
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Iodophthalimide as a halogen bond donor, a comparison with N-iodosuccinimide and N-iodosaccharin†
Ie-Rang
Jeon
 ,
Olivier
Jeannin
,
Antoine
Robert
,
Frédéric
Barrière
and
Marc
Fourmigué
*
,
Olivier
Jeannin
,
Antoine
Robert
,
Frédéric
Barrière
and
Marc
Fourmigué
*
UnivRennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), Campus de Beaulieu, Rennes, 35000, France. E-mail: marc.fourmigue@univ-rennes.fr
First published on 25th September 2024
Abstract
We investigate here the ability of N-iodophthalimide (NIPht) to act as a halogen bond (HaB) donor, in comparison with well-known HaB donors N-iodosuccinimide (NISucc) and N-iodosaccharin (NISacc). The structure of NIPht itself is reported, together with those of neutral adducts with 4-dimethylaminopyridine (DMAP), 4,4′-bipyridine and 2,2′-bipyridine derivatives. Comparison with analogous adducts involving NISucc and NISacc shows that NIPht behaves essentially like NISucc as a HaB donor, both forming weaker adducts than NISacc with a given Lewis base. A symmetric anionic complex [NPht–I–NPht]− is isolated in the presence of [K(18-crown-6)]+. It exhibits N–I distances very close to those observed in the known [NSucc–I–NSucc]− and [NSacc–I–NSacc]− species (2.24–2.26 Å), confirming the 3-center–4-electron (3c–4e) character of the bonding in these species. This similarity confirms the peculiar character of the only other reported salt of [NPht–I–NPht]−, namely [Me4N][NPht–I–NPht], where the longer N–I distances (2.29 Å) are a consequence of a specific solid-state arrangement and C–H⋯O hydrogen bonds.
Introduction
Because of its strength and directionality, halogen bonding (HaB) is currently considered as the prototypical σ-hole interaction, where the halogen atom acts as the electrophile.1 This interaction plays a crucial role in many areas of supramolecular chemistry and crystal engineering and has been shown to be the strongest in the halogen series in the order I > Br ≫ Cl ≫ F.2 Covalently linked iodine atoms are more prone to exhibit such strong σ-holes when properly activated by electron-withdrawing groups, as in perfluoroalkyl, perfluoroaryl or alkynyl iodide derivatives. When iodine is covalently linked to a nitrogen atom, strong σ-holes can also be found, for example in cationic N-iodopyridinium species, providing the symmetric [Py–I–Py]+ cation.3,4 Neutral halogen-bonded adducts are found with N-iodoimides (Fig. 1a) such as N-iodosuccinimide (NISucc)5,6 or N,N′-diiodohydantoins (DIH),7 or with N-iodosulfamides such as N-iodosaccharin (NISacc) as HaB donors.8 Each of them gives rise to many reported examples of halogen-bonded adducts, essentially with pyridines9,10 and pyridine N-oxides.11 Symmetric (or close-to-symmetric) anionic systems were also described upon the interaction of a HaB donor such as, for example, NISacc with the corresponding saccharinate anion to give the symmetric [NSacc–I–NSacc]− anion (Fig. 1a).12,13 | ||
| Fig. 1 Chemical structures of (a and b) the investigated HaB donors NISucc, NISacc and NIPht and their anionic complexes and (c) the investigated HaB acceptors. | ||
Surprisingly, among the different N-iodoimides, N-iodophthalimide (NIPht, Fig. 1b), a well-known iodinating agent in organic chemistry,14 has not been reported to act as an HaB donor in the solid state, perhaps because it is not commercially available. Only one acetonitrile adduct of the perfluoro-N-iodophthalimide has been reported to date,15 while, very recently, the crystal structure of the symmetric anion [NPht–I–NPht]− was described as a Me4N+ salt and characterized by a short N–I bond (2.293(11) Å) and a linear geometry (N–I–N angle at 180°).13 Although these structural characteristics are very close to those reported for other HaB donors such as NISucc and NISacc in similar symmetric [NSucc–I–NSucc]− and [NSacc–I–NSacc]− anions,12,13 they do not allow, in a first approximation, for a ranking of the HaB donor strength between these different HaB donors.
In order to evaluate the ability of NIPht to enter in such halogen-bonded cocrystals, we have investigated their formation with different pyridines as Lewis bases, and we were able to crystallize and structurally characterize three different halogen-bonded adducts of NIPht with, respectively, 4-dimethylaminopyridine (DMAP), 4,4′-bipyridine and 2,2′-bipyridine (Fig. 1c), allowing thus for useful comparisons of the structural characteristics of the adducts with those obtained with NISacc or NISucc and the same pyridine-based HaB acceptors. Besides, we were also able to isolate and structurally characterize a second example of the symmetric anionic [NPht–I–NPht]− species as its [(18-crown-6)K+] salt, at variance with the only reported Me4N+ salt.13 The evolution of the N–I distances within the [NPht–I–NPht]− species as a function of the nature of the cation counterbalancing its charge will also be analyzed.
