
Design of pyrazine cocrystals of enzalutamide: a lead from 1,4-dioxane solvates†
Jupally
Prashanth
ab,
Alexander P.
Voronin
 c,
Artem O.
Surov
*c and
Sridhar
Balasubramanian
*ab
c,
Artem O.
Surov
*c and
Sridhar
Balasubramanian
*ab
aCentre for X-ray Crystallography, Department of Analytical & Structural Chemistry, CSIR-Indian Institute of Chemical Technology, Tarnaka, Uppal Road, Hyderabad-500007, Telangana, India. E-mail: bsridhar@iict.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Uttar Pradesh-201 002, India
cG.A. Krestov Institute of Solution Chemistry RAS, 153045, Ivanovo, Russia. E-mail: aos@isc-ras.ru
First published on 12th June 2024
Abstract
Enzalutamide (Enz), sold under the brand name “XTANDI”, belongs to a group of drugs known as androgen receptor inhibitors and functions by suppressing androgen's actions, which in turn helps in treating castrate-resistant prostate cancer. The current study focuses on the solvates and cocrystals of enzalutamide. Firstly, we obtained single crystals of 1,4-dioxane (Dox) solvate in two different stoichiometric ratios, Enz–Dox 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 and Enz–Dox 2:0.5. Based on this, we chose the coformer pyrazine (Pyrz), which is structurally similar to the Dox solvate. Crystallization of Pyrz with Enz also yielded 1:0.5 and 2:0.5 stoichiometries like the Dox solvate, but these existed as cocrystals (Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5). Despite the solvate and cocrystal of the same stoichiometry (1:0.5/2:0.5) exhibiting identical sorts of interactions, the inclusion of a coformer in the cocrystal prompted a few additional bonds to form, but the overall crystal environment is still preserved. From the packing similarity analysis, it was found that Enz–Dox 1:0.5 and Enz–Pyrz 1:0.5 have similar packing indexes; however, the former generates a 2D hydrogen bonded network and the latter leads to a 3D network. The same trend is observed in the cases of Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5. Hirshfeld analysis was performed to evaluate the prominence of various intermolecular interactions. Void map analysis enabled us to understand the roles of solvent and coformer in the crystal packing. Periodic DFT computations and DSC studies were performed to correlate thermal stabilities of solvates and cocrystals. Accelerated stability experiments revealed the better stability of cocrystals over solvates. This study provides valuable insights into solvate formation, which can be used as an effective strategy for attaining cocrystals with desired entities.
:0.5 and Enz–Dox 2:0.5. Based on this, we chose the coformer pyrazine (Pyrz), which is structurally similar to the Dox solvate. Crystallization of Pyrz with Enz also yielded 1:0.5 and 2:0.5 stoichiometries like the Dox solvate, but these existed as cocrystals (Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5). Despite the solvate and cocrystal of the same stoichiometry (1:0.5/2:0.5) exhibiting identical sorts of interactions, the inclusion of a coformer in the cocrystal prompted a few additional bonds to form, but the overall crystal environment is still preserved. From the packing similarity analysis, it was found that Enz–Dox 1:0.5 and Enz–Pyrz 1:0.5 have similar packing indexes; however, the former generates a 2D hydrogen bonded network and the latter leads to a 3D network. The same trend is observed in the cases of Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5. Hirshfeld analysis was performed to evaluate the prominence of various intermolecular interactions. Void map analysis enabled us to understand the roles of solvent and coformer in the crystal packing. Periodic DFT computations and DSC studies were performed to correlate thermal stabilities of solvates and cocrystals. Accelerated stability experiments revealed the better stability of cocrystals over solvates. This study provides valuable insights into solvate formation, which can be used as an effective strategy for attaining cocrystals with desired entities.
Introduction
In the solid state, the active components of drugs (APIs) have been extensively used in dosage forms such as tablets, capsules, and powders in clinical circumstances. The first preference for the pharmaceutical industries is to attain a novel polymorph1 (existence of a compound in several crystalline forms due to various packing configurations of its molecules in the crystal structure) of an API for the earlier entry into the market. Other possibilities include cocrystals2 (crystalline substances made up of multiple molecules arranged stoichiometrically in a single crystal lattice) and solvate/hydrates3,4 (crystallized solids in which a solvent/water fills the structural gaps in a crystal), all of which require exploration and profiling within the scope of the drug development process. The emergence of multiple solid forms is an important strategy for modifying the physicochemical properties of organic components such as the permeability and bioavailability,5 hygroscopicity,6 solubility7 and dissolution characteristics,8,9 melting temperatures,10 and physical and chemical stabilities.11 Cocrystals, one of the many potential solid forms, have recently received a lot of interest12,13 since they can significantly increase the dissolution rate of nearly insoluble APIs.14–16 At present, numerous types of drugs are marketed that were designed to function as cocrystals, viz., Suglat®,17 Entresto®,18 and Steglatro®.19 The accidental formation of undesirable solvates can also be significant as such outcomes are frequently unanticipated and might alter a product's physiochemical traits.20–23 Nonetheless, a few notable marketed solvate medications are also available, including warfarin sodium 2-propanol solvate,24 darunavir ethanolate,25 dapagliflozin propanediol monohydrate,26 trametinib dimethyl sulfoxide solvate,27 and indinavir sulfate ethanolate.28 In recent years, researchers have displayed interest towards solid forms that are easily solvated; one prominent example is sulfathiazole.29,30 In certain situations, the desolvation of solvates may result in the formation of unique polymorphs that cannot be achieved by standard crystallization procedures.31–33During the development process, co-crystals/solvates are mixed with various types of additives and subjected to varying humidity and temperature conditions. In rare cases, stability may be compromised, resulting in a sudden phase transition or disintegration into its parent components, namely, the API and co-former (physical mixture or desolvated form).34,35 Cocrystal dissociation occurs in the solid state at high temperature and relative humidity (RH).36,37 Investigating the stabilities of cocrystals and solvates is an important process, since it helps to prevent unexpected solid forms from forming once a drug is accepted.38
This study is an extension of a continual attempt to find more pharmaceutically relevant solid forms of enzalutamide and to investigate a potentially vast solid-phase environment of the drug. Enzalutamide39 (a BCS class II medication) is a nonsteroidal antiandrogen (NSAA) medicine marketed under the trade name Xtandi and used to treat prostate cancer.40–42 After chemotherapy, men with metastatic castration-resistant prostate cancer who take the oral AR (androgen receptor) inhibitor enzalutamide are predicted to live longer.43 Enzalutamide has been described in a variety of solid forms, including polymorphs,39 solvates8,44 (ethanol and methanol), and as a cocrystal with saccharin.8 In this contribution, we study the structural features of enzalutamide with solvate and cocrystal. First, we utilized 1,4-dioxane solvent to obtain Enz solvate crystals. Later, we selected the coformer pyrazine which is similar to 1,4-dioxane (O atom is substituted by N) for crystallization with Enz (Fig. 1). The pyrazine base is a part of many physiologically active chemicals, including several licensed pharmaceuticals and drugs that are now in advanced phases of clinical trials,45 and it may also be employed as a flavoring component.46 Pyrazines and pyrazine-based compounds are often found in nature.47 The incorporation of isostructural additives (1,4-dioxane/pyrazine) yielded different outcomes which were fully investigated in the current study. The current study focuses on the design of cocrystals of enzalutamide based on the formation of solvates with a molecular replacement strategy.
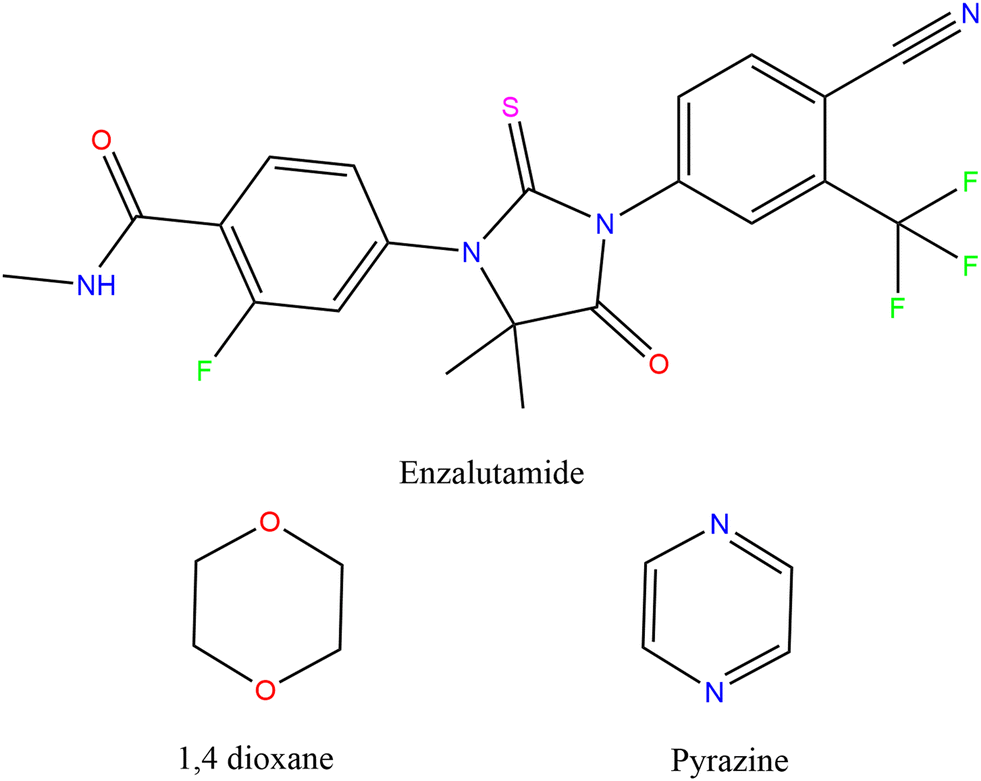 | ||
| Fig. 1 Chemical structures of enzalutamide, 1,4-dioxane and pyrazine. | ||
Materials and methods
Compounds and solvents
Enzalutamide (C21H16F4N4O2S, 98%) was procured from M/s. Spectevo Technologies Pvt., Ltd., Hyderabad, India, and pyrazine (C4H4N2, >99%) was purchased from Sigma-Aldrich, India. The solid compounds and solvents were of analytical or chromatographic grade and were utilized as received without any additional purification.Crystallization experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.5.
