
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urothermal synthesis of metal–organic frameworks
Michaël Teixeira
 and
Stéphane A. Baudron*
and
Stéphane A. Baudron*
Université de Strasbourg, CNRS, CMC UMR 7140, 4 rue Blaise Pascal, F-67000 Strasbourg, France. E-mail: sbaudron@unistra.fr
First published on 1st October 2024
Abstract
While ionothermal synthesis using deep eutectic solvents based on the combination of choline chloride and urea derivatives has been widely explored for metal–organic framework (MOF) synthesis, the alternative approach consisting in using urea derivatives on their own as solvents, albeit promising, remains comparatively underemployed. This highlight article aims to review the field of urothermal synthesis, covering the state of the art of this approach and its potential development. The use of e-urea (2-imidazolidinone, ethyleneurea), the most extensively employed species in this context, is detailed, showing its ability to play diverse roles in MOF construction. Beyond its role as solvent and soft regulator of solution acidity, it can be present in the pore or as a ligand, most commonly in a bridging mode with divalent metal cations via coordination of the carbonyl group assisted by hydrogen bonding of the NH moieties, or yield ethylenediamine as a decomposition product incorporated in the MOF. Furthermore, urothermal synthesis has demonstrated potential for the preparation of chiral architectures and their enantio-enrichment. Alternatives to e-urea in pure form or as a hemihydrate are also presented. The combination of e-urea with other organic solvents or the use of co-ligands have been shown to modulate its tendency to act as a bridging ligand, while fully N-alkylated urea derivatives represent appealing solvents. They have low melting point or can even be liquid at room temperature, making them media of choice, prone to ligation to the metal center in a terminal fashion given the absence of hydrogen bonding donor, favoring removal towards activation. The structures of the materials reported under urothermal conditions are described as well as their properties and applications.
 Michaël Teixeira (left) and Stéphane A. Baudron (right) | Michaël Teixeira obtained his Master's Degree in Green Chemistry at the University of Strasbourg (France), where he is currently a PhD student. His main research interest is the synthesis of metal–organic frameworks in deep eutectic solvents and urea derivative-based solvents. After his PhD from the University of Angers (France), Stéphane A. Baudron did a post-doctoral stay at U.C. Berkeley (USA) under the Lavoisier Fellowship Programme. He was appointed CNRS researcher in Strasbourg (France) in 2004, where he obtained his habilitation in 2009 and was promoted Research Director in 2019. He has been the recipient of a JSPS fellowship (Kyoto) and of a USIAS fellowship. His research focusses on coordination and supramolecular chemistry and on the development of reaction media for material synthesis. |
Introduction
In solution chemistry, the solvent plays a key role in determining the nature, purity, yield and potentially properties of the product. This holds true in many fields such as organic, supramolecular, organometallic and coordination chemistry as well as in materials science. For example, metal–organic frameworks (MOFs)1–4 are predominantly prepared following a solvothermal method consisting in heating a mixture of building blocks in a solvent under autogenous pressure.5–7 The latter medium can impact the final composition, purity, yield, crystallinity, crystal morphology and textural properties of porous materials. As MOFs have many applications spanning from gas sorption and storage, to catalysis and biology, their preparation is the object of intense research.5–7 In particular, efforts are being made to improve the sustainability and environmental impact of the synthetic strategy,8–15 with special attention towards the replacement of toxic and/or flammable solvents used in the solvothermal method by alternative media. In this context, the ionothermal method consisting in the use of an ionic medium as a solvent has been proposed.16–24 Extensively employed with ionic liquids, deep eutectic solvents (DESs)25–31 have been also considered. DESs result from the combination of two or more solid materials, commonly a hydrogen bond donor/acceptor, leading to a fluid mixture featuring a freezing temperature substantially lower than the one of its components and the one expected for the ideal mixture. Since they were first described in 2001,25 DESs have been widely studied, taking advantage of their low vapor pressure, relatively wide liquid range, non-flammability and ability to dissolve polar species.27–31 While many mixtures of diverse compounds have been reported to form DESs, the combinations of choline chloride (ChCl) with urea derivatives (Fig. 1) have particularly demonstrated their ability to act as solvents for MOF preparation.24 They have been shown to play different roles such as structure-directing agents acting as ligands or occupying the pores,32 stabilizing materials with metal-halide bonds prone to hydrolysis33,34 or water-sensitive MOFs,35 to induce the conversion from porous to non-porous architectures36 and to impact morphology and porosity.37 Interestingly, some urea derivatives have a low to moderate melting point in their pure form or as a hydrate allowing their use as solvents on their own rather than as a DES mixture with choline chloride. This solvothermal approach mediated by urea derivatives, coined urothermal synthesis, has been introduced by Bu and coworkers who reported, in a seminal article, the preparation of a wide range of MOFs.38 As for the ionothermal method, the urea derivative impacts the preparation, structure and properties of the MOFs synthesized under urothermal conditions. In particular, its decomposition that has been demonstrated to proceed via diverse and complex pathways39–42 allows the release of ammonia or amines, hence modifying the pH of the reaction medium. Its role as a soft regulator of solution acidity is key for the deprotonation of organic ligands, commonly carboxylic acid derivatives favoring metal coordination, and the subsequent formation of MOFs, paralleling solvothermal synthesis in DMF or DEF for example. This synthetic approach has been applied to a diversity of urea species, organic ligands and metal centers demonstrating its scope and the aim of this highlight article is to provide an overview of this field. The preparation of MOFs with di- or trivalent metal cations is reviewed, highlighting the impact of the solvent on the structure and the properties of the material. In particular, the influence of its nature and potential functionalization on coordination and on the resulting stability and potential porosity of MOFs is detailed (Table 1). | ||
