
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of supramolecular complexation of alkali hydrogenselenates with crown ethers and solid-solutions with their hydrogensulfate counterparts on the solid-to-solid phase transition behaviors†
Samet
Ocak‡
,
Dario
Braga
 and
Simone
d'Agostino
*
and
Simone
d'Agostino
*
The University of Bologna – Department of Chemistry “Giacomo Ciamician”, Via F. Selmi 2, 40126, Bologna (BO), Italy
First published on 27th July 2024
Abstract
This study investigates the structural and phase transition characteristics of supramolecular complexes composed of 18-crown-6 ether and hydrogen selenate (HSeO4−) anions with various cations (K+, Rb+, Cs+). Single crystals of [18-crown-6·K]HSeO4·2H2O, [18-crown-6·Rb]HSeO4·H2O, [18-crown-6·Cs]HSeO4·H2O, [18-crown-6·K]HSeO4, and [18-crown-6·K](HSeO4)0.5(HSO4)0.5 were grown and their structures determined via single-crystal X-ray diffraction. Differential scanning calorimetry and variable-temperature powder X-ray diffraction were employed to analyse dehydration and phase transition behaviors. The inclusion of 18-crown-6 ether significantly lowered the superprotonic phase transition temperatures by approximately 40 °C compared to pure solid acids. Additionally, substituting HSO4− with HSeO4− decreased phase transition temperatures for K and Cs-complexes and modified the phase transition behavior of the Rb-complex from two-step to single-step isostructural phase transition. Attempts to form solid solutions between the HSeO4− and HSO4− complexes yielded mixed results, with notable success in modulating phase transition temperatures in K-complexes.
Introduction
Solid electrolytes, which are known for their fast ion conduction, have attracted much attention in recent years for their various applications in electronic devices such as batteries, supercapacitors, molecular sensors, and fuel cells.1–8 One example of a solid electrolyte is Nafion, an organic polymer that contains sulfonic acid residues. It has a conductivity range of 10−1 to 10−5 S cm−1, which is affected by factors like temperature, hydration state, thermal history, and processing conditions.9–15 Current research is, thus, focused on finding materials that can conduct protons at high temperatures (100–300 °C) in dry or anhydrous environments.7,16–19 Many alternatives have been proposed, including polymers, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), metal oxides, glasses, and plastic crystals (PCs), all of which have shown promising characteristics for proton conductivity.5,11,16,18,20–32 Since they all have their pros and cons depending on different working conditions (temperature intervals, hydration levels, etc.), searching for alternative materials is still ongoing.Solid acids with the chemical formulas MHAO4 and MH2BO4, where M represents alkali cations, A represents S or Se, and B represents P or As, have been found to exhibit high protonic conductivity.33,34 This is due to the presence of dynamically disordered hydrogen-bond networks associated with reversible first-order solid-to-solid transitions, which are referred to as superprotonic phase transitions.2,35–40 Solid acids are more advantageous than polymeric electrolytes because they operate at higher temperatures, do not require humidification, and are impermeable to other ions or radical species thanks to their solid form.41,42 Moreover, studies have shown that for solid acids proton conductivity is further enhanced in the presence of larger cations, such as Cs+ compared to Rb+ and K+.37,43 In general, the crystal lattice of alkali hydrogen sulfates and selenates undergoes significant structural changes at high temperatures resulting in a solid-to-solid transition associated with an increase in crystal symmetry, and generation of dynamic disorder of the H-bond network, causing both protons and H-bonds to be de-localized over the entire lattice. Proton conduction in these crystals occurs through a structure diffusion mechanism, known as the Grotthuss mechanism,44 and are termed superprotonic phases.35,45 The hydrogen selenates can be expected to be more advantageous than the hydrogen sulfates because their superprotonic phase transitions take place at lower temperatures (see below).37,46,47
In our previous work,46 we discussed how the crystal structure of supramolecular complexes, comprising 18-crown-6 as the ligand and either potassium or rubidium as the cations, alongside hydrogen sulfate as the anion, influence their superprotonic conduction properties. These compounds were found to undergo enantiotropic solid-to-solid transitions associated with the onset of a dynamical process affecting both the crown ether ligand and the hydrogensulfate anion.