20 mg of Enz was dissolved in 1 ml of Dox in a small sample vial which was later placed in a larger weighing bottle containing 3 ml of cyclohexane and closed with a lid. This lid was sealed with a parafilm sheet. Crystallization was facilitated by the vapor diffusion of cyclohexane into the Dox solution. Single crystals suitable for data collection appeared after 7 days. The following procedure was used to prepare the pure bulk material: 100 mg of Enz was added to 150 μl of Dox in a 10 ml culture tube and was later left as slurry at room temperature for 2 days. After drying for 12 hours, the sample was subjected to PXRD analysis to confirm phase purity.
:0.5.
Enz (20 mg) was dissolved in 1 ml of Dox under hot conditions at 80 °C and left for slow evaporation at room temperature with a parafilm cover. Crystals formed within 4 days. Phase-pure bulk material preparation: 100 mg of Enz was dissolved in 3 ml of Dox, and then Millipore water (an antisolvent) was added dropwise to it, which led to the formation of a white precipitate in a 10 ml RBF. This was stirred at room temperature for 48 hours. Then it was filtered, and the residue was dried for 12 hours at RT and characterized by PXRD.
:0.5.
:0.5.
20 mg of Enz was dissolved in 1 ml of Dox in a small sample vial which was later placed in a larger weighing bottle containing 3 ml of cyclohexane and closed with a lid. This lid was sealed with a parafilm sheet. Crystallization was facilitated by the vapor diffusion of cyclohexane into the Dox solution. Single crystals suitable for data collection appeared after 7 days. The following procedure was used to prepare the pure bulk material: 100 mg of Enz was added to 150 μl of Dox in a 10 ml culture tube and was later left as slurry at room temperature for 2 days. After drying for 12 hours, the sample was subjected to PXRD analysis to confirm phase purity.
:0.5.
Enz (20 mg) was dissolved in 1 ml of Dox under hot conditions at 80 °C and left for slow evaporation at room temperature with a parafilm cover. Crystals formed within 4 days. Phase-pure bulk material preparation: 100 mg of Enz was dissolved in 3 ml of Dox, and then Millipore water (an antisolvent) was added dropwise to it, which led to the formation of a white precipitate in a 10 ml RBF. This was stirred at room temperature for 48 hours. Then it was filtered, and the residue was dried for 12 hours at RT and characterized by PXRD.
:0.5.
Preparation of phase-pure bulk material and single crystals. Enz (100 mg) and Pyrz (51.73 mg) were taken in a stoichiometric ratio of 1
:3, added to 200 μl of acetone, and left stirring at room temperature for 18 h. The slurry material was then dried for 12 hours. A few single crystals were observed in the powder, which was used for low-temperature data collection. The powder was subjected to PXRD analysis to confirm phase purity.
:0.5.
Enz (100 mg) and Pyrz (17.24 mg) were taken in an equimolar ratio and dissolved in 3 ml of acetone. Good-quality single crystals ideal for data collection were obtained in 3 days. The following procedure was chosen to obtain a phase-pure bulk material: 100 mg of Enz and 17.24 mg of Pyrz were taken in a 10 ml culture tube and 150 μl of acetone was added. The mixture was stirred under ambient conditions for 24 hours. The slurry material was dried for 12 hours and subjected to PXRD analysis.
Single crystal and powder X-ray diffraction
The grown crystals were screened using the polarizing microscope (LEICA S8APO), and a good-quality crystal was selected for data collection. A Bruker D8 QUEST instrument with an IμS Mo micro source (λ = 0.7107 Å) and a PHOTON-III detector was used to collect data at a low temperature (100(2) K). The raw data frames were processed, and absorption corrections were applied using the Bruker Apex 3 software suite programs.48–50 The intrinsic phasing method was implemented to determine the structure, and refinement was carried out by the SHELXL program.51 H atoms attached to the N atoms of Enz and Pyrz were found from the Fourier density map, and their site coordinates and isotropic thermal parameters were refined. C-bound H atoms were located in difference density maps but were positioned geometrically and included as riding atoms, with C–H = 0.93–0.98 Å, Uiso(H) = 1.5Ueq(C) for methyl H atoms and 1.2Ueq(C) for all other H atoms. The fluorine atoms (F2/F3) of the –CF3 group of Enz–Dox 1:0.5 were disordered over two positions, and their occupational factors were refined to 0.535(18) and 0.465(18). In Enz–Pyrz 1:0.5, the F1/F3 atoms of the –CF3 group were disordered over two positions, and their occupational factors were refined to 0.50(3) and 0.50(3). The C10A/F1A/F2A/F3A atoms of the –CF3 groups of Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5 were disordered over two positions whose occupational factors were refined to 0.572(3)/0.428(3) and 0.843(4)/0.157(4), respectively. The fluorine atom (F4A) of the benzyl ring of Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5 was disordered over two sites, and their occupational factors were refined to 0.553(3)/0.445(3) and 0.574(3)/0.42(3), respectively. Crystal data information is mentioned in Table S1.† PLATON software52 was used to calculate the hydrogen bonds, which are tabulated in Table S2.† Figures were generated using DIAMOND 4 software.53 CCDC numbers 2350853–2350856 for Enz–Dox 1:0.5, Enz–Dox 2:0.5, Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5.
Powder X-ray diffraction (PXRD) measurements were performed in Bragg–Brentano (reflection) geometry on a Bruker D8 Advance Davinci with a Cu X-ray source and LYNXEYE XE-T detectors. Plate sample holders were utilized to place the samples, and 15 rpm rotation was used during the measurements. Simulated PXRD patterns were obtained using Mercury Software54 from the room-temperature fast scan single crystal data and compared with bulk PXRD patterns.
Hot Stage Microscopy (HSM)
A polarizing microscope (model LEICA S8APO) attached to a Linkam hot stage (model LTS420) was used to observe the melting patterns of the obtained solid forms of Enz. The heating temperatures (30 °C to 250 °C) and heating rate (5 °C min−1) were maintained according to DSC experimental conditions using Link software. As the temperature rose, the observable changes in the crystal's morphology were recorded.Differential scanning calorimetry (DSC)
The DSC analysis of the cocrystal and solvate was performed using a TA Instruments Discovery DSC 250 and Mettler Toledo DSC-1 STARe instrument. The sample was placed in an aluminum sample pan and heated over a temperature range of 30–250 °C at a heating rate of 5 °C per minute.Thermogravimetric analysis (TGA)
A Hitachi TG/DTA 7300 instrument was used for the analysis in the 30–250 °C temperature range with a ramp of 5 °C min−1 under continuously purged nitrogen flow (50 mL min−1) for the solvates.Fourier Transform Infrared Spectroscopy (FT-IR)
The IR analysis of cocrystals, solvates, Enz and Pyrz was performed using a Jasco FT/IR-4600 equipped with attenuated total reflectance (ATR) along with zinc selenide crystals. The finely ground samples were used for analysis from 4000 cm−1 to 600 cm−1 with a resolution of 4 cm−1.Hirshfeld surface analysis
Crystal Explorer v.17.5 (ref. 55) software was utilized to carry out the Hirshfeld surface analysis.56 The surface resolution was set to ‘Very High’. The distances from the Hirshfeld surface to the nearest nuclei outside and inside the surface (de and di, respectively) were plotted into a 2D fingerprint map, and the contributions from the contacts between different atom pairs were evaluated.Physical stability of solvates and cocrystals at different RH values
Powders of the solvates and cocrystals were analyzed on the day of preparation and kept in closed glass desiccators containing saturated salt solutions of sodium chloride (70–75% RH) and potassium nitrate (90–95% RH) at 40 ± 2 °C.Computational studies
Periodic DFT computations were performed using CRYSTAL17 software57 at the B3LYP-D3 (BJ,ABC)/6-31G(d,p) level of theory.58–61 It has been demonstrated that this level of theory provides reliable and consistent results in studying the non-covalent interactions in organic crystals.62–64 Atomic positions from the SC-XRD experiments were used for SCF computations and quantum topology analysis of periodic electron density, with hydrogen atom positions normalized to the standard X–H distances from neutron diffraction data. The CRYSTAL parameters describing the level of accuracy in evaluating the Coulomb and Hartree–Fock exchange series were set to 7 7 7 7 15. The tolerance on energy controlling the self-consistent field convergence was set to 10−10 hartree. The mixing coefficient of Hartree–Fock/Kohn–Sham matrices was set to 25%. The shrinking factor of the reciprocal space net was set to 4.The crystal lattice energy (Elatt) of the multi-component crystal was estimated as the difference between the sum of total electronic energies of the isolated molecules Emoli and the total energy of the multi-component crystal Ecryst calculated per asymmetric unit with respect to basis set superposition error (BSSE).65
 | (1) |
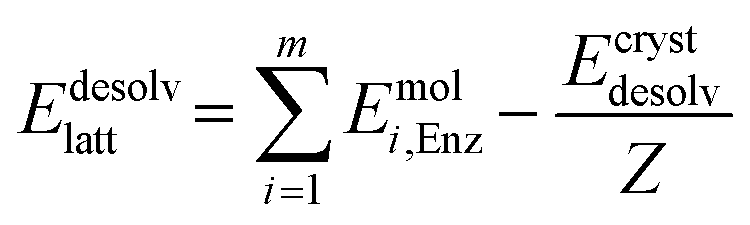 | (2) |
| Ebind = Elatt − Edesolvlatt | (3) |
Quantum topology analysis of non-covalent interactions in the considered solvates was performed in Topond software66 currently implemented into the CRYSTAL suite. The search for (3, −1) critical points was performed using a standard algorithm, and the following quantities were computed in the critical point: the electron density ρb, its Laplacian ∇2ρb, and the positively defined local electronic kinetic energy Gb. The interaction energy of a particular hydrogen bond, Eint, was estimated using the correlation equation proposed by Mata et al.67
| Eint (kJ mol−1) = 1124·Gb (atomic units) | (4) |
Results and discussion
Development and description of cocrystals and solvates
The slow evaporation and vapour diffusion methods were employed to obtain multi-component crystalline forms of enzalutamide using Dox and Pyrz which resulted in solvates Enz–Dox 1:0.5 and Enz–Dox 2:0.5 and cocrystals Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5, respectively. All these forms were successfully reproduced in bulk scale (Fig. S1–S4†). Both the solvent and coformer have two acceptors (O and N). Even though one is aromatic (1,4-dioxane) and the other is non-aromatic (pyrazine), the final outcomes, viz., space group, PXRD patterns and stoichiometries, are similar for the newly obtained solvates and cocrystals.