Results and discussion
NIPht was prepared, as previously described, from phthalimide, PhI(OAc)2 and I2 in 99% yield.16 It crystallizes in the orthorhombic system, space group P212121 with one molecule in the general position in the unit cell. As shown in Fig. 2, the molecules are associated into chains running along b through the 21 screw axis, with a short and linear HaB with an I⋯O distance at 2.726(4) Å (RR = 0.78), N–I⋯O angle at 166.8(1)° and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯I angle at 146.4(3)°. These values can be compared with those reported in the structure of NISucc,5 [I⋯O: 2.580(6) Å (RR = 0.74), N–I⋯O: 175.7(2)°, and CO⋯I: 121.6°(5)], indicating that NIPht appears as a slightly weaker HaB donor than NISucc, with, nevertheless a good HaB ability characterized with this RR = 0.78 value for the N–I⋯OC interaction. This is also confirmed by the evolution on the covalent N–I bond length, observed here at 2.034(4) Å, while it is found slightly more elongated in NISucc at 2.060(6) Å, another signature of a stronger HaB interaction in NISucc. Note that a similar comparison with the structure of NISacc itself cannot be conducted as its structure is reported as a solvate with one water molecule acting as the HaB acceptor, rather than a carbonyl or sulfonyl oxygen atom.8
O⋯I angle at 146.4(3)°. These values can be compared with those reported in the structure of NISucc,5 [I⋯O: 2.580(6) Å (RR = 0.74), N–I⋯O: 175.7(2)°, and CO⋯I: 121.6°(5)], indicating that NIPht appears as a slightly weaker HaB donor than NISucc, with, nevertheless a good HaB ability characterized with this RR = 0.78 value for the N–I⋯OC interaction. This is also confirmed by the evolution on the covalent N–I bond length, observed here at 2.034(4) Å, while it is found slightly more elongated in NISucc at 2.060(6) Å, another signature of a stronger HaB interaction in NISucc. Note that a similar comparison with the structure of NISacc itself cannot be conducted as its structure is reported as a solvate with one water molecule acting as the HaB acceptor, rather than a carbonyl or sulfonyl oxygen atom.8
 | ||
| Fig. 2 Details of the HaB interaction (as pink dotted lines) in the structure of NIPht. | ||
Calculations of the ESP extremum of the σ-hole on each of these three HaB donors, namely, NIPht, NISucc and NISacc were conducted to possibly propose a ranking of the three HaB donors. Indeed, since halogen bonding is primarily electrostatic in nature,17,18 a measure of the halogen bond donor character can be given by the maximum value of the molecular surface electrostatic potential at the iodine atom (VS,max). As shown in Fig. 3, it appears that NIPht exhibits the smallest calculated Vs,max value (+41.1 kcal mol−1) of the three HaB donor molecules. Nevertheless, this Vs,max value is very close to that found for NISucc (+41.8 kcal mol−1) and exceeds those calculated under the same conditions for well-known HaB donors such as C6F5–I (+35.1 kcal mol−1) or Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–I (+34.6 kcal mol−1) (Fig. S1†), indicating that NIPht should definitively act as a good HaB donor.
C–I (+34.6 kcal mol−1) (Fig. S1†), indicating that NIPht should definitively act as a good HaB donor.
 | ||
| Fig. 3 Molecular electrostatic potential surface at an isovalue of 0.001 for (a) NIPht, (b) NISucc and (c) NISacc (B3LYP/6-31G**-LANLdp, Gaussian16 Rev A.03) with extrema values at oxygen and iodine atoms indicated. Red indicates a negative charge density and blue, a positive charge density. The full-scale range is −0.0522/+0.0725 au (Hartrees), i.e. −32.8/+45.5 kcal mol−1 for the three compounds. | ||
Cocrystal structures within neutral adducts
Co-crystallization experiments of NIPht were accordingly conducted with 4-dimethylaminopyridine (DMAP), 4,4′-bipyridine, 2,2′-bipyridine, and its substituted analog 5,5′-dimethyl-2,2′-bipyridine, abbreviated, respectively, as p-bipy, o-bipy and Me2-o-bipy (Fig. 1c). The use of these geometrically constrained o-bipy or Me2-o-bipy molecules, furthermore blessed with a decreased Lewis base character compared with p-bipy, was indeed considered to test the evolution of the HaB strength. Crystals amenable to X-ray diffraction were obtained with the four pyridine derivatives, with the following composition, NIPht·DMAP, (NIPht)2·(p-bipy), (NIPht)2·(o-bipy) and (NIPht)2·(Me2-o-bipy).NIPht·DMAP crystallizes in the monoclinic system, space group P21/n. As shown in Fig. 4, the iodine atom is engaged in a short and linear HaB interaction with the pyridinic nitrogen atom. Note also that the molecular planes of both partners are almost perpendicular to each other, with a plane-to-plane angle of 85.7(4)°. Structural characteristics of the HaB interaction are collected in Table 1, together with the reported data for similar DMAP adducts of NISacc and NISucc. We note here that, despite a slightly weaker σ-hole on iodine, the HaB interaction with NIPht is stronger than that with NISucc, with a shortening of the I⋯NDMAP HaB length and a concomitant lengthening of the “covalent” N–I bond length.