| Fig. 1 The different urea derivatives and the organic ligands used in MOF urothermal synthesis discussed in this highlight. | ||
| MOF formula | Reaction conditions (solvent, T, time) | Dimensionality | Coordinating mode of urea derivative | Ref. |
|---|---|---|---|---|
| [Cd(bdc)(e-urea)] | e-urea, 110 °C, 3 days | 3 | Bridging | 43 |
| [M(1,4-ndc)(e-urea)] (M = Cd, Co, Mn) | e-urea, 120 °C, 3 days | 3 | Bridging | 45 |
| [Cd(1,3-bdc)(e-urea)] (URO-110) | e-urea, 140 °C, 4 days | 3 | Bridging | 38 |
| [Cd(1,3-bdc-NO2)(e-urea)] | (e-urea)(H2O)0.5, 120 °C, 3 days | 3 | Bridging | 46 |
| [Cd(1,3-bdc-OH)(e-urea)]2(en) | (e-urea)(H2O)0.5, 120 °C, 3 days | 3 | Bridging | 46 |
| [Cd2(tdc)2(en)(e-urea)2](e-urea)2 (URO-162) | e-urea, 140 °C, 4 days | 2 | Terminal/free | 38 |
| [Co(bdc)(e-urea)](e-urea) | (e-urea)(H2O)0.5, 140 °C, 4 days | 3 | Bridging/free | 50 |
| [Mn2(bptc)(e-urea)2] | e-urea/DMA/H2O, 100 °C, 2 days | 3 | Bridging | 51 |
| [Zn3(Hbptc)2(e-urea)2](e-urea)2 | e-urea, 120 °C, 2.5 days | 3 | Terminal/free | 59 |
| [Cd3(btc)2(e-urea)4] | e-urea/DMF/H2O, 110 °C, 2 days | 3 | Bridging/terminal | 60 |
| [Cd3(bdc)2(e-murea)(DMF)](e-murea) | e-murea/DMF, 110 °C, 3 days | 3 | Terminal/free | 61 |
| [Cd(bdc)(p-murea)2] (URO-42) | p-murea, 120 °C, 4 days | 1 | Terminal | 38 |
| [Zn2(bdc)2(H2O)2](tm-urea) (URO-40) | tm-urea/H2O, 120 °C, 4 days | 2 | Free | 38 |
| [Zn2(bdc)2(p-murea)2] (URO-41) | p-murea, 120 °C, 4 days | 2 | Terminal | 38 |
| [Zn2(1,4-ndc)2(tm-urea)2] (URO-151) | tm-urea, 120 °C, 4 days | 2 | Terminal | 38 |
| [Zn4(OH)2(1,2,4-btc)2(H2O)](p-murea)(URO-132) | p-murea/H2O, 140 °C, 3 days | 3 | Free | 38 |
| [Zn4O(btb)2](p-murea)x (U-MOF-177) | p-murea/H2O, 120 °C, 4 days | 3 | Free | 38 |
| [Mn3(tpa)2(H2O)(e-murea)2](MeCN)7(e-murea)9 (FIR-36) | e-murea/MeCN, 140 °C, 5 days | 3 | Terminal/free | 65 |
| [Zn2(1,3-bdc)(trz)2](e-urea)2 | e-urea/DMF, 110 °C, 3 days | 3 | Free | 68 |
| [Zn2(bdc-NH2)(trz)2](e-urea)2 | e-urea, 120 °C, 3 days | 3 | Free | 69 |
| [Zn4(bdc)2(trz)4(H2O)(e-urea)](e-urea) | e-urea, 120 °C, 3 days | 3 | Terminal | 69 |
| [Co5(OH)2(H2O)2(3,4′-bpdc)4(bib)2](H2O)2 | p-murea/H2O, 160 °C, 3 days | 3 | — | 70 |
| [Co2(OH)(3,4′,5-bpdc)(bib)]H2O | p-murea/H2O, 160 °C, 3 days | 3 | — | 70 |
| [Zn3I2(tib)2(1,4-ndc)2](e-murea)2(H2O)2 | e-murea/H2O, 160 °C, 3 days | 3 | Free | 71 |
| [Co3O(tib)(1,3-bdc-NH2)2(e-murea)](e-murea)2(H2O)2 | e-murea/H2O, 180 °C, 3 days | 3 | Terminal/free | 71 |
| [Co3(tib)2(1,3-bdc-NH2)2(ox)]2(e-murea)(H2O) | e-murea/H2O, 180 °C, 3 days | 3 | Free | 71 |
| [Co(bdc)(3-pybid)2(H2O)2](e-urea)4 | e-urea, 120 °C, 4 days | 1 | Free | 72 |
| [Zn3(1,3-bdc-SO3)2(tmdpy)2(e-urea)4(H2O)2](e-urea)2 | e-urea, 120 °C, 3 days | 1 | Terminal/free | 73 |
| [Y2(bdc)3(e-urea)4] | (e-urea)(H2O)0.5, 120 °C, 4 days | 3 | Terminal | 38 |
| [Ln2(bdc)3(e-urea)4] | ||||
| (Ln = Ce, Sm, Eu, Gd, Dy, Ho, Er, Yb) | ||||
| [Y2(bdc)3(e-urea)2(H2O)2] | (e-urea)(H2O)0.5, 140 °C, 4 days | 3 | Terminal | 38 |
| [Ln2(bdc)3(e-urea)2(H2O)2] | ||||
| (Ln = Ce, Sm, Eu, Gd, Dy, Ho, Er, Yb) | ||||
| [Y2(bdc)3(p-urea)2] | p-urea/DEF, 120 °C, 4 days | 3 | Terminal | 38 |
| [Y2(bdc)3(e-urea)2(p-murea)2] | e-urea/(p-murea, DMF, DEF), 120 °C, 4 days | 3 | Terminal | 38 |
| [Y2(bdc)3(e-urea)2(DMF)2] | ||||
| [Y2(bdc)3(e-urea)2(DEF)2] | ||||
| [Ln2(bdc)3(e-urea)2] (Ln = La, Pr, Nd) | e-urea, 140 °C, 4 days | 3 | Bridging | 38 |
| [Ce2(bdc)3(H2O)4] | (e-urea)(H2O)0.5/H2O, 120 °C, 5 days | 3 | — | 78 |
| [La2(2,6-ndc)3(e-urea)3] | e-urea, 120 °C, 3 days | 3 | Bridging/terminal | 80 |
| [La2(2,6-ndc)3(e-urea)2] | e-urea, 140 °C, 3 days | 3 | Terminal | 80 |
| [Yb2(obb)3(e-urea)] (URO-130) | e-urea, 120 °C, 4 days | 3 | Terminal | 38 |
| [Ln2(obb)3(e-urea)(H2O)] (Ln = Er, Yb, Ho) | (e-urea)(H2O)0.5, 140 °C, 5 days | 3 | Terminal | 81 |
| [Ln2(obb)3(e-urea)(H2O)3] (Ln = Tb, Dy, Eu) | (e-urea)(H2O)0.5, 140 °C, 5 days | 2 | Terminal | 81 |
| [La2(obb)3(e-urea)(H2O)1.5] | (e-urea)(H2O)0.5, 140 °C, 5 days | 2 | Terminal | 81 |
| [Sm2(obb)3(H2O)5](H2O) | (e-urea)(H2O)0.5, 140 °C, 5 days | 3 | — | 81 |
MOFs based on divalent metal cations
MOFs prepared from e-urea
Among the different urea-based species explored in the literature for urothermal synthesis, e-urea (2-imidazolidinone, ethyleneurea, Fig. 1) has been the most extensively used, either in solution, or in pure (m.p. = 131 °C) or hemihydrate (m.p. = 58 °C) form. The lower melting point of this latter form makes it a practical solvent for MOF synthesis. The key features of e-urea are the coordinating carbonyl group and the two NH moieties oriented in such a way that they can assist coordination via a hydrogen bond, as a result of the presence of the ethylene bridge.Cd-MOFs incorporating carboxylate ligands have been particularly investigated via urothermal synthesis in e-urea. For example, Liu and coworkers reported the synthesis of [Cd(bdc)(e-urea)] (Fig. 2a) by reaction of Cd(NO3)2·6H2O with H2bdc in e-urea at 110 °C for 3 days.43 In this MOF, the Cd(II) center is hepta-coordinated, bound to the carboxylate moieties of the ligand and to the bridging carbonyl group of e-urea molecules forming a one-dimensional secondary building unit (SBU).44 Coordination of e-urea is assisted by hydrogen bonding of the NH groups to the neighboring carboxylates. This leads to a three-dimensional network with channels occupied by the coordinated e-urea molecules. It is worth noting that, although the solvent molecules act as bridging ligands, their removal upon heating the material at 350 °C under vacuum for 48 h did not lead to a loss of crystallinity as demonstrated by X-ray powder diffraction. A similar behavior could be observed for [Cd(1,4-ndc)(e-urea)] (Fig. 2b) prepared from e-urea hemihydrate, featuring a SBU based on octahedral Cd(II) cations with bridging carboxylates and solvent molecules.45 Interestingly, while this compound could be thermally activated leading