DOI:
10.1039/D4CE00723A
(Paper)
CrystEngComm, 2024,
26, 5099-5107
Designed synthesis of Co(II) coordination polymers for evaluation of structural transformations†
Received
20th July 2024
, Accepted 13th August 2024
First published on 13th August 2024
Abstract
Five coordination polymers constructed from Co(II) salts, N,N′-bis(3-pyridylmethyl)oxalamide (L) and 4,4′-methylenedibenzoic acid (H2MBA), {[Co(L)(MBA)(H2O)]·H2O}n, 1, {[Co(L)(MBA)]·4H2O}n, 2, {[Co(L)(MBA)]·0.25H2O}n, 3, [Co(L)(MBA)(H2O)2]n, 4, and {[Co(L)0.5(MBA)(H2O)2]·CH3OH}n, 5, are reported, which have been structurally characterized by using single crystal X-ray crystallography. Complexes 1 and 4 have the same formula, but differ in the number of coordinated and cocrystallized water molecules, whereas 2 and 3 form a pair of supramolecular isomers with different numbers of cocrystallized water molecules. Complexes 1 and 4 show interdigitated 2D layers with a (64·8·10)(6)-2,4L3 topology, which differ in the ligand conformations of L and bonding modes of MBA2−. Complex 2 shows 2-fold interpenetrated 2D layers with a (64·8·10)(6)-2,4L3 topology and 3 displays a 2-fold interpenetrated 3D framework with a (4·62)(4·66·83)-fet topology, whereas 5 is a 1D ladder with a (62·10)(6)-2,3C1 topology, indicating that the coordination and/or cocrystallization of solvent molecules are important in determining their structural diversity. The structural transformations of 1–4 are also evaluated.
Introduction
Coordination polymers (CPs) with intriguing structural diversity and properties have been intensively investigated due to their potential applications in sensing, selective adsorption, gas storage, heterogeneous catalysis, magnetism, and luminescence.1–11 The structural diversity of CPs thus prepared is governed by the identity of the metal ion, and the length, softness, coordination ability and donor-atom direction of the ligands as well as the reaction conditions such as the solvent system, temperature and time. On the other hand, structural transformations of CPs, which can be initiated by the removal and exchange of cocrystallized solvent or exposure to reactive vapors and external stimuli such as heat, light, and mechanical energy, are important phenomena for application in switches and sensors.12–17
Although diverse structures can be observed for CPs, those showing structural transformation are hardly expected. Great effort remains necessary to enlighten the factors that may direct the structural diversity and structural transformations. Structural transformations have been observed in several bis-pyridyl-bis-amide (bpba)-based CPs supported by polycarboxylate ligands,18–24 demonstrating that the flexible nature of the bpba ligands and the various bonding modes of the polycarboxylate anions play important roles in promoting the reversible and irreversible structural transformations therein. Moreover, it is essential to prepare diverse CPs containing the same metal ion and organic ligand to investigate the structural transformations of CPs. Therefore, designed synthesis of the target products by careful evaluation of the metal to ligand ratio and reaction conditions is attempted.
Herein, we report the synthesis and structural characterization of five new CPs constructed from Co(II) salt, N,N′-bis(3-pyridylmethyl)oxalamide (L), Fig. 1, and 4,4′-methylenedibenzoic acid (H2MBA), {[Co(L)(MBA)(H2O)]·H2O}n, 1, {[Co(L)(MBA)]·4H2O}n, 2, {[Co(L)(MBA)]·0.25H2O}n, 3, [Co(L)(MBA)(H2O)2]n, 4, and {[Co(L)0.5(MBA)(H2O)2]·CH3OH}n, 5. The factors that govern the structural diversity and structural transformation of these CPs are also illuminated.
 |
| | Fig. 1 Structure of N,N′-bis(3-pyridylmethyl)oxalamide (L). | |
Experimental details
General procedures
IR spectra (KBr disk) were obtained on a JASCO FT/IR-460 plus spectrometer. Elemental analyses were performed using an Elementar Vario EL III or an Elementar Vario EL cube type analyzer. Thermal gravimetric analysis (TGA) measurements were carried out on a SII Nano Technology Inc. TG/DTA 6200 over the temperature range of 30 to 900 °C at a heating rate of 10 °C min−1 under N2. Powder X-ray diffraction patterns were measured by using a Bruker D2 PHASER diffractometer with CuKα (λα = 1.54 Å) radiation.
Materials
The reagent Co(OAc)2·4H2O was purchased from J. T. Baker Co., and H2MBA from Matrix Scientific Co. The ligand N,N′-bis(3-pyridylmethyl)oxalamide (L) was prepared according to a published procedure.25
Preparation of {[Co(L)(MBA)(H2O)]·H2O}n, 1.
A mixture of Co(OAc)2·4H2O (0.050 g, 0.20 mmol), L (0.027 g, 0.10 mmol) and H2MBA (0.026 g, 0.10 mmol) was placed in a 23 mL Teflon reaction flask containing 8 mL H2O and 2 mL MeOH, which was sealed and heated at 80 °C for 96 h under autogenous pressure and then the reaction system was cooled to room temperature at a rate of 2 °C per hour. Violet crystals suitable for single-crystal X-ray diffraction were obtained. Yield: 0.025 g (20%). Anal. calcd for C29H28CoN4O8 (MW = 619.48): C, 56.21; H, 4.56; N, 9.05%. Found: C, 55.79; H, 4.40; N, 8.88%. FT-IR (cm−1): 3505 (m), 3225 (w), 3043 (w), 1657 (s), 1607 (s), 1513 (s), 1430 (m), 1392 (m), 1355 (m), 1034 (m), Fig. S1.†
Preparation of {[Co(L)(MBA)]·4H2O}n, 2.