The first crystallographic studies of Enz were investigated by Lucia Maini et al.44 and deal with Enz form I, a solvate (formic acid, methanol, or water) and a substitution impurity (O-enzalutamide). Apart from this, solvate (EtOH), cocrystal and coamorphous forms with saccharin have also been determined by our group,8 paving the way for the first-ever cocrystal and coamorphous forms of Enz.
Crystal structure analysis
:0.5.
The asymmetric unit of Enz–Dox 1:0.5 consists of one molecule of Enz and half a molecule of Dox solvate (occupies an inversion centre) and crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 2). The amide group of Enz is connected to the Dox solvate through the N3–H3N⋯O3 bond. Two sets of homo dimers, namely C–H⋯O and C–H⋯S, were formed via (C12–H12C⋯O2, C18–H18⋯O1) and (C5–H5⋯S1) interactions, and their motifs are R22(10) and R22(14), respectively. The C–H⋯O and C–H⋯S dimers of Enz are interlinked together, which in turn forms a tetrameric unit of motif R44(28). On the other hand, C–H⋯O dimers of Enz are connected to Dox molecules, which together form a hexameric unit of motif R66(38) (Fig. 3). In the crystal packing, Enz molecules are interlinked with each other, forming a one-dimensional chain along the b-axis. The Dox solvate bridges this one-dimensional chain with the adjacent chain, thereby extending them into a two-dimensional hydrogen-bonded sheet.
space group (Fig. 2). The amide group of Enz is connected to the Dox solvate through the N3–H3N⋯O3 bond. Two sets of homo dimers, namely C–H⋯O and C–H⋯S, were formed via (C12–H12C⋯O2, C18–H18⋯O1) and (C5–H5⋯S1) interactions, and their motifs are R22(10) and R22(14), respectively. The C–H⋯O and C–H⋯S dimers of Enz are interlinked together, which in turn forms a tetrameric unit of motif R44(28). On the other hand, C–H⋯O dimers of Enz are connected to Dox molecules, which together form a hexameric unit of motif R66(38) (Fig. 3). In the crystal packing, Enz molecules are interlinked with each other, forming a one-dimensional chain along the b-axis. The Dox solvate bridges this one-dimensional chain with the adjacent chain, thereby extending them into a two-dimensional hydrogen-bonded sheet.
 | ||
| Fig. 2 ORTEP diagram of Enz–Dox 1:0.5 showing the atom-labelling scheme. Displacement ellipsoids were drawn at 50% probability level. Hydrogen bonds are shown as dashed lines. Unlabeled atoms were generated by the symmetry −x, −y + 1, −z + 1. | ||
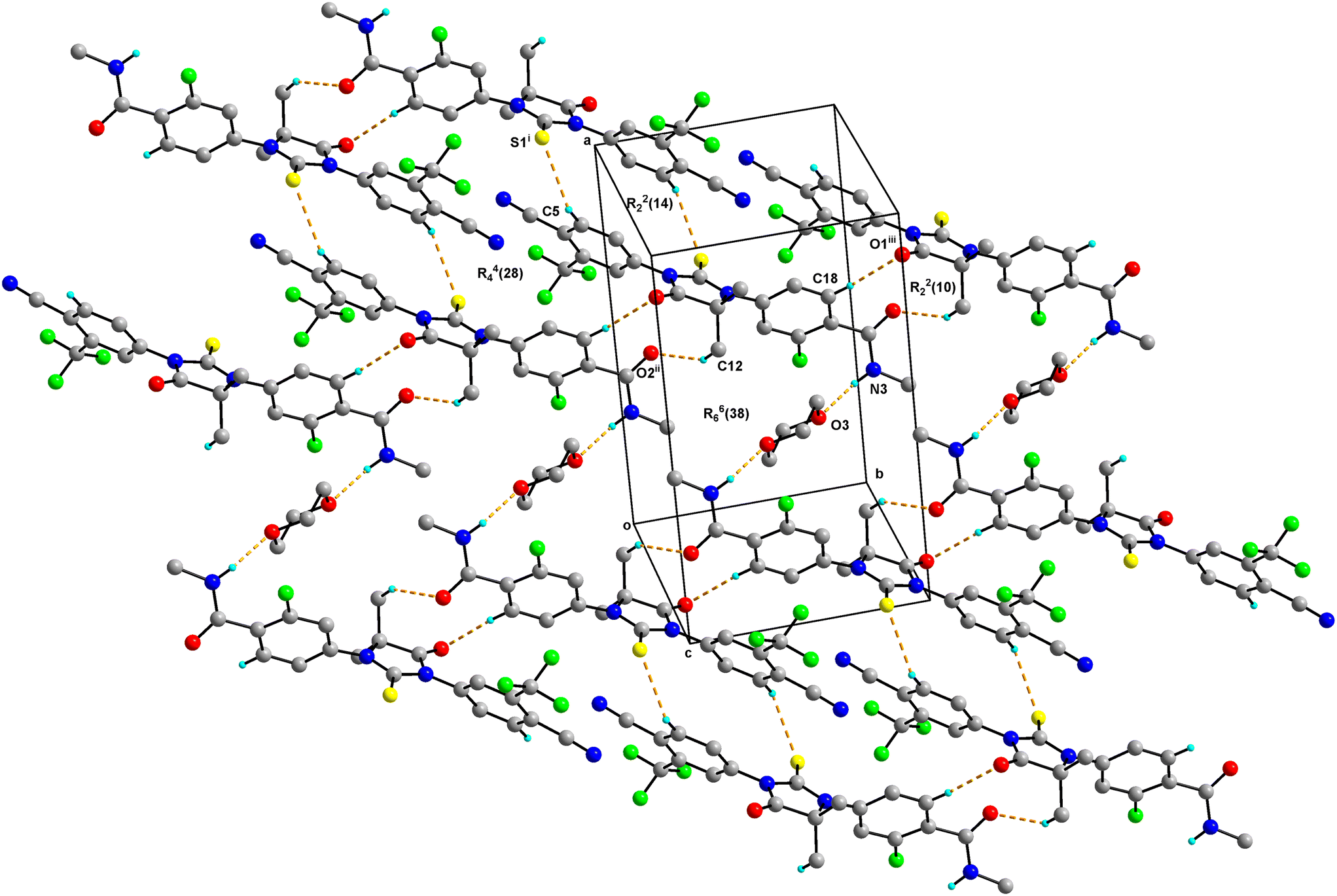 | ||
| Fig. 3 Crystal packing of Enz–Dox 1:0.5 showing dimeric and hexameric units which form a two-dimensional hydrogen-bonded network. H atoms that are not involved in hydrogen bonding have been omitted for clarity. Hydrogen bonds are shown as dashed lines. | ||
:0.5.
Enz–Dox 2:0.5 has two molecules of Enz (suffixed with A and B) and a half-molecule of Dox solvent (occupies an inversion center) in the asymmetric unit and crystallizes in the triclinic P space group (Fig. 4). The amide group of Enz molecule A is connected to the Dox solvate via N3A–H3NA⋯O3. Three sets of heterodimers were observed between Enz molecules A and B via (N3B–H3NB⋯O2A, C15B–H15B⋯S1A), (C15B–H15B⋯S1A, C6A–H6A⋯O1B) and (C12A–H12C⋯O2B, C18B–H18B⋯O1A) interactions whose motifs are R22(16), R22(13) and R22(10), respectively (Fig. S5†). Additionally, Enz molecules A and B are connected via the C2A–H2A⋯O2B interaction. Enz molecule B is connected to its symmetry-related molecule via C13B–H13F⋯N4B, which is further extended as an infinite chain along the c-axis. In the crystal packing, Enz molecules A and B are interlinked with each other and form a one-dimensional chain along the a-axis. Further, Dox solvate bridges Enz molecule A and helps to extend it into a two-dimensional hydrogen-bonded network (Fig. 5).
