
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal engineering and sorption studies on CN- and dipyridyl-bridged 2D coordination polymers†
Valoise Brenda
Nguepmeni Eloundou
ab,
Patrice
Kenfack Tsobnang
*a,
Theophile
Kamgaing
a,
Chiranjib
Gogoi
b,
Nieves
Lopez-Salas
c and
Susan A.
Bourne
 *b
*b
aDepartment of Chemistry, University of Dschang, PO Box 67, Dschang, Cameroon. E-mail: patrice.kenfack@univ-dschang.org
bCentre for Supramolecular Chemistry Research, University of Cape Town, Rondebosch 7701, South Africa. E-mail: susan.bourne@uct.ac.za
cSustainable Materials Chemistry, Department of Chemistry, Paderborn University, Warburger Str. 100, 33098, Paderborn, Germany
First published on 4th July 2024
Abstract
Two new cyano-bridged coordination polymers, {[Ni(bpe)(H2O)2][Ni(CN)4]·2H2O}n (1) and {[Cu(bpe)(H2O)2][Ni(CN)4]·ethanol}n (2) (bpe = 1,2-bis(4-pyridyl)ethane) were synthesized and characterized. This was part of a study attempting to rationalize the formation of Hofmann coordination polymers over their aqua-derivatives, in which we found that the length of the ligand does not control the outcome of the synthesis. In the initial phase of thermogravimetric analysis, compound 1 lost the guest water molecule while compound 2 lost its guest ethanol. Single crystal X-ray diffraction revealed that both compounds crystallize in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with similar unit cell parameters. In both compounds, the Ni(II) ion is coordinated with four cyanide–carbon atoms in a square planar geometry and Cu(II) or the second Ni(II) ion adopts a distorted octahedral coordination by two bpe ligands, two bridging cyano groups, and two water molecules. In both structures, chains composed of adjacent [Ni(C12H12N2)(H2O)2]2+ (for 1) or [Cu(C12H12N2)(H2O)2]2+ (for 2) are further linked together via [Ni(CN)4]2− units and extend to generate a two-dimensional framework. The presence of disordered terminal cyanide moieties in 1 could be attributed to a larger volume available to this moiety in the structure of 1. The BET surface areas of the activated phases 1′ of 1 and 2′ of 2 are 28 m2 g−1 and 5 m2 g−1, respectively, and originally, they have no pores to accommodate gas molecules at low pressure (1 atm). 1′ does not adsorb carbon dioxide at 195 K (0.30 mmol g−1), 273 K (0.086 mmol g−1) and 298 K (0.056 mmol g−1), or hydrogen at 77 K, but it shows an affinity to adsorb nitrogen (around 2.5 mmol g−1) at 77 K and water vapor molecules (around 0.95 mmol g−1) at 298 K with typical type-II isotherms. 2′ adsorbs a small quantity of water vapor (around 0.20 mmol g−1) at 298 K and does not adsorb carbon dioxide at 195 K (0.125 mmol g−1), 273 K (0.069 mmol g−1) and 298 K (0.062 mmol g−1), or hydrogen at 77 K, but shows more affinity to adsorb nitrogen (around 1.2 mmol g−1) at 77 K.