Our goal in advancing the search for new materials capable in conducting small ions such as protons entails merging the principles of Crystal Engineering48–50 with solid-solution formation techniques,51,52 to craft a versatile molecular toolkit for creating crystalline solids that exhibit phase transitions at varying temperatures.53–55
The aim of the work presented herein was essentially twofold. First, we were interested in investigating the impact of a different anion, specifically hydrogen selenate (HSeO4−), while using Cs+, Rb+, and K+ as the cations hosted in the cavity of 18-crown-6 ether. Crown ethers exhibit a pronounced affinity for alkali metal cations whose ionic radii align with the dimensions of their binding cavities, defined by the surrounding O-atoms.56,57 As a matter of fact, 18-crown-6 demonstrates a distinct preference for binding with K+; though, on passing to the larger cations such as Rb+ and Cs+, it can still form complexes, albeit with a reduced binding affinity. As a result, K+ seamlessly accommodates itself within, whereas Rb+ and Cs+ ions, depicted in Scheme 1a, position themselves above the midplane established by the crown ether molecule, extending beyond the cavities according to the ionic radii, and leaving larger cations partly “naked”. These cations must inevitably engage O-atoms from anions or water molecules to fulfill their coordination sphere. This is expected to influence metal coordination significantly and will shape the resulting crystalline materials' distinctive structural, thermal, and conduction characteristics.
 | ||
| Scheme 1 (a) Representation showing how the alkali cation (blue sphere) fits inside the 18-crown-6 ether cavity (black ring), remaining “naked” to the extent of their sizes. Distance (d) is expected to increase with cationic radius, and (b) general scheme for solid solution formation; orange and blue tetrahedra represent HSeO4− and HSO4− anions, respectively. | ||
Second, we wanted to explore the possibility of preparing crystalline solid solutions with their hydrogensulfate analogs (Scheme 1b) and studying how the composition of the resulting materials further affects the phase transition compared to the pure parent systems.
To achieve these objectives, we have grown single crystals of the supramolecular complexes made up of 18-crown-6 and MHSeO4 (where M+ = K+, Rb+, Cs+). Subsequently, we employed single-crystal X-ray diffraction data to elucidate their structures, thereby discerning the distinctive structural variations induced by diverse cations. The determined structures were identified as 18-crown-6·KHSeO4·2H2O (1·2H2O), 18-crown-6·RbHSeO4·H2O (2·H2O), and 18-crown-6·CsHSeO4·H2O (3·H2O). Next, we have used the differential scanning calorimetry (DSC) technique to explore their dehydration and phase transition behaviors which in general could be summarized as:
| HYDRATED COMPLEX ⇌ ANHYDROUS COMPLEX ⇌ HT ANHYDROUS COMPLEX. |
Experimental
Synthesis
KHSeO4, RbHSeO4, and CsHSeO4 were synthesized by combining related alkali metal carbonate salt, i.e., K2CO3 (Sigma-Aldrich ≥98%), Rb2CO3 (Sigma-Aldrich ≥98%), and Cs2CO3 (Merck ≥99%) with an excess amount of H2SeO4 (40 wt% in H2O, 99.95%) dissolved in water. The hydrogen selenate crystals were grown after approximately 2 days. The resulting crystals were filtered and subsequently washed with methanol. Then, they were dissolved again in the water together with a slight excess amount of 18-crown-6 (ca. 1.2 equivalents) to synthesize the hydrated supramolecular crown complex of each salt. After slow evaporation, the resulting crystals were washed with octane to remove the excess 18-crown-6 ether. In order to grow anhydrous single crystals of 18-crown-6·KHSeO4, the hydrated complex is left in the oven for 3 hours at 80 °C and then dissolved in dry acetone and left in a desiccator. The single crystals of solid solution 18-crown-6·K(HSO4)0.5(HSeO4)0.5 were formed by dissolving the same stoichiometric amounts of anhydrous 18-crown-6·KHSeO4 and 18-crown-6·KHSO4 in dry acetone which was then left in the desiccator. The powder-formed solid solutions in all ranges of molecular fractions for all the complexes were prepared by hand grinding the appropriate amount of parent compounds.X-Ray diffraction
The single-crystal data for 18-crown-6·KHSeO4·2H2O, 18-crown-6·RbHSeO4·H2O, 18-crown-6·CsHSeO4·H2O, 18-crown-6·KHSeO4, and 18-crown-6·K(HSO4)0.5(HSeO4)0.5 were obtained using an Oxford X'Calibur S CCD diffractometer with a graphite monochromator. The diffractometer used Mo-Kα radiation with a wavelength of 0.71073 Å and an Oxford CryoStream800 cryostat. Twinning was observed in each crystal, and the reflection data were integrated using the default configuration for twinned crystals in the CrysAlisPro Software. The subsequent structural solution and refinement processes were conducted using the HKLF4 file, which included non-overlapping reflections. The structural solutions were obtained through intrinsic phasing using SHELXT58 and subsequently refined on F2 with SHELXL.59 The refinement method used was the full-matrix least-squares refinement method within the Olex2 software.60 The position of HHSeO4− atoms was either directly determined or inserted at calculated positions. HCH atoms associated with all compounds were introduced at calculated positions and refined while riding on their respective carbon atoms. Anisotropic refinement was applied to all non-hydrogen atoms, accompanied by the implementation of rigid-body RIGU restraints.61To identify phases and investigate variable-temperature behavior, powder X-ray diffractograms were obtained within the 2θ range of 5–40°. This was done using a Panalytical X'Pert PRO automated diffractometer equipped with an X'Celerator detector and operated in Bragg–Brentano geometry. The diffractometer used Cu Kα radiation without a monochromator. The step size was 0.02°, time per step was 20 s, the 0.04 rad soller was used and the operating current was 40 mA × 40 kV. All powder diffraction patterns of the hydrated forms were compared with the calculated patterns for controlling the purity of the bulk samples (Fig. ESI-1†).