 | ||
| Fig. 4 ORTEP figure of Enz–Dox 2:0.5 showing the atom-labelling scheme. Displacement ellipsoids were drawn at 50% probability level. H atoms that are not involved in hydrogen bonding have been omitted for clarity. Hydrogen bonds are shown as dashed lines. Unlabeled atoms were generated by the symmetry −x + 1, −y + 2, −z + 1. | ||
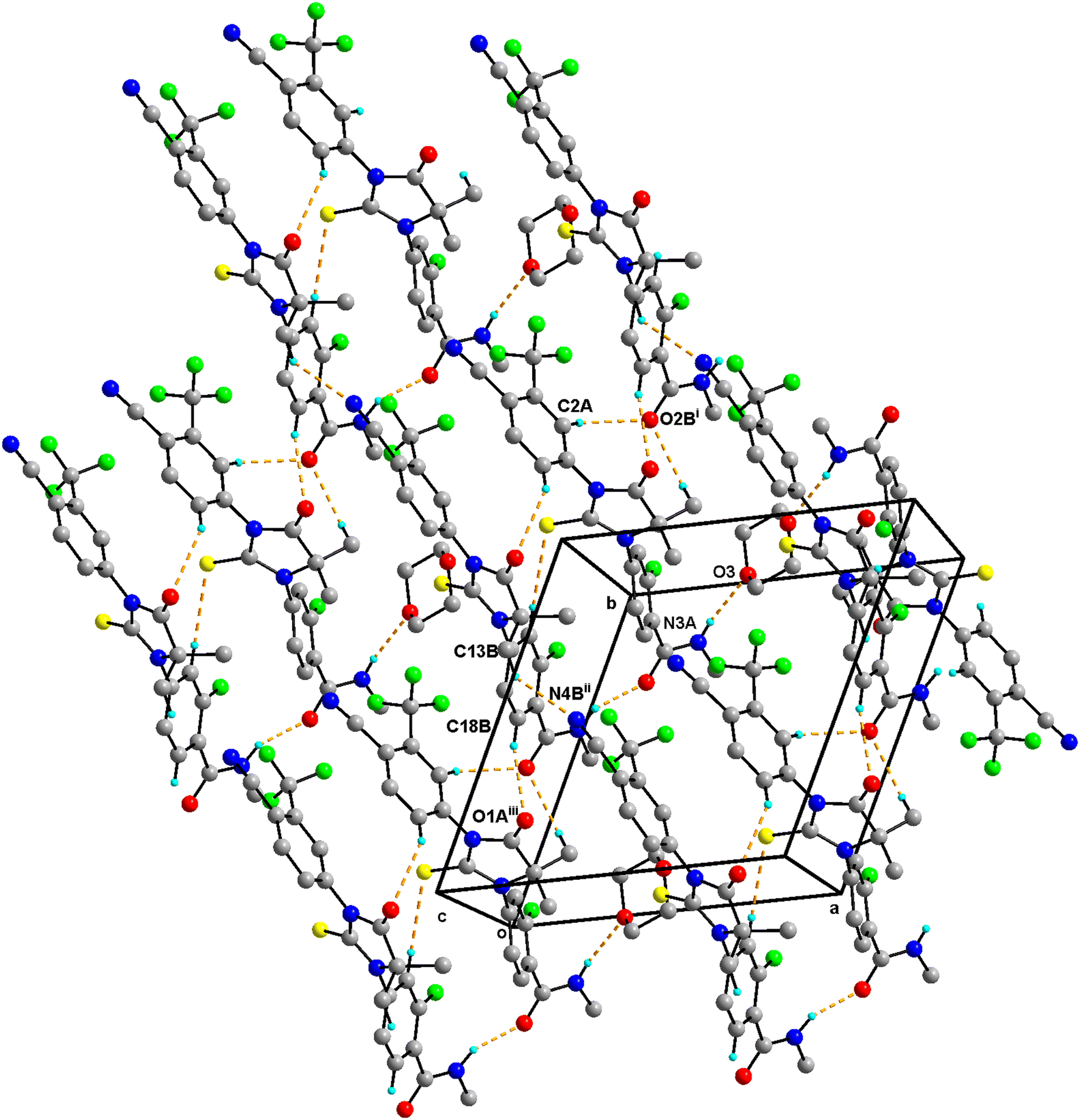 | ||
| Fig. 5 Crystal packing of Enz–Dox 2:0.5 showing dimeric units and interactions of solvate molecules which aid in forming two two-dimensional hydrogen-bonded networks. H atoms that are not involved in hydrogen bonding have been omitted for clarity. Hydrogen bonds are shown as dashed lines. | ||
:0.5.
Enz–Pyrz 1:0.5 crystallizes in the triclinic P space group and contains one molecule of Enz and half a molecule of Pyrz (occupies an inversion center) in the asymmetric unit (Fig. 6). The unit cell dimensions (Table S1†) and PXRD patterns (Fig. S6†) are similar to those of the Enz–Dox 1:0.5 structure.
 | ||
| Fig. 6 ORTEP diagram of Enz–Pyrz 1:0.5 showing the atom-labelling scheme. Displacement ellipsoids were drawn at 50% probability level. Hydrogen bonds are shown as dashed lines. Unlabeled atoms were generated by the symmetry −x + 2, −y + 1, −z + 1. | ||
The amide group of Enz is connected to the Pyrz molecule through the N3–H3N⋯N3 bond. Three sets of homodimers, viz., C–H⋯O (C12–H12C⋯O2 and C18–H18⋯O1), C–H⋯S (C5–H5⋯S1), and C–H⋯F (C15–H15⋯F4), were observed between symmetry-related Enz molecules with motifs R22(10), R22(14), and R22(8), respectively (Fig. S7†). Two sets of tetramer units were formed by the interlinkage of (C–H⋯O, C–H⋯S) and (C–H⋯O, C–H⋯F) dimers, whose motifs were found to be R44(28) and R44(24) respectively (Fig. S8†). These dimers and tetramers aggregate the Enz molecules into a two-dimensional hydrogen-bonded sheet. The Pyrz molecule connects Enz molecule dimers formed by the C–H⋯O hydrogen bond, in turn forming a hexameric unit with motif R66(38) encapsulated between C–H⋯F dimers (Fig. S9†). In the crystal packing, both the Enz and Pyrz molecules are interlinked by N–H⋯N, C–H⋯O, C–H⋯S, and C–H⋯F hydrogen bonds, which leads to the formation of a three-dimensional hydrogen-bonded network (Fig. 7).
 | ||
| Fig. 7 Crystal packing of Enz–Pyrz 1:0.5 showing the fusion of Pyrz molecules in between C–H⋯F dimers and the generated hexameric unit, which helps in forming a three-dimensional hydrogen-bonded network. H atoms that are not involved in hydrogen bonding have been omitted for clarity. Hydrogen bonds are shown as dashed lines. The green color represents the Enz molecule, and the blue color represents the Pyrz molecule. | ||
:0.5.
The asymmetric unit of Enz–Pyrz 2:0.5 has two molecules of Enz (suffixed with A and B) and a half molecule of Pyrz (occupies an inversion center) and crystallizes in the triclinic P space group (Fig. 8). The unit cell lengths (Table S1†) and PXRD pattern (Fig. S10†) of Enz–Pyrz 2:0.5 are similar to those of the Enz–Dox 2:0.5 structures. However, their unit cell angles are different. The amide group of Enz molecule A is connected to the Pyrz molecule via the N3A–H3NA⋯N5 bond. Three sets of heterodimers were formed between Enz molecules A and B via (N3B–H3NB⋯O2A and C15B–H15B⋯S1A), (C15B–H15B⋯S1A and C6A–H6A⋯O1B) and (C12A–H12C⋯O2B, C18B–H18B⋯O1A) interactions with motifs R22(16), R22(13), and R22(10), respectively (Fig. S11†). Additionally, Enz molecules A and B are connected via the C2A–H2A⋯O2B bond. Enz molecule B is connected to its symmetry-related molecule via C13B–H13F⋯N4B, further extending as an infinite chain along the b-axis. The Pyrz molecule bridges two symmetry-related Enz A molecules, which in turn leads to the formation of a tetramer of motif R44(38) via C22–H22⋯F2A and N3A–H3NA⋯N5 hydrogen bonds (Fig. S12†).
 | ||
| Fig. 8 ORTEP diagram of Enz–Pyrz 2:0.5, showing the atom-labelling scheme. Displacement ellipsoids were drawn at 50% probability level. Hydrogen bonds are shown as dashed lines. Unlabeled atoms were generated by the symmetry −x, −y, −z + 1. | ||
These dimers and tetrameric units are aggregated to form a two-dimensional hydrogen-bonded sheet. In the crystal packing, Enz molecule A is connected to Enz molecule B via C–H⋯O and C–H⋯S bonds, and the Pyrz molecule being encapsulated connects to Enz molecule A through N–H⋯N and C–H⋯F bonds. These interactions aggregate into a three-dimensional hydrogen-bonded network (Fig. 9).
 | ||
| Fig. 9 Part of the crystal packing of Enz–Pyrz 2:0.5 showing N–H⋯O, C–H⋯O, C–H⋯F, C–H⋯S interactions between Enz and Pyrz molecules, which in turn aid in forming a 3D network. H atoms that are not involved in hydrogen bonding have been omitted for clarity. Hydrogen bonds are shown as dashed lines. | ||
Crystal packing similarity and structural comparison
Solvates68–72 and cocrystals73–75 of organic compounds frequently exhibit the phenomenon of isostructurality. In the present work, the packing similarity is determined using Enz as a base molecule. The CrystalCMP76 program was used to assess the molecular packing of the Enz solvates and cocrystals (Fig. S13†). From the analysis, it is very clear that packing similarity is observed between the Enz–Dox 1:0.5 and Enz–Pyrz 1:0.5 crystal structures. Similarly, the crystal structures of Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5 show identical packing. However, there is no packing similarity between the Enz–Dox 1:0.5/2:0.5 (solvate) and Enz–Pyrz 1:05/2:0.5 (cocrystal) systems.