with similar unit cell parameters. In both compounds, the Ni(II) ion is coordinated with four cyanide–carbon atoms in a square planar geometry and Cu(II) or the second Ni(II) ion adopts a distorted octahedral coordination by two bpe ligands, two bridging cyano groups, and two water molecules. In both structures, chains composed of adjacent [Ni(C12H12N2)(H2O)2]2+ (for 1) or [Cu(C12H12N2)(H2O)2]2+ (for 2) are further linked together via [Ni(CN)4]2− units and extend to generate a two-dimensional framework. The presence of disordered terminal cyanide moieties in 1 could be attributed to a larger volume available to this moiety in the structure of 1. The BET surface areas of the activated phases 1′ of 1 and 2′ of 2 are 28 m2 g−1 and 5 m2 g−1, respectively, and originally, they have no pores to accommodate gas molecules at low pressure (1 atm). 1′ does not adsorb carbon dioxide at 195 K (0.30 mmol g−1), 273 K (0.086 mmol g−1) and 298 K (0.056 mmol g−1), or hydrogen at 77 K, but it shows an affinity to adsorb nitrogen (around 2.5 mmol g−1) at 77 K and water vapor molecules (around 0.95 mmol g−1) at 298 K with typical type-II isotherms. 2′ adsorbs a small quantity of water vapor (around 0.20 mmol g−1) at 298 K and does not adsorb carbon dioxide at 195 K (0.125 mmol g−1), 273 K (0.069 mmol g−1) and 298 K (0.062 mmol g−1), or hydrogen at 77 K, but shows more affinity to adsorb nitrogen (around 1.2 mmol g−1) at 77 K.
Introduction
Air pollution stands as a critical global challenge affecting both developed and developing nations. The continuous release of potentially harmful gases and particles into the atmosphere poses severe threats to human health and the environment.1 As we confront an imminent climate crisis, it becomes imperative to achieve rapid and substantial reduction in anthropogenic emissions, particularly focusing on carbon dioxide (CO2).2 The primary source of CO2 emissions is the combustion of fossil fuels like coal, oil, and natural gas.3 In response to the pressing environmental concerns, industrialized countries have committed to reducing greenhouse gas emissions through various protocols and agreements.4 In addition, various processes have been developed, including absorption,5 membrane separation,6 cryogenic separation,7 and adsorption on solid surfaces.8 Among these processes, adsorption stands out as highly cost-effective and non-destructive due to readily available adsorbents and minimal equipment requirements. Adsorbed CO2 can be utilized or transformed, and the adsorbent can be regenerated.9 For this process, the adsorbent must possess key properties such as high capacity, selectivity, thermal and mechanical stability, and cost-effectiveness.10 Coordination polymers, especially porous coordination polymers such as metal–organic frameworks, have emerged as a promising material in this context. Coordination polymers, a novel class of crystalline materials, feature metal centers bridged by multidentate ligands in infinite network structures, offering substantial promise in capturing and separating small molecules due to their high internal surface areas.11Cyano-bridged coordination polymers are a class of coordination compounds characterized by the presence of cyanide (CN−) ligands that act as bridging units between metal ions, forming extended polymeric structures.
Hofmann coordination polymers, also known as Hofmann-type MOFs, are a subset of cyano-bridged coordination polymers and are themselves a widely studied class. In these compounds, the molecular framework referred to as the “host structure” encapsulates a distinct species known as the “guest” within a cage-like structure.12 The general chemical formula for Hofmann-type coordination polymers is [ML][M′(CN)4], where M can include Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Mn2+ or Cd2+; M′ can be Ni2+, Pt2+ or Pd2+; and L represents bidentate organic ligands which act as bridges between metal ions.13 In the Cambridge Structural Database (CSD, version 2023.3.0)14 there are 410 structures reported in this class. The [M][M′(CN)4] layers are interconnected by organic ligands (L) through coordination bonds. Several Hofmann coordination polymers have shown promise for CO2 adsorption, including Ni(bpene)[Ni(CN)4] (where bpene is 1,2-bis(4-pyridyl)ethylene) which captures 495.6 cm3 g−1 at 273 K15 and NiL[Ni(CN)4]·3H2O (where L represents 1,3,6,8,11,14-hexaazatricyclo[12.2.1.18,11]octadecane) which captures 56 cm3 g−1 at 273 K.16 Additionally, polymers with pyrazine linkers, such as CoL[Ni(CN)4], NiL[Ni(CN)4], and FeL[Ni(CN)4] (where ‘L’ represents pyrazine), exhibit CO2 uptakes of 140 cm3 g−1, 130 cm3 g−1, and 120 cm3 g−1 respectively at 195 K.17 Culp et al. reported a Hofmann-type structure with 1,2-bis(4-pyridyl)ethane via a new synthesis method and showed that it had good sorption properties toward nitrogen and carbon dioxide.18
While attempting to synthesize this family of compounds, several authors have obtained a new packing mode of the constituent with the chemical formula [M(L)(H2O)n][M′(CN)4], where M can include transition metal ions,19–24 or lanthanides,25–27 M′ can be Ni2+, Pt2+ or Pd2+; and L represents bridging organic ligands. Currently, in the Cambridge Structural Database (CSD, version 2023.3.0) there are 47 structures of this family of compounds. The main differences between the packing mode of the Hofmann-type polymers (M(L)][M′(CN)4]·G) and the modified packing mode ([M(L)(H2O)2][M′(CN)4]·G) are the following: Hofmann-type polymers do not include aqua ligands in their structure, whereas the modified packing mode includes them. In Hofmann-type polymers, the metal ions are coordinated to the ligands (L) and the cyanide units (M′(CN)4), while in the modified packing mode, the metal ions are coordinated to both the ligands (L) and additional aqua ligands (H2O)2, along with the cyanide units (M′(CN)4). Then Hofmann-type polymers typically form three-dimensional networks where [M][M′(CN)4] layers are interconnected by organic ligands (L) through coordination bonds, while the modified packing mode forms two-dimensional units that are connected with hydrogen bonds.
In this work, with a view to contributing to the rationalization of the synthesis of these Hofmann polymers, we used a previously studied ligand (1,2-bis(4-pyridyl)ethane, (bpe)) along with nickel(II) and copper(II) ions with the (Ni(CN)4) units to synthesize the modified packing mode of Hofmann-type polymers. The crystal structures, the thermal stability, and the gas adsorption properties of both compounds are determined, and studies are performed to correlate all these properties to the structures of the compounds.
Experimental
Materials
For the synthesis of 1 and 2 the following reagents were purchased and used without further purification: 1,2-bis(4-pyridyl)ethane (99%, Sigma Aldrich), nickel(II) chloride hexahydrate (99.9%, Merck); copper(II) chloride dihydrate (99%, Sigma Aldrich), potassium cyanide (98%, BHD). The solvents used for synthesis were deionized water, N,N′-dimethylformamide (≥99% purity), ethanol (99%, Merck), and ammonia solution 25% (Merck). Paratone N oil (99%, Sigma Aldrich) was used to fix the crystal in place for diffraction analysis.Synthesis
K2[Ni(CN)4]·H2O was prepared with a yield of 85% as reported previously28–30 – following the procedure given in section 1 of the ESI.†{[Ni(bpe)(H2O)2][Ni(CN)4]·2H2O}n (1) and {[Cu(bpe)(H2O)2][Ni(CN)4]·ethanol}n (2) were prepared at room temperature by mixing in 2 mL of deionized water, K2[Ni(CN)4]·H2O (0.0259 g; 0.1 mmol) and NiCl2·6H2O (0.0237 g, 0.1 mmol) for 1 and CuCl2·2H2O (0.017 g, 0.1 mmol) for, with continuous stirring approximately for 15 min. To the resulting solutions, 2 mL of ethanol solution of 1,2-bis(4-pyridyl)ethane (0.0184 g; 0.1 mmol), was added with continuous stirring for approximately 15 min. The precipitate obtained was dissolved in 2 mL ammonia solution (25% in H2O) with continuous stirring for approximately 30 min. The resulting solution was filtered and kept at room temperature for crystallization. Light purple crystals of 1 were obtained after three weeks with a yield of 70% and for 2, light blue crystals were obtained after one week with a yield of 67%.