Complex 2 was prepared by following the similar procedures for 1, except that a mixture of Co(OAc)2·4H2O (0.050 g, 0.20 mmol), L (0.027 g, 0.10 mmol) and H2MBA (0.026 g, 0.10 mmol) in 5 mL H2O and 5 mL MeOH was heated at 60 °C for 24 h. Dark purple crystals were obtained. Yield: 0.030 g (23%). Anal. calcd for C29H32CoN4O10 (MW = 655.51): C, 53.12; H, 4.92; N, 8.55%. Found: C, 53.18; H, 4.54; N, 8.84%. FT-IR (cm−1): 3503.54 (m), 3222.47 (w), 3043.60 (w), 1658.96 (s), 1631.48 (m), 1607.38 (s), 1512.40 (s), 1430.92 (w), 1397.17 (w), 1359.09 (w), 1034.62 (m), Fig. S1.†
Preparation of {[Co(L)(MBA)]·0.25H2O}n, 3.
Complex 3 was prepared by following the similar procedures for 1, except that a mixture of Co(OAc)2·4H2O (0.050 g, 0.20 mmol), L (0.027 g, 0.10 mmol) and H2MBA (0.026 g, 0.10 mmol) in 10 mL H2O was heated at 120 °C for 48 h. Light purple crystals were obtained. Yield: 0.027 g (23%). Anal. calcd for C29H24.5CoN4O6.25 (MW = 587.95): C, 59.22; H, 4.20; N, 9.53%. Found: C, 59.30; H, 3.94; N, 9.40%. FT-IR (cm−1): 3503 (w), 3349 (w), 3259 (w), 1657 (m), 1596 (s), 1513 (m), 1430 (w), 1384 (w), 1351 (m), 1034 (m), Fig. S1.†
Preparation of [Co(L)(MBA)(H2O)2]n, 4.
Complex 4 was prepared by following the similar procedures for 1, except that a mixture of Co(OAc)2·4H2O (0.050 g, 0.20 mmol), L (0.027 g, 0.10 mmol) and H2MBA (0.026 g, 0.10 mmol) in 10 mL H2O was heated at 80 °C for 48 h. Light pink crystals were obtained. Yield: 0.026 g (21%). Anal. calcd for C29H28CoN4O8 (MW = 619.12): C, 56.21; H, 4.56; N, 9.05%. Found: C, 56.05; H, 4.43; N, 9.31%. FT-IR (cm−1): 3503 (m), 3239 (w), 3238 (w), 1657 (s), 1598 (s), 1514 (m), 1481 (w), 1391 (m), 1352 (w), 1035 (m), Fig. S1.†
Preparation of{[Co(L)0.5(MBA)(H2O)2]·CH3OH}n, 5.
Complex 5 was prepared by following the similar procedures for 1, except that a mixture of Co(OAc)2·4H2O (0.050 g, 0.20 mmol), L (0.027 g, 0.10 mmol) and H2MBA (0.06 g, 0.10 mmol) in 5 mL H2O and 5 mL MeOH was heated at 60 °C for 96 h. Violet crystals were obtained. Yield: 0.017 g (16%). Anal. calcd for C23H25CoN2O8 (MW = 516.38): C, 53.50; H, 4.88; N, 5.42%. Found: C, 53.75; H, 4.45; N, 6.29%. FT-IR (cm−1): 3491 (w), 3397 (w), 3266 (w), 1683 (m), 1595 (s), 1545 (m), 1508 (w), 1435 (m), 1387 (s), 1190 (m), 1027 (s), Fig. S2.†
While the above-mentioned procedures afforded the major products 1–5, some minor products can also be obtained, Table S1.†
X-ray crystallography
The diffraction data for complexes 1–5 were collected on a Bruker AXS SMART APEX II CCD diffractometer or Bruker D8 QUEST PHOTON III CPAD at 296 K, which was equipped with a graphite-monochromated Mo Kα (λα = 0.71073 Å) radiation source. Data reduction was performed by using standard methods and well-established computational procedures.26 The structure factors were obtained after Lorentz and polarization corrections. An empirical absorption correction based on “multi-scan” was applied to the data for all complexes. The positions of some of the heavier atoms were located by the direct method. The remaining atoms were found in a series of alternating difference Fourier maps and least-squares refinements, while the hydrogen atoms except those of the water molecules were added by using the HADD command in SHELXTL 6.1012.27Table 1 lists the crystal data for complexes 1–5.
Table 1 Crystallographic data for complexes 1–5
| Complex |
1
|
2
|
3
|
4
|
5
|
|
R
1 = ∑∥Fo| − |Fc∥/∑|Fo|.
wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. w = 1/[σ2(Fo2 + (ap)2 + (bp)], p = [max(Fo2 or 0) + 2(Fc2)]/3. a = 0.03694, b = 0.4742 for 1; a = 0.0467, b = 0 for 2; a = 0.0425, b = 1.926 for 3; a = 0.0425, b = 1.6437 for 4; a = 0.0380, b = 0 for 5.