In the crystal packing of Enz–Dox 1:0.5 solvate, dimeric units (C–H⋯O and C–H⋯S), tetrameric units, and hexamers lead to the formation of a 2D network, whereas in the case of Enz–Pyrz 1:0.5 cocrystal, dimers (C–H⋯O, C–H⋯F and C–H⋯S), tetramers (C–H⋯O/C–H⋯S and C–H⋯O/C–H⋯F) and hexamers aid in forming a 3D network. The crystal structure of Enz–Dox 2:0.5 consists of dimers formed by N–H⋯O, C–H⋯S, and C–H⋯O interactions which form a 2D network. On the other hand, the Enz–Pyrz 2:0.5 cocrystal system has dimers and tetramers formed by N–H⋯O, C–H⋯O, C–H⋯S, and C–H⋯F which are aggregated together to form a 3D network. In conclusion, the solvated forms (Enz–Dox 1:0.5 and Enz–Dox 2:0.5) generate a two-dimensional hydrogen-bonded network, whereas the cocrystal systems (Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5) are aggregated into a three-dimensional network. Although similar types of interactions were observed in both multi-component crystals, a few additional bonds in the Enz–Pyrz cocrystals make a significant difference in the crystal packing.
Void map analysis
Voids constitute distinct cavities that are commonly observed in host–guest combinations. These cavities are sometimes empty but are more frequently filled with solvent or other guest molecules.77,78 The computations of void map analysis (probe radius: 1.2 Å, grid spacing: 0.3 Å) in the crystals confirmed that in all the newly obtained structures, solvent/coformer molecules were trapped in the closed cavities, forming a continuous chain of closed voids along the b-axis. In the cases of Enz–Dox 1:0.5 and Enz–Pyrz 1:0.5, the voids constitute 10.6% and 10.4% of the cell volumes, respectively. This fact indicates that the cavities occupy a sizable portion of the cell volume, which hints that the solvent/coformer molecules have an easy path of escape (Fig. S14 and S15†). On the other hand, only 6.1% and 6.6% of the unit cell volumes were occupied by solvent/coformer molecules in the Enz–Dox 2:0.5 and Enz–Pyrz 2:0.5 systems, respectively. In these cases, the excess space was occupied by extra enzalutamide molecules, making it difficult for the solvent/coformer molecules to escape (Fig. S16 and S17†).
Conformational analysis
The torsion angles C6–C1–N1–C9 (t1) and C2–C1–N1–C7 (t2) provide insight into the orientation of the trifluoromethyl substituted phenyl ring (C1–C6) when compared to the thioxoimidazolidine ring (C7–C9/N1/N2); all the molecules in the newly obtained crystals exhibit syn-clinal orientation (±30–90°) except Enz–Pyrz 2:0.5 molecule A, which adopts anti-clinal (±90–150°) and syn-clinal conformations (Table S3†). The conformation of the thioxoimidazolidine ring with respect to the fluorine-substituted phenyl ring (C14–C19) can be seen from the torsion angles C15–C14–N2–C8 (t3) and C19–C14–N2–C7 (t4). For all the newly obtained solid forms, t3 and t4 exhibit anti-clinal conformations except for Enz–Pyrz 2:0.5 molecule A, which adopts syn-clinal orientation. The C16–C17–C20–N3 (t5) torsion angle can be used to determine the amide group's conformation in relation to the fluorine atom; all the solvate and cocrystal molecules acquire syn-clinal orientation, except for Enz–Pyrz 1:0.5 molecule A adopting anti-clinal conformation. The overlay illustrates clear differences in the molecular orientations of the newly obtained forms, viz., Enz–Dox (1:0.5, 2:0.5) and Enz–Pyrz (1:0.5, 2:0.5), which were compared with previously reported forms,8,39,44viz., Enz parent, Enz–R2, Enz–EtOH, and [Enz + Schr](cr) (1:1) (Fig. S18†).
Thermal analysis
DSC, TGA, and HSM analyses were performed to understand the thermal behavior of the newly obtained solvate and cocrystal systems. The corresponding results are tabulated (Table S4†). The DSC thermograms of solvates Enz–Dox 1:0.5 and Enz–Dox 2:0.5 showed desolvation at peak temperatures of 113.20 °C and 141.48 °C and complete melting at 199.93 °C and 200.24 °C, respectively (Fig. 10). The HSM data also comply with the above findings, in which the desolvation process ranges from 94 °C to 114 °C and 130 °C to 142 °C, with complete melting at 203 °C and 205 °C, respectively (Fig. S19†). For additional confirmation of these desolvation processes, single crystals of the solvated forms were heated to their corresponding desolvation temperatures and subjected to unit cell determinations. This step provided valuable insights into the thermal characterization of the solvates and revealed that the desolvated crystals covert into their stable parent form without any other phase transition. The TGA results of Enz Dox 1:0.5 and Enz–Dox 2:0.5 revealed experimental weight losses of 8.20% and 4.77%, respectively, which are similar to the theoretical values of 8.66% and 4.73% (Fig. 11).
 | ||
| Fig. 10 DSC data of newly obtained solvates/cocrystals. | ||
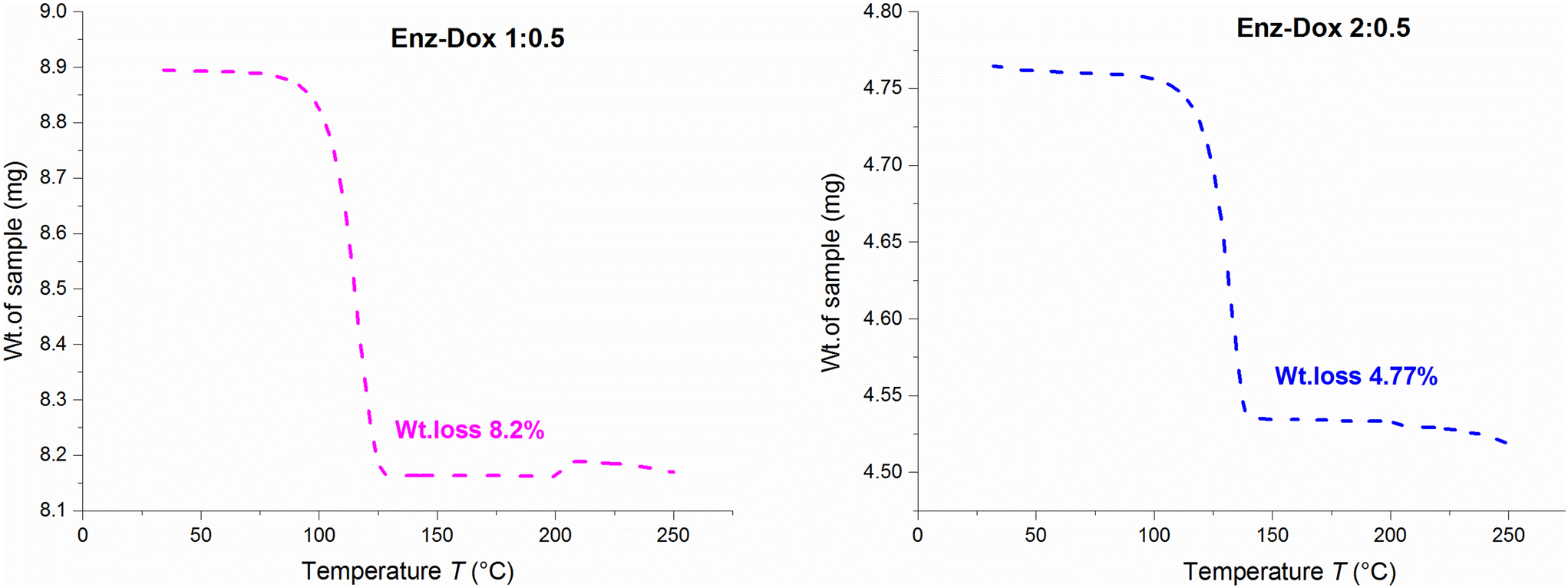 | ||
| Fig. 11 TGA data of solvates. | ||
The DSC data of the cocrystals Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5 showed endothermic peaks around 118.88 °C and 151.68 °C (Tpeak) and complete melting at 199.34 °C and 200.55 °C, respectively. The DSC plot of the Enz parent form showed a single endothermic peak at 200.48 °C (undergoes direct melting), whereas Pyrz showed two endothermic events, i.e., melting at 53.23 °C and evaporation at 107.85 °C. Hence, in the case of the cocrystal, the above findings indicate the occurrence of cocrystal dissociation at the first endotherm, where Pyrz is sublimed and the remaining traces of Enz are melted in the latter event. In order to gain additional evidence for the cocrystal dissociation process, single crystals of Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5 were heated to 120 °C and 155 °C under the control of HSM and subjected to unit cell determinations which confirmed the conversion of the cocrystal into the Enz parent. PXRD data (performed after cocrystal dissociation) also comply with the above findings (Fig. S20†).
In accordance with the above-discussed thermal investigations, the following conclusions can be drawn: in the case of solvate/cocrystal systems, viz., Enz–Dox 1:0.5/Enz–Pyrz 1:0.5, the solvate/coformer desolvates/sublimes faster compared than in Enz–Dox 2:0.5/Enz–Pyrz 2:0.5 due to the presence of loosely packed channels.
Physical stability of cocrystals and solvates
Samples were kept under accelerated stability conditions (70–75% RH, 90% RH) at 40 °C for four weeks to determine the physical stability of the cocrystal (Enz–Pyrz 1:0.5, Enz–Pyrz 2:0.5) and solvate (Enz–Dox 1:0.5, Enz–Dox 2:0.5) systems. PXRD analysis was utilized to evaluate the sample stability at predetermined intervals of time (7 days, 14 days, and 30 days).
It was revealed that the solvated form Enz Dox 1:0.5 was unstable and converted to Enz form I at both 70–75% and 90% RH conditions within a week (Fig. S21†). Enz–Dox 2:0.5 was stable for at least 1 week at 70–75% RH and started to show a few peaks (2θ = 13.083) of Enz form I within two weeks; after that, it remained concomitant throughout the experiment (four weeks). The same trend was followed for the 90% RH condition (Fig. S22†).
