
Impurity-induced acceleration of polymorphic conversion via crystalline solid solutions and the T–X phase diagrams of salicylic acid and 3-hydroxybenzoic acid†
Tao
Zhang
a,
Francesco
Ricci
a,
Fateme
Molajafari
 b,
Seyed Sepehr
Mohajerani
c,
Mitchell
Paolello
d and
Fredrik L.
Nordstrom
*a
b,
Seyed Sepehr
Mohajerani
c,
Mitchell
Paolello
d and
Fredrik L.
Nordstrom
*a
aMaterial & Analytical Sciences, Boehringer-Ingelheim, Ridgefield, 06877, Connecticut, USA. E-mail: fredrik.nordstrom@boehringer-ingelheim.com
bDepartment of Chemical Engineering, Texas Tech University, Lubbock, 79409, TX, USA
cDepartment of Physics, Stevens Institute of Technology, Hoboken, 07030, New Jersey, USA
dDepartment of Chemical Engineering, Rowan University, Glassboro, 08028, New Jersey, USA
First published on 4th October 2024
Abstract
Two T–X binary phase diagrams have been constructed between salicylic acid (SA) and two monotropic polymorphs of the isomer 3-hydroxybenzoic acid (3HBA). Crystalline solid solutions (CSS) were formed at all extremes of the phase diagrams. The solid-state miscibilities ranged from 0.5% up to 6% of the second component. The thermodynamically stable form I of 3HBA exhibited a higher solid state miscibility than form II of 3HBA across all investigated temperatures. The solubility changes induced by the different CSS were measured experimentally in 40 w% methanol in water at 25 °C and are presented in two ternary phase diagrams. The SA-rich CSS phase exhibited the highest solubility increase corresponding to 160% up to the solvus at 0.7% 3HBA in SA. The changes in solubility of the CSS phases belonging to the two 3HBA polymorphs were found to diverge with increasing incorporation of SA in the respective crystal lattices. This thermodynamic divergence in combination with the monotropic stability relationship caused the driving force for polymorphic conversion to increase with increasing SA content. This unusual scenario was demonstrated experimentally through the use of solution-mediated phase transformation (SMPT) experiments analyzed in situ by Raman. It was found that the incorporation of 0.5% SA in the crystal lattice of 3HBA form II caused the polymorphic conversion rate to form I to double, in comparison to when 3HBA is chemically pure. The current example thus demonstrates the thermodynamic context for how solid-state miscible impurities can expedite polymorphic conversions. This and other contributions showcase how the rates of crystallization can be enhanced or reduced solely based on formation of CSS with an impurity or additive, without accounting for any surface adsorption effects.
Introduction
Crystalline solid solutions (CSS) are an intriguing class of crystal forms that are ubiquitous in metals, inorganic systems and in nature.1,2 CSS of exclusively organic compounds similar to pharmaceuticals have not been subjected to the same level of research and scrutiny as in related sciences dealing with crystalline materials. CSS differ from the discrete crystal forms (anhydrates, hydrates, solvates, salts and co-crystals) in that they contain a second component in non-stoichiometric amounts and can persist over a continuous range of compositions. There are two types of CSS, viz. substitutional CSS and interstitial CSS. The incorporation of a second component (guest) of similar molecular size commonly takes place in the crystal lattice of the product (host) via molecular substitution. The molecular exchange alters the lattice environment of the host, which can be observed by e.g. solvent solubility enhancements and melting point depressions. This class of crystalline materials thus provides a way to modify the physical properties of compounds over a certain compositional range, making it potentially an attractive option for active pharmaceutical ingredients (APIs). Examples include modifications in solubility and dissolution rate, as well as in mechanical properties, which may impact e.g. milling performance and drug product processability.1–6At the same time, synthetic organic impurities that are structurally similar to the API (or intermediates) are also frequently entrapped in the solid phase via formation of CSS with the API.7 Thus, it can be expected that APIs are often isolated as multicomponent CSS, where the effects of the impurities on the crystal lattice are not always clear. In API processing, impurities are known to negatively impact the crystallization leading to a number of problems, e.g. seeds dissolving or not growing, yield losses and encrustation. Lot to lot variability in the material properties of an API produced during pharmaceutical development phases is also a known issue, which impacts drug product processing and performance.
Another issue stemming from CSS formation with impurities relates to compounds that are polymorphic. The thermodynamic change imparted by the CSS formation on the lattice arrangement of the product is polymorphically selective. Different polymorphs and their corresponding CSS phases may exhibit different solvi and free energy changes, dependent on how well the impurity molecule fits inside the structural packing arrangements of the host. This structural selectivity can give rise to thermodynamic stability shifts and polymorphic coexistence domains in polymorphic systems that are observable in T–X phase diagrams.8–11 In the former case, a previously metastable polymorph may become thermodynamically stable, through its CSS phase, at certain ranges in compositions and temperatures. In the latter case, the two polymorphs can become equally stable (i.e., coexist) across a range of compositions and temperatures at constant pressure. Since the XRPD patterns of the CSS phases remain largely unchanged as compared to the chemically pure polymorphs, the thermodynamic stability relationships can be misinterpreted, leading to incorrectly assigned monotropic or enantiotropic relationships.
There are no reasons why two polymorphs forming CSS with a second component (viz. as impurity, additive or dopant) must result in a thermodynamic shift in their stability relationships, as demonstrated in a recent contribution.11 The opposite may of course also happen. The two polymorphs can diverge in thermodynamic stability with increasing lattice incorporation. This can cause a polymorph that's metastable when chemically pure, to become even more metastable as an impurity is incorporated in the solid phase. Presented in this contribution is an example of this latter thermodynamic scenario using the polymorphs of 3-hydroxybenzoic acid (3HBA) in combination with salicylic acid (SA). The changes in the thermodynamic stability with incorporation of SA is shown to translate to changes in the driving force for polymorphic conversion. This leads to the very unusual scenario wherein the addition of an impurity accelerates the polymorphic conversion, rather than the often-reported opposite effect.
SA and 3HBA are isomers differing only in the hydroxy group position (ortho versus meta). Their chemical structures are shown in Fig. 1. The molecular weights are thus the same and mol% and w% are equivalent. Only one crystal form of SA is known, and it is based on dimers in a monoclinic structure.12–14
 | ||
| Fig. 1 Chemical structure of salicylic acid, SA (left) and 3-hydroxybenzoic acid, 3HBA (right). | ||
Three anhydrous crystal forms of 3HBA are known. The crystal structures of the monoclinic (form I) and orthorhombic (form II) forms of 3HBA were resolved by Gridunova et al.15 Form I (P21/c) has been found to melt at an onset temperature of around 201–202 °C exhibiting a melting enthalpy of close to 36 kJ mol−1.16–18 The melting properties of form II of 3HBA (Pna21) are difficult to measure during heating as it readily converts to form I prior to melting. However, using melt-quench in DSC experiments, Svard and Rasmuson measured the onset melting point of form II to 194.3 °C with a melting enthalpy of 32.7 kJ mol−1.18
A third anhydrous polymorph of 3HBA was recently discovered by Braun in 2021 and is referred to as form III.17 This polymorph was obtained via quench cooling from the melt in a DSC study and was reported to have a melting point of around 196 °C and a melting enthalpy of 33.85 kJ mol−1. Furthermore, based on DSC, solution calorimetry and in silico calculations, it was shown that form III is metastable relative to form I, and possibly also relative form II.17
In this contribution, only form I and II of 3HBA were observed. As a single component, form I is known to be thermodynamically stable whereas form II is metastable and monotropically related to form I.16–18 The solubility ratios between form I and II were also evaluated in several solvents showing that form II exhibits approximately 20–40% higher solubility than form I, at 25 °C.16,18 Form II can be considered the kinetically favoured polymorph and readily crystallizes in evaporative or anti-solvent crystallization. Over time form II converts to form I via solution-mediated phase transformation (SMPT). The rate of this polymorphic conversion is slow enough to enable determination of solubility data of the metastable form in different solvents.
The objectives of the current contribution are to i) construct the T–X binary phase diagram of SA with form I of 3HBA using experimental data, ii) establish the T–X binary phase diagram of SA with the metastable and partially experimentally inaccessible form II of 3HBA using thermodynamic modelling, iii) determine ternary phase diagrams at 25 °C with a solvent and iv) give an example of how addition of a second component can non-monotonically increase the solubility ratios between two polymorphs and accelerate the rate of solution-mediated polymorphic conversion.