to retained crystallinity upon removal of e-urea as for [Cd(bdc)(e-urea)], the isostructural Mn(II) and Co(II) MOFs were found to decompose under these conditions.45 For both [Cd(bdc)(e-urea)] and [Cd(1,4-ndc)(e-urea)], an intraligand emission is observed in the solid state upon excitation at 342–350 nm.43,45 In their thermally activated form, these two MOFs showed a CO2 uptake at 273 K under 1 atm of 16.00 and 13.6 cm3 g−1 respectively. The Bu group has reported [Cd(1,3-bdc)(e-urea)], named URO-110, obtained by reaction in pure e-urea at 140 °C for 4 days (Fig. 2c),38 while Yang, Ling and coworkers have later described the isostructural [Cd(1,3-bdc-NO2)(e-urea)] prepared from e-urea hemihydrate.46 The SBUs for both these MOFs are analogous to the one observed in [Cd(bdc)(e-urea)] leading to the same overall 3D organization (Fig. 2). The thermogravimetric analysis and emission properties of [Cd(1,3-bdc-NO2)(e-urea)]46 are consistent with the two previous MOFs.43,45 [Cd(1,3-bdc)(e-urea)] and [Cd(1,3-bdc-NO2)(e-urea)] are isostructural to the α polymorph of [Ca(1,3-bdc)(e-urea)] prepared from the ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :e-urea (1:2) DES.35
:e-urea (1:2) DES.35
 | ||
| Fig. 2 View of the crystal structures and SBU of [Cd(bdc)(e-urea)] (a), [Cd(1,4-ndc)(e-urea)] (b) and [Cd(1,3-bdc)(e-urea)] (c). | ||
When substituting 1,3-bdc-OH for 1,3-bdc-NO2, [Cd(1,3-bdc-OH)(e-urea)]2(en) was isolated.46 This MOF is isostructural to the two previous analogues based on 1,3-bdc type ligands but incorporates ethylenediamine (en) (Fig. 3). Decomposition of urea derivatives has been well documented upon heating DESs47 and has been shown to impact the formation of MOFs.37,48 Under urothermal conditions, e-urea has been found to lead to the release of en that may be present in the final MOF structure, playing different roles.38,48,49 It can be a guest in the pores as in [Cd(1,3-bdc-OH)(e-urea)]2(en), or be coordinated to the metal center either as a bridging or chelating ligand (Fig. 3a). For example, Bu and coworkers reported URO-162, formulated as [Cd2(tdc)2(en)(e-urea)2](e-urea)2, where the Cd(II) cations are bridged in one direction by chelating carboxylates of the tdc ligand and en in another direction leading to a 2D arrangement (Fig. 3b).38 The coordination sphere is completed by an e-urea molecule. In [Cu(1,4-ndc)(en)](e-urea)(H2O) prepared from e-urea hemihydrate at 120 °C for 3 days, en chelates the metal cation and the solvent molecule is present in the pores of this 3D MOF (Fig. 3c).49
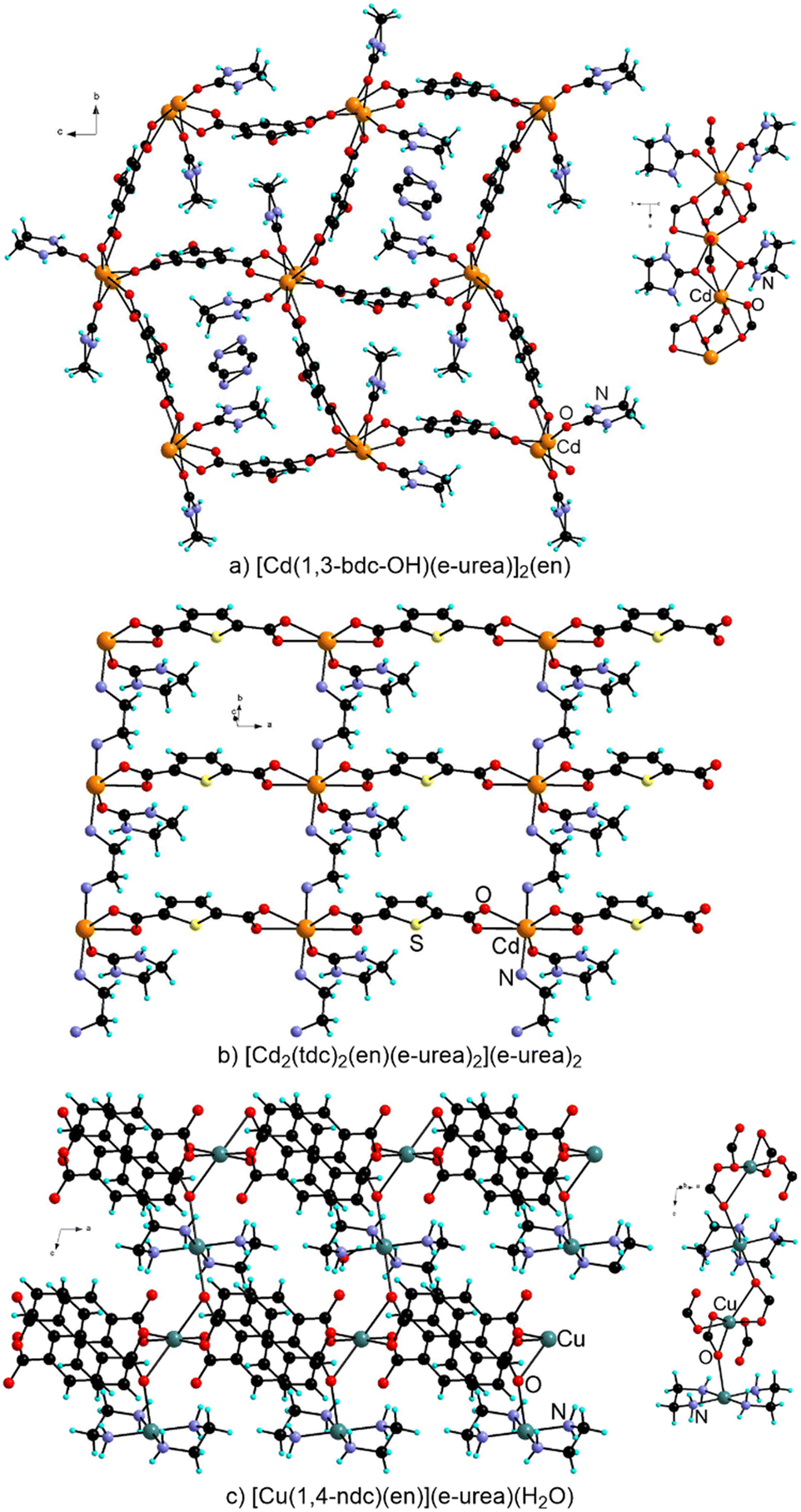 | ||
| Fig. 3 View of the crystal structure and metal nodes in MOFs prepared under urothermal conditions and incorporating en resulting from decomposition of e-urea: [Cd(1,3-bdc-OH)(e-urea)]2(en) (a), [Cd2(tdc)2(en)(e-urea)2](e-urea)2 (b) and [Cu(1,4-ndc)(en)](e-urea)(H2O) (c). For the latter structure, the e-urea and water solvate molecules have been omitted for clarity. | ||
Beyond just decomposition, the reactivity of e-urea has been observed with D-camphoric acid in the presence of CuI at 120 °C for 4 days.38 The product of this reaction is a molecular compound, named URO-502, based on a cubane like Cu4I4 core coordinated to a chiral ligand formed in situ (Fig. 4). While the chirality of this molecular complex emerges in this case from the reaction of the enantiopure ligand with e-urea, asymmetric induction has been also observed in the MOF via urothermal synthesis.50,51
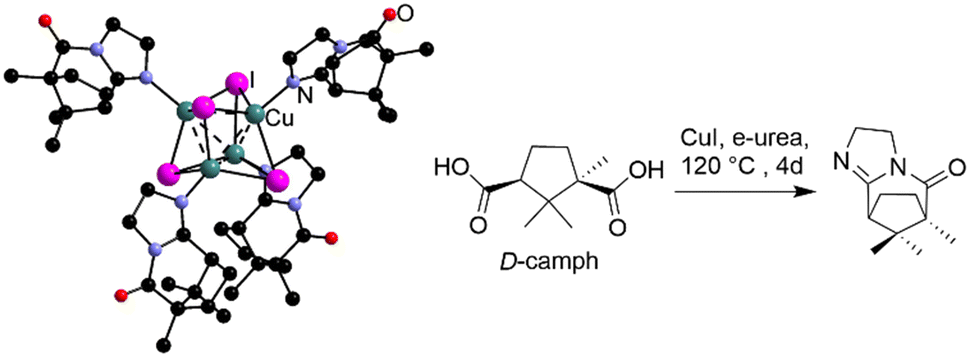 | ||
| Fig. 4 Representation of the molecular Cu compound obtained by reaction of CuI with D-camphoric acid in e-urea and featuring a chiral ligand formed in situ. Hydrogen atoms have been omitted for clarity. | ||
The Bu group has reported [Co(bdc)(e-urea)](e-urea) prepared from e-urea hemihydrate at 140 °C and found to crystallize in the P4122 chiral space group.50 In this material, the Co(II) cations are bridged by carboxylates and e-urea molecules leading to the 41 helical arrangement (Fig. 5a). While each crystal is enantiopure, the overall batch is a conglomerate. In order to favor chiral induction, enantiopure additives can be employed or, alternatively, chiral solvents can be considered, as demonstrated for hydro-, solvo- and ionothermal syntheses.18,52–58 Considering e-urea as a template in the MOF formation, Bu and coworkers have proposed to employ achiral urea derivatives with enantiopure species incorporating an analogous coordinating motif such as camphor or carvone as a solvent for induction under urothermal conditions.50 Chirality transfer was indeed found to operate successfully upon using a 4.3:1 mixture of e-urea with (−)-carvone, as confirmed by structure determination of five randomly selected single-crystals showing the same handedness as well as by solid state circular dichroism by comparing with the material obtained when using (+)-carvone. Interestingly, Wang and Li have reported [Mn2(bptc)(e-urea)2] obtained from e-urea, crystallizing in the same P4122 chiral space group and featuring analogous metallo-organic helical chains with bridging carboxylates and solvent molecules (Fig. 5b).51 While chiral induction for this MOF has not been further investigated, its textural properties were characterized. A BET specific surface area of 729 m2 g−1 was calculated from the N2 sorption–desorption isotherm at 77 K. At 273 K, adsorption values of 52.2 and 13.9 cm3 g−1 were found respectively for CO2 and CH4, with a selectivity of 7.8 for the former gas over the latter.51 Furthermore, cytotoxicity against HeLa and Hep 32 cancer cells was studied by the MTT assay method showing an activity comparable to cisplatin, whereas neither the ligand nor MnCl2 showed any significant activity. It is worth noting that using the same ligand, Zhou et al. obtained the centrosymmetric [Zn3(Hbptc)2(e-urea)2](e-urea)2.59 In this achiral MOF, e-urea is coordinated to the metal cation and present in the pores. This material could be thermally activated at 420 °C under vacuum while retaining its crystal structure and showed a BET specific surface area of 425 m2 g−1 by analysis of its N2 sorption isotherm at 77 K. Other gases were explored indicating no uptake for H2 at 273 K and a moderate uptake of 65 cm3 g−1 for CO2.59 Furthermore emission at 415 nm was observed upon excitation of the solid at 340 nm, corresponding to ligand-centered luminescence.