Characterization

Results and discussion
Characterisation of {[Ni(bpe)(H2O)2][Ni(CN)4]·2H2O}n (1) and {[Cu(bpe)(H2O)2][Ni(CN)4]·ethanol}n (2)
Viewing the synthesized compounds under a microscope reveals that 1 forms prismatic light-purple crystals, while 2 crystallizes as light-blue blocks (Fig. S1†).The Fourier-transformed infrared (FTIR) spectrum of the as-synthesized compounds over the range 4000–400 cm−1 is shown in Fig. S2 while Table S1† summarises the absorption bands of 1 and 2. In the FTIR spectrum of those compounds, the broad band observed at 3359 cm−1 in compound 1 or at 3354 cm−1 in 2 is assigned to the valence vibration mode of the hydroxyl group (OH) of water molecules. The appearance of this band at a lower value than that observed in the literature (expected around 3500 cm−1) and its width suggest that this group participates in hydrogen bonding. Further FTIR analysis and UV-vis analysis results are provided in the ESI† (see both spectra in Fig. S5†).
Structural analysis of {[Ni(bpe)(H2O)2][Ni(CN)4]·2H2O}n (1) and {[Cu(bpe)(H2O)2][Ni(CN)4]·ethanol}n (2)
Single-crystal X-ray diffraction analyses revealed that 1 and 2 are similar, and both crystallized in the P space group (see Table 1). The asymmetric units (ASUs) of 1 and 2 consist of half of the cationic component [M(C12H12N2)(H2O)2]2+ (M = Ni1 or Cu1), half of the anionic component [Ni(CN)4]2−, and one crystallization water molecule for 1 (Fig. 1a, see Fig. S6† for the complete labelling scheme) or a half-occupied uncoordinated ethanol molecule for 2 (Fig. 1b, see Fig. S7† for the complete labelling scheme). Within the [M(C12H12N2)(H2O)2]2+ fragment, the central Ni1 or Cu1 ion adopts a distorted octahedral coordination arrangement. In this configuration, the six coordination positions are occupied by four nitrogen atoms, two nitrogen atoms from two cyanide groups and two other nitrogen atoms from two 1,2-bis(4-pyridyl)ethane ligands and two oxygen atoms from coordinated water molecules. The average Ni1–Nbpe bond distance (2.125(2) Å) is slightly longer than the Ni1–NCN bonds (2.051(2) Å). This bond distance difference was also observed in the literature in similar compounds.18,23 Additionally, both M–O bonds have similar lengths of 2.103(2) Å (for Ni–O in 1) and 2.030(2) Å (Cu–O in 2), and the two Ni1–NC angles exhibit a nearly linear arrangement with identical angles of 175.1(2)°. These bond lengths and angle values agree with the data reported in the CSD for similar materials with formula M(H2O)2(L)[Ni(CN)4].21,23 As expected, the anionic moiety [Ni(CN)4]2− adopts a square planar geometry, with two cyanide groups acting as bridging linkers to octahedral nickel (Ni1) sites while the others are terminal ligands. In 1, these terminal cyano groups are disordered over three positions with equal site occupancies (see Fig. 1). The mean bond lengths for square planar nickel Ni2–C and C–N are 1.866(11) Å and 1.150(15) Å, respectively, and the square planar nickel Ni2–CN bonds display nearly linear angles, ranging between 177.5(11)° and 179.6(10)°, in agreement with the data reported for similar tetracyanonickelate salts.21,23