Experimental
The experimental procedures were similar to what was used in previous reports, with the exception of the experimental work regarding the metastable form II of 3HBA.4,5,9–11 Hence, previously reported methods are summarized while the new methods are provided in more detail.Materials
SA and 3HBA were purchased from Sigma-Aldrich having a purity exceeding 99%. The sourced 3HBA was obtained as a mixture comprising form I and II. Methanol, MeOH (HPLC grade), and acetonitrile, ACN (HPLC grade), were purchased from Millipore Sigma and Fisher Scientific, respectively. Deionized and distilled water was obtained from in-house source.Methods
X-ray powder diffraction (XRPD)
Samples were analyzed by an Inel Equinox 3000 and a Rigaku MiniFlex 600. The Inel XRPD comprised a curved position sensitive X-ray Detector, CPS120 providing simultaneous acquisition across all 2θ angles. The Rigaku XRPD contained a HyPix-400 MF detector and graphite monochromator to acquire diffraction through all 2θ angles. Samples were placed on the XRPD holder and evened out, before initiating acquisition while being continuously rotated.Differential scanning calorimetry (DSC)
Solid samples prepared via procedures below, were added to hermetically sealed pans and heated at 1 °C min−1 until complete melting in a TA Instruments Discovery 2500.Thermogravimetric analysis (TGA)
A Discovery TGA from TA Instruments was used for TGA analysis. Approx. 5–10 mg of the sample was placed in an open pan and heated at 10 °C min−1 past the melting point.Hot-stage polarized light microscopy (HS-PLM)
Samples were placed on a glass slide and heated at a heating rate of 3 °C min−1 passed the melting point, using a THMS 600 thermal stage from Linkam. Videos were collected via an Olympus BX51 microscope equipped with a FLIR Grasshoppe3 camera.High-performance liquid chromatography (HPLC)
Samples comprising mixtures of SA and 3HBA were separated by an Agilent Eclipse XDB-C8 column (4.6 × 100 mm, 1.8 mm) on an Agilent 1290 Infinite II UHPLC, at a column temperature of 30 °C, a flow rate of 1 mL min−1, a run time of 5 min, an injection volume of 3 mL, and 240 nm wavelength of detection. The mobile phase of 85 vol% A (0.1% H3PO4 in H2O) and 15 vol% B (acetonitrile) was injected at start and switched to 30 vol% A/70 vol% B at 4 min for 30 s, followed by returning to 85 vol% A/15 vol% B for the rest of the run. Response factors for SA and 3HBA were 6086.22 and 6149.28, respectively. The linear ranges for quantitatively analysis were between 0.02–0.68 mg g−1 (R2 = 0.9999) for 3HBA and 0.02–0.7 mg g−1 (R2 = 0.9998) for SA.Online Raman
A HyperFluxTM PRO Plus Raman Spectrometer (Tornado) equipped with a process ball probe (MARQMETRIX) was used for obtaining the inline Raman spectra. The exposure time was set at 100 milliseconds and each spectrum was the average of 5 exposures. The peak area between 1685 and 1705 cm−1 of 3HBA form II was used as reference peak to monitor the form conversion rate.Preparation of samples for construction of T–X phase diagram
Two different experimental approaches were utilized to prepare samples of SA-3HBA form I vs. SA-3HBA form II. Samples comprising SA and form II of 3HBA were prepared by evaporative crystallization from MeOH or acetone. Approx. 1 g total of SA and 3HBA was added to a vial in desired composition. Around 5 g MeOH was added creating a clear solution. The solutions were allowed to slowly evaporate in a lab hood until dryness. The resulting solids were then gently ground using a mortar and pestle and annealed in a vacuum oven at 70–80 °C overnight. No vacuum was used, and all vials were sealed to avoid changes in composition due to sublimation.As form II of 3HBA is the kinetically favoured polymorph, samples containing form I were made using an approach that did not rely on recrystallization from solution. Samples of SA and 3HBA form I were prepared by physically combining solid samples in desired proportions. The samples were ground in a mortar and pestle and then annealed in sealed vials at 80–90 °C for several days. Intermittent mixing was occasionally used to homogenize the samples. The final samples were analyzed by XRPD.
Construction of ternary phase diagram at 25 °C
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm for around 5 min. The filtrate was removed from the centrifuge filter and a second round of centrifugation was carried out to effectively deliquor the filter cake. The solid phase was analyzed by XRPD and the liquid and solid phases were analyzed by HPLC.
200 rpm for around 5 min. The filtrate was removed from the centrifuge filter and a second round of centrifugation was carried out to effectively deliquor the filter cake. The solid phase was analyzed by XRPD and the liquid and solid phases were analyzed by HPLC.
3HBA form II to form I SMPT studies
A saturated solution with respect to 3HBA form II was prepared by equilibration of a suspension in 40 w% MeOH in H2O at 25 °C for a few hours. The suspension was filtered (0.2 μm, PTFE) into 20 mL vials equipped with magnetic stir bars (Fisherbrand, Cat No:14-512-125) yielding a total of 8.0 g solution (7.1 g solvent and 0.92 g 3HBA). XRPD was used to ensure that no form conversion occurred prior to filtration.Form II of 3HBA was prepared separately a few days before the experiment. 3HBA form II (as pure 3HBA or containing 1.5 w% SA in 3HBA) was obtained after evaporative crystallization from acetone. The dried solids were ground using a mortar and pestle and annealed at around 70 °C for 1–2 days while being sealed.
0.50 g of the premade 3HBA form II (with or without SA) was then added to the 8.0 g clear solution to produce a suspension. In the experiment containing SA, the addition of the premade form II solids created a liquid phase comprising 0.5 w% SA in 3HBA and a solid phase, also containing 0.5 w% SA in 3HBA. The vial was then moved to a magnetic stirrer plate (Chemglass, AREX 3, CG-1995-V) having a stirrer speed setting of 3. A Raman probe was added so that the solid form content could be determined (Tornado, HyperFlux Pro Plus Raman Spectrometer equipped with a Marqmetrix ball probe). Finally, 25 mg form I was added to the suspension. This constituted the start of the experiment. The solid form content was monitored throughout the experiment with Raman. The final solids (as 3HBA form I) were also analyzed by XRPD and PLM. Each experiment was repeated once, with or without SA to afford a total of four experiments.
Results and discussion
Thermal properties of pure components
The thermal events obtained in the DSC curves at a heating rate of 1 °C min−1 were characterized using four thermal parameters, as also carried out in previous contributions.4,5,9–11 The inset temperature, Tinset represents the first deviation from the baseline and is used for the solidus. The offset temperature, Toffset denotes the return to the baseline and is used for the creation of the liquidus, solvus and the eutectic melting, based on the observations by Bhatnagar et al., 2005, using sucrose and water.19 The onset temperature, Tonset, and the peak temperature, Tpeak, are also recorded and available in ESI.† The enthalpy of the endotherm was determined based on linear integration from Tinset to Toffset.As previously reported by the same authors, SA exhibits an onset melting temperature of 158.6 °C at a heating rate of 1 °C min−1 and its sublimation tendency has also been evaluated by TGA.9 The onset melting point and melting enthalpy of 3HBA form I was measured at 201.4 °C and 248.94 J g−1, which is close to previous reports.16–18 The sublimation tendency of 3HBA form I was determined by TGA in an open pan at a heating rate of 10 °C min−1, and is presented in Fig. 2. Sublimation occurs above ∼150 °C and a weight loss of 1.58% was obtained up to its melting point. All DSC experiments comprising SA and 3HBA were carried out in hermetically sealed Al pans.
 | ||
| Fig. 2 TGA of 3HBA form I at a heating rate of 10 °C min−1 in open pan. | ||
The eutectic melting diagrams
 | ||
| Fig. 3 DSC curves of select compositions of SA/3HBA form I, at a heating rate of 1 °C min−1. | ||
With addition of 3HBA form I to SA, only one endotherm was obtained at X(3HBA) = 0.0031 and 0.0050 and the first eutectic endotherm appeared at X(3HBA) = 0.0099. A terminal CSS, henceforth labelled CSS α, is thus suggested below this composition. Based on the first emergence of the eutectic endotherm, it would be expected that the location of the solvus at the eutectic temperature, Xα–βI–L, would be located in-between X(3HBA) = 0.0050 and 0.0099. Another approach based on thermal data is to estimate the solvus from where the solidus meets the eutectic temperature. However, accurate determination of Tinset can be difficult and only 3 data points could be used in this system for CSS α. Nevertheless, trending of the solidus towards the eutectic temperature suggests a solvus closer to approx. X(3HBA) = 0.010 than to 0.005. At X(3HBA) = 0.0099, the two endotherms partially overlap, which again supports that the solvus is located below this composition.
