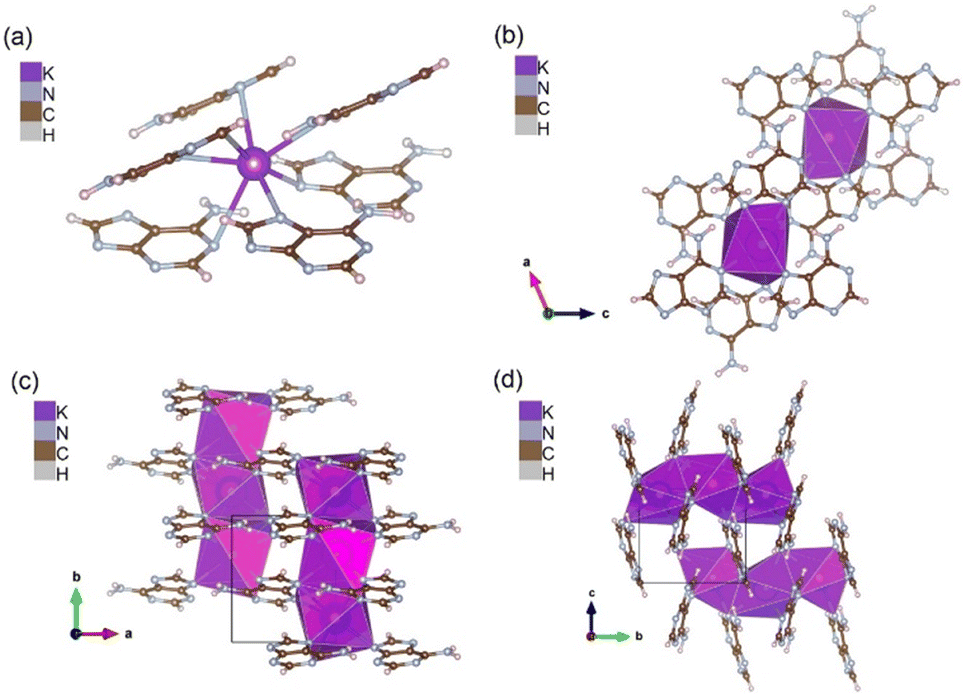 | ||
| Fig. 4 K-adenine (a and b) K+, hepta-coordinated by heterocycle nitrogen in the K+C5H4N5− crystal. Each potassium is surrounded by six adenine anions in a capped trigonal prismatic geometry, (c and d) layers of tilted adenine anions in the ac-plane coordinating to face-sharing chains of potassium-cations in b-direction. | ||
The coordination geometry around potassium is complex and is best described as a distorted capped trigonal prism (Fig. 4b). Potassium is surrounded by six adenine anions, whereof one is coordinating by N9 and N3 to the same cation, forming an edge of the upper triangle of the prism. Except for N3, all nitrogen atoms link coordination polyhedra in b-direction, resulting in face-sharing chains of potassium polyhedra (Fig. 4a and c).
The crystal packing can be described by layers of tilted molecules in the ac-plane, which coordinate with all 4 ring-nitrogen atoms to potassium-ions above and below the molecules (Fig. 4d). N3 only connects to one cation, while N1, N7 and N9 connect cations above and below, creating the chains of potassium-cations in b-direction (Fig. 4d). The expression of chains of cation–polyhedra, is somewhat reminiscent of the mono-potassium guanine, but in contrast the adenine salt contains no water, and the amine functions of nucleobases do not engage in helical channel formation as in the case of the potassium guanine salt. Instead, the amine function appears to stabilize two neighboring molecules in the organic layers by hydrogen–N distances of 2.3 and 2.5 Å to N9 and N1, respectively.
In its potassium salt, adenine makes use of all the possible binding sites (N1, N3, N7 and N9) that are known for alkali metal cation coordination to neutral or anionic adenine (Fig. 4a and c).54,68 The distance between K+ and its ligands is on average 3.03 Å (list of bond distances and angles in Tables S5 and S6†).21,54,69,70 Only the –NH2 amine group of adenine does not participate in coordination with potassium but stabilizes the arrangement by hydrogen bonding to neighboring molecules. Adenine molecules within the same and between layers in the ac-plane are tilted with respect to each other, which makes stabilization by π–π stacking improbable.55 This structure seems to be entirely dominated by cation–anion interaction.