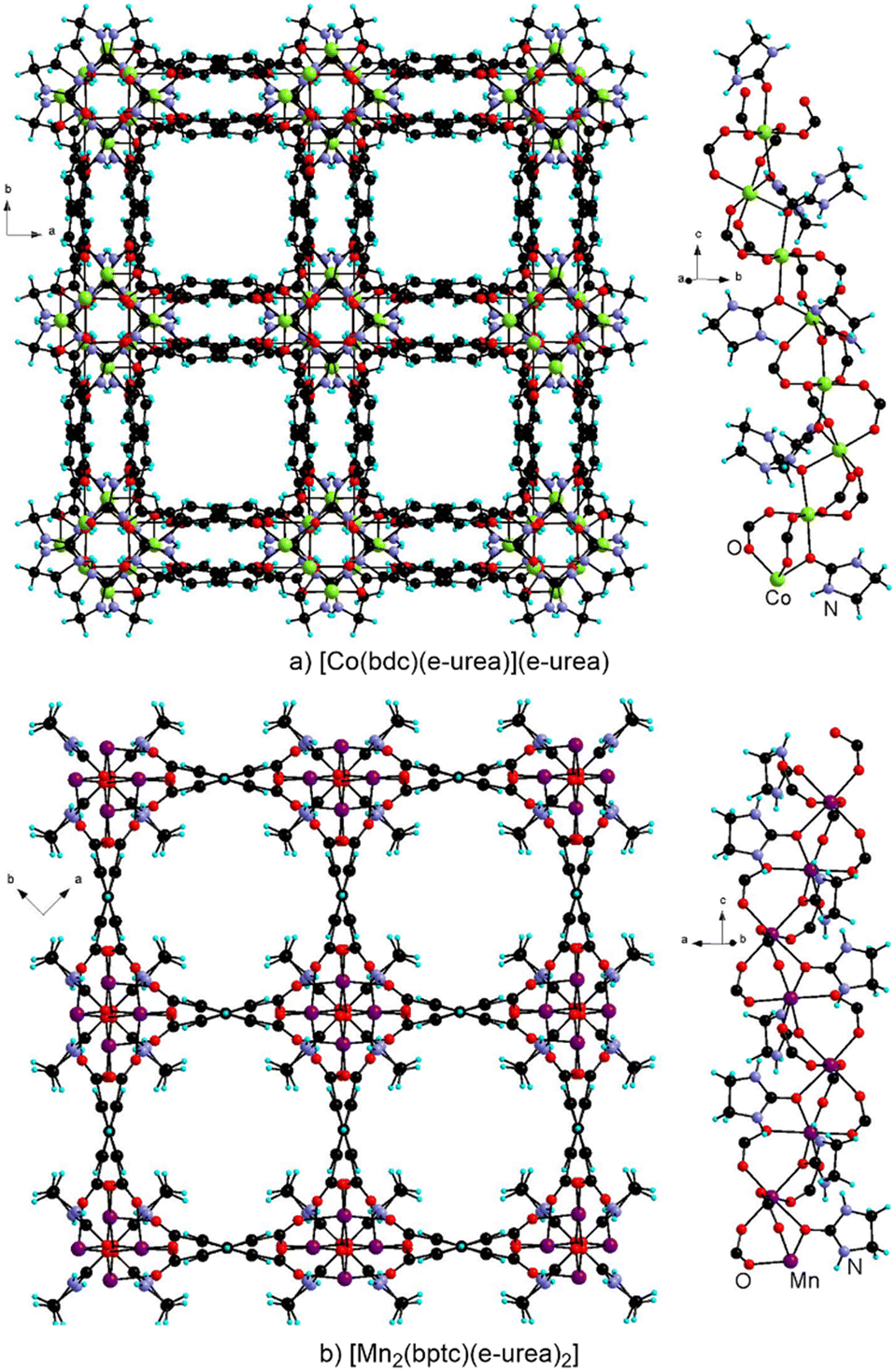 | ||
| Fig. 5 Crystal structure and view of the helical chains in [Co(bdc)(e-urea)](e-urea) (a) and [Mn2(bptc)(e-urea)2] (b). | ||
MOFs prepared from N-alkylated urea derivatives
It is remarkable that, in the vast majority of the examples of MOFs prepared by urothermal synthesis in e-urea presented so far, the latter solvent is coordinated to the metal cation, most often as a bridging ligand. This coordination motif assisted by hydrogen bonding is recurrent and, while it has a positive structuring impact, it can sometimes lead to issues when trying to activate the materials to access their porosity. Other SBUs can be observed upon modifying the synthetic conditions, such as the addition of an organic solvent or water and use of N-alkylated urea derivatives or of co-ligands. These MOFs will now be detailed.Using a mixture of e-urea, DMF and water as a solvent, Zhang and coworkers have prepared the [Cd3(btc)2(e-urea)4] MOF, emitting at 413 nm upon photoexcitation at 325 nm.60 It is based on a trinuclear SBU incorporating e-urea molecules as bridging ligands as well as in the terminal position (Fig. 6a). The bridging position is quite favored, as it is also stabilized by hydrogen bonding of the NH moieties with the neighboring carboxylates. N-Alkylated urea derivatives (Fig. 1) are particularly interesting as solvents, as they have low melting points (tm-urea, m.p. = −1 °C; e-murea, m.p. = 8 °C; p-murea, m.p. = −24 °C) and do not incorporate hydrogen bond donors, thus favoring a terminal position in the coordination sphere of the metal cation.
 | ||
| Fig. 6 Crystal structure and SBU of the [Cd3(btc)2(e-urea)4] (a), [Cd3(bdc)2(e-murea)(DMF)](e-murea) (b), [Cd(bdc)(p-murea)2] (c) MOFs. | ||
For example, using a mixture of e-murea and DMF as a solvent led to [Cd3(bdc)2(e-murea)(DMF)](e-murea) (Fig. 6b).61 It is built around a hexanuclear SBU with bridging carboxylates and terminal DMF and e-murea molecules. Removal of the latter towards the study of their textural properties has not been described for this emissive material (λem = 523 nm upon excitation at 330 nm).61 The Bu group has described [Cd(bdc)(p-murea)2] (Fig. 6c) prepared from pure p-murea at 120 °C.38 This compound features chains with mononuclear Cd(II) cations coordinated to carboxylates and terminal p-murea. These last two MOFs can be put in perspective with [Cd(bdc)(e-urea)] (Fig. 2) prepared with the same ligand and metal cation from e-urea but featuring bridging solvent molecules.43
The impact of the additional solvent and alkylation of the urea derivatives can be further highlighted in a series of MOFs based on Zn(II) and the bdc or 1,4-ndc ligands (Fig. 7).38 [Zn2(bdc)2(H2O)2](tm-urea), URO-40, [Zn2(bdc)2(p-murea)2], URO-41, and [Zn2(1,4-ndc)2(tm-urea)2], URO-151, all feature a two-dimensional layer reminiscent of MOF-262 but with varying solvent molecules in the axial position of the paddlewheel unit (Fig. 7). While URO-41 and URO-151 feature the urea derivative used in pure form as a solvent, water, used in a mixture with tm-urea as a reaction medium, is present in this position for URO-40 (Fig. 7).