The overlap between the endotherms progressively increases with increasing 3HBA content until an almost complete overlap at X(3HBA) = 0.2378 and X(3HBA) = 0.2993. The DSC curves at these two compositions exhibit the lowest Toffset values, which is an indication of close proximity to the eutectic composition. Above X(3HBA) = 0.2993, a steep increase can be seen in the liquidus and the two endotherms continuously become further apart up to X(3HBA) = 0.8987, where the eutectic endotherm has decreased in magnitude. At X(3HBA) = 0.9504, only the high-temperature endotherm remains. The absence of an eutectic endotherm signals the formation of a second CSS phase, labelled βI (where I denotes form I of 3HBA). Based on the thermal data, the solvus of CSS βI at the eutectic temperature, XβI–α–L, can be expected to be located between X(3HBA) = 0.8987 and X(3HBA) = 0.9504. Around this range of compositions, the liquidus remains fairly constant and similar to the melting point of 3HBA form I. In terms of the solidus, the acquired Tinset data of the high-temperature endotherms significantly exceeded the eutectic temperature and the trend towards XβI–α–L is less clear.
The collection of thermal data in the SA-3HBA form I system produces the outline of the experimental melting diagram, presented in Fig. 4. Note that this set of experimental data does not show the required convergence of the liquidus and solidus at the melting of the pure components, due to limitations of the DSC measurements.
 | ||
| Fig. 4 Eutectic T–X phase diagram of SA-3HBA form I based on experimental DSC data at 1 atm. | ||
The melting enthalpies of the phase transitions were also determined from the DSC curves by integration from Tinset to Toffset. When overlapping endotherms were present, a split was inserted at the local heat flow maximum, so that the endotherm was divided into two measurable portions. This allowed for the construction of a Tammann diagram across all compositions. The low-temperature endotherms (assigned M) represent the eutectic melts and are always linear with composition.20 The diagram, shown in Fig. 5, has been complimented with the high-temperature endotherms, which are not required thermodynamically to exhibit linearity with composition. Nevertheless, in this system a high degree of linearity still appears, similar to what was observed in other previously reported systems involving SA. The phase transitions shown in Fig. 5 are listed in Table 1.
 | ||
| Fig. 5 Extended Tammann diagram of SA-3HBA form I. | ||
| Line | Low temp. ΔH (line T) | High temp. ΔH (line M) |
|---|---|---|
| 0 | α → L | |
| 1 | α + βI → α + L | α + L → L |
| 2 | α + βI → βI + L | βI + L → L |
| 3 | βI → L |
The Tammann diagram is highly useful as a tool to further determine thermodynamic loci, beyond what can be inferred from the thermal data in the T–X phase diagram. Obviously, both sets of data should align to provide consistent values of critical boundaries and points. In reality, slight deviations and inconsistencies do appear as a result of experimental errors. This is also the case in this system, where a larger variability in the enthalpy values occurred due to the applied experimental approach in preparing the samples for DSC. As described in the Experimental method section, a recrystallization step had to be omitted from the sample preparation procedure as it generated the metastable form II of 3HBA.
The composition of the α solvus can be estimated in the extended Tammann diagram from where line T1 reaches zero, and where lines M0 and M1 intersect. Unfortunately, the linear regression and extrapolation of T1 to zero yielded negative values, which obviously is incorrect. Experimental errors in the enthalpies from the divided endotherms close to the eutectic are especially responsible for this error, due to difficulties in integration of the separate DSC signals. Removing the mostly overlapping endotherms (X(3HBA) > 0.15), which are associated with higher uncertainty yields an α solvus of X(3HBA) = 0.002. This is still far below the anticipated α solvus of X(3HBA) = 0.005–0.010, based on the thermal data above, and the composition where the first eutectic melt was first observed. When determining the composition at which M0 and M1 intersect, a more reasonable value is obtained, viz. X(3HBA) = 0.008, which aligns with the thermal data. This value is henceforth used as the α solvus (±0.002) at the eutectic temperature, which in turn was determined to 149.4 °C on average (using composition of X(3HBA) from 0.0099 to 0.8987).
The eutectic composition can be determined from both low- and high-temperature endotherms in Fig. 5. Traditionally and strictly thermodynamically,20 the intersection between line T1 and T2 provides the eutectic composition, which in this system amounted to X(3HBA) = 0.254. Using the high-temperature endotherms, the eutectic composition can be estimated either from where line M1 and M2 intersect, where line M1 reaches zero, or where line M2 reaches zero. This extrapolation is then based on linear regression. The intersection between line M1 and M2 yields an eutectic composition of X(3HBA) = 0.264, whereas the latter individual extrapolations of line M1 and M2 give X(3HBA) = 0.239 and 0.308, respectively. Overall, given the data quality in the Tammann diagram combined with the thermal data (specifically the liquidus), the eutectic composition was estimated to X(3HBA) = 0.25 (±0.02).
Similar to the α solvus, the composition of the solvus of the βI phase at the eutectic temperature can be estimated from where line T2 equals zero, or alternatively, at the intersection between line M2 and M3. Regression and extrapolation of line T2 to zero yields an α solvus at X(3HBA) = 0.940. This composition aligns with the thermal data and composition corresponding to the disappearance of the eutectic (low-temperature) endotherm. The second approach gives a slightly lower α solvus of X(3HBA) = 0.924. However, line M3 is regressed over a relatively narrow composition range and hence fraught with higher uncertainty. Therefore, the α solvus is estimated to X(3HBA) = 0.940 (±0.01).
Hot-stage PLM was carried out at three compositions, i.e. X(3HBA) = 0.0927, 0.2378 and 0.6005, and are available in ESI.† The recordings at 3 °C min−1 show both the eutectic melting and final high-temperature melting, consistent with the T–X phase diagram in Fig. 4.
 | ||
| Fig. 6 DSC curves of select compositions of SA/3HBA form II, at a heating rate of 1 °C min−1. | ||
Starting on the SA-rich side, only one endotherm corresponding to the melting of the SA-phase was obtained up to X(3HBA) = 0.0079. The first eutectic melting event in the DSC curves was observed at X(3HBA) = 0.0103, which suggests an α solvus in-between these compositions. The eutectic melting is close to 3 °C lower when form II of 3HBA is present as compared to form I, and on average equalled 146.1 °C. The solidus of CSS α decreases quickly towards the eutectic temperature and at X(3HBA) = 0.0103 the two endotherms partially overlap. With additional 3HBA the liquidus drops fairly linearly with composition and at X(3HBA) = 0.2489, the two endotherms significantly overlap. This substantial overlap in the thermal events is also present at X(3HBA) = 0.3050 suggesting closeness to the eutectic composition. From this composition and at increasing 3HBA content, the two endotherms again separate. However, the second endotherm is preceded by an exotherm, which represent a conversion of 3HBA form II to form I (or βII to βI). These exotherms can be clearly observed in the DSC curves from X(3HBA) = 0.4889 and higher (an example is provided in ESI†). Therefore, only the eutectic melting events could be used to create the experimental T–X phase diagram. On the 3HBA-rich side, the eutectic melting events persist up to and including X(3HBA) = 0.9754 but has disappeared at X(3HBA) = 0.9904. A second CSS phase is thus expected, henceforth assigned as CSS βII, having a solvus in-between these two compositions. The partially constructed experimental T–X phase of SA and 3HBA form II is presented in Fig. 7. The missing liquidus and solidus on the right side of the diagram is highlighted with dot-dashed curves. Again, note that the required convergence of the liquidus and solidus at the melting of SA is not fulfilled, due to limitations of the DSC measurements.
 | ||
| Fig. 7 Incomplete eutectic T–X phase diagram of SA-3HBA form II based on experimental DSC data at 1 atm. Note that this was created using the metastable 3HBA form II and hence lacks the solidus and liquidus (dot-dashed orange and red curves) right of the eutectic composition due to exothermic form conversion to 3HBA form I. The melting point of pure 3HBA form II was obtained from Svard & Rasmuson.18 | ||
Similar to the SA-3HBA form I system, the extended Tammann diagram was also created for SA-3HBA form II, based on the integrated enthalpies in the DSC data of the low-temperature (T) and high-temperature (M) endotherms. This diagram is presented in Fig. 8 and the phase transitions represented by the M and T lines are listed in Table 2.
 | ||
| Fig. 8 Extended Tammann diagram of SA-3HBA form II. | ||
| Line | Low temp. ΔH (line T) | High temp. ΔH (line M) |
|---|---|---|
| 0 | α → L | |
| 1 | α + βII → α + L | α + L → L |
| 2 | α + βII → βII + L | βII + L → L (not exp. accessible) |
| 3 | βII → L (not exp. accessible) |
Due to the form conversions taking place at around 150–160 °C to the stable phases comprising 3HBA form I, the extended Tammann diagram in Fig. 8 is incomplete. Nevertheless, some thermodynamic loci can still be determined. The solvus of α can be determined from where line T1 reaches zero. This takes place close to X(3HBA) = 0.009 (±0.002), which is very similar to the α solvus in the SA-3HBA form I system. The eutectic composition can be estimated from where line T1 and T2 intersect, which in this case takes place at X(3HBA) = 0.270. The other approach, explained above, is to use linear extrapolation of line M1 to zero. This resulted in X(3HBA) = 0.262. However, this latter value assumes linearity in the enthalpies, which is not required thermodynamically and hence may carry some uncertainty. Therefore, the eutectic composition in the SA-3HBA form II system was estimated to X(3HBA) = 0.27 (±0.01). The βII solvus could only be estimated based on the composition where line T2 reaches zero, as no M2 line could be constructed. Linear regression yielded a βII solvus of X(3HBA) = 0.980.