Differential scanning calorimetry
Differential Scanning Calorimetry (DSC) analyses were performed on 18-crown-6·KHSeO4·2H2O, and 18-crown-6·RbHSeO4·H2O using a PerkinElmer DSC-7 instrument, equipped with a PII intracooler. Additionally, DSC measurements for 18-crown-6·CsHSeO4·H2O and all the solid solutions were carried out using a Q10 instrument from TA Instruments, fitted with a Refrigerated Cooling System (RCS90, TA instruments). The samples, with weights ranging between 3–5 mg, were heated in aluminum pans at a rate of 5 °C min−1 within the temperature range of 30–160 °C under a nitrogen (N2) atmosphere.Results and discussion
In the following sections, we present the structures, as determined from single-crystal analysis, of three hydrated supramolecular complexes: [18-crown-6·K]HSeO4·2H2O (1·2H2O), [18-crown-6·Rb]HSeO4·H2O (2·H2O), [18-crown-6·Cs]HSeO4·H2O (3·H2O). Subsequently, we analyse and discuss their dehydration and phase transition behaviors associated with increasing temperature, employing Differential Scanning Calorimetry (DSC) and variable-temperature powder X-ray diffraction (PXRD) analyses. Finally, we discuss our findings about solid solution formation with their hydrogensulfate analogous and present the structures of the anhydrous form of the K-complex, [18-crown-6·K]HSeO4, and its solid solution [18-crown-6·K](HSeO4)0.5(HSeO4)0.5.Hydrated complexes
All compounds crystallize in the monoclinic system (see Table ESI-1† for details) with the metal cations coordinated by the crown ether. In compounds 1·2H2O and 2·H2O, the coordination spheres are satisfied by two and three O-atoms originating from the hydrogenselenate anions, respectively. In the case of 3·H2O, an extra water molecule contributes to the coordination of the cesium cation, and, as expected, M+ cations protrude above the midplane of the crown ether molecule according to their growing ionic radii (K+ = 151 pm, Rb+ = 161 pm, and Cs+ = 167 pm). See Fig. 1 and Table 1 for distances. Due to the different coordination geometry, the HSeO4− anions form hydrogen-bonded dimers, either among themselves or mediated by water molecules. 1·2H2O is characterized by the presence of chains of HSeO4− anions linked by hydrogen bonds via the water molecules to form supramolecular tapes [O–Hw⋯OHSeO4− = 2.60(1)–2.79(1) Å and O–Hw⋯Ow = 2.70(1) Å], as depicted in Fig. 2a.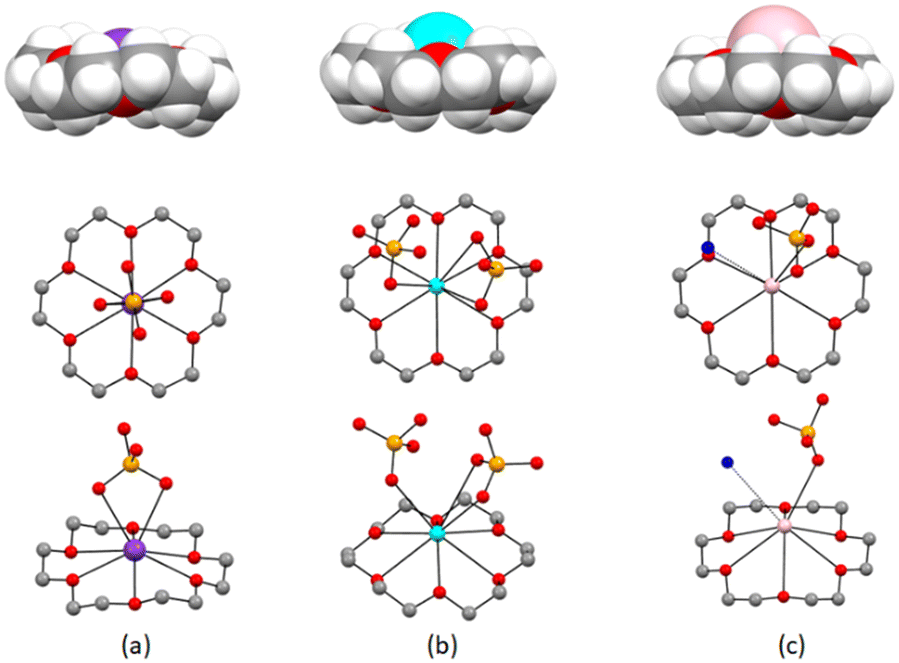 | ||
| Fig. 1 Space filling representation showing the protrusion of the cations, according to their ionic radii, from the crown ether, and top and side views showing the coordination geometry of the around the K+, Rb+, and Cs+ ions in crystalline 1·2H2O (a), 2·H2O (b), and 3·H2O (c). H-atoms omitted for clarity. | ||
| M+ | d (Å) | M+⋯Ocrown (Å) | M+⋯OHSeO4− (Å) | M+⋯Owater (Å) |
|---|---|---|---|---|
| a Distance between the metal cation M+ and the midplane of the crown ether ligand as defined in Scheme 1. | ||||
| K+ | 0.694(1) | 2.772(7)–2.976(7) | 2.901(9)–2.932(7) | — |
| Rb+ | 1.098(2) | 2.927(5)–3.044(3) | 3.094(3)–3.297(4) | — |