Quality-of-fit = [∑w(|Fo2| − |Fc2|)2/(Nobserved − Nparameters)]1/2.
|
| Formula |
C29H28CoN4O8 |
C29H32CoN4O10 |
C29H24.5CoN4O6.25 |
C29H28CoN4O8 |
C23H25CoN2O8 |
| Formula weight |
619.48 |
655.51 |
587.95 |
619.48 |
516.38 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pc
|
P21/n |
C2/c |
P |
|
a, Å |
10.1343(9) |
8.9391(9) |
11.4825(4) |
27.7041(11) |
9.834(4) |
|
b, Å |
10.7473(9) |
9.8609(10) |
21.8344(6) |
9.3364(4) |
11.041(5) |
|
c, Å |
13.9008(12) |
17.5100(18) |
11.8159(4) |
10.6200(4) |
11.166(5) |
|
α, ° |
71.026(2) |
90 |
90 |
90 |
85.008(10) |
|
β, ° |
88.874(2) |
93.993(2) |
114.8619(9) |
95.3715(13) |
76.029(11) |
|
γ, ° |
82.993(2) |
90 |
90 |
90 |
85.758(11) |
|
V, Å3 |
1420.7(2) |
1539.7(3) |
2687.86(15) |
2734.87(19) |
1170.3(9) |
|
Z
|
2 |
2 |
4 |
4 |
2 |
|
D
calc, Mg m−3 |
1.448 |
1.414 |
1.453 |
1.505 |
1.465 |
|
F(000) |
642 |
682 |
1214 |
1284 |
536 |
|
μ(Mo Kα), mm−1 |
0.662 |
0.619 |
0.691 |
0.687 |
0.784 |
| Range (2θ) for data collection, ° |
3.09 ≤ 2θ ≤ 56.90 |
4.13 ≤ 2θ ≤ 52.00 |
3.73 ≤ 2θ ≤ 56.81 |
2.95 ≤ 2θ ≤ 56.55 |
3.70 ≤ 2θ ≤ 52.00 |
| Independent reflections |
7105 [R(int) = 0.0652] |
5787 [R(int) = 0.0529] |
6710 [R(int) = 0.0486] |
3382 [R(int) = 0.0512] |
4604 [R(int) = 0.0718] |
| Data/restraints/parameters |
7105/0/379 |
5787/2/397 |
6710/0/370 |
3382/0/200 |
4604/0/307 |
| Quality-of-fit indicatorc |
1.009 |
1.005 |
1.022 |
1.027 |
1.007 |
| Final R indices [I > 2σ(I)]a,b |
R
1 = 0.0511, wR2 = 0.0948 |
R
1 = 0.0515, wR2 = 0.0997 |
R
1 = 0.0461, wR2 = 0.1010 |
R
1 = 0.0343, wR2 = 0.0795 |
R
1 = 0.0446, wR2 = 0.0843 |
|
R indices (all data) |
R
1 = 0.0914, wR2 = 0.1083 |
R
1 = 0.0942, wR2 = 0.1134 |
R
1 = 0.0731, wR2 = 0.1126 |
R
1 = 0.0575, wR2 = 0.0895 |
R
1 = 0.0815, wR2 = 0.0953 |
Results and discussion
Synthesis of complexes 1–5
By careful evaluation of the metal to ligand ratio and reaction conditions, five distinct CPs constructed from Co(II) salts, L and H2MBA can be achieved. Table S1† lists the reaction conditions and yields of each attempt, indicating that the yields of these complexes are dependent on the reaction time, temperature and solvent. While crystals of complexes 3, 4 and 5 can be isolated directly from the reaction mixtures by the manipulation of the reaction temperatures, those of 1 and 2 were obtained by manual separation based on the shapes and colours of the crystals.
Crystal structure of 1.
Crystals of 1 conform to the triclinic space group P and each asymmetric unit consists of one Co(II) ion, one L ligand, one MBA2− ligand, one coordinated water molecule and one cocrystallized water molecule. Fig. 2(a) shows the coordination environment about the Co(II) metal center, which is six-coordinated by two nitrogen atoms from two L ligands [Co–N = 2.114(2) and 2.136(2) Å], three oxygen atoms from two MBA2− ligands [Co–O = 2.0652(17)–2.2381(17) Å] and one oxygen atom from coordination water [Co–O = 2.1188(17) Å]. The Co(II) central metal atoms are bridged by MBA2− and L to form a 2D layer. Topologically, if the Co(II) atoms are regarded as 4-connected nodes and the MBA2− ligands as 2-connected nodes, whereas the L ligands are considered as linkers, the structure of 1 can be simplified as a 2,4-connected 2D net with a (64·8·10)(6)-2,4L3 topology, Fig. 2(b). Fig. 2(c) shows a pair of the 2D layers that are interdigitated to each other. On the other hand, if the MBA2− ligands are also considered as linkers, the structure of 1 can be further simplified as a 2D net with a (44·62)-sql topology.28
 |
| | Fig. 2 (a) Coordination environment of the Co(II) ion in 1 showing the (64·8·10)(6)-2,4L3 topology. Symmetry transformations used to generate equivalent atoms: (A) x, y + 1, z − 1; (B) x − 1, y, z. (b) Topological structure of 1, showing the (64·8·10)(6)-2,4L3 topology. (c) Two drawings showing a pair of interdigitated 2D layers in different directions. | |
Crystal structure of 2.