While the solidus and liquidus on the right side in Fig. 7 are experimentally inaccessible, the exotherms associated with the 3HBA form II/βII to form I/βI conversion still provide valuable information. In Fig. 9 is presented the temperature of conversion at different compositions (top), as well as the enthalpy of the exothermic conversions across the investigated compositions (bottom). From the temperature of conversion, two distinct compositional regions can be observed. From X(3HBA) = 0.4889 to X(3HBA) = 0.9513, the temperature of conversion increases slightly with increasing composition. Above X(3HBA) = 0.9513, there is steep drop and the Tonset of the form conversion decreases from 163.11 to 143.41 °C at X(3HBA) = 1, i.e. a reduction of almost 20 °C. The apparent boundary between these two regions aligns with the solvus of βI. This can be explained by the environment in which the form conversion takes place. On the left side of the βI solvus, α + βII convert to α + βI, whereas on the right side α + βII convert to only βI. These conversions can thus be useful as a complementary approach in pinpointing the location of the βI solvus.
 | ||
| Fig. 9 Temperature and enthalpy of exothermic form conversions from βII to βI as measured by DSC at a heating rate of 1 °C min−1 (average of triplicates). Top: Temperature of conversion (Tinset: blue circles, Tonset: purple triangles, Toffset: red squares). Bottom: Enthalpy of exothermic conversions. The dashed line was created using the lever rule in conjunction with 2.5 J g−1 for βII to βI conversion and 28 J g−1 for the α to βI conversion. | ||
Furthermore, the magnitude of the exotherm observed in Fig. 9 (bottom) differs significantly at different compositions. When the form conversion proceeds from α + βII to βI (or βII to βI above X(3HBA) = 0.980), the exotherm is weaker and around 2–3 J g−1 on average. Below X(3HBA) = 0.9513, the exotherm becomes amplified and increases up to around 15 J g−1. These changes are related to the enthalpy of transition based on the involved solid phases and can be estimated using the lever rule.1 The βII to βI conversion averaged around 2.5 J g−1 across compositions from X(3HBA) = 0.98 to 1.00. The α to βI conversion was not measured independently. However, using an approximate value of 28 J g−1 yields the dashed line shown in Fig. 9 bottom, which appear to fit quite well with the experimental data. The obtained significant difference in the exothermic enthalpies between the two types of phase transitions may not be surprising as the α to βI conversion completely alters the crystal lattice wherein 3HBA moves from the minor component to the predominant. In contrast, in the βII to βI conversion the SA molecules remain the minor component, just distributed in two separate crystal lattices of the 3HBA polymorphs.
In addition to the DSC and XRPD data present above, hot-stage PLM was also carried out at a heating rate of 3 °C min−1 of samples comprising SA and βII, at X(3HBA) = 0.0959 and 0.4889, which are available in ESI.† The hot-stage PLM recordings confirm the observed transition in Fig. 7.
Ternary phase diagrams in 40 w% MeOH in H2O
Ternary phase diagrams were created for SA with both polymorphs of 3HBA at 25 °C in the same solvent system. The selected solvent system, 40 w% MeOH in H2O, was maintained at fixed composition throughout the two studies. Nevertheless, we note that the system is a de facto quaternary system but is treated as a pseudo-ternary system. The construction of the ternary phase diagram for SA and 3HBA form I was straightforward as it only involved two stable phases, i.e. CSS α and βI. The metastable form II of 3HBA however required a different experimental approach, given its propensity to convert to form I of 3HBA over time. It should be noted that the time for equilibration of CSS also requires equilibration of the solid phase. Therefore, it was desired to extend the equilibration time as much as possible, while avoiding form conversion. From previous studies working with exclusively 3HBA in different solvents, polymorphic conversion took place in around 5–10 h.16 During initial experimentation it became clear that the introduction of shear forces in the experiment greatly accelerated the form conversion. For example, the use of larger magnetic stirrers and high agitation rate was found to expedite the time for form conversion to just a few hours, while the reduction of shear using e.g. overhead stirrers at low rpm could maintain the metastable form II for 1–2 weeks. Ultimately, the latter approach was selected wherein the composition of the solid and liquid phases were monitored over time to ensure that equilibration had been achieved. The two ternary phase diagrams are presented in Fig. 10. All data that were used in the construction of the ternary phase diagrams are available in ESI.† | ||
| Fig. 10 Ternary phase diagram of SA-3HBA form I (left) and SA-3HBA form II (right) in mol%. The upper diagrams show a magnification of the top corners of the two ternary phase diagrams, respectively. | ||
The ternary phase diagrams demonstrate some differences as the solid phase of 3HBA changes. The solubility of 3HBA form II/βII is considerably higher than 3HBA form I/βI, which causes the invariant point to shift to the lower right in the diagram. Similarly, the slope of the βII solubility line is steeper as compared to the slope of the βI solubility line, which causes the two solubility curves to diverge with increasing SA content.
The data presented in Fig. 10 can be replotted in a Roozeboom plot (solvent-free solid–liquid equilibria diagram) in order to determine the solvi at the respective CSS phases at 25 °C.4 This is presented in Fig. 11, based on the two polymorphs of 3HBA.
 | ||
| Fig. 11 Roozeboom diagrams (solvent-free SLE diagram) of SA-3HBA form I (left side) and SA-3HBA form II (right side). The dashed lines are the invariant points (X(3HBA) = 0.70 vs. 0.73). The bottom figures show the right sides of the top diagrams and the location of the βI (left) and βII (right) solvi. | ||
The Roozeboom plot for the SA-3HBA form I system at 25 °C (left in Fig. 11) discloses an α solvus at X(3HBA) = 0.006. This can be compared to X(3HBA) = 0.008 at 149.4 °C, indicating a fairly temperature-insensitive α solvus. In contrast, the βI solvus increases dramatically from X(SA) = 0.006 at 25 °C to X(SA) = 0.060 at 149.4 °C, i.e. a 10-fold increase in solid state miscibility.
The SA-3HBA form II system (right in Fig. 11) exhibit several similarities to the SA-form I system. There is a smaller increase in the invariant point (from X(3HBA) = 0.70 to 0.73). This slight change causes the α solvus to rise to X(3HBA) = 0.007 (cf. X(3HBA) = 0.009 at 146.1 °C). On the right side, the βII phase almost overlap with the βI phase. However, because of the change in the invariant point, the solid state miscibility of the βII phase is slightly smaller in comparison to the βI phase (solvus at X(3HBA) = 0.995 vs. 0.994). Thus, βI phase maintains a higher solid state miscibility over the βII phase at all investigated temperatures.