 | ||
| Fig. 4 Molecular structure of the NIPht·DMAP adduct. The HaB interaction is depicted as a pink dotted line. | ||
| T(K) | N–I dist. | I⋯NDMAP dist. | N–I⋯NDMAP angle | N⋯NDMAP dist. | Ref. | |
|---|---|---|---|---|---|---|
| NISucc·DMAP | RT | 2.146(4) | 2.407(4) | 178.9(1) | 4.553(6) | 9a |
| RT | 2.138(5) | 2.403(6) | 178.7(2) | 4.541(8) | 6 | |
| NIPht·DMAP | 150(2) | 2.177(3) | 2.325(3) | 177.68(5) | 4.501(6) | This work |
| NISacc·DMAP | 150(2) | 2.292(2) | 2.218(2) | 178.5(1) | 4.509(3) | 9a |
| RT | 2.292(1) | 2.228(1) | 178.8(1) | 4.520(1) | 9a |
Cocrystallization of NIPht with 4,4′-bipyridine (p-bipy) afforded two different phases (see the ESI†). Both adopt a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, α-(NIPht)2(p-bipy) in the monoclinic P21/n space group and β-(NIPht)2(p-bipy) in the monoclinic P21/c space group, with, in both structures, the adduct located on an inversion center (Fig. 5). As shown in Table 2, characteristic N–I and I⋯Nbipy bond distances are, within a 3σ tolerance, the same in both phases. The main difference is found in the plane-to-plane angle between the NIPht and o-bipy moieties, at θ = 20.4(2)° in α-(NIPht)2(p-bipy) (Fig. 5b) but at θ = 52.5(1)° in β-(NIPht)2(p-bipy) (Fig. 5d). This difference brings important consequences on the solid-state association of the adducts in the crystal, with a face-to-face stacking of the close-to-planar adducts in the α-phase, but a more complex organization in the β-phase (Fig. S2†).
:1 stoichiometry, α-(NIPht)2(p-bipy) in the monoclinic P21/n space group and β-(NIPht)2(p-bipy) in the monoclinic P21/c space group, with, in both structures, the adduct located on an inversion center (Fig. 5). As shown in Table 2, characteristic N–I and I⋯Nbipy bond distances are, within a 3σ tolerance, the same in both phases. The main difference is found in the plane-to-plane angle between the NIPht and o-bipy moieties, at θ = 20.4(2)° in α-(NIPht)2(p-bipy) (Fig. 5b) but at θ = 52.5(1)° in β-(NIPht)2(p-bipy) (Fig. 5d). This difference brings important consequences on the solid-state association of the adducts in the crystal, with a face-to-face stacking of the close-to-planar adducts in the α-phase, but a more complex organization in the β-phase (Fig. S2†).