Crystals of 2 conform to the monoclinic space group Pc and each asymmetric unit consists of one Co(II) ion, one L ligand, one MBA2− ligand and four cocrystallized water molecules. Fig. 3(a) shows the coordination environment about the Co(II) metal center, which is six-coordinated by two nitrogen atoms from two L ligands [Co–N = 2.082(5) and 2.086(6) Å] and four oxygen atoms from two MBA2− ligands [Co–O = 2.047(5)–2.285(5) Å]. The Co(II) central metal atoms are bridged by MBA2− and L to form a 2D layer. Topologically, if the Co(II) atoms are regarded as 4-connected nodes and the MBA2− ligands as 2-connected nodes, whereas the L ligands are considered as linkers, the structure of 2 can be simplified as a 2,4-connected 2D net with a (64·8·10)(6)-2,4L3 topology, Fig. 3(b), showing a 2-fold interpenetration, Fig. 3(c). On the other hand, if the MBA2− ligands are also considered as linkers, the structure of 2 can be further simplified as a 2-fold interpenetrated 2D net with a (44·62)-sql topology.
 |
| | Fig. 3 (a) Coordination environment of the Co(II) ion in 2. Symmetry transformations used to generate equivalent atoms: (A) x − 1, y − 1, z; (B) x − 1, y + 1, z. (b) Topological structure of 2, showing the (64·8·10)(6)-2,4L3 topology. (c) Two drawings showing the 2-fold interpenetration in different directions. | |
Crystal structure of 3.
Crystals of 3 conform to the monoclinic space group P21/n and each asymmetric unit consists of one Co(II) ion, one L ligand, one MBA2− ligand and a quarter of a cocrystallized water molecule. Fig. 4(a) shows the coordination environment about the Co(II) metal center, which is six-coordinated by two nitrogen atoms from two L ligands [Co–N = 2.152(2) and 2.165(2) Å] and four oxygen atoms from three MBA2− ligands [Co–O = 1.9995(17)–2.2228(17) Å]. The Co(II) central metal atoms are bridged by MBA2− to form dinuclear units, which are extended by L to form a 3D structure. Topologically, if the Co(II) atoms are regarded as 5-connected nodes and the MBA2− ligands as 3-connected nodes, whereas the L ligands are considered as linkers, the structure of 3 can be simplified as a 3,5-connected net with a (4·62)(4·66·83)-fet topology (standard representation), Fig. 4(b), showing a 2-fold interpenetration, Fig. 4(c). If the Co(II) dinuclear units are regarded as 6-connected nodes, while the organic ligands are considered as linkers, the structure of 3 can be simplified as a 6-connected net with a {412·63}-pcu topology (cluster representation), Fig. 4(d), showing a 2-fold interpenetration, Fig. 4(e).
 |
| | Fig. 4 (a) Coordination environment of the Co(II) ion in 3. Symmetry transformations used to generate equivalent atoms: (A) x − 1/2, −y + 3/2, z + 1/2; (B) −x + 5/2, y + 1/2, −z + 1/2; (C) x − 1, y, z − 1; (D) −x + 2, −y + 2, −z + 1; (E) −x + 3, −y + 2, −z + 2. (b) Topological structure of 3, showing a (4·62)(4·66·83)-fet topology. (c) A drawing showing the 2-fold interpenetration with the fet topology. (d) Topological structure of 3, showing the {412·63}-pcu topology. (e) A drawing showing the 2-fold interpenetration with the pcu topology. | |
Structure of 4.
Crystals of 4 conform to the monoclinic space group C2/c and each asymmetric unit consists of one Co(II) ion, one L ligand, one MBA2− ligand and two coordinated water molecules. Fig. 5(a) shows the coordination environment about the Co(II) metal center, which is six-coordinated by two nitrogen atoms from two L ligands, [Co–N = 2.2474(15) Å], two oxygen atoms from two MBA2− ligands [Co–O = 2.0598(12)–2.0599(12) Å] and two oxygen atoms from coordination water [Co–O = 2.0890(14) Å]. The Co(II) central metal atoms are bridged by MBA2− and L to form a 2D layer. Topologically, if the Co(II) atoms are regarded as 4-connected nodes and the MBA2− ligands as 2-connected nodes, whereas the L ligands are considered as linkers, the structure of 4 can be simplified as a 2,4-connected 2D net with a (64·8·10)(6)-2,4L3 topology, Fig. 5(b). Fig. 5(c) shows a pair of the 2D layers that are interdigitated to each other. If the MBA2− ligands are considered as linkers, the structure of 4 can be further simplified as a 4-connected net with a {44·62}-sql topology.
 |
| | Fig. 5 (a) Coordination environment of the Co(II) ion in 4. Symmetry transformations used to generate equivalent atoms: (A) −x + 1/2, −y + 1/2, −z; (B) −x + 1, y, −z − 1/2; (C) −x, y, −z − 1/2. (b) Topological structure of 4, showing the (64·8·10)(6)-2,4L3 topology. (c) Two drawings showing a pair of interdigitated 2D layers in different directions. | |
Although complexes 1 and 4 adopt the same (64·8·10)(6)-2,4L3 topology with some degree of interdigitation, they are different in the evenness of the planes due to the different ligand conformations of L and bonding modes of MBA2−, vide infra.
Structure of 5.