Thermodynamic modelling and the T–X binary phase diagrams
The thermodynamic loci determined from the experimental data collected from the DSC studies at melting and ternary phase diagram at 25 °C are summarized in Tables 3 and 4.| Thermodynamic boundaries and points | Exp. method | X(3HBA) mol 3HBA/(mol 3HBA + SA) | T °C |
|---|---|---|---|
| Eutectic (α + βI) | DSC | 0.25 | 149.4 |
| Invariant (α + βI) | SLE (40% MeOH in H2O) | 0.6993 | 25.0 |
| Solvus of α relative to βI (Xα–βI–L) | DSC | 0.008 | 149.4 |
| Solvus of α relative to βI | SLE (40% MeOH in H2O) | 0.006 | 25.0 |
| Solvus of βI (XβI–α–L) | DSC | 0.940 | 149.4 |
| Solvus of βI | SLE (40% MeOH in H2O) | 0.994 | 25.0 |
| Thermodynamic boundaries and points | Exp. method | X(3HBA) mol 3HBA/(mol 3HBA + SA) | T °C |
|---|---|---|---|
| Eutectic (α + βII) | DSC | 0.27 | 146.1 |
| Invariant (α + βII) | SLE (40% MeOH in H2O) | 0.7296 | 25.0 |
| Solvus of α relative to βII (Xα–βII–L) | DSC | 0.009 | 146.1 |
| Solvus of α relative to βII | SLE (40% MeOH in H2O) | 0.007 | 25.0 |
| Solvus of βII (XβII–α–L) | DSC | 0.980 | 146.1 |
| Solvus of βII | SLE (40% MeOH in H2O) | 0.995 | 25.0 |
The T–X phase diagrams for both the βI and βII systems at 1 atm were predicted via the NRTL activity coefficient model, using the same methods described4 and demonstrated4,5 in earlier publications. However, unlike those previous works, in this case we are examining a system for which one of the species exhibits a monotropic polymorphism between two solid phases. As discussed extensively above, this polymorphism leads to two distinct CSS phases which are also monotropically related (βI and βII). Therefore, we must separately determine the solid-state NRTL binary interaction parameters (BIPs) ΔgS12 and ΔgS21 for the βI and βII cases. To do this, we performed two separate sets of regressions: one for the α-βI phase diagram (herein referred to as the α-βI model), and the other for the α-βII diagram (herein referred to as the α-βII model), as described below.
For the ΔgS12 and ΔgS21 parameters we adopt an ad hoc linear temperature-dependence given below:
| ΔgS12 = c12 + d12T | (1a) |
| ΔgS21 = c21 + d21T | (1b) |
To determine the solid-state NRTL BIPs for the α-βI and α-βII models, we separately regressed the solid–solid equilibrium (SSE) data (i.e., the solvi at 25 °C and the eutectic temperature) for the α-βI and α-βII phase equilibria, respectively. We also fixed the non-randomness parameters αS,I12 and αS,II12 to be constant and equal to 0.2, as is often done.
With the solid-state NRTL models in hand, we proceeded with determining the liquid-state models. We note that, for the liquid-state NRTL model, the BIPs ΔgL12 and ΔgL21 should theoretically be independent of any information regarding solid phases, as these terms are accounting for the 1–2 interaction energetics in the liquid state of aggregation. However, in the practical interest of obtaining two separate models capable of reproducing the α-βI and α-βII phase diagrams as accurately as possible, we determined liquid-state NRTL BIPs {ΔgL,I12,ΔgL,I21} and {ΔgL,II12,ΔgL,II21} separately for each model. These were determined by regressing to the solid–solid–liquid equilibrium (SSLE) data at the eutectic temperature for the α-βI and α-βII cases, respectively. As in the earlier publications,4,5 the T-dependence of the liquid-state parameters was neglected. The non-randomness parameters αL,I12 and αL,II12 were assumed constant and equal to 0.2. The regressed values of all model parameters can be found in the ESI.†
The predicted T–X phase diagrams, as well as their overlay, are shown in Fig. 12 and 13. Note that the α-βII model provides a prediction of the βII solidus and liquidus curves, which is experimentally inaccessible due to the fast conversion to βI upon heating in DSC experiments.
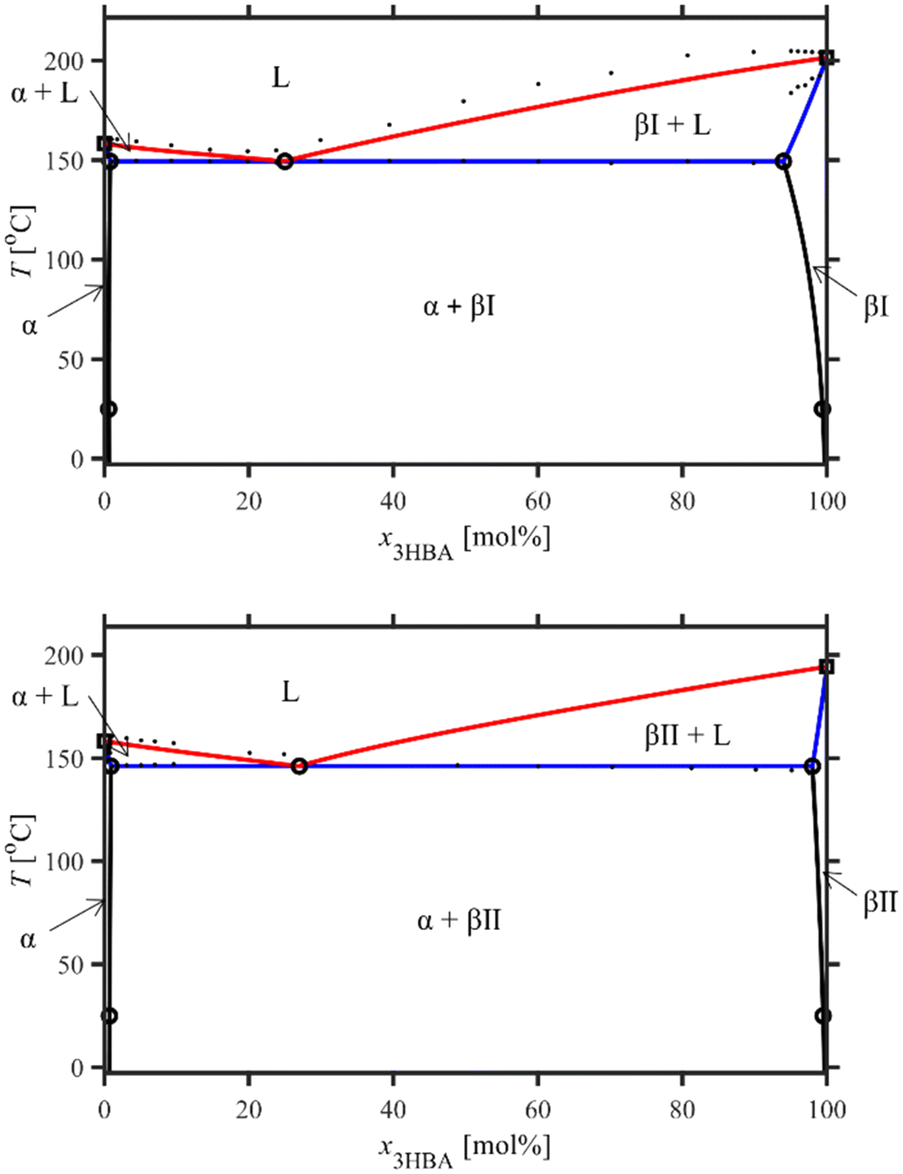 | ||
| Fig. 12 NRTL model prediction of T–X phase diagrams of SA-3HBA form I (top) and SA-3HBA form II (bottom) at 1 atm. The predictions include the solvus (black), solidus (blue) and liquidus (red) curves. The open circles are experimental data used to fit the solid state and liquid state model parameterizations via the SSE and SSLE equations, respectively, as descried in the text. The open squares are the melting points of pure SA and 3HBA, and the dots are the experimental solidus and liquidus data. | ||
 | ||
| Fig. 13 Overlay of T–X phase diagrams predicted via NRTL. Solid curves represent predicted α-βI equilibria, and dashed curves represent α-βII equilibria. The filled circles and squares are the experimental data from the α-βI equilibria, and the open circles and squares are the experimental data from the α-βII equilibria. We here use the labelling βI(II) to emphasize that βII equilibria are metastable with respect to βI equilibria. Coloration of the solvus, solidus, and liquidus curves are the same as Fig. 12. | ||
The solubility of the CSS phases in the solvent system 40 w% MeOH in H2O was predicted by the same methods described in a previous study.5 Namely, this requires the regression of the NRTL liquid-state BIPs to solubility data. In this particular instance, we once again make the practical decision to treat the liquid BIPs as distinct parameters for the α-βI and α-βII systems, and regress the missing parameters for each model to their corresponding solubility dataset: {ΔgL,I13, ΔgL,I31, ΔgL,I23, ΔgL,I32} and {ΔgL,II13, ΔgL,II31, ΔgL,II23, ΔgL,II32} where 3 = solvent. In this way, we leverage our extensive regression dataset to obtain the most accurate phase diagrams predictions. We also assume these parameters are temperature-independent, which is necessitated in this case since solubility was only measured at 25 °C. As done previously, we assume the corresponding non-randomness parameters to be equal to 0.2 throughout.