Crystal structure of Cs+C5H4N5−
Similar to the K-adenine, the cesium salt (referred as Cs-adenine hereon) is anhydrous. The structure crystallizes in the orthorhombic space group Pbcn (Table S1†). The asymmetric unit comprises a singly deprotonated adenine anion and two cesium counter-ions on special positions i.e. Cs1 on the 2-fold axis (Wyckoff site 4c) and Cs2 on the inversion centre (Wyckoff site 4a). Similar to K-adenine, both Cs1 and Cs2 directly coordinate with heterocycle nitrogen of adenine anions (at N1, N7, N3 and N9; Fig. 5a). Both cesium ions are in coordination geometries of face-sharing, distorted cubes (Fig. 5b). These are roughly aligned with the unit cell, with one set of faces parallel with the bc-plane, the other two face sets normal to [011] and [01−1] (Fig. 5b and c). Both cubes, denoted as Cs1c and Cs2c, are generated by 6 adenine anions. Cs2c in the cell origin shares opposite faces ((110)-planes) with Cs1c. Cubes of Cs1c share neighboring faces of the same Cs2c cube, consequently sharing an edge (Fig. 5b). This results in zig-zag chains of face sharing polyhedra along the c-direction of the unit cell (Fig. 5c). One edge of the shared faces consists of N9 and N3 of the same anion. In this, N9 forms the vertex of the polyhedral chain, shared by 3 cubes, 2 Cs2c and 1 Cs1c (Fig. 5b). This is causing a significantly distorted coordination environment of Cs2c along the diagonal in space (N9–C2 = 3.9 Å), resulting in an elongation of the thermal ellipsoid of Cs2 in this direction (Fig. S2†). | ||
| Fig. 5 Cs-Adenine (a and b) coordination pattern of Cs1 and Cs2 with the adenine nitrogens in the Cs+C5H4N5− crystal where cesium forms face-sharing, distorted cubes, Cs1c and Cs2c, (c) Cs-cubes in a zig-zag arrangement in bc-plane and other two normal to [011] and [01−1] plane (blue and red lines) (d) a N3–N10 hydrogen bond (3.102(10) Å) stabilizes an edge of a shared face between C1c and C2c and a second one (3.092(11) Å) connects N7 atom to N10 of the neighboring chain. | ||
Adenine anions bridge neighboring zig-zag chains, N3 and N9 interact with the same chain while N7 coordinates to corners of Cs1c of neighboring chains above and below (Fig. 5b and c). Similar as in the potassium salt, the amine function of adenine is involved in hydrogen bonding. One hydrogen bond stabilizes an edge of a shared face between Cs1c and Cs2c, the second is connecting to a N7 atom on a neighboring chain (Fig. 5d). Nominally, N10 is in interaction distance also to both Cs cations (3.668(8) and 3.989(9) Å; list of bond distances and angles in Tables S11 and S12†). However, the geometry of the amine function, and its orientation makes its participation in a true coordinative bond to either of the metal-cations rather improbable.
Comparison with previously-known purine alkali salts
In the di-sodium salt of guanine, the vertices of edge- and corner-sharing polyhedra of sodium are solely formed by the oxygen of water molecules, which in turn hydrogen-bond to the carbonyl-group of guanine (Fig. 2).33 No ring- or amine nitrogen atoms are involved in consolidating the crystal. The absence of a carbonyl group in adenine and high affinity of the strongly polarizing sodium-ion for oxygen coordination may be the reason, no crystalline sodium salt of adenine was obtained in this work.Also the potassium salt of guanine contains water molecules. However, in this structure the cation shows mixed coordination of water and guanine molecules (Fig. 3). It is quite remarkable that one of the two symmetry-independent potassium ions entirely coordinates to oxygen either from water or the guanine carbonyl-group (O6), while the other engages oxygen from water and nitrogen of the heterocycle (N3, N9). While the oxygen atoms from both guanine anions coordinate to cations, the nitrogen atom behavior is different, with one anion coordinating the cation through N3 and N9, while the other only uses N3 for cation coordination. All nitrogen functions of the imidazolium ring (N7, N9) which are not directly coordinating to a potassium ion are involved in hydrogen bonding with the water ligands. The amine function of the guanine is not involved in cation coordination but instead hydrogen-bonding to neighboring guanine amine functions resulting in hydrogen-bonded channels of approximately 4-fold symmetry.34 The potassium and cesium salts of adenine are both anhydrous, implying a low propensity of the adenine anion to form hydrogen bonds with water molecules, unlike what is often observed for guanine. Instead, all heterocycle nitrogen atoms are coordinating potassium ions, forming columns of coordination polyhedra in b-direction (Fig. 4 and 5). Similarly to the case of guanine, N3 and N9 are coordinating to the same cation, N1 and N7 connect to neighboring columns. While the guanine potassium salt is a compromise between affinity of guanine for hydrogen bonding and optimized cation-coordination, the adenine salt is dominated by the latter, while water inclusion is avoided.