| 1 | 2 | |
|---|---|---|
| ASU formula | C8H10N3NiO2 | C18H16CuN6NiO |
| Formula weight (g mol−1) | 238.90 | 486.62 |
| Crystal color | Light purple | Light blue |
| Crystal size (mm) | 0.172 × 0.131 × 0.08 | 0.04 × 0.12 × 0.18 |
| Temperature (K) | 100(2) K | 100(2) K |
| Crystal system | Triclinic | Triclinic |
| Space group |
P |
P |
| a/Å | 6.3253(13) | 5.9234(5) |
| b/Å | 9.2032(18) | 9.5597(8) |
| c/Å | 10.202(2) | 10.2625(8) |
| α/° | 64.48(3) | 114.847(3) |
| β/° | 74.18(3) | 96.831(3) |
| γ/° | 78.74(3) | 95.811(3) |
| Z | 2 | 2 |
| Volume/Å3 | 513.6(2) | 516.16(7) |
| Density/g cm−3 | 1.545 | 1.566 |
| Absorption coefficient μ (mm−1) | 1.866 | 1.973 |
| F(000) | 246.0 | 247.0 |
| Radiation | 0.71073 | 0.71073 |
| 2θ range for data collection (°) | 2.264 to 28.310 | 2.223 to 28.301 |
| Index ranges | −8 ≤ h ≤ 8, −12 ≤ k ≤ 12, −13 ≤ l ≤ 13 | −7 ≤ h ≤ 7, −12 ≤ k ≤ 12, −13 ≤ l ≤ 13 |
| Reflections collected | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 727 727 |
38726 |
| Independent reflections | 2563 [Rint = 0.0802, Rsigma = 0.0404] | 2565 [Rint = 0.0719, Rsigma = 0.0281] |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0347, wR2 = 0.0763 | R 1 = 0.0327, wR2 = 0.0827 |
| Final R indexes [all data] | R 1 = 0.0615, wR2 = 0.0848 | R 1 = 0.0448, wR2 = 0.0882 |
| Data/restraints/parameters | 2563/6/153 | 2565/4/177 |
| Goodness-of-fit on F2 | 1.066 | 1.109 |
| Largest diff. peak/hole (e Å−3) | 0.578/−0.561 | 0.606/−0.666 |
 | ||
| Fig. 1 A view of the extended (1 − x, −y, 1 − z, −1 + x, y, z) asymmetric unit in the crystal structures of 1 (a) and 2 (b) showing the atom numbering scheme and displacement ellipsoids drawn at the 50% level. | ||
As illustrated in Fig. 2, the structure of 1 extends along both the bpe and cyano directions, generating a 2-D grid (compounds 1 and 2 have the same framework structure, see Fig. S8† for 2). With octahedral nickel (Ni1) considered as the nodes, the linkers are either bpe linkers or [Ni(CN)4] groups, generating a planar sql topology. Neighbouring networks are layered without interpenetration, and with Ni⋯Ni distances of 6.325 Å in 1 and 5.923 Å in 2. Within the 2D framework (depicted in Fig. 3), the octahedral nickel (Ni1) units are interconnected through a distorted pseudo-octahedral coordination with water molecules extending above and below the plane. The long edge of the rectangle is given with a Ni1⋯N1 distance of 13.389 Å, while the short edge runs through the [Ni2(CN)4]2− anions and results in a octahedral nickel⋯octahedral nickel (Ni1⋯Ni1) distance of 10.098 Å. Corresponding distances in 2 are 13.239 Å and 10.724 Å.