| Cs+ | 1.506(1) | 3.075(9)–3.25(1) | 3.19(1) | 3.67(2) |
 | ||
| Fig. 2 Hydrogen bond patterns detected within crystalline: (a) 1·2H2O, (b) 2·H2O, and (c) 3·H2O. Water molecules in blue and H-atoms were omitted for clarity. | ||
On the other hand, 2·H2O features hydrogen bonded HSeO4− pairs [O–HHSeO4−⋯OHSeO4− = 2.601(5) Å] connecting two [18-crown-6·Rb]+ cationic units to form supramolecular dimers (Fig. 2b) which, in turn, are bridged via hydrogen bonds with water molecules [O–Hw⋯OHSeO4− = 2.875(8) Å], whereas, in 3·H2O, the water molecule, unlike its counterparts in the series, directly coordinates with the alkali metal ion captured within the crown ether ring, as illustrated in Fig. 1. Furthermore, water molecules establish hydrogen bonds with the oxygen atoms of the anions [O–Hw⋯OHSeO4− = 2.74(3) Å], which also engage in interactions with each other, leading to the formation of a hydrogen-bonded supramolecular dimer [OHSeO4−⋯OHSeO4− = 2.64(3) Å], as depicted in Fig. 2c. The thermal characteristics of all complexes were examined through a combination of differential scanning calorimetry (DSC) and variable-temperature X-ray powder diffraction (VT-XRPD). DSC measurements for 1·2H2O, shown in Fig. ESI-2,† indicate that the complex undergoes dehydration at around 60 °C followed by a reversible solid-to-solid phase transition at 96 °C, with 19 °C hysteresis. The VT-PXRD experiments shown in Fig. 3 also confirm this. Fig. ESI-3† reveals that the powder diffractograms of [18-crown-6·K]HSeO4 and [18-crown-6·K]HSO4 exhibit striking similarity post-dehydration and after the solid-to-solid phase transition. This observation suggests that both compounds behave similarly from a structural standpoint. However, the replacement of HSO4− with HSeO4− seems to be responsible for a decrease of approximately 20 °C in the phase transition temperature, which is noteworthy.
 | ||
| Fig. 3 VT-PXRD patterns recorded at different temperatures for: (1·2H2O), (2·H2O), and (3·H2O). | ||
2·H2O also experiences dehydration in the temperature range of 60–80 °C, as evidenced by Differential Scanning Calorimetry (DSC) measurements (Fig. ESI-4†). Subsequently, a reversible solid-to-solid phase transition occurs at approximately 125 °C. The structural transformations associated with these phenomena are closely monitored through variable-temperature powder XRD, as depicted in Fig. 3. It is noticeable that the dehydration process results in a clear change of peak positions from RT to 100 °C. However, the neat phase change is not observed at 125 °C. The broadening of diffraction peaks, and the lowering of their intensities are most consistent with an isostructural phase transition, which likely results in a higher level of mobility within the structure above 125 °C. Fig. ESI-5† shows a comparison of the powder diffractograms of rubidium HSO4− and HSeO4− complexes in each phase. Although the structures are not isomorphous in their hydrated forms, they become isomorphous in their anhydrous and high temperature phases.