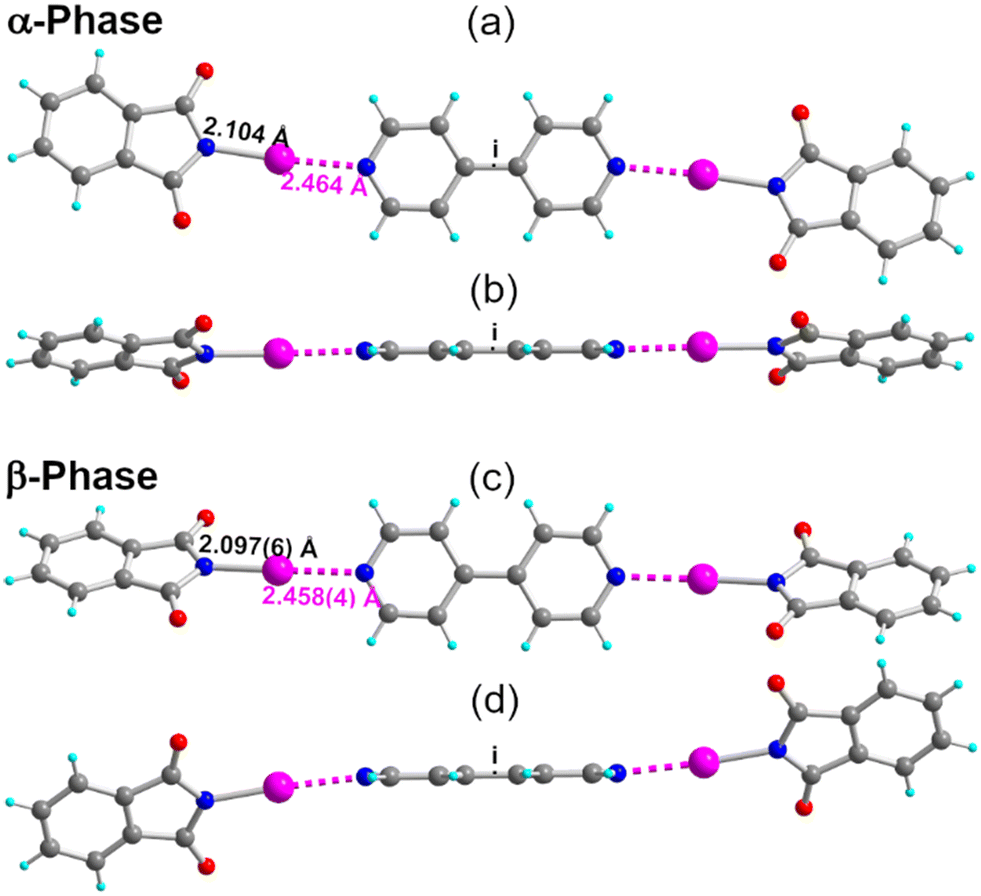 | ||
| Fig. 5 Details of the α-(NIPht)2(p-bipy) adduct, viewed (a) perpendicular to the p-bipy plane and (b) parallel to the p-bipy plane, and of the β-(NIPht)2(p-bipy) adduct, viewed (c) perpendicular to the p-bipy plane and (d) parallel to the p-bipy plane. The HaB interaction is depicted as pink dotted lines. | ||
| T(K) | N–I dist. | I⋯NPy dist. | N–I⋯NPy angle | N⋯NPy dist. | θ angle | |
|---|---|---|---|---|---|---|
| (NIPht)2(p-bipy): α-phase | 296(2) | 2.104(5) | 2.464(5) | 176.2(2) | 4.565(7) | 20.4(2) |
| β-phase | 296(2) | 2.097(6) | 2.458(4) | 176.79(16) | 4.553(7) | 52.5(1) |
| (NISacc)2(p-bipy) | 296(2) | 2.173(6) | 2.346(6) | 176.4(2) | 4.517(8) | 56.8(2) |
In order to allow for a comparison of NIPht with the other HaB donors, NISucc and NISacc, we also attempted the preparation of their respective adducts with 4,4′-bipyridine. Our trials were unsuccessful with NISucc, but crystals of good quality could be obtained with NISacc. (NISacc)2(p-bipy) adopts a very similar geometry (Fig. 6), with notably stronger HaB interactions than those with NIPht (Table 2), and with a plane-to-plane angle between NISacc and the pyridine moieties of θ = 56.8(2)° (Fig. 6b).
 | ||
| Fig. 6 Molecular structure of the (NISacc)2(p-bipy) adduct, viewed (a) perpendicular to the p-bipy plane and (b) parallel to the p-bipy plane. The HaB interaction is depicted as pink dotted lines. | ||
Cocrystals with the sterically demanding 2,2′-bipyridine were also investigated but our efforts to isolate good quality cocrystals with NIPht for satisfactory refinements were unsuccessful (unit cell parameters are reported in the ESI†). We therefore also considered the dimethyl analog, 5,5′-dimethyl-2,2′-bipyridine (o-Me2bipy) and were able to isolate good quality crystals of the 2:1 adduct with NIPht as a CH2Cl2 solvate, formulated as (NIPht)2(o-Me2bipy)·(CH2Cl2). It crystallizes in the orthorhombic system, space group Pba2, with the adduct located on a 2-fold axis (Fig. 7). The o-Me2bipy molecule itself is not planar with a Nbipy–C–C–Nbipy torsion angle ψ that amounts to +143.0(7)° (Table 3). Also, as already observed in the para analog (NIPht)2(p-bipy), the NIPht moiety is not coplanar with the pyridinyl ring but makes an angle θ of 28°. The HaB interaction with the I⋯NPy distance at 2.624(4) Å is also notably weaker than that with the para analogs α/β-(NIPht)2(p-bipy), where it is found at ≈2.46 Å.