Enz–Pyrz 1:0.5 was found to be stable for at least two weeks under both conditions; however, by four weeks, it had converted to Enz form I (Fig. S23†). On the other hand, Enz–Pyrz 2:0.5 was stable upon storage for up to 4 weeks at 70–75% and 90% RH conditions and was later found to have few traces of Enz form I under both conditions (Fig. S24†).
From the above findings, it can be stated that the solvated form Enz–Dox 1:0.5 converts to Enz form I in the early stages (within a week), whereas the Enz–Dox 2:0.5 form requires a longer period for the conversion (two weeks) under both RH conditions. The Enz–Pyrz 1:0.5 cocrystal system was stable for up to two weeks and later converted to Enz form I by the end of the stability experiment (four weeks), whereas Enz–Pyrz 2:0.5 was stable for up to four weeks. Hence, it can be concluded that Enz–Pyrz 2:0.5 possesses longer stability than the Enz–Dox (1:0.5 and 2:0.5) and Enz–Pyrz 1:0.5 systems.
IR spectroscopy
Vibrational spectroscopy provided more details on the structural characteristics of the cocrystals, Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5 and solvates Enz–Dox 1:0.5 and Enz–Dox 2:0.5. It is evident from the IR spectra (Fig. S25 and S26†) that the most significant bands in Enz appeared at 3434 cm−1 (N–H stretching), 2236 cm−1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching), 1767 cm−1 (carbonyl C
N stretching), 1767 cm−1 (carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), and 1662 cm−1 (secondary amide CO stretching). Pyrz has frequency values of 1488 cm−1 (CN stretching), 1412 cm−1 (C–H bending), and 3082 cm−1 and 3004 cm−1 (C–H aromatic stretch). The N–H stretching value shifted to 3299 cm−1 in Enz–Pyrz 1:0.5 and 3351 cm−1/3301 cm−1 in Enz–Pyrz 2:0.5 (due to the presence of two N–H groups). In the case of the solvates, it shifted to 3352 cm−1 in Enz–Dox 1:0.5 and 3344 cm−1 in Enz–Dox 2:0.5. No significant changes were observed in the CN stretching in either cocrystals or solvates; it appeared at ∼2230 cm−1, as similar hydrogen bond interactions were found compared to the Enz parent. The carbonyl CO stretching values shifted to 1756 cm−1/1752 cm−1 in the cocrystals and 1750 cm−1/1752 cm−1 in the solvates from 1767 cm−1. Secondary amide CO stretching frequencies were observed at 1662 cm−1 in Enz–Pyrz 1:0.5 and 1664 cm−1 in Enz–Dox 1:0.5; due to the presence of distinct intermolecular interactions, it was shifted to 1653 cm−1 in both the Enz–Pyrz 2:0.5 and Enz–Dox 2:0.5 forms. The appearance of lower wave numbers in cocrystals/solvates (N–H stretching and CO stretching) is a clear indication of the participation of intermolecular hydrogen bonding interactions, which is in accordance with the crystal structure analysis.
O stretching), and 1662 cm−1 (secondary amide CO stretching). Pyrz has frequency values of 1488 cm−1 (CN stretching), 1412 cm−1 (C–H bending), and 3082 cm−1 and 3004 cm−1 (C–H aromatic stretch). The N–H stretching value shifted to 3299 cm−1 in Enz–Pyrz 1:0.5 and 3351 cm−1/3301 cm−1 in Enz–Pyrz 2:0.5 (due to the presence of two N–H groups). In the case of the solvates, it shifted to 3352 cm−1 in Enz–Dox 1:0.5 and 3344 cm−1 in Enz–Dox 2:0.5. No significant changes were observed in the CN stretching in either cocrystals or solvates; it appeared at ∼2230 cm−1, as similar hydrogen bond interactions were found compared to the Enz parent. The carbonyl CO stretching values shifted to 1756 cm−1/1752 cm−1 in the cocrystals and 1750 cm−1/1752 cm−1 in the solvates from 1767 cm−1. Secondary amide CO stretching frequencies were observed at 1662 cm−1 in Enz–Pyrz 1:0.5 and 1664 cm−1 in Enz–Dox 1:0.5; due to the presence of distinct intermolecular interactions, it was shifted to 1653 cm−1 in both the Enz–Pyrz 2:0.5 and Enz–Dox 2:0.5 forms. The appearance of lower wave numbers in cocrystals/solvates (N–H stretching and CO stretching) is a clear indication of the participation of intermolecular hydrogen bonding interactions, which is in accordance with the crystal structure analysis.
Hirshfeld analysis
To offer additional insights into the intermolecular interactions,79 a Hirshfeld surface analysis and fingerprint plot were generated for all the forms presented in this work using Crystal Explorer software (version 17.5).In Enz–Dox 1:0.5, F⋯H contacts account for the majority of the contribution, which is 20.2% in Enz and 28.4% in Dox molecules. This might be due to the C–H⋯F hydrogen bond interactions. The next notable contribution is from O⋯H, which occupies 14% (Enz) and 16.5% (Dox) of the Hirshfeld surface area, and this can be attributed to the major interactions involving C–H⋯O and N–H⋯O bonds. H⋯H/N⋯H contacts occupy 22.8%/9.4% of the surface in Enz and 41.9%/0.2% in Dox molecules, respectively. The lowest occupancy of N⋯H contacts in the Dox molecule is due to the absence of N–H⋯N interactions. C⋯H contacts occupy 11% and 9.2% of the surface area in both molecules. In Enz–Dox 2:0.5, major contributions come from F⋯H, O⋯H, and H⋯H contacts. For F⋯H contacts, Enz molecule A has 20.6%, Enz molecule B has 16.5%, and Dox has 29.6%, which indicates the significance of C–H⋯F interactions. For O⋯H and H⋯H contacts, Enz molecule A occupies 15.7%/23.8%, Enz molecule B occupies 12.7%/23.5%, and Dox occupies 15.9%/39.3% of the surface area. This is due to the participation of C–H⋯O and N–H⋯O interactions. N⋯H and C⋯H contacts occupy 7.6%/7.8% (Enz molecule A), 9.9%/13.3% (Enz molecule B), and 9.4%/0% (Dox).
In Enz–Pyrz 1:0.5, the Pyrz has more F⋯H (25.8%) and N⋯H (19.2%) compared to Enz, which has 20.5% and 11.8%. The prominent occupancy is due to the higher number of C–H⋯F and N–H⋯N interactions. H⋯H contacts occupy 20.9% in Enz and 27.4% in Pyrz molecules, respectively. C⋯H/O⋯H contacts occupy 11.8%/11.6% in the Enz molecule and 16.5% of C⋯H contacts in the Pyrz molecule. The lowest occupancy of O⋯H contacts is due to the absence of N–H⋯O bonds. In Enz–Pyrz 2:0.5, the notable contributions are coming from F⋯H (17.7%/14.7%/24.4), N⋯H (9.8%/9.6%/20.5%), and O⋯H (13.8%/12.7%/0%) contacts in Enz molecule A/ Enz molecule B/ Pyrz, respectively. H⋯H contacts occupy 21.9%/23.8/28.9% of the surface. C⋯H contacts contribute 9.7%/13%17.2% of the Hirshfeld surface area, due to the involvement of C–H⋯O, C–H⋯F, and N–H⋯N interactions.
In the Enz parent, F⋯H contacts occupy the majority of the surface area, i.e., 24.4%, which reveals the significance of C–H⋯F interactions. The next prominent occupancy is from H⋯H contacts (19.2%). Both C⋯H and N⋯H contacts occupy similar surface areas (14.3%). O⋯H contacts contribute 9.4% of the Hirshfeld surface area (Fig. S27†).
Computations of crystal lattice energy and its constituents
As shown by DSC, the multi-component crystals with Dox and Pyrz at the same stoichiometry demonstrate different thermal stabilities (Table S4,†Fig. 10). This observation implies that the difference in solvent/coformer molecules significantly impacts the packing forces in the crystals without changing the geometry of the crystal. In order to elucidate the origin of the difference in packing energies among the studied multicomponent crystals, a set of computational studies was performed to estimate the crystal lattice energy and contributions from non-covalent interactions between Enz and the solvent/coformer. Three complementary approaches were applied to fulfill this task, namely periodic DFT-D3 computations, QTAIMC analysis of individual non-covalent interactions and the parametrized CE-B3LYP force field from the Crystal Explorer framework.From the data provided in Table S5,† one can see that the results of the DFT-D3 and CE-B3LYP methods are consistent with each other, while the energies derived from QTAIMC differ considerably from the rest. This phenomenon may be due to the fact that QTAIMC is known to underestimate the interaction energy for weak non-directed dispersive interactions, which are responsible for at least 26% of the lattice energy in the considered crystals (bottom three rows in Table S8†). It is also worth noting that the molecules of the second component within the loosely packed chains are in fact separated by fragments of Enz molecules, since no intermolecular contacts between two Dox/Pyrz molecules were observed by QTAIMC.
According to DFT-D3 and CE-B3LYP calculations, the Enz–Dox 1:0.5 solvate has higher lattice energy compared to the Pyrz cocrystal of the same stoichiometry. Meanwhile, the packing energies of the 2:0.5 crystals differ by no more than 3 kJ mol−1, which is lower than the error of theoretical estimation.
As the data in Table S5† show, the binding energy of the second molecule is higher in Dox solvates than in Pyrz cocrystals. The decomposition of interaction energy into individual interaction energies within the QTAIMC framework reveals that the main contribution of the Enz–Dox/Pyrz interactions comes from C–H⋯X contacts between donor C–H groups of the second component and X = O/N/F/S/π acceptors of Enz (Table S6†). Since the Dox molecule contains twice as many C–H groups capable of forming H-bonds with nearby Enz molecules, the total energy of weak C–H⋯X interactions is higher in Dox solvates; however, the higher polarity of the C–H bonds in Pyrz results in higher energy per single interaction (3–7 vs. 3–9 kJ mol−1). Of the C–H⋯X contacts, C–H⋯F interactions are the most numerous and have the largest contribution to lattice energy in the 1:0.5 crystals, which agrees with the results of the Hirshfeld surface analysis. In the 2:0.5 crystals, this contribution is comparable with that from C–H⋯O/N H-bonds.