 | ||
| Fig. 7 View of the two-dimensional layers in [Zn2(bdc)2(H2O)2](tm-urea) (a), [Zn2(bdc)2(p-murea)2] (b) and [Zn2(1,4-ndc)2(tm-urea)2] (c). For [Zn2(bdc)2(H2O)2](tm-urea), the tm-urea solvate molecules present between the layers have been omitted for clarity. | ||
Interestingly, as the addition of water or use of methylated urea species as a reaction mixture leads to materials where the solvent is a terminal ligand, known MOF arrangements otherwise obtained under solvothermal conditions can also be isolated via urothermal synthesis, thereby providing an alternative synthetic approach. For example, [Zn4(OH)2(1,2,4-btc)2(H2O)](p-murea) is isostructural to a MOF prepared from DEF/EtOH/H2O and [Zn4O(btb)2](p-murea)x is isostructural to MOF-177 prepared from DEF.63,64
A recent study by Xu, Zhang and coworkers showed that reacting MnCl2·4H2O with H3tpa (Fig. 1) in pure e-urea or a 2:1 e-murea/MeCN mixture led to MOFs with either low porosity or mesopores respectively.65 Indeed, with e-urea, bridging solvent molecules are observed, whereas e-murea molecules are present as terminal ligands and in the pores in [Mn3(tpa)2(H2O)(e-murea)2](MeCN)7(e-murea)9 (Fig. 8). Upon activation by immersion in MeCN followed by evacuation under vacuum at 50 °C, a distortion of the structure is observed by X-ray powder diffraction. For the activated MOF, a BET specific surface of 595 m2 g−1 was calculated from the N2 sorption isotherm at 77 K and a H2 uptake capacity of 134.5 cm3 g−1, corresponding to 1.20 wt%, was measured at 77 K and 1 bar.65
 | ||
| Fig. 8 View of the mesopores and SBU in [Mn3(tpa)2(H2O)(e-murea)2](MeCN)7(e-murea)9. The solvate molecules have been omitted and only the oxygen atom of the coordinated e-murea is presented, for clarity. | ||
It can also be noted that the e-murea and p-murea have also been employed in the rare examples of Li- and Ca-MOFs prepared under urothermal conditions.38,66,67
Mixed ligand-MOFs
The preparation of MOFs based on the combination of different ligands has also been explored under urothermal conditions. In these materials, the presence of the additional coordinating unit greatly impacts the arrangement of the SBU, as expected, and the binding mode of the urea-based species is modified. Remarkably, the majority of these materials result from the combination of a carboxylic acid bearing ligand with an azole-based derivative.Qiao and An have described the preparation of [Zn2(1,3-bdc)(trz)2](e-urea)2, from e-urea and DMF.68 This three-dimensional MOF is based on a layer formed by coordination of the triazolate, while the carboxylate-based ligand acts as a pillar (Fig. 9a). A similar organization is observed in the crystal structure of [Zn2(bdc-NH2)(trz)2](e-urea)2 (Fig. 9b) prepared from pure e-urea at 120 °C for 3 days.69 For both MOFs, e-urea is present in the pores and not coordinated to the Zn(II) cation. Under the same conditions, [Zn4(bdc)2(trz)4(H2O)(e-urea)](e-urea) (Fig. 9c), with a pillar-layer type structure, albeit with a different organization of the two-dimensional sheet, is isolated.69 Indeed, in this case, both e-urea and water are coordinated as terminal ligands to the metal center. All three MOFs are luminescent in the solid state, with a ligand-centered emission ranging from 422 to 468 nm upon excitation around 325–330 nm.68,69
 | ||
| Fig. 9 View of the crystal structure and of the coordination sphere in [Zn2(1,3-bdc)(trz)2](e-urea)2 (a), [Zn2(bdc-NH2)(trz)2](e-urea)2 (b) and [Zn4(bdc)2(trz)4(H2O)(e-urea)](e-urea) (c). The uncoordinated e-urea solvate molecules have been omitted for clarity. | ||
Lin and coworkers have investigated the formation of Co-MOFs in H2O:p-murea mixtures using the bib and either the H23,4′-bpdc or H33,4′,5-bpdc carboxylic acid-based ligands (Fig. 1).70 For both materials, p-murea is not present, neither bound to the Co cation nor in the pore. [Co5(OH)2(H2O)2(3,4′-bpdc)4(bib)2](H2O)2 (Fig. 10) is built around a pentanuclear SBU where the metal cations are bridged by water molecules, hydroxide and carboxylate anions, and are coordinated to the azole from the bib ligand. A study of the magnetic susceptibility indicated the presence of antiferromagnetic interactions as χMT goes from 12.08 cm3 K mol−1 at 300 K to 0.86 cm3 K mol−1 at 2 K. It obeys the Curie–Weiss law with C = 14.62 cm3 K mol−1 and θ = −54.25 K. In the same report, MOF [Co2(OH)(3,4′,5-bpdc)(bib)]H2O was also described.70 The SBU for this MOF is a tetranuclear unit obtained by bridging of hydroxide and carboxylate anions that leads to two-dimensional layers pillared by the diazole ligand (Fig. 10). As for the previous material, the temperature dependence of magnetic susceptibility could be modelled using the Curie–Weiss law with C = 4.16 cm3 K mol−1 and θ = −81.75 K. This MOF was carbonized at 900 °C under argon and then etched to remove Co traces. The resulting heteroatom-doped material was found to be electrocatalytically active for the oxygen reduction reaction in alkaline medium. Using the tridentate tib co-ligand in 1:1 H2O:e-murea, the same authors have reported a series of Zn and Co-MOFs, for which the urea derivative does not act as a bridging moiety, [Zn3I2(tib)2(1,4-ndc)2](e-murea)2(H2O)2, [Co3O(tib)(1,3-bdc-NH2)2(e-murea)](e-murea)2(H2O)2 and [Co3(tib)2(1,3-bdc-NH2)2(ox)]2(e-murea)(H2O).71 The latter material, presented in Fig. 11, was activated by solvent exchange with MeOH followed by heating at 180 °C under vacuum. For this MOF, a SBET specific surface area of 700 m2 g−1 was determined from the N2 sorption isotherm at 77 K. A study of the sorption capacity of this compound towards small hydrocarbons indicated better performance towards C2H2 and C2H4 compared with C2H6 and CH4.71 These results were rationalized by DFT calculations suggesting the preferred interactions of the unsaturated compounds with the aromatic system of the 1,3-bdc-NH2 ligand.71
 | ||
| Fig. 10 View of the crystal structure and of the metal node in [Co5(OH)2(H2O)2(3,4′-bpdc)4(bib)2](H2O)2 (a) and [Co2(OH)(3,4′,5-bpdc)(bib)]H2O (b). Water solvate molecules have been omitted for clarity. | ||
 | ||
| Fig. 11 View along the c axis of the crystal structure of [Co3(tib)2(1,3-bdc-NH2)2(ox)]2(e-murea)(H2O) and its metal node. The molecules present in the pores are not presented for clarity. | ||
Using pyridine-based co-ligands in e-urea, two MOFs have been described, [Co(bdc)(3-pybid)2(H2O)2](e-urea)4 and [Zn3(1,3-bdc-SO3)2(tmdpy)2(e-urea)4(H2O)2](e-urea)2.72,73 In both systems, e-urea is present in the pores, and in the latter material, also coordinated as a terminal ligand to the Zn cation. The Co-MOF was found to allow the degradation of methylene blue,72 while the Zn-based material emits at 435 nm upon excitation at 350 nm.73
It is striking to note that, overall, in the presence of a competing co-ligand, the urea solvent does not act as a bridging ligand in any of these MOFs described in this section and, when present, occupies the pore or is bound as a terminal ligand.
MOFs based on trivalent metal centres
A similar behavior is observed when preparing MOFs with trivalent metal centers under urothermal conditions, with only scarce occurrences of bridging coordination for the urea derivatives in these materials.The Bu group reported a thorough and systematic study of the preparation of Y-MOFs using various urea species either in pure form or as mixtures with organic solvents such as DMF and DEF (Fig. 12).38 When performing the reaction in e-urea hemihydrate at 120 and 140 °C, [Y2(bdc)3(e-urea)4] and [Y2(bdc)3(e-urea)2(H2O)2] were obtained respectively. In both compounds, e-urea acts as a pendant ligand occupying the pores (Fig. 12). An increase of temperature leads to the presence of water rather than e-urea in the Y(III) coordination sphere.