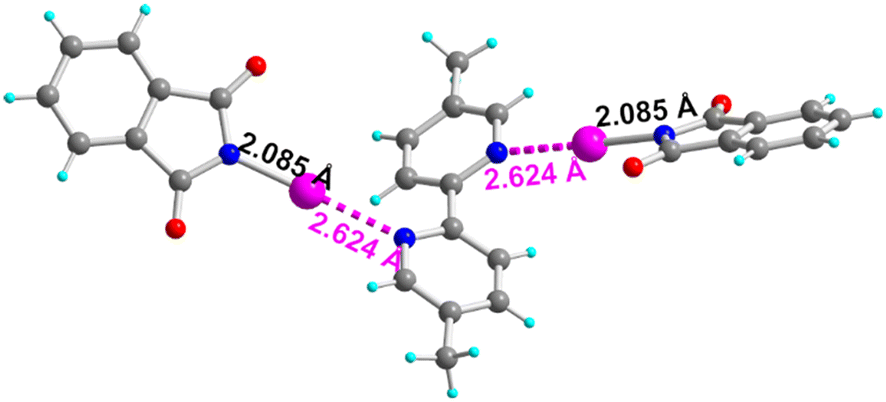 | ||
| Fig. 7 Details of the solid-state association in the (NIPht)2(o-Me2bipy) adduct. | ||
| T(K) | N–I dist. | I⋯NPy dist. | N–I⋯NPy angle | N⋯NPy dist. | ψ NpyCCNpy torsion angle | θ angle | |
|---|---|---|---|---|---|---|---|
| (NIPht)2(o-Me2bipy) | 296(2) | 2.085(6) | 2.624(6) | 178.9(2) | 4.709(9) | +143.0(7) | 28(1) |
| (NISucc)2(o-bipy) | 150(2) | 2.121(4) | 2.467(4) | 177.26(7) | 4.587(7) | −117.3(2) | 84(1) |
| 2.099(4) | 2.507(4) | 176.53(7) | 4.604(7) | 11(1) | |||
| (NISucc)2(o-Me2bipy) | 150(2) | 2.103(4) | 2.512(5) | 175.09(6) | 4.611(8) | +56.4(22) | 58.5(5) |
| 2.100(2) | 2.522(2) | 175.41(6) | 4.619(4) | 60.0(1) | |||
| (NISacc)2(o-bipy) | 296(2) | 2.139(5) | 2.438(5) | 175.4(1) | 4.573(7) | −132.7(4) | 80.0(1) |
| 2.131(5) | 2.454(5) | 174.2(1) | 4.580(9) | 79.7(1) |
To further strengthen our comparison with the two other HaB donors NISucc and NISacc, we also investigated their adducts with o-bipy and Me2-o-bipy and were able to isolate and structurally characterize three of them, namely, (NISucc)2(o-bipy), (NISucc)2(o-Me2bipy) and (NISacc)2(o-bipy). As shown in Fig. 8 and Table 3, they adopt different geometries, particularly with respect to the NpyCCNpy torsion angle ψ between the two pyridine rings, adopting an anticlinal conformation with three of them while the (NISucc)2(o-Me2bipy) exhibits a synclinal conformation with 30° < ψ < 90°. All adducts with NISucc and NISacc exhibit a stronger HaB interaction than that with NIPht, with the I⋯N Py distance in the 2.44–2.52 Å range, to be compared with the 2.624(6) Å value found with NIPht in (NIPht)2(o-Me2bipy).