This trend continues in the Cs-salt. The large and polarizable Cs-ion shows affinity for coordination by the nitrogen atoms of the heterocycles and expands its coordination number up to eight. All nitrogen atoms of the heterocycle are therefore engaged in direct coordination of cations. Like in the potassium case, in Cs-adenine, the amine function of adenine is involved in hydrogen bonding to N3 and N7 of neighboring molecules. Similar to the potassium salt, an optimization of cation-coordination under avoidance of water molecules is observed.
Quantum-chemical calculations to evaluate the preference of Na+, K+ and Cs+ cation with nucleobase anions
Quantum-chemical calculations were performed to provide qualitative insight into the apparent preference of the sodium ion to coordinate with water or guanylate oxygen atoms, while the potassium and cesium ions prefer to coordinate to nitrogen functions of adenylate directly. This order of preference could be justified in terms of the hardness and softness of the respective systems, with adenylate postulated to be softer than guanylate, while the sodium ion is obviously harder than the potassium or cesium ions. Interestingly, attempts to support this assumption based on computed intrinsic properties of the isolated anions did not show a marked difference between them, similar values of softness were obtained. Instead, to explain the observed differences in relative affinity of the three cations for the two anions interaction energies between the two anions and the Na+, K+ and Cs+ cations are computed and compared.In its neutral form, adenine has one endocyclic –NH group, which is deprotonated in the anion. Guanine has in its main tautomer two endocyclic –NH groups, either of which could in principle be deprotonated in the anion. Out of these possibilities, the guanylate anion formed by deprotonation at N9 is more stable and is the one that has been studied here. The optimized guanylate and adenylate anions are found to be nearly but not quite planar at this level of theory, with the exocyclic nitrogen atoms found to prefer a pyramidal structure. For adenine, the most stable form of the anion–cation complexes is found to be the one chelated by N3 and N9, a coordination encountered, among others, also in the K- as well as in the Cs-salt. For guanine, the most stable form involves chelation by N7 and the oxygen atom. Interestingly this coordination is present neither in the Na-, nor in the K-salts of guanine. Here direct coordination of potassium by the carbonyl group of guanine occurs, while N7 interacts with the K-polyhedra via hydrogen bonding involving a water molecule, while N3 and N9 chelate to a cation, similar as adenine. In the sodium salt no direct interaction between guanine and the cation is observed, all inorganic–organic interactions are mediated by water ligands of the sodium. In the theoretically studied configurations of guanine, the anion is only weakly distorted in the complexes, and the two cation distances to the chelating N and O atoms are roughly equal (Fig. S3†).
The cation affinities of the two anions for the Na+, K+ and Cs+ cations, at the B3LYP-D3BJ and DLPNO-CCSD(T) levels of theory are shown in Table 1.
| B3LYP-D3 | DLPNO-CCSD(T) | ||
|---|---|---|---|
| Adenylate | Na+ | −560.4 | −557.7 |
| K+ | −487.2 | −483.3 | |
| Cs+ | −418.2 | −439.5 | |
| Guanylate | Na+ | −581.9 | −581.8 |
| K+ | −503.8 | −502.5 | |
| Cs+ | −431.4 | −456.7 | |
| M1 = Na+ | M2 = K+ | −4.8 | −4.9 |
| M1 = Na+ | M2 = Cs+ | −8.3 | −6.9 |
| M1 = K+ | M2 = Cs+ | −3.5 | −2.0 |
Based on this, the following exchange reactions between the metal–adenylate complex and metal–guanylate complex have been studied (Table 1).
| Adenylate·M1 + Guanylate·M2 → Adenylate·M2 + Guanylate·M1 | (1) |
| M1 = Na+ or K+ and M2 = K+ or Cs+. |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anion affinities.
:anion affinities.
The exchange reaction (1) is exothermic by −5 kJ mol−1 for M1 = Na+ and M2 = K+ and this indicates that the harder Na+ prefers to be combined with guanylate. The same trend is found when exchanging Na+ and Cs+ or K+ and Cs+. We note that the calculated structures for the 1:1 complexes studied here do not encompass all the structure complexity of the crystals. For example, in the 1:1 ion pair, guanine has a coordination mode that is not found for the crystals, as noted above. The trend of relative affinities from Table 1 should not be sensitive to these detailed aspects.