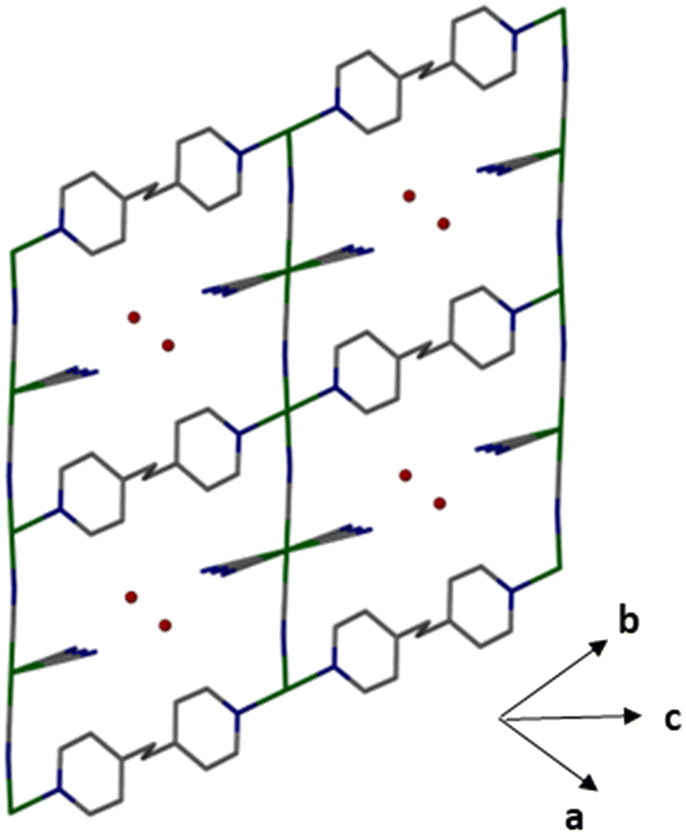 | ||
| Fig. 2 2-Periodic framework of 1 (cyan – Ni, blue – N, grey – C, red – O). | ||
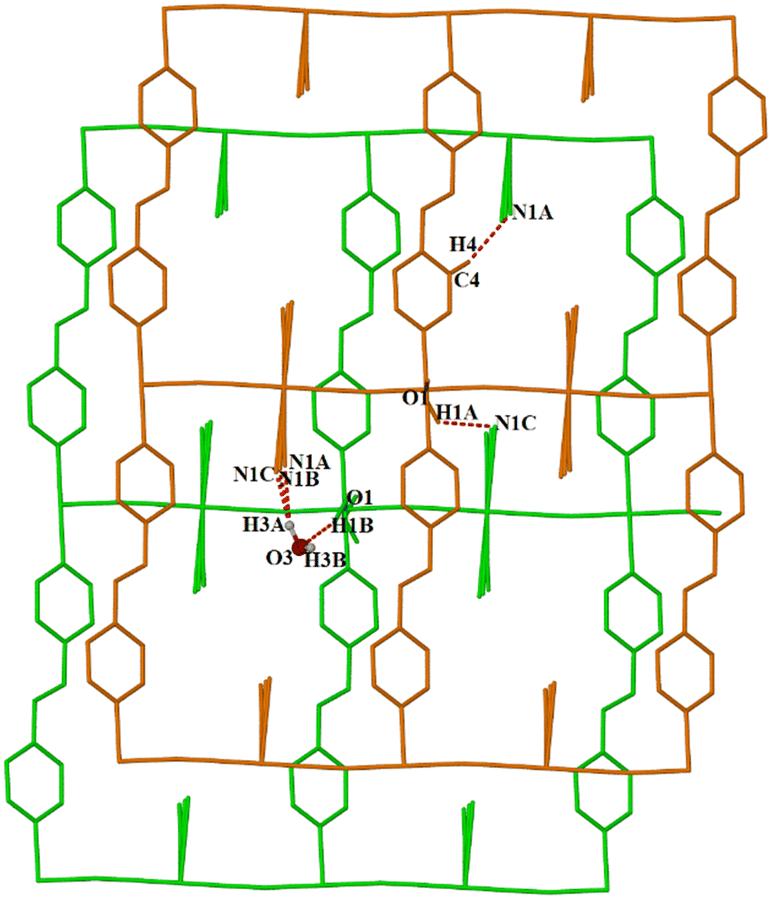 | ||
| Fig. 3 Two layers in 1 formed by neighboring 2D-sql frameworks and linked through the hydrogen bonds. Hydrogen atoms on the framework have been omitted for clarity. | ||
Terminal cyanides, ligands, and coordinated and uncoordinated solvent molecules all participate in the hydrogen-bonding network within 1 and 2. There are three distinct types of hydrogen bonds shown in Fig. 3 and Table 2. In 1, two terminal cyanides engage in O–H⋯N hydrogen bonds with coordinated O1–H1A⋯N1C and guest water molecules (O3–H3A⋯N1A; O3–H3A⋯N1B and O3–H3A⋯N1C); meanwhile in 2, only one cyanide engages in an O–H⋯N hydrogen bond with a coordinated water molecule (O1–H1A⋯N2). The bpe ligands interact with terminal cyanides through C–H⋯N hydrogen bonds (C4–H4⋯N1A) in 1 while in 2 these ligands interact rather with axial cyanide through C–H⋯N hydrogen bonds (C5–H5⋯N2). Lattice solvent (water or ethanol) molecules establish O–H⋯O hydrogen bonds with coordinated water molecules O1–H1B⋯O3 for 1 and O1–H1A⋯O3 for 2. Consequently, a single 2D-sql network is connected to its neighbour through hydrogen bonds as illustrated in Fig. 3 for 1 (see Fig. S8 and S9† for 2).