To account for the difference in the boiling temperature between pure Dox and Pyrz, we introduced the Td,corr quantity, which is equal to the difference between the desolvation temperature of a solvate observed by DSC (Tdesolv) and the boiling point of the pure second component (Tboil).
| Td,corr = Tdesolv − Tboil | (5) |
| Td,corr = Tdissoc − Tboil | (6) |
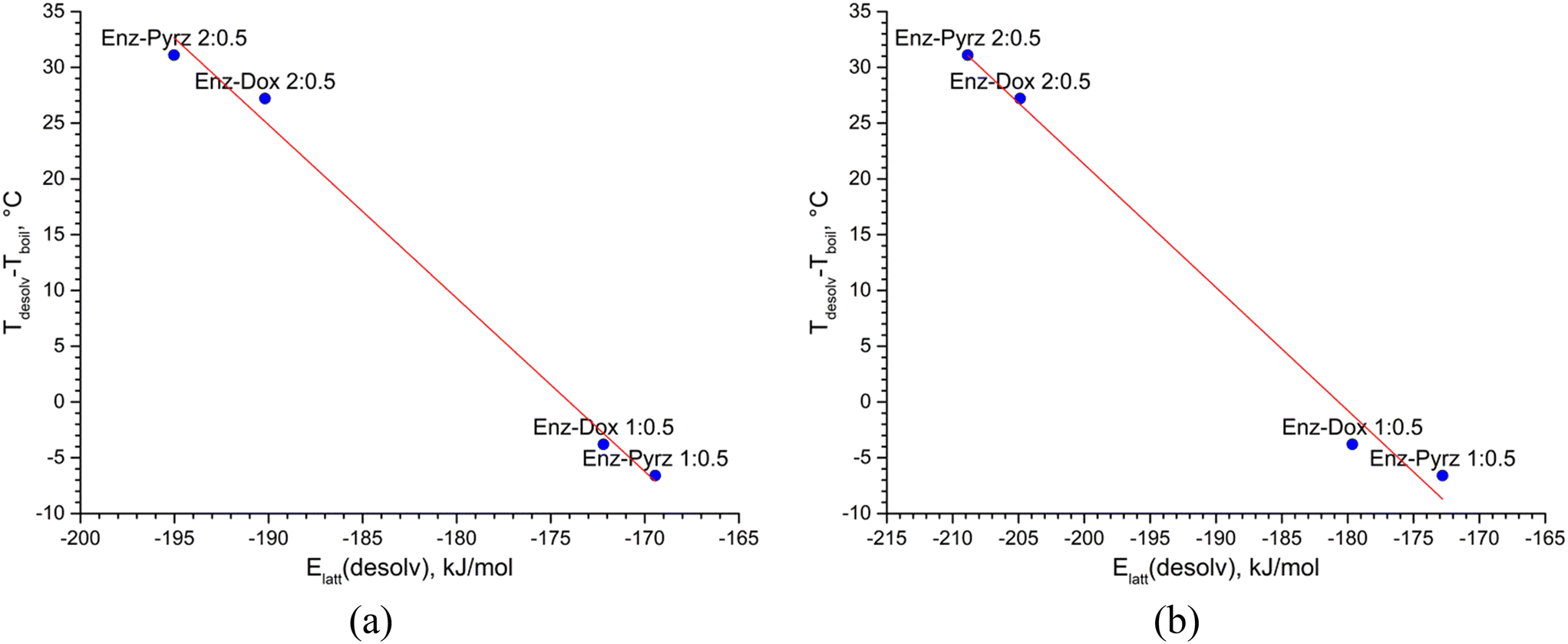 | ||
| Fig. 12 Correlation between the theoretical lattice energy of the desolvated multicomponent crystals of Enz estimated by DFT-D3 computations (a) or CE-B3LYP method (b) and the difference between the desolvation temperature of a crystal and boiling temperature of the pure second component. | ||
Conclusion
In this work, structurally similar compounds 1,4-dioxane (Dox) and pyrazine (Pyrz) were chosen to produce multi-component systems with enzalutamide (Enz), which resulted in two solvates, Enz–Dox 1:0.5 and Enz–Dox 2:0.5, and two cocrystals, Enz–Pyrz 1:0.5 and Enz–Pyrz 2:0.5. Structural analysis revealed that the solvated forms generate a two-dimensional hydrogen-bonded network, while the cocrystal systems aggregate into a three-dimensional network. The void analysis shows closed void spaces for both solvates and cocrystals. However, the presence of an additional Enz molecule in 2:0.5 and more intermolecular interactions make the solvate/cocrystal more binding compared to the 1:0.5 ratio. DSC results also show higher desolvation/dissociation temperatures for solvate/cocrystals in 2:0.5 systems. According to the stability studies performed under accelerated conditions (70–75%, 90–95% RH) at 40 °C, the Enz–Pyrz 2:0.5 form possesses longer physical stability compared to other forms. FT-IR spectroscopy disclosed major shifts in the frequency values of N–H stretching and CO stretching, which further confirmed the formation of multi-component systems via various intermolecular interactions. Thermal studies revealed the phase transformation of solvates/cocrystals to Enz form I after being desolvated or dissociated, which is in line with the HSM experiments. It is evident from the Hirshfeld analysis that the majority of the surface area was occupied by F⋯H, followed by N⋯H and O⋯H contacts, which indicates the involvement of C–H⋯F, N–H⋯O, and N–H⋯O interactions. Analysis of contributions to the lattice energy based on QTAIMC pair interaction energies revealed a correlation between the thermal stability of the multi-component crystal and the lattice energy of the hypothetical unsolvated crystal, suggesting that the desolvation process in isolated-type crystals depends mainly on the breaking of the Enz–Enz intermolecular bonds rather than on the breaking of the non-covalent bonds between Enz and the second component. The present study showcases the design of cocrystals based on the structural similarity concept. Further, this work provides valuable insight into the development of cocrystals on the basis of solvate formation.
Data availability
Single-crystal X-ray crystallographic information files (CIF's) were deposited with Cambridge Crystallographic Data Center with CCDC numbers 2350853–2350856 for solvates and cocrystals of enzalutamide.†Author contributions
Jupally Prashanth: investigation, writing-original draft. Alexander P. Voronin: computational experiments. Artem O. Surov: investigation, validation, writing original draft. Sridhar Balasubramanian: conceptualization, investigation, writing-original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr. D. Srinivasa Reddy, Director, CSIR-IICT, Hyderabad, India, is thanked for his kind encouragement (manuscript communication number: IICT/Pubs./2024/149). Dr. B. S. thanks DST for Grant INT/RUS/RFBR/374 and JP acknowledges for the fellowship. Dr. Jagadeesh Babu Nanubolu was thanked for his suggestions. Dr. N. Sunil Kumar was thanked for the support in plotting FTIR data and interpretation. Mr. P. Sivanarayanan, Mr. Lohith, Ms. S. V. S. D. Anuja were thanked for their support. Dr. AOS thanks the State Program of Fundamental Scientific Research (No. AAAA-A21-121011590019-8) for support.References
- A. O. Surov, A. P. Voronin, K. V. Drozd, M. S. Gruzdev, G. L. Perlovich, J. Prashanth and S. Balasubramanian, Phys. Chem. Chem. Phys., 2021, 23, 9695–9708 RSC.
- D. D. Gadade and S. S. Pekamwar, Adv. Pharm. Bull., 2016, 6, 479–494 CrossRef CAS PubMed.
- J. Prashanth, A. Sivalakshmi Devi, A. O. Surov, A. P. Voronin, A. V. Churakov, G. L. Perlovich and S. Balasubramanian, CrystEngComm, 2022, 24, 3511–3528 RSC.
- Polymorphism: in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley, 1st edn, 2006 Search PubMed.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC.
- L. Liu, Q. An, Y. Zhang, W. Sun, J. Li, Y. Feng, Y. Geng and G. Cheng, J. Drug Delivery Sci. Technol., 2022, 76, 103732 CrossRef CAS.
- K. V. Drozd, A. N. Manin, A. V. Churakov and G. L. Perlovich, Eur. J. Pharm. Sci., 2017, 99, 228–239 CrossRef CAS PubMed.
- J. Prashanth, K. V. Drozd, G. L. Perlovich, S. Balasubramanian and A. Surov, Cryst. Growth Des., 2022, 22, 6703–6716 CrossRef CAS.
- A. Manin, A. Surov, A. Churakov and G. Perlovich, Crystals, 2015, 5, 650–669 CrossRef CAS.
- J. B. Nanubolu and K. Ravikumar, CrystEngComm, 2016, 18, 1024–1038 RSC.
- A. Trask, W. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS PubMed.
- S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly, J. Am. Chem. Soc., 2004, 126, 13335–13342 CrossRef CAS PubMed.
- N. Issa, S. A. Barnett, S. Mohamed, D. E. Braun, R. C. B. Copley, D. A. Tocher and S. L. Price, CrystEngComm, 2012, 14, 2454 RSC.
- D. Maddileti, B. Swapna and A. Nangia, Cryst. Growth Des., 2014, 14, 2557–2570 CrossRef CAS.
- K. V. Drozd, A. N. Manin, A. P. Voronin, D. E. Boycov, A. V. Churakov and G. L. Perlovich, Phys. Chem. Chem. Phys., 2021, 23, 12456–12470 RSC.