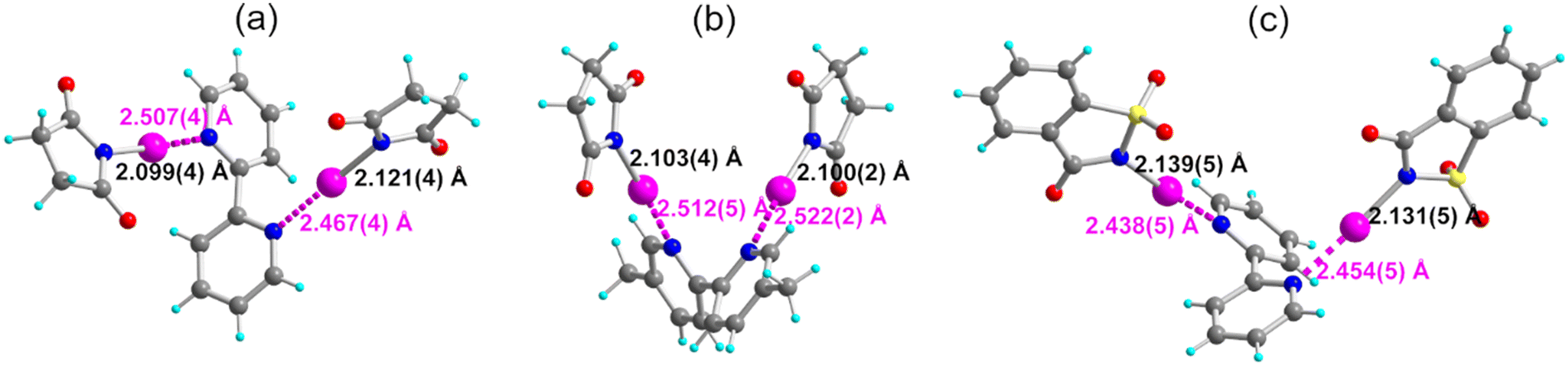 | ||
| Fig. 8 Molecular structure of the three adducts with 2,2′-bipyridines: (a) (NISucc)2(o-bipy), (b) (NISucc)2(o-Me2bipy) and (c) (NISacc)2(o-bipy). The HaB interaction is depicted as pink dotted lines. | ||
In conclusion of this part dedicated to neutral halogen-bonded adducts, the evolution of the HaB strength of NIPht vs. the different Lewis bases explored here, namely, DMAP, p-bipy and o-Me2bipy follows the trends observed with other HaB donors, with a ranking DMAP > p-bipy > o-Me2bipy. Indeed, the overall decreased Lewis base character of 2,2′-bipyridines was discussed by Pennington et al.19 in cocrystals of p-diiodotetrafluorobenzene with p-bipy,20o-bipy,21 and Me2-o-bipy.19 This ranking is also in line with the diiodine basicity scale defined by Laurence et al.22 who determined the pKBI2 values of 3.78 for DMAP, 2.22 for pyridine itself, 1.81 for p-bipy and 0.62 for o-bipy.
The different systems described above, in neutral adducts with pyridine-based Lewis bases, allow us to establish the HaB donor ability of NIPht, by comparison with other well-known HaB donors such as NISucc and NISacc. With such neutral pyridines, it appears that NIPht is always a weaker HaB donor than NISacc, as anticipated from the large difference of the calculated extrema of the electrostatic potential on iodine (Fig. 3). The comparison with NISucc is ambiguous, as NIPht appears to give a notably stronger HaB than NISucc with DMAP, but a weaker one with the other Lewis bases (o-bipy and o-Me2bipy) investigated here. Altogether both HaB donors NIPht and NISucc behave very closely.
The anionic [NPht–I–NPht]− species
As mentioned in the Introduction, symmetric (or close-to-symmetric) anionic systems involving NIPht (and other N-iodiimides) were already described upon interactions of an HaB donor such as NIPht with the corresponding phthalimidate anion to give the symmetric [NPht–I–NPht]− anion (Fig. 1b). In such systems, the covalent part of the N–I bond is enhanced up to the point that they have been described as 3-center–4-electron bonds. Several examples were reported with the bromine analog [NPht–Br–NPht]− while the iodine derivatives are known with NISucc in [Bu4N][NSucc–I–NSucc]23 and with NISacc in [Bu4N][NSacc–I–NSacc].12,13 One single example has been reported to date involving NIPht, namely, the tetramethylammonium salt [Me4N][NPht–I–NPht].13 We report here another example of this [NPht–I–NPht]− anion prepared directly from NIPht and potassium phthalimidate in the presence of 18-crown-6, allowing the isolation of [K(18-crown-6)][NPht–I–NPht]. It crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with both ions in the general position (Fig. 9). We have reported in Table 4 the structural characteristics of the anion, together with those reported for [Me4N][NPht–I–NPht], as well as the Bu4N+ salts of the analogous NISucc and NISacc derivatives.
with both ions in the general position (Fig. 9). We have reported in Table 4 the structural characteristics of the anion, together with those reported for [Me4N][NPht–I–NPht], as well as the Bu4N+ salts of the analogous NISucc and NISacc derivatives.