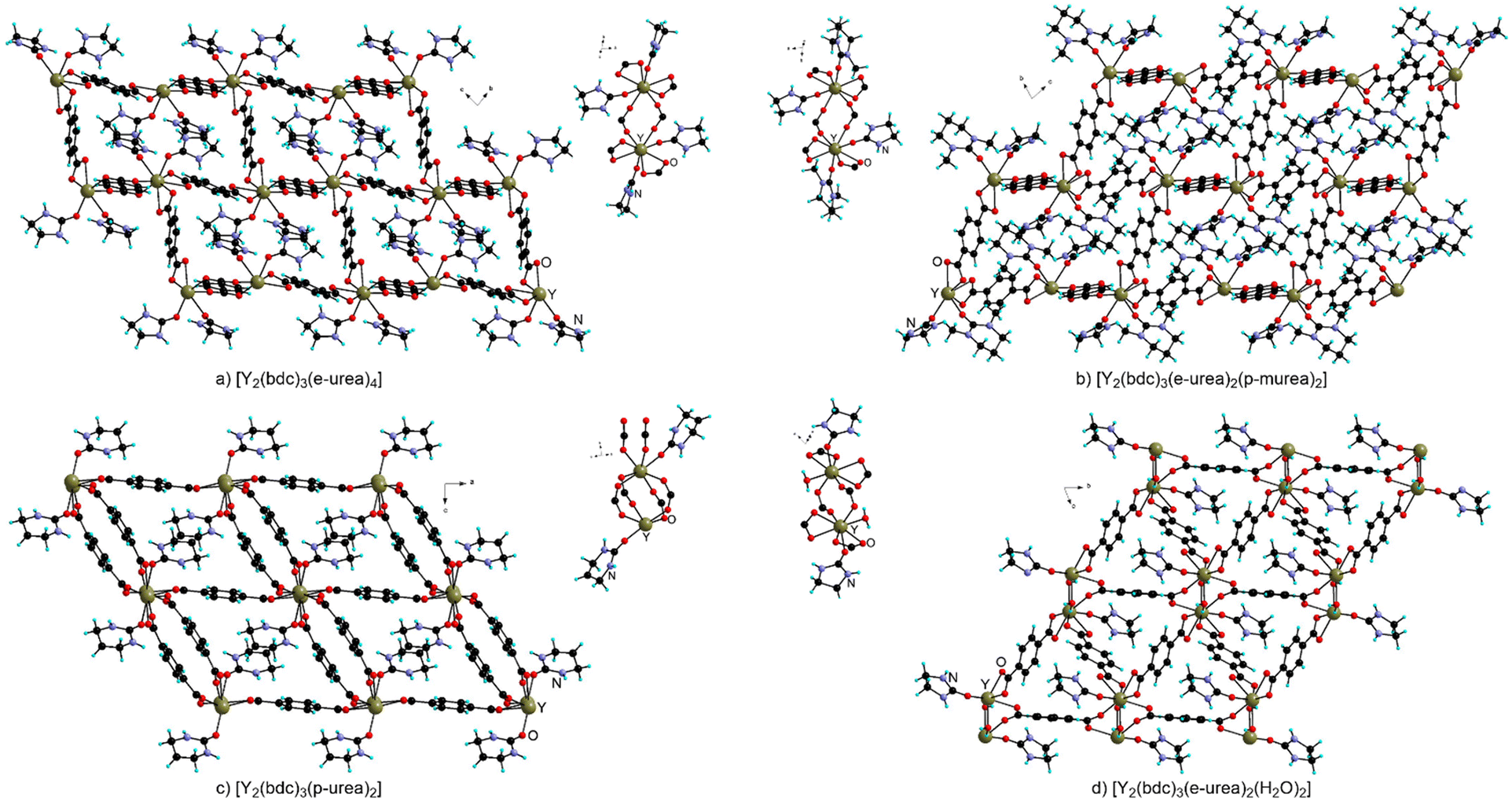 | ||
| Fig. 12 View of the crystal structure and of the metal node in [Y2(bdc)3(e-urea)4] (a), [Y2(bdc)3(e-urea)2(p-murea)2] (b), [Y2(bdc)3(p-urea)2] (c) and [Y2(bdc)3(e-urea)2(H2O)2] (d). | ||
Coordination is assisted by hydrogen bonding of the NH moieties to the carboxylate groups in these MOFs as well as in [Y2(bdc)3(p-urea)2] (Fig. 12a). Interestingly, when the reaction is performed in a mixture of e-urea with p-murea, DMF or DEF, the resulting MOFs, [Y2(bdc)3(e-urea)2(p-murea)2] (Fig. 12b), [Y2(bdc)3(e-urea)2(DMF)2] and [Y2(bdc)3(e-urea)2(DEF)2], feature both types of solvents coordinated to the metal cation in a terminal fashion. For these Y-MOFs, thermogravimetric analysis showed solvent loss between 200 and 300 °C and X-ray powder diffraction demonstrated that the crystal structure was retained. A study using the N2 sorption isotherm at 77 K on the materials activated at 200 °C under vacuum indicated a BET specific surface area ranging between 170 and 300 m2 g−1.38
The synthesis of lanthanide MOFs (Ln-MOFs)74–77 has also been explored under urothermal conditions. For example, Ln-MOFs isostructural to [Y2(bdc)3(e-urea)4] or [Y2(bdc)3(e-urea)2(H2O)2] were obtained with various lanthanides (Ln = Ce, Sm, Eu, Gd, Dy, Ho, Er, Yb) from e-urea hemihydrate.38 Remarkably another structure type, formulated as [Ln2(bdc)3(e-urea)2] (Ln = La, Pr, Nd), was also isolated (Fig. 13). This series is worth citing as it incorporates bridging e-urea molecules and is isostructural to the Sm analogue obtained under ionothermal conditions in the 1:2 ChCl:e-urea deep eutectic solvent.32
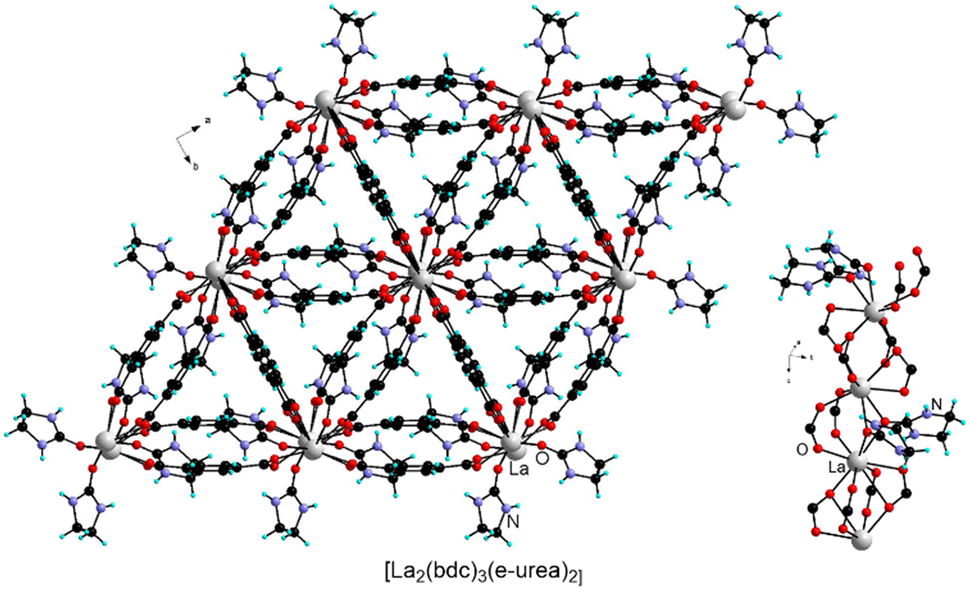 | ||
| Fig. 13 View of the crystal structure and of the metal node in [La2(bdc)3(e-urea)2]. | ||
Fu and coworkers also reported the [Ce2(bdc)3(H2O)4] MOF, prepared in a mixture of e-urea and water.78 This material is isostructural to the Eu and Tb analogues prepared under hydrothermal conditions.79 Jiang, Zhang and coworkers have investigated the formation of Ln-MOFs based on the 2,6-ndc ligand in e-urea (Fig. 14).80 While [La2(2,6-ndc)3(e-urea)3] formed upon reaction at 120 °C for 3 days, [La2(2,6-ndc)3(e-urea)2] was obtained at 140 °C. In the former MOF, e-urea acts as a bridging and pendant ligand, whereas it is bound exclusively in the terminal position in the latter phase (Fig. 14). For both materials, thermogravimetric analysis showed that the solvent could be removed upon heating, and that [La2(2,6-ndc)3(e-urea)2] was found to be stable at higher temperatures, up to 340 °C. Lanthanide MOFs are particularly interesting due to their porosity, luminescence, and potential catalytic activity.74–77 However, neither the textural nor the photophysical properties of these activated MOFs have been reported.80
 | ||
| Fig. 14 View of the crystal structure and of the metal node in [La2(2,6-ndc)3(e-urea)3] (a) and [La2(2,6-ndc)3(e-urea)2] (b). | ||
Another series of Ln-MOFs has been prepared from e-urea using the H2obb ligand (Fig. 1).38,81 The Bu group has first reported the formation of the [Yb2(obb)3(e-urea)] MOF (URO-130) prepared under urothermal conditions (Fig. 15a).38 In this three-dimensional material, the obb ligand bridges two-dimensional layers formed by coordination of the carboxylate moieties to the Yb cation with e-urea acting as a terminal ligand. The assembly of the obb ligand with lanthanides has been further explored by Zhang and coworkers, under the same conditions as in the previous work,38 yielding a variety of materials depending on the nature of the cation.81 The three-dimensional networks [Ln2(obb)3(e-urea)(H2O)] (Ln = Er, Yb, Ho) are built by obb-bridging of chains formed by coordination of the carboxylate to the Ln cation (Fig. 15b). Water and e-urea molecules are in terminal positions, further stabilized by hydrogen bonding. Another type of two-dimensional MOF with a similar SBU is obtained with Ln = Tb, Dy, Eu ([Ln2(obb)3(e-urea)(H2O)3]) and La ([La2(obb)3(e-urea)(H2O)1.5]) (Fig. 15c and d). Finally, [Sm2(obb)3(H2O)5](H2O) was also isolated (Fig. 15e). This three-dimensional MOF is formed by coordination of bridging carboxylate moieties. While water molecules are coordinated in a terminal fashion, no e-urea is present in this material. Thermogravimetric analysis on these MOFs showed a loss of solvent molecules (e-urea and/or H2O) between 100 and 200 °C and a decomposition above 360 °C.81 The textural properties of activated materials have not been described. The photophysical properties of [Ln2(obb)3(e-urea)(H2O)3] (Ln = Tb, Eu) have been investigated in the solid state. Both MOFs feature emission bands typical of their respective lanthanide ions upon excitation at 350 nn.81
 | ||
| Fig. 15 View of the crystal structure and of the metal node in [Yb2(obb)3(e-urea)] (a), [Yb2(obb)3(e-urea)(H2O)] (b), [Tb2(obb)3(e-urea)(H2O)3] (c), [La2(obb)3(e-urea)(H2O)1.5] (d) and [Sm2(obb)3(H2O)5](H2O) (e). | ||
Conclusions