Furthermore, the DSC (Fig. SI-6†) and VT-PXRD (Fig. SI-7†) measured on 3·H2O show two non-resolved processes, occurring at 76 and 86 °C, likely associated with dehydration followed by a solid-to-solid transition. These results suggest that the specific position of the water molecule, i.e., directly bound to the metal ion, is the main factor in stabilizing the structure. Finally, at around 140 °C, there is an endothermic peak belonging to the 18-crown-6 ether leaving the structure that is also confirmed by the VT-PXRD. This can be seen through the similarities in the powder pattern with the high-temperature phase of its Rb analogous (Fig. ESI-7†). On the other hand, unlike the other two selenate complexes in this series, 3·H2O has certain structural differences from its hydrogensulfate analogous43 as shown in Fig. ESI-7.† Compared to the parent compounds MHSeO4 (where M+ = K+, Rb+, and Cs+), the significant decrease in the solid-to-solid transition temperatures leading to superprotonic phases, as depicted in Table 2, can be attributed to both the supramolecular interactions weakening, i.e., breaking and formation of hydrogen bonds arising from re-orientational motions of the HSeO4− and the dynamic behavior affecting the 18-crown-6 ether ring. Also, the solid-to-solid phase transition behaviors are different in comparison to their hydrogensulfate counterparts. For K-complexes, the transition temperature is reduced from 116 °C (ref. 62) to 96 °C, and for Cs-complexes, it is changed from 90 °C (ref. 63) to 86 °C. In the sulfate counterpart of Rb-complex, it was a two-step first-order transition (116–122 °C);46 in the selenate counterpart, it became a one-step isostructural phase transition.
It is worth noting that rehydration in this class of compounds occurs quite rapidly; when exposed to open air, they readily absorb water from the moisture in a matter of minutes.
Solid solutions
Finally, given the high degree of isomorphism and/or isostructurality of the novel HSeO4− supramolecular complexes with their HSO4− counterparts,46,62,63 we also attempted solid-solution51,52 formation to achieve further modulation of phase transition behaviors.Solid solutions were prepared either mechanochemically or with slow evaporation of solution with equimolar amounts of parent compounds (see Experimental section for details). PXRD and DSC measurements were used to characterize the resulting solids and establish whether mixed phase formation was successful.
Despite the aforementioned structural similarities, our efforts solely yielded a real mixed phase in the cases of K+ and Cs+. It was observed that the complexes with K+ were isomorphous through all variations of molar fractions (Fig. ESI-8†) and demonstrated a decrease in the solid-to-solid phase transition temperature in a nearly linear manner as the amount of hydrogenselenate anion in the structure increases, up to 75%, reaching the same value as neat [18-crown-6·K]HSeO4 (Fig. 4 and ESI-9†). Furthermore, the VT-PXRD results show that the solid solutions are also isomorphous to the high-temperature phases of the parent compounds (Fig. ESI-10†).
 | ||
| Fig. 4 The trends of unit cell volume and the phase transition temperature vs. anion composition of the supramolecular solid solutions [18-crown-6·K](HSeO4)x(HSO4)1−x. | ||
Powder X-ray diffraction results revealed that the solid solutions of Cs-complexes formed by up to 50% hydrogensulfate crystallized as the [18-crown-6·Cs]HSeO4·H2O structure. At RT, the solid solutions of [18-crown-6·Cs](HSeO4)x(HSO4)1−x, where x ≥ 0.5, were all isomorphous (Fig. ESI-11†). The dehydration and phase transition processes were convoluted for all three compounds examined in this range and they all behaved the same which is shown by VT-PXRD and DSC (Fig. ESI-12 and ESI-13†). Increasing the hydrogen sulfate content by more than 50% in the structure caused a physical mixture of parent compounds that is monitored by PXRD at RT and high temperatures (Fig. ESI-11 and ESI-14†). Due to the concurrent nature of the dehydration and order–disorder phase transition processes, we were unable to conduct a detailed analysis of the shift in phase transition temperature.
The attempts made for Rb+ resulted in physical mixtures, likely due to their subtle structural differences in the hydrated/anhydrous forms, as evidenced by the DSC traces which showed the presence of endothermic peaks attributable to transitions of the respective parent compounds. In Fig. ESI-15,† we can see two endothermic peaks from the hydrogensulfate analogous of Rb+ and one from the hydrogenselenate. This clearly indicates the formation of a physical mixture.
The overall results agree with the structural considerations, crystalline [18-crown-6·K]HSeO4 and [18-crown-6·K]HSO4 are highly isomorphous and isostructural at each level (meant as hydrated, anhydrous, and high temperature phases), and the difference in size between the two anions is small (ca. 10%).