The other key difference between this and the previous study5 is that this solvent system (i.e., “component 3” in the modelling analysis) is not a single species, but rather a binary mixture. Therefore, as previously mentioned, we are treating a quaternary system as a quasi-ternary one by keeping the ratio of solvent composition constant throughout. A theoretically proper utilization of the NRTL model would require that methanol and water are treated as distinct species (i.e., species “3” and “4”), with additional BIPs ΔgL,I14, ΔgL,I41, etc. Moreover, these separate solvent BIPs would ideally be regressed with a more comprehensive dataset including solubility in pure MeOH and pure H2O to de-convolute the impact of each solvent. However, because we are primarily concerned with utilization of these models to perform predictions in the current 3HBA/SA/(40 w% MeOH in H2O) system, our simplifying assumptions are fit-for-purpose.
As described in the aforementioned study,5 for simplicity we restrict the solubility regression dataset to only include data in the two-phase regions (α + L & β + L), and do not incorporate data from the three-phase region (α + β + L), where the solubility is constant at the invariant point. Multiple regressions were attempted, including varying the number of datapoints included in the regression, and multiple initial guesses for the parameters were tried for each number of data points.
As shown in Fig. 14 and 15, the model predictions are in quite good agreement with experimental data.
 | ||
| Fig. 14 Ternary phase diagram for SA/3HBA (form I)/solvent (40 w% MeOH in H2O) predicted via NRTL at 25 °C and 1 atm. Top: Full phase diagram, the majority of which is the 3-phase α + βI + L region. Bottom: Zoomed view to focus on the top of the phase diagram, with experimental data superimposed. Green curves = predicted solubility, black lines = selected tie-lines connecting the predicted solubility to the corresponding compositions of the α and βI phases (chosen at regular intervals for illustration). Red circles = experimental solubility data used in the parameter regression, blue circles = experimental solubility data not used in the regression. The red and blue dashed lines are the corresponding experimentally measured tie-lines. The cluster of blue circles lowest in the diagram correspond to data measured in the 3-phase region (i.e., they represent the invariant point) – in this region the composition of the α, βI and L phases are fixed at the α solvus, βI solvus, and invariant point (predicted to occur at the intersection of the green curves), respectively. | ||
 | ||
| Fig. 15 Ternary phase diagram for SA/3HBA (form II)/solvent (40 w% MeOH in H2O) predicted via NRTL at 25 °C and 1 atm. Top: Full phase diagram, the majority of which is the 3-phase α + βII + L region. Bottom: Zoomed view to focus on the top of the phase diagram, with experimental data superimposed. Green curves = predicted solubility, black lines = selected tie-lines connecting the predicted solubility to the corresponding compositions of the α and βII phases (chosen at regular intervals for illustration). Red circles = experimental solubility data used in the parameter regression, blue circles = experimental solubility data not used in the regression. The red and blue dashed lines are the corresponding experimentally measured tie-lines. The cluster of blue circles lowest in the diagram correspond to data measured in the 3-phase region (i.e., they represent the invariant point) – in this region the composition of the α, βII and L phases are fixed at the α solvus, βII solvus, and invariant point (predicted to occur at the intersection of the green curves), respectively. | ||
Solvent solubilities and polymorphic stability relationships
The experimental solubilities of the respective solid phases in 40 w% MeOH in H2O and 25 °C are presented in Fig. 16a (SA/α-3HBA form I/βI) and Fig. 16b (SA/α-3HBA form II/βII) and are also available in ESI.† The solubility of SA increases from 23.4 mg to 52.4 SA per g solvent at the α solvus relative to βI. This corresponds to a solubility increase of 124.3% over just 0.6 w% 3HBA. When the solubility increase is compared to the α solvus relative βII, the solubility increase is even higher (23.4 to 60.8 mg SA per g solvent), corresponding to 160.3% over 0.7 w% 3HBA in the solid phase of SA/α. | ||
| Fig. 16 Solubility of respective solid phases in 40 w% MeOH in H2O at 25 °C for SA-3HBA form I (a) and SA-3HBA form II (b). The bottom figure c depicts the solubilities of 3HBA form I/βI and form II/βII at around the respective solvi (far right in figure a and b). The 1 and 2 arrows illustrate the direction of the polymorphic conversion from form II/βII to form I/βI when chemically pure and when containing 0.5 w% SA in 3HBA in the respective solid phases. Note that the concentrations of the two components are presented at the two far left and right side of the diagrams in a and b. | ||
The solubility curves of the two polymorphs of 3HBA can be observed to diverge with increasing SA content (highlighted region is shown in Fig. 16c). As SA is incorporated into form I of 3HBA, the solubility increases only moderately from 98.4 to 121.9 mg 3HBA per g solvent up to the solvus of βI (24% increase). For form II of 3HBA however, the solubility rise is slightly higher with an increase from 130.5 to 164.1 mg 3HBA per g solvent at the βII solvus (26% increase). The solubility ratio between form II and I amounts to 1.32 when 3HBA is pure, which is similar to reported systems for other solvents.16,18
The two polymorphs of 3HBA, form I and form II, exhibit distinct crystal structures and intermolecular interactions, influencing their response to the addition of SA (Fig. 17). Form I, the thermodynamically stable polymorph, crystallizes in a monoclinic structure featuring the carboxylic acid dimer motif packed in a common herring-bone pattern. These dimers form an (R22)8 motif, which is centrosymmetric and creates a robust network of intermolecular hydrogen bonds primarily between the carboxylic acid groups. This stable dimer-based lattice ensures that the structural integrity of form I is maintained upon the introduction of SA, as the hydrogen bonding interactions remain largely intact, and the lattice structure is not significantly disturbed.
 | ||
| Fig. 17 Crystal structure of 3HBA form I (left) and form II (right) with substituted SA molecules. | ||
In contrast, form II crystallizes in an orthorhombic structure and consists of intermolecular hydrogen bond chains involving alternating COOH and OH-groups, creating layers of ribbons. These ribbons interact through van der Waals forces between their non-polar regions. Form II is non-centrosymmetric, lacking the typical carboxylic acid dimer configuration seen in form I, making its lattice more dependent on the chain-like hydrogen bonding interactions for stability. The introduction of SA disrupts these hydrogen bonds, weakening both the hydrogen bonding network and the van der Waals interactions. This disruption leads to a less stable lattice structure, explaining why form II exhibits a more pronounced increase in solubility compared to form I. The differential impact of SA on the two polymorphs can thus be attributed to the inherent differences in their hydrogen bonding networks, with form I's robust dimer-based lattice resisting disruption, while form II's chain-based lattice becomes more susceptible to disturbance.
Analysing the solubility ratios between 3HBA form I/βI and form II/βII, as the SA content exceeds beyond the βII solvus, a divergence in thermodynamic stability is observed. At higher concentrations of SA, the lattice may stabilize by converting to another form. The presence of SA may facilitate the reorganization of the lattice and alter the balance of intermolecular interactions, converting form II to the more stable form I.
Solution-mediated polymorphic transformation
The driving force for a polymorphic conversion is based on the difference in chemical potential, Δμ, between the two states exhibited by the two polymorphs. For a pure system, this is straightforward and can be derived based on the solid-state activities, α, in equilibrium with their respective saturated solution: | (2) |
 | (3) |
 | (4) |
The ratio in solubilities of the two CSS belonging to form I and II are presented in Fig. 18.
 | ||
| Fig. 18 Solubility ratios between 3HBA form II/βII and form I/βI (as averages), with increasing content SA in the respective crystal lattices. | ||
Due to the different solvi of βI and βII, three distinct geometric regions are generated as the solubility ratios are plotted vs. solid phase composition. When the solid phase is pure (point 1), the solubility ratio is 1.32. As SA is incorporated in the two crystal lattices of 3HBA (region A), the solubility ratio increases non-linearly up to 1.38 (point 2). This constitutes the maximum solubility ratio and take place at the solvus of βII. As the SA content increases further (region B), the solubility ratio drops steeply down to the solvus of βI (point 3) where the solubility ratio is 1.34. This ratio is then maintained constant as the SA content is increased further (region 3).
The non-monotonic behaviour visible in Fig. 18 shows simplistically how the thermodynamic driving force for conversion from βII to βI can be enhanced by the addition of a solid-state miscible second component. Based on the solubility ratio, the largest driving force can be expected to be located at the solid phase composition belonging to the solvus of βII. The solubility ratio is always higher in comparison to when the material is pure, also at compositions above the solvi for both polymorphs. This increase is contingent on the fact that the solvus of the stable polymorph is higher than the solvus of the metastable form.
However, using the solubility ratios as a representation of the driving force for a CSS polymorphic conversion can be fraught with errors, depending on the extent of changes to the solid phase composition and deviation from equilibrium. Different polymorphs can have vastly different solvi, which would lead to considerable compositional changes as one polymorph converts to another. Hence the start and end points in Fig. 18 would not necessarily have the same solid phase composition, which refutes the basis of eqn (2).