Crystals of 5 conform to the triclinic space group P and each asymmetric unit consists of one Co(II) ion, half an L ligand, one MBA2− ligand, two coordinated water molecules and one cocrystallized MeOH molecule. Fig. 6(a) shows the coordination environment about the Co(II) metal center, which is six-coordinated by one nitrogen atom from the L ligand, [Co–N = 2.168(2) Å], two oxygen atoms from two MBA2− ligands [Co–O = 2.019(2) - 2.202(2) Å] and two oxygen atoms from coordination water molecules [Co–O = 2.073(2)–2.105(2) Å]. The Co(II) central metal atoms are bridged by MBA2− and L to form a 1D ladder. Topologically, if the Co(II) atoms are regarded as 3-connected nodes, and MBA2− as 2-connected nodes, whereas the L ligands are considered as linkers, the structure of 5 can be simplified as a 2,3-connected 1D net with a (62·10)(6)-2,3C1 topology, Fig. 6(b). If the MBA2− ligands are also considered as linkers, the structure can be further simplified as a 3-connected net with a (42·6)-(4,4)(0,2) topology. Fig. 6(c).
 |
| | Fig. 6 (a) Coordination environment of the Co(II) ion in 5. Symmetry transformations used to generate equivalent atoms: (A) x, y + 1, z − 1; (B) −x + 1, −y + 2, −z. (b) Topological structure of 5, showing the (62·10)(6)-2,3C1 topology. (c) Topological structure of 5, showing the (42·6)-(4,4)(0,2) topology. | |
Ligand conformations, bonding modes and structural types
For the ligand N,N′-bis(3-pyridylmethyl)oxalamide (L), the positions of the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups can be distinguished as trans or cis. When the two CO groups show opposite directions, the position is defined as trans, and when they show the same direction, it is cis. Because of the different orientations adopted by the pyridyl nitrogen atoms and the amide oxygen atoms, three more conformations can be expressed as anti–anti, syn–anti and syn–syn. Accordingly, the ligand conformations of L in 1–5 are listed in Table 2, as well as the coordination modes of the MBA2− ligands. Moreover, while the L ligands in 1–5 coordinate to the metal centers through the two pyridyl nitrogen atoms, the MBA2− ligands bridge two or three metal atoms, showing μ2-κ2O,O′:κO′′, μ2-κ2O,O′ κ2O′′,O′′′, μ3-κ2O,O′:κO′′:κO′′′, μ2-κO:κO′ and μ2-κ2O,O′:κO′′ coordination modes, respectively, and resulting in 1-, 2- and 3D structures subject to the changes of the coordinated and cocrystallized solvent molecules. Complexes 2 and 3 can be regarded as a pair of supramolecular isomers29 that cocrystallized with different numbers of water molecules, showing 2-fold interpenetrated 2D layers with a 2,4L3 topology and a 2-fold interpenetrated 3D framework with a fet topology, respectively.
O groups can be distinguished as trans or cis. When the two CO groups show opposite directions, the position is defined as trans, and when they show the same direction, it is cis. Because of the different orientations adopted by the pyridyl nitrogen atoms and the amide oxygen atoms, three more conformations can be expressed as anti–anti, syn–anti and syn–syn. Accordingly, the ligand conformations of L in 1–5 are listed in Table 2, as well as the coordination modes of the MBA2− ligands. Moreover, while the L ligands in 1–5 coordinate to the metal centers through the two pyridyl nitrogen atoms, the MBA2− ligands bridge two or three metal atoms, showing μ2-κ2O,O′:κO′′, μ2-κ2O,O′ κ2O′′,O′′′, μ3-κ2O,O′:κO′′:κO′′′, μ2-κO:κO′ and μ2-κ2O,O′:κO′′ coordination modes, respectively, and resulting in 1-, 2- and 3D structures subject to the changes of the coordinated and cocrystallized solvent molecules. Complexes 2 and 3 can be regarded as a pair of supramolecular isomers29 that cocrystallized with different numbers of water molecules, showing 2-fold interpenetrated 2D layers with a 2,4L3 topology and a 2-fold interpenetrated 3D framework with a fet topology, respectively.
Table 2 Drawings showing the ligand conformations of L and the coordination modes of MBA2− in 1–5
|
|
Ligand conformation |
Coordination mode |
|
1
|
 trans syn–syn
trans syn–syn |
 μ2-κ2O,O′:κO′′ μ2-κ2O,O′:κO′′ |
|
2
|
 trans anti–anti