One might also expect that the observed increased preference for Na+ relative to K+ to form mixed salts with additional water of hydration could be explained by considering that water as a ligand confers a harder character to the metal environment. Computing ligand exchange reactions such as reaction (2) where one of the ligands is a water molecule is however not particularly informative, as error cancellation is less favorable given the difference in charge between neutral water and the anionic purine-derived ligands.
| purine·M1 + M2(H2O) → purine·M2 + M1(H2O) | (2) |
| Na+(aq.) + purine−·K+ + → K+(aq.) + purine−·Na+ | (3) |
Conclusions
The previously unknown crystal structures of K-adenine and Cs-adenine were determined using crystals grown from alkali hydroxide (KOH and CsOH) solutions of adenine. The obtained crystal structures were further compared with sodium and potassium salts of guanine and adenine reported in the literature.31,32 Both K-adenine and Cs-adenine are anhydrous and counter ions (K+ and Cs+) directly coordinate to the ring nitrogens (N1, N3, N7 or N9) of adenine anions (Fig. 4a and b and 5a and b). Cation–anion interactions are dominant in stabilizing the crystal lattice and that the N–H⋯N hydrogen-bonding tendency between the amine and heterocycle nitrogen of the adenine anion aids in the stabilisation of the crystal structure. Both structures exhibit chains of face-sharing coordination polyhedra, with typical coordination numbers of Cs- and K-cations, inferring a structure directing effect of the cations.Both Cs-adenine and K-adenine are anhydrous in nature, which is in marked contrast to the known guanine alkali-salts, wherein hydrogen-bonding between water-protons and heterocycle nitrogen atoms is observed in the guanine potassium-, as well as in the sodium salt. These differences contribute to the unique structural properties of each compound. Moreover, quantum-chemical calculations were attempted to understand the inclination of the sodium, potassium and cesium cations to coordinate or avoid water and the influence of either of adenylate or guanylate on this behavior. Comparison of the cation exchange energies reveal sodium cations prefer to coordinate with water or guanylate whereas potassium and cesium cations prefer adenylate avoiding water interaction. This explains the successful crystallization of potassium and cesium salts of adenine from water, and the failure to obtain crystals of sodium adenylate, or the cesium salt of guanine. Further studies will show if these trends can be used to rationally design new crystalline compounds and co-crystals exploiting the here observed characteristics.
Data availability
CCDC 2364419 and CCDC 2364420 contain the supplementary crystallographic data for K-adenine and Cs-adenine, respectively, for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Interdisciplinary ID-N ESCHER project of KU Leuven. L. V. M. thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035. C. K. and S. K. acknowledge support from NMRCoRe – the NMR/X-ray platform for convergence research (FWO project I001321N).References
- G. A. Soukup, in Encyclopedia of Life Sciences, John Wiley and Sons, 2001, pp. 1–9 Search PubMed.
- A. C. Rios and Y. Tor, Isr. J. Chem., 2013, 53, 469–483 CrossRef CAS PubMed.
- M. Mirzaei, Lab-in-Silico, 2020, 1, 61–66 Search PubMed.
- J. Šponer, J. Leszczynski and P. Hobza, J. Biomol. Struct. Dyn., 1996, 14, 117–135 CrossRef PubMed.
- U. Thewalt, C. E. Bugg and R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2358–2363 CrossRef CAS.
- A. Hirsch, D. Gur, I. Polishchuk, D. Levy, B. Pokroy, A. J. Cruz-Cabeza, L. Addadi, L. Kronik and L. Leiserowitz, Chem. Mater., 2015, 27, 8289–8297 CrossRef CAS.
- K. Guille and W. Clegg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o515–o517 CrossRef PubMed.
- D. Gur, B. Leshem, D. Oron, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2014, 136, 17236–17242 CrossRef CAS PubMed.
- G. S. Oxford, in Proceedings of the 17th European Colloquium of Arachnology, Edinburgh, 1997, pp. 121–131 Search PubMed.
- M. Hanus, M. Kabelác, J. Rejnek, F. Ryjáček and P. Hobza, J. Phys. Chem. B, 2004, 108, 2087–2097 CrossRef CAS.
- M. Hanus, F. Ryjácek, M. Kabelác, T. Kubař, T. V. Bogdan, S. A. Trygubenko and P. Hobza, J. Am. Chem. Soc., 2003, 125, 7678–7688 CrossRef CAS PubMed.