To further prove solid solution formation, SCXRD analyses were carried out on single crystal specimens grown for the anhydrous hydrogen selenate complex [18-crown-6·K]HSeO4 (1) and the mixed-phase obtained with equimolar amounts of anions, [18-crown-6·K](HSeO4)0.5(HSO4)0.5 and comparing the unit cell metrics with those of the anhydrous [18-crown-6·K]HSeO4 complex. Fig. 5 showcases the anhydrous structures of [18-crown-6·K]HSO4,46 [18-crown-6·K]HSeO4, and their solid solution [18-crown-6·K](HSO4)0.5(HSeO4)0.5, (see Table ESI-2† for metal coordination geometries and hydrogen bonding interaction distances). All three structures are isomorphous and isostructural, as mentioned above, and crystallize in the monoclinic P21/n space group (see Table ESI-1† for crystallographic details). Notably, the solid solution's unit cell volume satisfies Vegard's rule64 by lying in the middle of the two parent compounds (Fig. 4) as well as the solid-to-solid transition temperature. In general, the linear response of the unit cell metrics and of physical properties with composition is typical of inorganic alloys but can be extended also to molecular materials.65
 | ||
| Fig. 5 Crystal structures of [18-crown-6·K]HSO4 (left), [18-crown-6·K](HSeO4)0.5(HSO4)0.5 (middle), and [18-crown-6·K]HSeO4 (right). HSeO4− and HSO4− depicted in orange and blue respectively, H-atoms omitted for clarity. | ||
Conclusions
This study explores the impact of the hydrogen selenate anion, HSeO4−, in conjunction with various cations (Cs+, Rb+, and K+) that are encapsulated within the cavity of 18-crown-6 ether through coordination interactions on structural arrangements and phase change characteristics of the resulting supramolecular complexes. Single crystals of [18-crown-6·K]HSeO4·2H2O, [18-crown-6·Rb]HSeO4·H2O, and [18-crown-6·Cs]HSeO4·H2O were grown, and their structures were elucidated through single-crystal X-ray diffraction analysis. The investigation also looked into their dehydration processes and phase transition behaviors using the differential scanning calorimetry (DSC) technique, with subsequent validation and analysis provided by variable-temperature powder X-ray diffraction.Notably, the inclusion of 18-crown-6 ether into the hydrogenselenates alkali structures resulted in ca 40 °C drop in the solid-to-solid phase transition temperatures compared to the neat solid acids, and at the same time, replacing HSO4− with HSeO4− induced a decrease of the same phase transition temperature of the anhydrous complexes in the series of ca 20 °C, while significantly altering the behavior of the Rb-complex. The two successive first-order phase transitions previously detected in [18-crown-6·Rb]HSO4 (ref. 46) turned into one isostructural phase transition.
Additionally, during the same study, we also explored the possibility of obtaining solid solutions between the novel HSeO4− supramolecular complexes synthesized in this study and the formerly reported HSO4− counterparts.46,62,63 Attempts made in the case of Rb+ resulted in physical mixtures, whereas for Cs+ and K+ solid solutions [18-crown-6·Cs](HSeO4)x(HSO4)1−x with x ≥ 0.5 and [18-crown-6·K](HSeO4)x(HSO4)1−x with x covering the entire compositional range were obtained. Only for latter, a modulated phase transition temperature was detected, indicating that this is a viable route to achieve fine tuning.
However, it should be stressed that all the anhydrous complexes in the series quickly uptake water from the moisture, posing a challenge to the electrochemical impedance measurements (EIS). Further research is ongoing to stabilize the anhydrous phases and study the ion conduction features of such materials, as well as to extend the same synthetic approach to other systems, and explore how the ions' structural diversity and size may affect the formation of solid solutions and phase transitions in terms of temperature and type.
Data availability
CCDC 2350122–2350126 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures. The datasets supporting this article have been uploaded as part of the supplementary material.Author contributions
SD, DB: conceptualization, manuscript preparation; and supervision; SO: experimentation and data collection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SO, DB, and SD thank the University of Bologna (RFO-Scheme) for financial support and the Ms Student Grega Venturini for helping with some synthesis. SD also acknowledges Italian MUR (Fondi PNRR-CNMS-Spoke 13-MOST Code: CN00000023) for financial support.References
- B. Hilczer, C. Pawlaczyk and F. E. Salman, Ferroelectrics, 1988, 81, 193–196 CrossRef.
- D. A. Boysen, T. Uda, C. R. I. Chisholm and S. M. Haile, Science, 2004, 303, 68–70 CrossRef CAS PubMed.
- S. M. Haile, K. D. Kreuer and J. Maier, Acta Crystallogr., Sect. B: Struct. Sci., 1995, B51, 680–687 CrossRef CAS.
- V. V. Sinitsyn, A. I. Baranov and E. G. Ponyatovsky, Solid State Ionics, 2000, 136, 167–171 CrossRef.
- L. S. Wang, S. V. Patel, S. S. Sanghvi, Y. Y. Hu and S. M. Haile, J. Am. Chem. Soc., 2020, 142, 19992–20001 CrossRef CAS PubMed.
- L. Fan, Z. Tu and S. H. Chan, Energy Rep., 2021, 7, 8421–8446 CrossRef.