trans anti–anti |
 μ2-κ2O,O′ κ2O′′,O′′′ μ2-κ2O,O′ κ2O′′,O′′′ |
|
3
|
 trans syn–syn
trans syn–syn |
 μ3-κ2O,O′:κO′′:κO′′′ μ3-κ2O,O′:κO′′:κO′′′ |
|
4
|
 trans anti–syn
trans anti–syn |
 μ2-κO:κO′ μ2-κO:κO′ |
|
5
|
 trans syn–syn
trans syn–syn |
 μ2-κ2O,O′:κO′′ μ2-κ2O,O′:κO′′ |
Table 3 lists the CPs containing L that have been reported, demonstrating that the structural types of CPs based on L are governed by the polycarboxylate ligands and metal ions. Moreover, combination of L with 5-tert-IPA or MBA2− may lead to the formation of entangled Co(II) CPs, revealing the importance of the identity of the supporting dicarboxylate ligands in the formation of the entangled CPs based on the semi-rigid L. In contrast, it is interesting to note that supported by the angular dicarboxylate ligand, the flexible bpba ligands may adopt the suitable conformations for the formation of the entangled Co(II) CPs.30 Structural comparisons of 1–5 with four CPs derived from Co(OAc)2·4H2O, L and 4,4′-sulfonyldibenzoic acid (H2SDA), revealing non-entangled 2D layers with sql, 2,6L1, (4,4)Ia, and 6L12 topologies, respectively,31 suggest that delicate change from the carbon atom to the SO2 group that bridges the pyridyl rings of the angular dicarboxylate ligand drastically modifies the structural diversity. On the other hand, five Cd(II) CPs constructed from 1,4-bis(2-methyl-imidazol-1-yl)butane and 5-bromoisophthalic acid have been reported.32
Table 3 CPs based on L in mixed ligand systems
| Complex |
Structure |
References |
| Abbreviations: H2OBA = 4,4-oxydibenzoic acid; H2SDA = 4,4-sulfonyldibenzoic acid; 1,4-NDC = 1,4-naphthalenedicarboxylic acid; H2FIPBB = 4,4′-hexafluoroisopropylidenebis(benzoic acid); 1,2,4,5-H4BTEC = 1,2,4,5-bezenetetracarboxylic acid; 1,3,5-H3BTC = 1,3,5-benzenetricarboxylic acid; 5-tert-H2IPA = 5-tert-butylbenzene-1,3-dicarboxylic acid; 5-NH2-H2IPA = 5-aminoisophthalic acid. |
| {[Ni(L)(OBA)(H2O)]·H2O}n |
Interdigitated 2D, 2,4L2 |
25
|
| {[Ni(L)(SDA)(H2O)2]·H2O·CH3OH}n |
2D, sql |
25
|
| {[Cd(L)(1,4-NDC)(H2O)]·5H2O}n |
1D looped chain, 2,4C5 |
33
|
| {[Co2(L)2(1,4-NDC)2(H2O)4]·3MeOH·H2O}n |
3D, cds |
33
|
| [Zn(L)0.5(FIPBB)·H2O]n |
2D, bey |
34
|
| [Co(L)(OBA)·2H2O]n |
2D, 3,5L2 |
34
|
| [Co(L)0.5(FIPBB)]n |
2D, bey |
34
|
| [Co2(L)2(FIPBB)2(H2O)5·4H2O]n |
1D looped chain, 2,2,2,4C10 |
34
|
| [Co(L)0.5(1,2,4,5-BTEC)0.5(H2O)]n |
3D, 4,5,6T11 |
35
|
| {[Co(L)(1,2,4,5-BTEC)0.5(H2O)]·H2O}n |
2D, bex |
35
|
| {[Co(L)1.5(1,2,4,5-BTEC)0.5(H2O)]·3H2O}n |
3D, dmp |
35
|
| [Zn(L)0.5(1,3,5-HBTC)(H2O)2]n |
1D looped chain |
36
|
| {[Cu(L)(1,3,5-HBTC)]·H2O}n |
2D, sql |
37
|
| {[Co(L)(SDA)(H2O)2]·H2O·CH3OH}n |
2D, sql |
31
|
| {[Co(L)0.5(SDA)]·2H2O·0.5L}n |
2D, 2,6L1 |
31
|
| {[Co(L)1.5(SDA)(H2O)]·H2O}n |
2D, (4,4)Ia |
31
|
| {[Co2(L)1.5(SDA)2(H2O)2]·4H2O}n |
2D, 6L12 |
31
|
| {[Co(L)(1,3,5-HBTC)]·H2O}n |
2D, 3,5L2 |
35
|
| {[Co(L)(5-tert-IPA)(H2O)2]·H2O}n |
3-fold interpenetrated 2D, hcb |
38
|
| [Co(L)0.5(5-NH2-IPA)(H2O)]n |
2D, bey |
38
|
| {[Co(L)(MBA)(H2O)]·H2O}n |
2D, 2,4L3 |
This work |
| {[Co(L)(MBA)]·4H2O}n |
2-fold interpenetrated 2D, 2,4L3 |
This work |
| {[Co(L)(MBA)]·0.25H2O}n |
2-fold interpenetrated 3D, fet |
This work |
| [Co(L)(MBA)(H2O)2]n |
2D, 2,4L3 |
This work |
| {[Co(L)0.5(MBA)(H2O)2]·CH3OH}n |
1D, 2,3C1 |
This work |
Powder X-ray analysis and thermal properties
In order to check the bulk purity of the products, powder X-ray diffraction (PXRD) experiments have been carried out for all complexes. As shown in Fig. S3–S7,† the peak positions of the experimental and simulated PXRD patterns are in agreement with each other, which demonstrates that the crystal structures are truly representative of the bulk materials. The differences in intensity may be owing to the preferred orientation of the powder samples.
Thermal gravimetric analyses (TGA) were carried out to examine the thermal decomposition of all complexes, which were recorded from about 30 to 800 °C at 10 °C min−1 under a N2 atmosphere, Fig. S8–S12.†Table 4 reveals that two-step decomposition involving loss of solvent molecules and loss of organic ligands is observed for all of the complexes.