Furthermore, and from a practical point of view, SMPTs are often carried out by suspending a metastable polymorph in a solvent and letting the form conversion take place over time, with or without seeding of the stable form. Hence, for a system involving CSS, there are multiple mechanisms taking place concurrently, which ultimately determines the overall rate of the polymorphic conversion. A metastable polymorph having a certain level of a solid-state miscible component will initially dissolve up to the concentration equivalent to the solubility of the metastable form. At that point, the system is not at equilibrium, even when not accounting for the stable polymorph. The compositions in the liquid and solid phases will start to adjust towards their equilibrium concentrations, as determined by the Roozeboom diagram of the metastable form. This typically means that the impurity level in the solid phase is reduced, while the liquid is enriched. In other words, the initial state and hence the thermodynamic driving force, is changing during this re-equilibration.
With the appearance of the stable polymorphs, the conversion can take place in different ways. If the stable polymorph is introduced as chemically pure seeds, the composition of the seeds will also begin to change over time as the impurity is incorporated into the seeds during subsequent growth and equilibration. If the stable polymorph emerges via nucleation, the composition will likely be different than when at equilibrium, which again results in gradual changes to the solid phase composition over time.
The kinetics of the various molecular transports in the solid state may be different as diffusion is dependent on the prevailing crystal lattice. Hence, there are multiple mass transfer mechanisms taking place during a SMPT of a CSS system, all of which with their own timelines and contributions to the overall free energy of the system. Still, there is nothing that negates why polymorphic conversions can't be accelerated in the presence of a solid-state miscible second component. The following experimental example provides the necessary evidence.
Suspensions were prepared at ambient temperature comprising saturated solutions of 3HBA in 40 w% MeOH in H2O with respect to form II, with 0.5 g excess 3HBA form II solids. Seeds of pure 3HBA form I were added to the suspensions. The solid phase was monitored by Raman throughout the SMPT, which provided discrimination between form I and II, also in the presence of SA (ESI†). In the first experiment, no SA was included. In the second experiment, SA was added so that the starting slurry initially exhibited a composition of 0.5 w% SA in 3HBA in both solid and liquid phases, prior to seeding with form I. This condition corresponds to point 2 in Fig. 18. Both conditions were repeated to assess the reproducibility of the polymorphic conversion. The results are shown in Fig. 19.
 | ||
| Fig. 19 Rate of polymorphic conversion for 3HBA form II to form I, when chemically pure (X(3HBA) = 1.000) and containing 0.5 w% SA in 3HBA. Raman peak area between 1685 and 1705 cm−1 was used to quantify the form content in situ. Reference spectra are available in ESI.† | ||
Without the addition of SA, the polymorphic conversion is completed in about 260 min. When SA has been incorporated in the form II material, the time for the SMPT has been reduced to approx. 125 min. This ∼50% reduction in time is only attributed to the addition of SA, as all other parameters were kept constant. While the two experiments that include SA do exhibit higher variability as compared to the one without SA, this is not enough to alter the conclusion that adding a solid-state miscible impurity caused a significant acceleration in the conversion rate between two monotropic polymorphs.
The underlying reasons for the acceleration by addition of an impurity (or dopant) can be rationalized by analysis of the determined solubility profiles of 3HBA form I/βI and form II/βII in conjunction with changes in the compositions of the respective solid phases. As discussed earlier, the thermodynamic driving force as estimated by the solubility ratio, is at its maximum value at 0.5% SA in 3HBA in the solid phase, i.e. at the starting composition in the SMPT experiment (point 2 in Fig. 18). While this is only slightly higher than the solubility ratio between form II and I of pure 3HBA (1.38 vs. 1.32), the βII phase containing 0.5% SA in 3HBA is in fact even further away from equilibrium than is indicated by Fig. 18. The intrinsic solubility of βII is at that point equal to 164.1 mg g−1 solvent (i.e. the invariant point for βII). When reaching equilibrium after a completed form conversion to βI, the final compositions in the solid and liquid phases equal 0.014% and 98.99 w% SA in 3HBA, respectively (allowing βII to equilibrate without converting to βI yields 0.011% and 99.20 w% in the solid and liquid phases, respectively). Comparing the initial condition with the final condition thus correspond to a solubility ratio of 1.67 (164.1/98.4). Thus the de facto thermodynamic driving force for the initial solid phase of βII can be expected to be even higher in the start of the SMPT experiments containing 0.5% SA in 3HBA. These changes in the solid phase compositions are illustrated with black arrows in Fig. 16c. Finally, the last influencing factor in the acceleration of the polymorphic conversion relates to the solubility level itself. Higher solubility has been shown to be an important factor in enhancing the rate of a SMPT.27 Incorporating SA into the crystal lattice of 3HBA form II obviously elevates its solubility, which would then help to expedite the polymorphic conversion.
CSS or surface adsorption?
The impurity-induced SMPT acceleration observed herein can be compared to prior literature dealing with impurity or additive effects on crystal growth. Impurities are often reported to inhibit crystal growth, while some examples also exist of the opposite.28–30 These effects are almost exclusively rationalized by effects relating to surface adsorption, e.g. by blockage of kinks or steps during growth. The Cabrera–Vermilyea model for immobile impurities has for example been used to describe zero-growth behaviour or terminal crystal growth.28 This type of surface adsorption arguments have also been used to explain stabilization of different polymorphs via tailor-made additives.31–35 Considering the example provided herein and elsewhere, it is clear that CSS-formation with an impurity or second component provides an alternative explanation to these phenomena. CSSs are able to alter the thermodynamic properties of the host crystals resulting in different free energy states of polymorphs. For a dimorphic system, this can either translate into two polymorphs converging in stability with increasing composition,10,11 or as shown in this example, diverging in stability with higher levels of the second component in the crystal lattices.Without regard for surface adsorption, the selective thermodynamic changes imposed by the CSS formation alone provide an explanation for the acceleration of the polymorphic conversion of 3HBA. Given the scarcity of reports demonstrating the existence and effects of CSS of organic materials, it is difficult to ascertain how often solid-state miscible additives or impurities are responsible for observed changes in crystal growth rates or polymorphic conversions. But certainly, CSS can be expected to be far more relevant than originally assumed, especially in the case of tailor-made additives, which tend to exhibit significant structural similarities with the product molecules.
Conclusions
Two separate T–X binary phase diagrams have been constructed of SA and the two monotropic polymorphs of 3HBA using a combination of experimental data and thermodynamic modelling via the NRTL equation. CSS were formed on both sides of the phase diagram for both 3HBA polymorphs. The limited solid-state miscibility of 3HBA in the crystal lattice of SA (0.6% and 0.7% 3HBA in SA relative 3HBA form I and II, respectively) produced significant solubility enhancements up to the respective solvi (124% and 160%) in 40 w% MeOH in H2O at 25 °C. The solubility curves for both the form I and form II systems were regressed to the NRTL equation, with quantitative accuracy. For the 3HBA-rich side, the thermodynamically more stable form I is shown to exhibit higher miscibility in the solid state with SA in relation to form II, from 25 °C to melting. By measurement of solvent solubilities of the two polymorphs of 3HBA and their CSS phases, it is shown that the solubility curves diverge with increasing incorporation of SA. This feature in concert with the higher solvus of form I vis-à-vis form II result in an expanding free energy gap between the polymorphic CSS phases as the SA content increases. This system thus provides an intriguing example of how CSS formation in a monotropic polymorphic system can make a metastable phase even more metastable through the addition of an impurity.One implication of the diverging thermodynamic stability relationship was demonstrated in an experimental study by evaluating the rate of solution-mediated phase transformation, from form II to form I of 3HBA. By incorporation of only 0.5% of SA into the crystal lattice of form II of 3HBA, the polymorphic conversion was found to accelerate at twice the rate, as compared to when 3HBA is chemically pure. The results rationalized by the CSS formation provide an alternative explanation to observed crystal growth enhancements or inhibitions induced via impurities or additives, over prior theories linked to surface adsorption.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Tao Zhang: investigation, writing – original draft preparation. Francesco Ricci: methodology, software, writing – original draft preparation, writing – review and editing. Fateme Molajafari: software, writing – original draft preparation. Seyed Sepehr Mohajerani: investigation. Mitchell Paolello: investigation. Fredrik L. Nordstrom: conceptualization, formal analysis, investigation, supervision, writing – original draft preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge members of the Material and Analytical Sciences department.Notes and references
- Alloy phase diagrams, ASM Handbook, ed. H. Baker, ASM International, USA, 2016, vol. 3 Search PubMed.