 | ||
| Fig. 9 Details of the solid-state association within [K(18-crown-6)][NPht–I–NPht] showing the chain-like motif formed by coordination with the [K(18-crown-6)]+ cation. | ||
| T(K) | N–I dist. | I–N dist. | N–I–N angle | N⋯N dist. | CN⋯NC torsion angle | Ref. | |
|---|---|---|---|---|---|---|---|
| a As the [K(18-crown-6)]+ salt. b As the Me4N+ salt. c As the Bu4N+ salt. d The lowest of the possible C–N⋯N–C torsion angles is reported. | |||||||
| [NPht–I–NPht]−a | 296(2) | 2.246(9) | 2.247(9) | 179.7(1) | 4.493(18) | 7.7(4)d | This work |
| [NPht–I–NPht]−b | 170(2) | 2.293(11) | 2.293(11) | 180.00 | 4.587(16) | 0.5(4)d | 13 |
| [NSucc–I–NSucc]−c | 100 | 2.264(2) | 2.244(2) | 179.57(6) | 4.508(2) | 36.7(2)d | 23 |
| [NSacc–I–NSacc]−c | 100 | 2.247(5) | 2.250(5) | 176.82(12) | 4.495(8) | 36.2(4) | 12 |
As shown in Fig. 9, the potassium cation completes its coordination sphere with two carbonyl oxygen atoms of the phthalimidate moieties in apical positions, leading to the formation of supramolecular chains with an alternation of cationic and anionic moieties. The two K+⋯O contact lengths are comparable, as well as the two N–I bond lengths in the [NPht–I–NPht]− anion (Table 4), giving an essentially symmetric complex (at the 3σ level).
By analogy with other reports on analogous structures with NISacc (see below), the angle between the two phthalimidate molecular planes is illustrated here by the C–N⋯N–C dihedral angle, here at 7.7(4)°, indicating a close-to-planar conformation. The N–I distances compare very well with those reported in other systems with NISucc and NISacc and appear therefore to be somehow independent of the nature of the imidato anions surrounding the iodonium moiety, an observation in line with the strong covalency of the N–I bonds in these 3-center–4-electron (3c–4e) systems. In that respect, the reported tetramethyammonium salt of [NPht–I–NPht]− appears as unusual, with notably longer N–I bonds (Table 4). This was attributed to the presence of hydrogen bonds with the Me4N+ cation, which leads to the formation of ⋯[Me4N]+⋯ [NPht–I–NPht]−⋯[Me4N]+⋯[NPht–I–NPht]−⋯ chains (Fig. S3†). However, a closer inspection of the two other reported systems with NISucc and NISacc as Bu4N+ salts also reveals the presence of similar (or even shorter) hydrogen bonds involving the hydrogen atoms of the α-methylene moieties in Bu4N+ (Fig. S4 and Table S3†). We believe that besides these HBs, it is also the rigidity of the Me4N+ cation that pushes away the two phthalimidate from the iodonium cation in [Me4N][NPh–I–NPht], while such an effect is absent in the Bu4N+ salts of [NSucc–I–NSucc]− and [NSacc–I–NSacc]− and in the [K(18-crown-6)]+ salt reported here, which all exhibit N–I distances in the same range.
Conclusions
We have demonstrated here with several examples that NIPht, like the well-known NISucc and NISacc compounds, can act as a powerful HaB donor through its σ-hole on iodine, whose amplitude is comparable to that of NISucc but notably smaller than that of NISacc. HaB interactions within neutral adducts with pyridines compare with those found with NISucc, allowing for a NIPht ≈ NISucc < NISacc ranking. A second example of the negatively charged, essentially symmetric [NPht–I–NPht]− species is isolated here as the [K(18-crown-6)]+ salt, with N–I distances comparable to those observed in other 3c–4e systems such as [NSucc–I–NSucc]− and [NSacc–I–Sacc]−. The carbonyl oxygen atoms of the phthalimidate moieties are involved in the coordination sphere of the [K(18-crown-6)]+ cation, allowing for the formation of a 1-D polymer in the solid state.Experimental
All experimental information (synthesis, characterization data, crystal growth conditions, X-ray crystallography and DFT calculations of ESP maps) are in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data have been deposited at the CCDC under CCDC numbers 2366101–2366110 for, respectively, compounds NIPht, (NIPht)·DMAP, α-(NIPht)2(p-bipy), β-(NIPht)2(p-bipy), (NIPht)2(o-Me2bipy)·CH2Cl2, (NISacc)2(p-bipy), (NISucc)2(o-bipy), (NISucc)2(o-Me2bipy), (NISacc)2(o-bipy) and [K(18-crown-6)][NPht–I–NPht] and can be obtained directly from https://www.ccdc.cam.ac.uk/products/csd/request/request.php4.
Author contributions
I.-R. J. and M. F. formulated the project. A. R. and I.-R. J. synthesized and crystallized the compounds. O. J. collected and solved X-ray diffraction data. F. B. performed the DFT calculations. M. F. wrote the paper and all the authors contributed to revising it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ANR (Paris) for financial support under grants ANR-17-ERC3-0003-01 and ANR-17-CE07-0025-02. This work was granted access to the HPC resources of TGCC/CEA/CINES/IDRIS under the allocation 2024 AD010814136R1 awarded by GENCI.References
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) M. H. Kolár and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed; (d) H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- (a) J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 229–229 RSC; (b) I. Haque and J. L. Wood, J. Mol. Struct., 1968, 2, 217–238 CrossRef CAS.