While ionothermal synthesis using deep eutectic solvents based on the combination of choline chloride and urea derivatives has been widely explored for MOF synthesis, an alternative approach consisting in using urea derivatives with low to moderate melting points as solvents remains comparatively underemployed. However, as highlighted in this contribution, urothermal synthesis can be an effective approach for the preparation of MOFs. E-urea has been the most extensively used derivative in this context and has demonstrated an ability to play several roles in MOF construction. Beyond being a solvent, it can be present in the pore or as a ligand, most commonly in a bridging mode with divalent metal cations via coordination of the carbonyl group assisted by hydrogen bonding of the NH moieties. Decomposition of e-urea acts as a soft regulator of solution acidity but, in some instances, the formed ethylenediamine can also be incorporated in the resulting MOF. While e-urea has been explored as a solvent of choice, its combination with other organic solvents and the use of co-ligands have been shown to favor MOFs where it binds the metal in a terminal fashion. N-Alkylated urea derivatives represent interesting alternative solvents, as they have low melting points or can even be liquid at room temperature, making them media of choice. They are more prone to ligation to the metal center in the terminal position given the absence of hydrogen bonding ability, favoring their potential removal towards activation of the materials.As shown in this highlight article, a wide variety of MOFs can be prepared under urothermal conditions, with transition metals as well as lanthanides. The resulting materials can possess interesting photophysical or textural properties. This makes urothermal synthesis an attractive method worth further investigation and with potential future development. For example, urothermal conditions for the preparation of chiral architectures and their enantio-enrichment are a promising area. Systematic comparison of urothermal versus ionothermal conditions using deep eutectic solvents based on the combination of choline chloride with urea derivatives would be of interest and would shed light on the potential of these two methods in the context of the development of alternative approaches for MOF synthesis. It is our hope that this review will elicit such investigation in the future.
Abbreviations
| e-urea | 2-Imidazolidinone, ethyleneurea |
| e-murea | 1,3-Dimethylimidazolidin-2-one |
| p-urea | Tetrahydro-2(1H)-pyrimidinone |
| p-murea | 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone |
| tm-urea | Tetramethylurea |
| ChCl | Choline chloride |
| H2bdc | Terephthalic acid |
| H2bdc-NH2 | 2-Aminoterephthalic acid |
| H21,3-bdc | Isophthalic acid |
| H21,3-bdc-NH2 | 5-Aminoisophthalic acid |
| H21,3-bdc-NO2 | 5-Nitroisophthalic acid |
| H21,3-bdc-OH | 5-Hydroxyisophthalic acid |
| H21,3-bdc-SO3 | 5-Sulfoisophthalic acid |
| H21,4-ndc | 1,4-Naphthalenedicarboxylic acid |
| H22,6-ndc | 2,6-Naphthalenedicarboxylic acid |
| H2tdc | 2,5-Thiophenedicarboxylic acid |
| H2obb | 4,4′-Oxybisbenzoic acid |
| H3btc | Trimesic acid |
| H31,2,4-btc | Trimellitic acid |
| H23,4′-bpdc | Biphenyl-2,4′-dicarboxylic acid |
| H33,4′,5-bpdc | Biphenyl-3,4′,5-tricarboxylic acid |
| H3btb | 1,3,5-Benzenetrisbenzoic acid |
| H3tpa | Tris((4-carboxyl)phenylduryl)amine |
| H4bptc | Biphenyl-3,3′,5,5′-tetracarboxylic acid |
| D-camph | D-Camphoric acid |
| 3-pybid | 2-(3-Pyridine)benzimidazole |
| Htrz | Triazole |
| tmdpy | 1,3-Di(4-pyridyl)propane |
| bib | 1,4-Bis(1-imidazolyl)benzene |
| tib | 1,3,5-Tris(1-imidazolyl)benzene |
| en | Ethylenediamine |
| ox | Oxalate |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Writing – original draft: M. T. and S. A. B.; writing – review & editing: M. T. and S. A. B.; funding acquisition: S. A. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Université de Strasbourg, the C.N.R.S., and the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation is gratefully acknowledged.References
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed , Metal-organic frameworks special issue.
- H. C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC , themed issue on metal-organic frameworks.
- M. Dincă and J. R. Long, Chem. Rev., 2020, 120, 8037 CrossRef PubMed , Porous framework chemistry special issue.
- Q. Wand and D. Astruc, Chem. Rev., 2020, 120, 1438 CrossRef PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed.
- J. Ren, X. Dyosiban, N. Musyoka, H. W. Langmi, M. Mathe and S. Liao, Coord. Chem. Rev., 2017, 352, 187 CrossRef CAS.
- Q. He, F. Zhan, H. Wang, W. Xu, H. Wang and L. Chen, Mater. Today Sustain., 2022, 17, 100104 Search PubMed.
- T. Stolar and K. Užarević, CrystEngComm, 2020, 22, 4511 RSC.
- N. A. Khan and S. H. Jhung, Coord. Chem. Rev., 2015, 285, 11 CrossRef CAS.
- H. Reinsch, Eur. J. Inorg. Chem., 2016, 4290 CrossRef CAS.