Table 4 Thermal properties of complexes 1–5
| Complex |
Weight loss of solvent |
Weight loss of ligand |
| °C (calc/found), % |
°C (calc/found), % |
|
1
|
2H2O |
L + (MBA2−) |
| ∼150 (5.80/7.19) |
200–800 (84.91/83.23) |
|
2
|
4H2O |
L + (MBA2−) |
| ∼200 (10.98/10.85) |
200–800 (80.24/79.82) |
|
3
|
0.25 H2O |
L + (MBA2−) |
| ∼100 (0.77/0.31) |
250–800 (89.46/90.79) |
|
4
|
2H2O |
L + (MBA2−) |
| ∼200 (5.81/7.34) |
290–800 (84.90/85.46) |
|
5
|
2H2O + MeOH |
0.5L + (MBA2−) |
| ∼130 (13.17/14.30) |
250–800 (75.72/72.63) |
Chemical stability and structural transformation of complexes 1–4
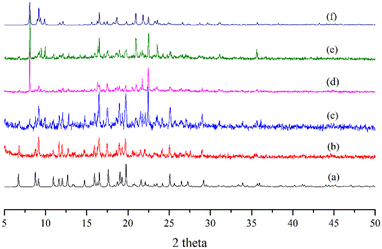
Complexes 1–4 provide a unique opportunity to study structural transformation due to the water removal and adsorption because they comprise the same metal center and organic ligand. We first checked their chemical stability in organic solvents. For each experiment, 10 mg of the complex was immersed in 10 mL of the solvent for a week, involving water, methanol (MeOH), ethanol (EtOH), ether, hexane, tetrahydrofuran (THF), acetonitrile (ACN), dichloromethane (DCM), dimethylacetamide (DMAC), and dimethylformamide (DMF), which was then filtered and then dried under vacuum. The PXRD patterns of these solid samples, Fig. S13–S16,† reveal that complexes 1–3 were not stable upon immersion in MeOH, DMF and water, respectively, whereas 4 retained its structure in all of the solvents.
The stabilities of complexes 1–4 upon removal of the cocrystallized/coordinated water molecules were then investigated. Complexes 1–4 were heated at 150, 200, 100 and 200 °C, respectively, for two hours to obtain fully desolvated samples, which were then immersed in various solvents. Fig. S17† shows the photos and PXRD patterns of complex 1 heated at 150 °C for 2 hours and then immersed in a variety of organic solvents, whereas Fig. 7 indicates that 1 is partially transformed into 3 upon desolvation and the desolvated sample can be effectively transformed into 3 in THF and ACN. Fig. S18 and S19† indicate that the framework of complex 2 decomposed upon desolvation and the desolvated sample of 3 retained its structure in the organic solvents. Moreover, Fig. S20† and 8 show that complex 4 can be transformed into 3 upon water removal, but the transformation is irreversible.
 |
| | Fig. 7 PXRD patterns of (a) simulation of 1, (b) as-synthesized 1, (c) 1 heated at 150 °C for 2 h, (d) desolvated 1 in THF, (e) desolvated 1 in ACN and (f) simulation of 3. | |
 |
| | Fig. 8 PXRD patterns of (a) simulation of 4, (b) as-synthesized 4, (c) 4 heated at 200 °C for 2 h, (d) desolvated 4 in water and (e) simulation of 3. | |
To check the possibility of structural transformation under hydrothermal conditions, complexes 1–4, respectively, were heated in water at 120 °C for 2 days. Fig. S21† displays the PXRD patterns after the attempts, indicating that complexes 1–4 were all transformed into the same complex that was not structurally characterized by using single crystal X-ray diffraction. Supported by the IR spectra, Fig. S22,† and the elemental analysis, Fig. S23,† this unknown complex probably comprises only the Co(II) ion and MBA2− ligand. Fig. 9 summarizes the synthetic pathways and structural transformations for complexes 1–4.
 |
| | Fig. 9 Synthetic pathways and structural transformations for complexes 1–5. | |
Conclusions
The synthesis and structural characterization of five diverse Co(II) CPs comprising L and MBA2− have been successfully accomplished. 1D, 2D and 3D structures can be shown for these CPs, demonstrating that cocrystallized and coordinated solvent molecules govern the structural diversity. The yields of these CPs were dependent on the metal-to-ligand ratio, solvent system, and reaction temperature. Modification of the bridging group of the angular dicarboxylate ligand from the SO2 group31 to the carbon atom has significantly altered the structural diversity of the Co(II) CPs thus prepared. Investigations on the chemical stability and structural transformation of 1–4 indicate that the desolvated sample of 1 can be transformed into 3 in THF and ACN, while 4 can be irreversibly transformed into 3 upon water removal. Designed synthesis of diverse CPs for the evaluation of structural transformations can thus be achieved if proper combination of the mixed ligands as well as adequate reaction conditions can be manipulated.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for complexes 1–4 have been deposited at the CCDC with no. 2369132–2369136.
Author contributions
Investigation, C.-Y. L.; data curation, M. U., S.-W. W. and K. B. T.; review and supervision, T.-R. C. and J.-D. C. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Science and Technology Council of the Republic of China for support.
References
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrstrom, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- D. J. Wales, J. Grand, V. P. Ting, R. D. Burke, K. J. Edler, C. R. Bowen, S. Mintova and A. D. Burrows, Chem. Soc. Rev., 2015, 44, 4290–4321 RSC.
-
E. R. T. Tiekink and J. J. Vittal, Frontiers in Crystal Engineering, John Wiley & Sons, Ltd., England, 2006 Search PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed.
- X.-K. Yang, W.-T. Lee, J.-H. Hu and J.-D. Chen, CrystEngComm, 2021, 23, 4486–4493 RSC.
- S. Mondal and P. Dastidar, Cryst. Growth Des., 2020, 20, 7411–7420 CrossRef CAS.
-
S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, Cambridge, UK, 2009 Search PubMed.
- Sh. Li, B. Wang, G. Liu, X. Li, C. Sun, Z. Zhang and X. Wang, Inorg. Chem. Front., 2024, 11, 1561–1572 RSC.