- E. Nir, C. Janzen, P. Imhof, K. Kleinermanns and M. S. De Vries, J. Chem. Phys., 2001, 115, 4604–4611 CrossRef CAS.
- T. Bartl, Z. Zacharová, P. Sečkářová, E. Kolehmainen and R. Marek, Eur. J. Org. Chem., 2009, 2009, 1377–1383 CrossRef.
- J. J. Christensen, J. H. Rytting and R. M. Izatt, Biochemistry, 1970, 9, 4907–4913 CrossRef CAS PubMed.
- M. K. Shukla and J. Leszczynski, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2014 Search PubMed.
- C. Colominas, F. J. Luque and M. Orozco, J. Am. Chem. Soc., 1996, 118, 6811–6821 CrossRef CAS.
- P. O. P. Ts'o, Basic Principles in Nucleic Acid Chemistry V2, Elsevier, 2012, vol. 2 Search PubMed.
- R. M. C. Dawson, D. C. Elliott, W. H. Elliott and K. M. Jones, Data for biochemical research, Clarendon press, 2002 Search PubMed.
- D. Shugar and J. J. Fox, Biochim. Biophys. Acta, 1952, 9, 199–218 CrossRef CAS PubMed.
- D. R. Lide, CRC handbook of chemistry and physics, CRC press, 2004, vol. 85 Search PubMed.
- C. V. Sonntag, in Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective, Springer Science & Business Media, 2006, pp. 211–334 Search PubMed.
- N. S. Venkataramanan, A. Suvitha and Y. Kawazoe, J. Mol. Graphics Modell., 2017, 78, 48–60 CrossRef CAS PubMed.
- T. Eicher, S. Hauptmann and A. Speicher, in The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, 2003, pp. 381–416 Search PubMed.
- P. Kumar, M. K. Cabaj, A. Pazio and P. M. Dominiak, IUCrJ, 2018, 5, 449–469 CrossRef CAS PubMed.
- D. Matković-Čalogović and K. Sanković, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 467–469 CrossRef.
- K. Bouchouit, N. Benali-Cherif, L. Benguedouar, L. Bendheif and H. Merazig, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o1397–o1399 CrossRef CAS.
- J. M. Adams and R. W. H. Small, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2191–2193 CrossRef.
- A. Cherouana, N. Benali-Cherif and L. Bendjeddou, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o180–o182 CrossRef CAS.
- P. Bose, B. N. Ahamed and P. Ghosh, Org. Biomol. Chem., 2011, 9, 1972–1979 RSC.
- M. Li, Y. Zhang, H. Gao, Y. Peng, S. Tang, L. Yu, R. Chen and W. Huang, J. Phys. Chem. C, 2021, 125, 2866–2874 CrossRef CAS.
- D. Gur, M. Pierantoni, N. Elool Dov, A. Hirsh, Y. Feldman, S. Weiner and L. Addadi, Cryst. Growth Des., 2016, 16, 4975–4980 CrossRef CAS.
- N. Nagapradeep, S. Sharma and S. Verma, Cryst. Growth Des., 2013, 13, 455–459 CrossRef CAS.
- D. Gur and L. J. W. Shimon, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, 281–283 CrossRef CAS PubMed.
- A. A. Gaydamaka, S. G. Arkhipov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2021, 77, 808–818 CrossRef CAS.
- D. Dutta, N. Nagapradeep, H. Zhu, M. Forsyth, S. Verma and A. J. Bhattacharyya, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- C. M. Mikulski, L. Mattucci, L. Weiss and N. M. Karayannis, Inorg. Chim. Acta, 1985, 108, L35–L37 CrossRef CAS.
- C. M. Mikulski, L. Mattucci, L. Weiss and N. M. Karayannis, Inorg. Chim. Acta, 1985, 107, 147–152 CrossRef CAS.
- V. Langer, K. Huml and L. Lessinger, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1978, 34, 2229–2234 CrossRef.
- V. Langer and K. Huml, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1978, 34, 1157–1163 CrossRef.
- L. Bendjeddou, A. Cherouana, S. Dahaoui, N. Benali-Cherif and C. Lecomte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o649–o651 CrossRef CAS.
- T. J. Kistenmacher and T. Shigematsu, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1974, 30, 1528–1533 CrossRef.
- G. L. Hardgrove, J. R. Einstein, B. E. Hingerty and C. H. Wei, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 88–90 CrossRef.
- V. Langer, K. Huml and J. Zachova, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1979, 35, 1148–1152 CrossRef.