- J. Chen, K. Shen and Y. Li, ChemSusChem, 2017, 10, 3165 CrossRef CAS PubMed.
- S. Wang and C. Serre, ACS Sustainable Chem. Eng., 2019, 7, 11911 CAS.
- S. Emad Hooshmand, R. Afshari, D. J. Ramón and R. S. Varma, Green Chem., 2020, 22, 3668 RSC.
- E. R. Engel and J. L. Scott, Green Chem., 2020, 22, 3693 RSC.
- S. Kumar, S. Jain, M. Nehra, N. Dilbaghi, G. Marrazza and K.-H. Kim, Coord. Chem. Rev., 2020, 420, 214307 CrossRef.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012 CrossRef CAS PubMed.
- E. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- R. E. Morris, Chem. Commun., 2009, 2990–2998 RSC.
- D. Freudenmann, S. Wolf, M. Wolff and C. Feldmann, Angew. Chem., Int. Ed., 2011, 50, 11050–11060 CrossRef CAS PubMed.
- B. Zhang, J. Zhang and B. Han, Chem. – Asian J., 2016, 11, 2610–2619 CrossRef CAS PubMed.
- T. P. Vaid, S. P. Kelley and R. D. Rogers, IUCrJ, 2017, 4, 380–392 CrossRef CAS PubMed.
- P. Li, F.-F. Cheng, W.-W. Xiong and Q. Zhang, Inorg. Chem. Front., 2018, 5, 2693 RSC.
- R. A. Maia, B. Louis and S. A. Baudron, CrystEngComm, 2021, 23, 5016 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheeda and V. Tambyrajah, Chem. Commun., 2001, 2010 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70 RSC.
- Q. Zhang, K. de Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS PubMed.
- B. Gurkan, H. Squire and E. Pentzer, J. Phys. Chem. Lett., 2019, 10, 7956 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232 CrossRef CAS PubMed.
- D. Yu, Z. Xue and T. Mu, Chem. Soc. Rev., 2021, 50, 8596 RSC.
- J. Zhang, T. Wu, S. Chen, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2009, 48, 3486 CrossRef CAS PubMed.
- E. A. Drylie, D. S. Wragg, E. R. Parnham, P. S. Wheatley, A. M. Z. Slawin, J. E. Warren and R. E. Morris, Angew. Chem., Int. Ed., 2007, 46, 7839 CrossRef PubMed.
- Q.-Q. Xu, B. Liu, L. Xu and H. Jiao, J. Solid State Chem., 2017, 247, 1 CrossRef CAS.
- M. Teixeira, R. A. Maia, L. Karmazin, B. Louis and S. A. Baudron, CrystEngComm, 2022, 24, 601 RSC.
- R. A. Maia, B. Louis and S. A. Baudron, Dalton Trans., 2021, 50, 4145 RSC.
- M. Teixeira, R. A. Maia, S. Shanmugam, B. Louis and S. A. Baudron, Microporous Mesoporous Mater., 2022, 343, 112148 CrossRef CAS.
- J. Zhang, J. T. Bu, S. Chen, T. Wu, S. Zheng, Y. Chen, R. A. Nieto, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2010, 49, 8876 CrossRef CAS PubMed.
- F. Wöhler, Ann. Phys., 1829, 12, 619 CrossRef.
- W. H. Shaw and J. J. Bordeaux, J. Am. Chem. Soc., 1955, 77, 4729 CrossRef CAS.
- P. M. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131 CrossRef CAS.
- S. Tischer, M. Börnhorst, J. Amsler, G. Schoch and O. Deutschmann, Phys. Chem. Chem. Phys., 2019, 21, 16785 RSC.
- Y. Dong, P. Zhou and H. Liu, J. Inorg. Organomet. Polym., 2014, 24, 874 CrossRef CAS.
- D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- F. Wang, Y.-X. Tan and J. Zhang, Solid State Sci., 2012, 14, 1263 CrossRef CAS.
- E. Yang, H.-Y. Li, Z.-S. Liu and Q.-D. Ling, Inorg. Chem. Commun., 2013, 30, 152 CrossRef CAS.
- S. P. Simeonov and C. A. M. Alfonso, RSC Adv., 2016, 6, 5485 RSC.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 4962 CrossRef CAS PubMed.
- H. Yang, T. H. Li, Y. Kang and F. Wang, Inorg. Chem. Commun., 2011, 14, 1695 CrossRef CAS.
- Y. Kang, S. Chen, F. Wang, J. Zhang and X. Bu, Chem. Commun., 2011, 47, 4950 RSC.
- Y. Wang and C. Li, Inorg. Chem. Commun., 2018, 96, 180 CrossRef CAS.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 343 RSC.
- S. Xue, P. Xing, J. Zhang, Y. Zeng and Y. Zhao, Chem. – Eur. J., 2019, 25, 7426 CrossRef CAS PubMed.
- Z. Lin, A. Slawin and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 4880 CrossRef CAS PubMed.
- J. Zhang, S. Chen, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2008, 130, 12882 CrossRef CAS PubMed.
- J. Zhang, T. Wu, S. Chen, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2009, 48, 3486 CrossRef CAS PubMed.
- Q.-Y. Liu, W.-L. Wong, C.-M. Liu, Y.-L. Wang, J.-J. Wei, Z.-J. Xiahou and L.-H. Xiong, Inorg. Chem., 2013, 53, 6773 CrossRef PubMed.
- R. A. Maia, A. Fluck, C. Maxim, B. Louis and S. A. Baudron, Green Chem., 2023, 25, 9103 RSC.
- P. Zhou, X. Li and Y. Dong, Z. Anorg. Allg. Chem., 2015, 641, 596 CrossRef CAS.
- Y. Dong, L. Zhang and Y. Zhang, Inorg. Nano-Met. Chem., 2017, 47, 73 CrossRef CAS.
- X. Li, Q. Liu and Y. Dong, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1202 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS.
- Z. Zhang, S. Xiang, X. Rao, Q. Zheng, F. R. Fronczek, G. Qian and B. Chen, Chem. Commun., 2010, 46, 7205 RSC.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS PubMed.
- Y.-P. He, L.-B. Yuan, H. Xu and J. Zhang, Cryst. Growth Des., 2017, 17, 290 CrossRef CAS.
- Z.-Q. Jiang, Y.-L. Tan, S.-Y. Wang, B. Li, D. Teng, C. Liao, D.-J. Zhou and Y. Kang, J. Inorg. Organomet. Polym., 2017, 27, 1583 CrossRef CAS.
- M. Kang, D. Luo, Y. Deng, R. Li and Z. Lin, Inorg. Chem. Commun., 2014, 47, 52 CrossRef CAS.
- Y.-H. Qiao and Z. An, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 936 CrossRef CAS.
- J. Li and J. Li, Z. Anorg. Allg. Chem., 2020, 646, 1683 CrossRef CAS.
- M.-X. Yang, L.-J. Chen, R. Ma, J.-N. Cai, Y.-D. Shia and S. Lin, Dalton Trans., 2019, 48, 13369 RSC.
- M.-X. Yang, L.-J. Chen, J. Gao, Y. Gao, G.-T. Yu and S. Lin, Dalton Trans., 2022, 51, 4862 RSC.
- Y.-M. Dai and M.-J. Liu, Inorg. Nano-Met. Chem., 2022, 52, 614 CrossRef CAS.
- Z. S. Liu, E. Yang, Y. Kang and J. Zhang, Inorg. Chem. Commun., 2011, 14, 355 CrossRef CAS.
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76 CrossRef CAS.
- C. Pagis, M. Ferbinteanu, G. Rothenberg and S. Tanase, ACS Catal., 2016, 6, 6063 CrossRef CAS.
- F. Saraci, V. Quezeda-Novoa, P. R. Donnarumma and A. Howarth, Chem. Soc. Rev., 2020, 49, 7949 RSC.
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590 RSC.
- M. Zhu, W. Fu and G. Zou, J. Coord. Chem., 2012, 65, 4108 CrossRef CAS.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS.
- D.-C. Hou, G.-Y. Jiang, Z. Zhao and J. Zhang, Inorg. Chem. Commun., 2013, 29, 148 CrossRef CAS.
- Z.-Q. Jiang, G.-Y. Jiang, D.-C. Hou, F. Wang, Z. Zhao and J. Zhang, CrystEngComm, 2013, 15, 315 RSC.
| This journal is © The Royal Society of Chemistry 2024 |