- M. Zeng, W. Liu, H. Guo, T. Li, Q. Li, C. Zhao, X. Li and H. Li, ACS Appl. Energy Mater., 2022, 5, 9058–9069 CrossRef CAS.
- H. Tang, J. Li, Z. Wang, H. Zhang, M. Pan and S. P. Jiang, ACS Symp. Ser., 2013, 1140, 243–263 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- Y. Sonef, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef.
- H. Gao and K. Lian, RSC Adv., 2014, 4, 33091–33113 RSC.
- T. Yamada, M. Sadakiyo and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 3144–3145 CrossRef CAS PubMed.
- M. Schuster, W. H. Meyer, G. Wegner, H. G. Herz, M. Ise, K. D. Kreuer and J. Maier, Solid State Ionics, 2001, 145, 85–92 CrossRef CAS.
- G. H. Li, C. H. Lee, Y. M. Lee and C. G. Cho, Solid State Ionics, 2006, 177, 1083–1090 CrossRef CAS.
- B. Karadedeli, A. Bozkurt and A. Baykal, Phys. B, 2005, 364, 279–284 CrossRef CAS.
- P. Jhariat, P. Kumari and T. Panda, CrystEngComm, 2020, 22, 6425–6443 RSC.
- T. Kajita, H. Tanaka, Y. Ohtsuka, T. Orido, A. Takano, H. Iwamoto, A. Mufundirwa, H. Imai and A. Noro, ACS Omega, 2023, 8, 1121–1130 CrossRef CAS PubMed.
- S. Ocak, F. Poli, D. Braga, T. Salzillo, F. Tarterini, G. Carì, E. Venuti, F. Soavi and S. d'Agostino, Cryst. Growth Des., 2023, 23, 4336–4345 CrossRef CAS.
- Z. Zheng, M. Li, Q. Zhou, L. Cai, J. F. Yin, Y. Cao and P. Yin, ACS Appl. Nano Mater., 2021, 4, 811–819 CrossRef CAS.
- M. Rautenberg, B. Bhattacharya, C. Das and F. Emmerling, Inorg. Chem., 2022, 61, 10801–10809 CrossRef CAS PubMed.
- N. Ma, N. Horike, L. Lombardo, S. Kosasang, K. Kageyama, C. Thanaphatkosol, K. Kongpatpanich, K. I. Otake and S. Horike, J. Am. Chem. Soc., 2022, 144, 18619–18628 CrossRef CAS PubMed.
- Z. Yang, P. Chen, W. Hao, Z. Xie, Y. Feng, G. Xing and L. Chen, Chem. – Eur. J., 2021, 27, 3817–3822 CrossRef CAS PubMed.
- Y. Y. Enakieva, A. A. Sinelshchikova, M. S. Grigoriev, V. V. Chernyshev, K. A. Kovalenko, I. A. Stenina, A. B. Yaroslavtsev, Y. G. Gorbunova and A. Y. Tsivadze, Chem. – Eur. J., 2019, 25, 10552–10556 CrossRef CAS PubMed.
- X. Meng, H. N. Wang, S. Y. Song and H. J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- M. T. Caldes, K. V. Kravchyk, M. Benamira, N. Besnard, V. Gunes, O. Bohnke and O. Joubert, Chem. Mater., 2012, 24, 4641–4646 CrossRef CAS.
- P. Winiarz, K. Dzierzgowski, A. Mielewczyk-Gryń, M. Gazda and S. Wachowski, Chem. – Eur. J., 2021, 27, 5393–5398 CrossRef CAS PubMed.
- S. M. Ahn, J. E. Park, G. Y. Jang, H. Y. Jeong, D. M. Yu, J. K. Jang, J. C. Lee, Y. H. Cho and T. H. Kim, ACS Energy Lett., 2022, 7, 4427–4435 CrossRef CAS.
- T. Scholz, C. Schneider, M. W. Terban, Z. Deng, R. Eger, M. Etter, R. E. Dinnebier, P. Canepa and B. V. Lotsch, ACS Energy Lett., 2022, 7, 1403–1411 CrossRef CAS PubMed.
- S. C. Pal, D. Mukherjee, R. Sahoo, S. Mondal and M. C. Das, ACS Energy Lett., 2021, 6, 4431–4453 CrossRef CAS.
- R. Sahoo, S. C. Pal and M. C. Das, ACS Energy Lett., 2022, 7, 4490–4500 CrossRef CAS.
- J. Luo, O. Conrad and I. F. J. Vankelecom, J. Mater. Chem. A, 2013, 1, 2238–2247 RSC.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- S. M. Haile, Mater. Today, 2003, 1369, 24–29 CrossRef.
- A. B. Papandrew, C. R. I. Chisholm, R. A. Elgammal, M. M. Özer and S. K. Zecevic, Chem. Mater., 2011, 23, 1659–1667 CrossRef CAS.