- (a) L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC; (b) S. B. Hakkert and M. Erdélyi, J. Phys. Org. Chem., 2015, 28, 226–233 CrossRef CAS; (c) L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed; (d) L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed; (e) R. Puttreddy, P. Kumar and K. Rissanen, Chem. – Eur. J., 2024, 30, e202304178 CrossRef CAS PubMed.
- K. Padmanabhan, I. C. Paul and D. Y. Curtin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 88–92 CrossRef.
- V. Stilinovic, G. Horvat, T. Hrenar, V. Nemec and D. Cincic, Chem. – Eur. J., 2017, 23, 5244–5257 CrossRef CAS PubMed.
- I. Nicolas, F. Barrière, O. Jeannin and M. Fourmigué, Cryst. Growth Des., 2016, 16, 2963–2971 CrossRef CAS.
- D. Dolenc and M. Modec, New J. Chem., 2009, 33, 2344–2349 RSC.
- (a) O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS; (b) E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigué, Faraday Discuss., 2017, 203, 389–406 RSC; (c) V. V. Syamala, E. Aubert, M. Deutsch, E. Wenger, A. Dhaka, M. Fourmigué, M. Nespolo and E. Espinosa, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2022, 78, 436–449 CrossRef PubMed; (d) E. Aubert, I. Nicolas, O. Jeannin, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2023, 23, 7798–7810 CrossRef CAS.
- (a) K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC; (b) K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235–1239 RSC; (c) G. Anyfanti, A. Bauzá, L. Gentiluomo, J. Rodrigues, G. Portalone, A. Frontera, K. Rissanen and R. Puttreddy, Front. Chem., 2021, 9, 623595 CrossRef CAS PubMed.
- (a) R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC; (b) R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed; (c) R. Puttreddy, J. M. Rautiainen, S. Yu and K. Rissanen, Angew. Chem., Int. Ed., 2023, 62, e202307372 CrossRef CAS PubMed.
- N. Lucchetti, A. Tkacheva, S. Fantasia and K. Muñiz, Adv. Synth. Catal., 2018, 360, 3889–3893 CrossRef CAS.
- S. Yu, K. N. Truong, M. Siepman, A. Siiri, C. Schumacher, J. S. Ward and K. Rissanen, Cryst. Growth Des., 2023, 23, 662–669 CrossRef CAS.
- (a) K. Mori, M. Kobayashi, T. Itakura and T. Akiyama, Adv. Synth. Catal., 2015, 357, 35–40 CrossRef CAS; (b) V. Pace, G. Vilkauskaite, A. Sackus and W. Holzer, Org. Biomol. Chem., 2013, 11, 1085–1088 RSC.
- N. Savjani, S. J. Lancaster, S. Bew, D. L. Hughes and M. Bochmann, Dalton Trans., 2011, 40, 1079–1090 RSC.
- A. Artaryan, A. Mardyukov, K. Kulbitski, I. Avigdori, G. A. Nisnevich, P. R. Schreiner and M. Gandelman, J. Org. Chem., 2017, 82, 7093–7100 CrossRef CAS PubMed.
- (a) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC; (b) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- (a) C. H. Suresh and S. Anila, Acc. Chem. Res., 2023, 56, 1884–1895 CrossRef CAS PubMed; (b) E. Aubert, I. Nicolas, O. Jeannin, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2023, 23, 7798–7810 CrossRef CAS.
- A. J. Peloquin, C. D. McMillen and W. T. Pennington, CrystEngComm, 2021, 23, 6098–6106 RSC.
- R. B. Walsh, C. W. Padgett, P. Metrangolo, G. Resnati, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2001, 1, 165–175 CrossRef CAS.
- J.-L. Syssa-Magale, K. Boubekeur, P. Palvadeau, A. Meerschaut and B. Schollhorn, CrystEngComm, 2005, 7, 302–308 RSC.
- C. Laurence, J. Graton, M. Berthelot and M. J. El Ghomari, Chem. – Eur. J., 2011, 17, 10431–10444 CrossRef CAS PubMed.
- A. J. Guzmán Santiago, C. A. Brown, R. D. Sommer and E. A. Ison, Dalton Trans., 2020, 49, 16166–16174 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data (syntheses and crystal growth); X-ray crystallographic information; DFT calculations; additional figures (Fig. S1–S14) and Tables (Tables S1–S2). CCDC 2366101–2366110. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00654b |
| This journal is © The Royal Society of Chemistry 2024 |