- Molecular mixed crystals, Physical chemistry in action, ed. M. A. Cuevas-Diarte and H. A. J. Oonk, Springer, Switzerland, 2021 Search PubMed.
- A. I. Kitaigorodsky, Mixed crystals, Solid-state sciences 33, Springer-Verlag, Berlin Heidelberg, 1984 Search PubMed.
- S. S. Mohajerani, F. Ricci and F. L. Nordstrom, Solubility enhancements through crystalline solid solutions, the non-linear Tammann diagram and the T-X phase diagram of salicylic acid-benzoic acid, CrystEngComm, 2023, 25, 2607 RSC.
- Y. Wang, F. Ricci and F. L. Nordstrom, Terminal crystalline solid solutions and T-X phase diagram of salicylic acid – 4-hydroxybenzoic acid, Phys. Chem. Chem. Phys., 2024, 26, 3069 RSC.
- L. Keshavarz, R. R. E. Steendam, M. A. R. Blijlevens, M. Pishnamazi and P. J. Frawley, Influence of Impurities on the Solubility, Nucleation, Crystallization, and Compressibility of Paracetamol, Cryst. Growth Des., 2019, 19(7), 4193–4201 CrossRef CAS.
- F. L. Nordstrom, E. Sirota, C. Hartmanshenn, T. T. Kwok, M. Paolello, H. Li, V. Abeyta, T. Bramante, E. Madrigal, T. Behre and G. Capellades, Prevalence of impurity retention mechanisms in pharmaceutical crystallizations, Org. Process Res. Dev., 2023, 27(4), 723–741 CrossRef CAS.
- G. Baaklini, M. Schindler, L. Yuan, C. De Saint Jores, M. Sanselme, N. Couvrat, S. Clevers, P. Negrier, D. Mondieig, V. Dupray, Y. Cartigny, G. Gbabode and G. Coquerel, Critical influence of water on the polymorphism of 1,3-dimethylurea and other heterogenous equilibria, Molecules, 2023, 28, 7061 CrossRef CAS PubMed.
- F. L. Nordstrom, S. S. Mohajerani, B. Linehan and F. Ricci, Enantiotropic inconstancy, crystalline solid solutions and co-crystal in the salicylic acid-anthranilic acid system, Phys. Chem. Chem. Phys., 2022, 24, 26485 RSC.
- M. Paolello, S. S. Mohajerani, B. Linehan, F. Ricci, G. Capellades and F. L. Nordstrom, Polymorphic stability shifts, Co-crystals and Crystalline Solid Solutions. The T-X Phase Diagram of Salicylic acid – Salicylamide, Cryst. Growth Des., 2024, 24, 2188–2201 CrossRef CAS.
- S. S. Mohajerani, M. Paolello, B. Linehan, F. Ricci, G. Capellades and F. L. Nordstrom, Thermodynamic stability transitions and coexistence of two polymorphs via crystalline solid solutions: The T-X phase diagram of salicylic acid – 2,3-dihydroxybenzoic acid, Cryst. Growth Des., 2024, 24(11), 4847–4861 CrossRef CAS.
- W. Cochran, The Crystal and Molecular Structure of Salicylic Acid, Acta Crystallogr., 1953, 6, 260–268 CrossRef CAS.
- M. Sundaralingam and L. H. Jensen, Refinement of the Structure of Salicylic Acid, Acta Crystallogr., 1965, 18, 1053–1058 CrossRef CAS PubMed.
- G. E. Bacon and R. J. Jude, Neutron-diffraction Studies of Salicylic Acid and R-Resorcinol, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1973, 138, 19–40 CrossRef CAS.
- G. V. Gridunova, N. G. Furmunova, Y. T. Struchkov, Z. I. Ezhkova, L. P. Grigoreva and B. A. Chayanov, Crystal structures of two polymorphic modifications of m-hydroxybenzoic acid, Kristallografiya, 1982, 27(2), 267 CAS.
- F. L. Nordstrom and A. C. Rasmuson, Polymorphism and thermodynamics of m-hydroxybenzoic acid, Eur. J. Pharm. Sci., 2006, 28, 377–384 CrossRef PubMed.
- D. E. Braun, The trimorphism of 3-hydroxybenzoic acid: an experimental and computational study, CrystEngComm, 2021, 23, 2513 RSC.
- M. Svard and A. C. Rasmuson, m-Hydroxybenzoic acid: Quantifying thermodynamic stability and influence of solvent on the nucleation of a polymorphic system, Cryst. Growth Des., 2013, 13, 1140–1152 CrossRef.
- B. S. Bhatnagar, S. Cardon, M. J. Pikal and R. H. Bogner, Reliable determination of freeze-concentration using DSC, Thermochim. Acta, 2005, 425, 149–163 CrossRef CAS.
- L. Rycerz, Practical remarks concerning phase diagrams determination on the basis of differential scanning calorimetry measurements, J. Therm. Anal. Calorim., 2013, 113, 231–238 CrossRef CAS.
- I. A. Kasatkin, A. E. Glikin, H. Bradaczek and W. Franke, Kinetics of mixed crystal K2(SO4, CrO4) growth from aqueous solution, Cryst. Res. Technol., 1995, 30(5), 659–666 CrossRef CAS.
- J. M. Astilleros, C. M. Pina, L. Fernandez-Diaz and A. Putnis, Supersaturation functions in binary solid solution – aqueous solution systems, Geochim. Cosmochim. Acta, 2003, 67(9), 1601–1608 CrossRef CAS.
- M. Prieto, F. Heberling, R. M. Rodriguez-Galan and F. Brandt, Crystallization behavior of solid solutions from aqueous solutions: An environmental perspective, Prog. Cryst. Growth Charact. Mater., 2016, 62, 29–68 CrossRef CAS.
- M. Prieto, Thermodynamics of solid solution – aqueous solution systems, Rev. Mineral. Geochem., 2009, 70, 47–85 CrossRef CAS.
- A. G. Shtukenberg, Y. O. Punin and P. Y. Azimov, Crystallization in solid solution – aqueous solution systems: Thermodynamic and kinetic approaches, Kristallografiya, 2010, 55(2), 344–358 CrossRef.
- G. A. Pal'yanova, K. V. Chudnenko and T. V. Zhuravkova, Thermodynamic properties of solid solutions in the system Ag2S-Ag2Se, Thermochim. Acta, 2014, 575, 90–96 CrossRef.
- C.-H. Gu, V. Young Jr and D. J. W. Grant, Polymorph screening: Influence of solvents on the rate of solvent-mediated polymorphic transformation, J. Pharm. Sci., 2001, 90(11), 1878–1890 CrossRef CAS PubMed.
- P. A. Meenan, S. R. Anderson and D. L. Klug, in Handbook of Industrial Crystallization, ed. A. S. Myerson, Butterworth-Heinemann, Boston, 2nd edn, 2002, ch. 3, pp. 67–100 Search PubMed.
- H. J. Meyer, The influence of impurities on the growth rate of calcite, J. Cryst. Growth, 1984, 66, 639–646 CrossRef CAS.
- K. Sangwal, Effects of impurities on crystal growth processes, Prog. Cryst. Growth Charact. Mater., 1996, 32, 3–43 CrossRef CAS.
- C. Cashell, D. Corcoran and B. K. Hodnett, Effect of amino acid additives on the crystallization of L-glutamic acid, Cryst. Growth Des., 2005, 5(2), 593–597 CrossRef CAS.
- E. Staab, L. Addadi, L. Leiserowitz and M. Lahav, Control of polymorphism by “tailor-made” polymeric crystallization auxiliaries. Preferential precipitation of a metastable polar form for second harmonic generation, Adv. Mater., 1990, 2(1), 40–43 CrossRef CAS.
- R. J. Davey, N. Blagden, G. D. Potts and R. Docherty, Polymorphism in molecular crystals: Stabilization of a metastable form by conformational mimicry, J. Am. Chem. Soc., 1997, 119, 1767–1772 CrossRef CAS.
- C.-H. Gu, K. Chatterjee, V. Young Jr and D. J. W. Grant, Stabilization of a metastable polymorph of sulfamerazine by structurally related additives, J. Cryst. Growth, 2002, 235, 471–481 CrossRef CAS.
- R. Dowling, R. D. Davey, R. A. Curtis, G. Han, S. K. Poornachary, P. S. Chow and R. B. H. Tan, Acceleration of crystal growth rates: an unexpected effect of tailor-made additives, Chem. Commun., 2010, 46, 5924–5926 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce00784k |
| This journal is © The Royal Society of Chemistry 2024 |