- A. V. Belushkint, C. J. Carlile and L. A. Shuvalov, Ferroelectrics, 1995, 167, 21–31 CrossRef.
- A. V. Belushkint, C. J. Carlilet and L. A. Shuvalov, J. Phys.: Condens. Matter, 1992, 4, 389–398 CrossRef.
- M. Friesel, B. Baranowski and A. Lunden, Solid State Ionics, 1989, 35, 85–89 CrossRef.
- Z. Hassanzadeh Fard, N. E. Wong, C. D. Malliakas, P. Ramaswamy, J. M. Taylor, K. Otsubo and G. K. H. Shimizu, Chem. Mater., 2018, 30, 314–318 CrossRef CAS.
- A. Ikeda, D. A. Kitchaev and S. M. Haile, J. Mater. Chem. A, 2014, 2, 204–214 RSC.
- Y. Matsuda, M. Yonemura, H. Koga, C. Pitteloud, M. Nagao, M. Hirayama and R. Kanno, J. Mater. Chem. A, 2013, 1, 15544–15551 RSC.
- C. Dreßler, G. Kabbe and D. Sebastiani, J. Phys. Chem. C, 2016, 120, 19913–19922 CrossRef.
- R. B. Merle, C. R. I. Chisholm, D. A. Boysen and S. M. Haile, Energy Fuels, 2003, 17, 210–215 CrossRef CAS.
- K. D. Kreuer, Th. Dippel, N. G. Hainovsky and J. Maier, Ber. Bunsenges. Phys. Chem., 1992, 96, 1736–1742 CrossRef CAS.
- A. Pawłowski and M. Połomska, Solid State Ionics, 2005, 176, 2045–2051 CrossRef.
- D. A. Boysen, T. Uda, C. R. I. Chisholm and S. M. Haile, Science, 2004, 303, 68–70 CrossRef CAS PubMed.
- S. Ocak, S. d'Agostino, G. Venturini, F. Soavi, S. Bordignon, M. R. Chierotti and D. Braga, J. Phys. Chem. C, 2024, 128, 4789–4795 CrossRef CAS.
- O. Checa, R. A. Vargas and J. E. Diosa, Ionics, 2014, 20, 545–550 CrossRef CAS.
- D. Braga, F. Grepioni, L. Maini and S. D'Agostino, IUCrJ, 2017, 4, 369–379 CrossRef CAS PubMed.
- O. Shemchuk, S. D'Agostino, C. Fiore, V. Sambri, S. Zannoli, F. Grepioni and D. Braga, Cryst. Growth Des., 2020, 20, 6796–6803 CrossRef CAS.
- D. Braga, Chem. Commun., 2023, 59, 14052–14062 RSC.
- M. Lusi, CrystEngComm, 2018, 20, 7042–7052 RSC.
- M. Lusi, Cryst. Growth Des., 2018, 18, 3704–3712 CrossRef CAS.
- S. D'Agostino, L. Fornasari, F. Grepioni, D. Braga, F. Rossi, M. R. Chierotti and R. Gobetto, Chem. – Eur. J., 2018, 24, 15059–15066 CrossRef PubMed.
- S. D'Agostino, L. Fornasari and D. Braga, Cryst. Growth Des., 2019, 19, 6266–6273 CrossRef.
- S. Ocak, R. Birolo, G. Carì, S. Bordignon, M. R. Chierotti, D. Braga, R. Gobetto, T. Salzillo, E. Venuti, O. Yaffe and S. d'Agostino, Mol. Syst. Des. Eng., 2022, 7, 950–962 RSC.
- C.-C. Liou and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1992, 3, 543–548 CrossRef CAS PubMed.
- K. Frensdorff, J. Am. Chem. Soc., 1967, 89, 600–606 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Thorn, B. Dittrich and G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 448–451 CrossRef CAS.
- D. Braga, M. Gandolfi, M. Lusi, D. Paolucci, M. Polito, K. Rubini and F. Grepioni, Chem. – Eur. J., 2007, 13, 5249–5255 CrossRef CAS PubMed.
- D. Braga, E. Modena, M. Polito, K. Rubini and F. Grepioni, New J. Chem., 2008, 32, 1718–1724 RSC.
- A. Denton and N. Ashcroft, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 3161–3164 CrossRef CAS PubMed.
- A. I. Kitaigorodsky, Mixed Crystals, Springer, 1984, vol. 33, pp. 75–81 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DSC traces, VT-PXRD results and crystallographic information. CCDC 2350122–2350126. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00514g |
| ‡ Current address: Institute for Microelectronics and Microsystems - Research National Council (IMM-CNR), Via Pietro Gobetti 101, 40129, Bologna (BO), Italy. |
| This journal is © The Royal Society of Chemistry 2024 |