- J. Prashanth, A. O. Surov, K. V. Drozd, G. L. Perlovich and S. Balasubramanian, CrystEngComm, 2023, 25, 3501–3513 RSC.
- T. Takasu, S. Takakura and S. Kaku, Folia Pharmacol. Jpn., 2015, 145, 36–42 CrossRef CAS PubMed.
- A. M. Feldman, J. A. Haller and S. T. DeKosky, JAMA, J. Am. Med. Assoc., 2016, 315, 25 CrossRef CAS PubMed.
- J. Hu, A. Deng and Y. Zhao, Expert Opin. Pharmacother., 2018, 19, 1841–1847 CrossRef CAS PubMed.
- B. Zhu, Q. Zhang, G. Ren and X. Mei, Cryst. Growth Des., 2017, 17, 5994–6005 CrossRef CAS.
- B. Zhu, X. Fang, Q. Zhang, X. Mei and G. Ren, Cryst. Growth Des., 2019, 19, 3060–3069 CrossRef CAS.
- G. Zhang, X. Xiao, L. Zhang, G. Ren and S. Zhang, Cryst. Growth Des., 2019, 19, 768–779 CrossRef CAS.
- C. Weiss, P. McLoughlin, P. Manesiotis, W. Redington and H. Cathcart, Cryst. Growth Des., 2018, 18, 5832–5844 CrossRef CAS.
- H. S. Shah, K. Chaturvedi, R. H. Dave and K. R. Morris, Mol. Pharmaceutics, 2021, 18, 1779–1791 CrossRef CAS PubMed.
- V. P. Chavda, N. Gajjar, N. Shah and D. J. Dave, Eur. J. Med. Chem. Rep., 2021, 3, 100013 CAS.
- G. Sodeifian, H. Nateghi and F. Razmimanesh, J. CO2 Util., 2024, 80, 102687 CrossRef CAS.
- Z. Zheng, B. Hou, X. Cheng, W. Liu, X. Huang, Y. Bao, T. Wang, Z. Wang and H. Hao, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 343–352 CrossRef CAS PubMed.
- B. D. Johnson, A. Howard, R. Varsolona, J. McCauley and D. K. Ellison, in Analytical Profiles of Drug Substances and Excipients, Elsevier, 1999, vol. 26, pp. 319–357 Search PubMed.
- G. J. Kruger and G. Gafner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 326–333 CrossRef.
- A. L. Bingham, D. S. Hughes, M. B. Hursthouse, R. W. Lancaster, S. Tavener and T. L. Threlfall, Chem. Commun., 2001, 603–604 RSC.
- A. Kons, A. Bērziņš, A. Actiņš, T. Rekis, S. Van Smaalen and A. Mishnev, Cryst. Growth Des., 2019, 19, 4765–4773 CrossRef CAS.
- D. E. Braun, T. Gelbrich, V. Kahlenberg, R. Tessadri, J. Wieser and U. J. Griesser, Cryst. Growth Des., 2009, 9, 1054–1065 CrossRef CAS.
- S. Gundlapalli, R. Devarapalli, R. R. Mudda, R. Chennuru and R. Rupakula, CrystEngComm, 2021, 23, 7739–7749 RSC.
- N. K. Duggirala, A. Vyas, J. F. Krzyzaniak, K. K. Arora and R. Suryanarayanan, Mol. Pharmaceutics, 2017, 14, 3879–3887 CrossRef CAS PubMed.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 691–699 RSC.
- S. Koranne, A. Sahoo, J. F. Krzyzaniak, S. Luthra, K. K. Arora and R. Suryanarayanan, Mol. Pharmaceutics, 2018, 15, 3297–3307 CrossRef CAS PubMed.
- M. Arhangelskis, G. O. Lloyd and W. Jones, CrystEngComm, 2012, 14, 5203 RSC.
- S. L. Morissette, S. Soukasene, D. Levinson, M. J. Cima and Ö. Almarsson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2180–2184 CrossRef CAS PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2020, 24, 347–362 CrossRef CAS.
- C. D. Chen, D. S. Welsbie, C. Tran, S. H. Baek, R. Chen, R. Vessella, M. G. Rosenfeld and C. L. Sawyers, Nat. Med., 2004, 10, 33–39 CrossRef PubMed.
- M. E. Jung, S. Ouk, D. Yoo, C. L. Sawyers, C. Chen, C. Tran and J. Wongvipat, J. Med. Chem., 2010, 53, 2779–2796 CrossRef CAS PubMed.
- C. Tran, S. Ouk, N. J. Clegg, Y. Chen, P. A. Watson, V. Arora, J. Wongvipat, P. M. Smith-Jones, D. Yoo, A. Kwon, T. Wasielewska, D. Welsbie, C. D. Chen, C. S. Higano, T. M. Beer, D. T. Hung, H. I. Scher, M. E. Jung and C. L. Sawyers, Science, 2009, 324, 787–790 CrossRef CAS PubMed.
- T. M. Beer, A. J. Armstrong, D. E. Rathkopf, Y. Loriot, C. N. Sternberg, C. S. Higano, P. Iversen, S. Bhattacharya, J. Carles, S. Chowdhury, I. D. Davis, J. S. De Bono, C. P. Evans, K. Fizazi, A. M. Joshua, C.-S. Kim, G. Kimura, P. Mainwaring, H. Mansbach, K. Miller, S. B. Noonberg, F. Perabo, D. Phung, F. Saad, H. I. Scher, M.-E. Taplin, P. M. Venner and B. Tombal, N. Engl. J. Med., 2014, 371, 424–433 CrossRef PubMed.
- L. Maini, D. Braga, F. Farinella, E. Melotto, M. Verzini, R. Brescello, I. Michieletto and I. Munari, Cryst. Growth Des., 2018, 18, 3774–3780 CrossRef CAS.
- M. Dolezal and J. Zitko, Expert Opin. Ther. Pat., 2015, 25, 33–47 CrossRef CAS PubMed.
- A. Woolfson and M. Rothschild, Proc. R. Soc. London, Ser. B, 1990, 242, 113–119 CAS.
- T. B. Adams, J. Doull, V. J. Feron, J. I. Goodman, L. J. Marnett, I. C. Munro, P. M. Newberne, P. S. Portoghese, R. L. Smith, W. J. Waddell and B. M. Wagner, Food Chem. Toxicol., 2002, 40, 429–451 CrossRef CAS PubMed.
- AXS Bruker, APEX3 Package, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- W. T. Pennington, J. Appl. Crystallogr., 1999, 32, 1028–1029 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, WIREs Comput. Mol. Sci., 2018, 8, e1360 CrossRef.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. S. D. Surampudi, S. Rajendrakumar, J. B. Nanubolu, S. Balasubramanian, A. O. Surov, A. P. Voronin and G. L. Perlovich, CrystEngComm, 2020, 22, 6536–6558 RSC.
- Q. Tao, Q.-Q. Hao, A. P. Voronin, X.-L. Dai, Y. Huang, G. L. Perlovich, T.-B. Lu and J.-M. Chen, Cryst. Growth Des., 2019, 19, 5636–5647 CrossRef CAS.
- A. P. Voronin, N. A. Vasilev, A. O. Surov, A. V. Churakov and G. L. Perlovich, CrystEngComm, 2021, 23, 8513–8526 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- C. Gatti, V. R. Saunders and C. Roetti, J. Chem. Phys., 1994, 101, 10686–10696 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Chem. Phys. Lett., 2011, 507, 185–189 CrossRef CAS.
- G. Zhang, X. Xiao, L. Zhang, G. Ren and S. Zhang, Cryst. Growth Des., 2019, 19, 768–779 CrossRef CAS.
- R. Thakuria and A. Nangia, Cryst. Growth Des., 2013, 13, 3672–3680 CrossRef CAS.
- A. Bērziņš, T. Rekis and A. Actiņš, Cryst. Growth Des., 2014, 14, 3639–3648 CrossRef.
- D. E. Braun, T. Gelbrich and U. J. Griesser, CrystEngComm, 2019, 21, 5533–5545 RSC.
- H. Yang, Y. Yang, L. Jia, W. Tang, S. Xu, S. Du, M. Li and J. Gong, Cryst. Growth Des., 2019, 19, 1550–1558 CrossRef CAS.
- J. C. Bennion, L. Vogt, M. E. Tuckerman and A. J. Matzger, Cryst. Growth Des., 2016, 16, 4688–4693 CrossRef CAS.
- S. K. Gohel, V. Palanisamy, P. Sanphui, M. Prakash, G. P. Singh and V. Chernyshev, RSC Adv., 2021, 11, 30689–30700 RSC.
- D. Cinčić, T. Friščić and W. Jones, Chem. – Eur. J., 2008, 14, 747–753 CrossRef PubMed.
- J. Rohlíček, E. Skořepová, M. Babor and J. Čejka, J. Appl. Crystallogr., 2016, 49, 2172–2183 CrossRef.
- M. J. Turner, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystEngComm, 2011, 13, 1804–1813 RSC.
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, J. Mol. Graphics Modell., 2013, 44, 208–219 CrossRef CAS PubMed.
- J. J. McKinnon, F. P. A. Fabbiani and M. A. Spackman, Cryst. Growth Des., 2007, 7, 755–769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal X-ray crystallographic information files (CIF's) were deposited with Cambridge Crystallographic Data Center with CCDC numbers 2350853–2350856 for solvates and cocrystals of enzalutamide. The supporting information section also contains details of preparation procedures of the solid forms, results of the PXRD analysis of the Enz cocrystals and solvates, crystal packing figures, crystal packing similarity, void map analysis, conformational overlay, hot stage microscopic images, PXRD overlay of physical stability experiments, FT-IR spectra, Hirshfeld surface analysis, DFT-D3 computations, crystallographic data and hydrogen bonding details. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00566j |
| This journal is © The Royal Society of Chemistry 2024 |