- T. Murashige, Annu. Rev. Plant Physiol., 1974, 25, 135–166 CrossRef CAS.
- J. H. Miller and E. S. Kempner, Biochim. Biophys. Acta, Spec. Sect. Nucleic Acids Relat. Subj., 1963, 76, 333–340 CrossRef CAS.
- J.-Y. Kohno, F. Mafuné and T. Kondow, Eur. Phys. J. D, 2002, 20, 339–345 CrossRef CAS.
- S. Mahapatra, S. K. Nayak, S. J. Prathapa and T. N. Guru Row, Cryst. Growth Des., 2008, 8, 1223–1225 CrossRef CAS.
- T. Stolar, S. Lukin, J. Požar, M. Rubčić, G. M. Day, I. Biljan, D. Š. Jung, G. Horvat, K. Užarević, E. Meštrović and I. Halasz, Cryst. Growth Des., 2016, 16, 3262–3270 CrossRef CAS.
- S. M. Tret'yak, V. V. t Mitkevich and L. F. Sukhodub, Crystallogr. Rep., 1987, 32, 1268–1771 Search PubMed.
- Z. Li, Z. Mao and Z. Chen, Microchim. Acta, 2019, 186, 1–8 CrossRef PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed.
- P. X. Rojas-González, A. Castiñeiras, J. M. González-Pérez, D. Choquesillo-Lazarte and J. Niclós-Gutiérrez, Inorg. Chem., 2002, 41, 6190–6192 CrossRef PubMed.
- D. M. S. Buyens, L. A. Pilcher and E. Roduner, ChemPhysChem, 2021, 22, 2025–2033 CrossRef CAS PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed.
- M. K. Mishra, S. P. Kelley, V. Smetana, D. A. Dixon, A. S. McNeill, A.-V. Mudring and R. D. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18224–18230 CrossRef CAS PubMed.
- M. Rasmussen and J. M. Hope, Aust. J. Chem., 1982, 35, 525–534 CrossRef CAS.
- M. Rasmussen and J. M. Hope, Aust. J. Chem., 1982, 35, 535–542 CrossRef CAS.
- E. Boichot, J. L. Wallace, N. Germain, M. Corbel, C. Lugnier, V. Lagente and J.-J. Bourguignon, J. Pharmacol. Exp. Ther., 2000, 292, 647–653 CAS.
- D. L. Riley, D. R. Walwyn and C. D. Edlin, Org. Process Res. Dev., 2016, 20, 742–750 CrossRef CAS.
- D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Beilstein J. Org. Chem., 2019, 15, 801–810 CrossRef CAS PubMed.
- S. J. Sowerby, W. M. Heckl and G. B. Petersen, J. Mol. Evol., 1996, 43, 419–424 CrossRef CAS PubMed.
- D. Winter and G. Zubay, Origins Life Evol. Biospheres, 1995, 25, 61–81 CrossRef CAS PubMed.
- J. P. T. Baú, S. A. Villafañe-Barajas, A. C. S. da Costa, A. Negrón-Mendoza, M. Colín-Garcia and D. A. M. Zaia, Astrobiology, 2020, 20, 26–38 CrossRef PubMed.
- E. Largy, J.-L. Mergny and V. Gabelica, The alkali metal ions: their role for life, 2016, pp. 203–258 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, Gaussian 16, Revision A. 03, Gaussian, Wallingford, CT, USA, 2016 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- S. Verma, A. K. Mishra and J. Kumar, Acc. Chem. Res., 2010, 43, 79–91 CrossRef CAS PubMed.
- A. Stachowicz-Kuśnierz and J. Korchowiec, Struct. Chem., 2016, 27, 543–555 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Anisotropic distortion and coordination sphere of Cs2 in Cs-Adenine; ORTEP drawings showing 50% probability ellipsoids of asymmetric units of K-Adenine and Cs-Adenine; optimized geometries of alkali metal nucleobase salts; fractional atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103), anisotropic displacement parameters (Å2 × 103), bond lengths and angles, torsion angles, hydrogen atom coordinates (Å × 104), isotropic displacement parameters (Å2 × 103) for K-Adenine and Cs-Adenine; Cartesian coordinates of adenylate anion, Na-adenylate, K-adenylate, Cs-adenylate, 9H-guanylate anion, 1H-guanylate anion, Na-guanylate, K-guanylate and Cs-guanylate complex. CCDC 2364419 and 2364420. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00892h |
| This journal is © The Royal Society of Chemistry 2024 |