- X.-D. Zhang, J.-A. Hua, J.-H. Guo, Y. Zhao and W.-Y. Sun, J. Mater. Chem. C, 2018, 6, 12623–12630 RSC.
- X. Wu, D. Li, L. Xu, Y.-F. Jiang, Y. Zhao and J. Zhao, CrystEngComm, 2023, 25, 3222–3228 RSC.
- Z.-P. Chen, D. Li, L. Xu, Y.-F. Jiang, K. Lin, Y. Zhao and J. Zhao, New J. Chem., 2022, 46, 12994–13000 RSC.
- J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS.
- G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC.
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS PubMed.
- D. Liu, Z.-G. Ren, H.-X. Li, J.-P. Lang, N.-Y. Li and B. F. Abrahams, Angew. Chem., Int. Ed., 2010, 49, 4767–4770 CrossRef CAS PubMed.
- F.-L. Hu, Y. Mi, C. Zhu, B. F. Abrahams, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2018, 57, 12696–12701 CrossRef CAS PubMed.
- M.-F. Wang, Y. Mi, F.-L. Hu, Z. Niu, X.-H. Yin, Q. Huang, H.-F. Wang and J.-P. Lang, J. Am. Chem. Soc., 2020, 142, 700–704 CrossRef CAS PubMed.
- C.-H. Hsu, W.-C. Huang, X.-K. Yang, C.-T. Yang, P. M. Chhetri and J.-D. Chen, Cryst. Growth Des., 2019, 19, 1728–1737 CrossRef CAS.
- X.-K. Yang and J.-D. Chen, CrystEngComm, 2019, 21, 7437–7446 RSC.
- Y.-F. Liu, J.-H. Hu, W.-T. Lee, X.-K. Yang and J.-D. Chen, Cryst. Growth Des., 2020, 20, 7211–7218 CrossRef CAS.
- C.-J. Chen, W.-T. Lee, J.-H. Hu, P. M. Chhetri and J.-D. Chen, CrystEngComm, 2020, 22, 7565–7574 RSC.
- C.-J. Chen, C.-L. Chen, W.-T. Lee, J.-H. Hu, P. M. Chhetri and J.-D. Chen, J. Mol. Struct., 2021, 1244, 131252 CrossRef CAS.
- J.-H. Hu, Y.-C. Liu and J.-D. Chen, CrystEngComm, 2021, 23, 7471–7478 RSC.
- J.-H. Hu, Y.-C. Liu, Y.-H. Liu and J.-D. Chen, CrystEngComm, 2022, 24, 4120–4127 RSC.
- W.-T. Lee, T.-T. Liao and J.-D. Chen, Int. J. Mol. Sci., 2022, 23, 3603 CrossRef CAS PubMed.
-
Bruker AXS, APEX2, V2008.6, SAD ABS V2008/1, SAINT+V7.60A, SHELXTL V6.14, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385–2396 RSC.
- W.-C. Huang, W.-H. Chen, C.-L. Chen, T.-T. Liao, Y.-W. Chen and J.-D. Chen, CrystEngComm, 2023, 25, 5575–5587 RSC.
- C.-Y. Lee, Y.-H. Ye, S.-W. Wang and J.-D. Chen, Molecules, 2024, 29, 1748 CrossRef CAS PubMed.
- Y.-F. Hou, B. Liu, K.-F. Yue, C.-S. Zhou, Y.-M. Wang, N. Yan and Y.-Y. Wang, CrystEngComm, 2014, 16, 9560–9567 RSC.
- W.-J. Chen, C.-Y. Lee, Y.-H. Huang and J.-D. Chen, Polyhedron, 2022, 223, 115991 CrossRef CAS.
- V. Lakshmanan, C.-Y. Lee, Y.-W. Tseng, Y.-H. Liu, C.-H. Lin and J.-D. Chen, CrystEngComm, 2022, 24, 6076–6086 RSC.
- T.-T. Liao, S.-Y. Lin and J.-D. Chen, CrystEngComm, 2023, 25, 1723–1730 RSC.
- J.-H. Hu, H.-H. Hsu, Y.-W. Chen, W.-H. Chen, S.-M. Liu and J.-D. Chen, J. Mol. Struct., 2023, 1289, 135896 CrossRef CAS.
- S.-Y. Lin, Y.-L. Shen and W.-H. Chen, Molecules, 2024, 29, 311 CrossRef CAS PubMed.
- J.-H. Hu, Y.-H. Ye, Y.-W. Chen, S.-M. Liu and J.-D. Chen, J. Mol. Struct., 2024, 1312, 138544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra (Fig. S1, S2 and S22). Yields (Table S1). PXRD patterns (Fig. S3–S7 and Fig. S13– S21). TGA curves (Fig. S8–S12). Elemental analysis (Fig. S23). CCDC no. 2369132–2369136 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00723a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a






 trans syn–syn
trans syn–syn μ2-κ2O,O′:κO′′
μ2-κ2O,O′:κO′′ trans anti–anti
trans anti–anti μ2-κ2O,O′ κ2O′′,O′′′
μ2-κ2O,O′ κ2O′′,O′′′ trans syn–syn
trans syn–syn μ3-κ2O,O′:κO′′:κO′′′
μ3-κ2O,O′:κO′′:κO′′′ trans anti–syn
trans anti–syn μ2-κO:κO′
μ2-κO:κO′ trans syn–syn
trans syn–syn μ2-κ2O,O′:κO′′
μ2-κ2O,O′:κO′′