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| Symmetry operators: 1x, y − 1, z; 2 −x, −y + 1, −z + 1; 3 −x + 1, −y + 1, −z + 1; 4 −x, + 1, −y + 1, −z; 5x − 1, y, z; 6 −x + 1, −y, −z + 1. | ||||
| 1 | ||||
| O1–H1A⋯N1C1 | 0.980 (17) | 2.50 (3) | 3.192 (8) | 127 (2) |
| O1–H1B⋯O3 | 0.997 (17) | 2.091 (19) | 3.041 (4) | 159 (2) |
| O3–H3A⋯N1A2 | 1.037 (18) | 1.0.99 (3) | 2.979 (8) | 158 (6) |
| O3–H3A⋯N1B2 | 1.037 (18) | 2.50 (3) | 3.518 (13) | 166 (6) |
| O3–H3B⋯N1B1 | 0.896 (19) | 2.04 (3) | 2.865 (10) | 153 (6) |
| O3–H3B⋯N1A1 | 0.896 (19) | 2.30 (4) | 3.059 (7) | 143 (5) |
| O3–H3B⋯N1C1 | 0.896 (19) | 1.95 (3) | 2.796 (8) | 158 (6) |
| C4–H4⋯N1A3 | 0.95 | 2.55 | 3.467 (8) | 163 |
| C4–H4⋯N1B3 | 0.95 | 2.65 | 3.574 (11) | 164.1 |
| C6–H6⋯N2 | 0.95 | 2.63 | 3.103 (4) | 111.2 |
| C3–H3⋯N24 | 0.95 | 2.64 | 3.102 (4) | 110.3 |
| 2 | ||||
| O1–H1A⋯N25 | 0.993 (17) | 2.453 (19) | 3.436 (3) | 163 |
| O1–H1B⋯O36 | 0.90 (2) | 2.28 (2) | 3.183 (10) | 148 |
| C5–H5⋯N26 | 0.97 (3) | 2.61 (3) | 3.160 (3) | 116 (2) |
The potential void space assuming no collapse of the network (without coordinated and uncoordinated solvent molecules) for 1 is 12.6% (64.80 Å3) while for 2 it is 25.4% (131.11 Å3). They were calculated with Mercury31 using a probe radius of 1.2 Å with a grid spacing of 0.7 Å. Using this calculation, the void space observed in 1 (Fig. S10†) consists of discrete pockets while in 2 (Fig. S11†) there are one-dimensional continuous channels.
To investigate the occurrence of disorder of the cyanide ligand in 1, we calculated the volume of space occupied by terminal cyanide ligands in 1 and 2 using Mercury (probe radius of 1.2 Å and grid spacing of 0.7 Å). The volume occupied by the disordered terminal cyanide in 1 (Fig. S13†) is 48.93 Å3, whereas in 2 (Fig. S14†) the same terminal cyanide occupies a smaller volume of 35.72 Å3. It appears that the larger volume available in 1 results in the disorder observed. We also noted that the dihedral Cu1–N–C–Ni2 is substantially more bent than the equivalent dihedral Ni1–N–C–Ni2 with a Cu1–N–C angle of 157.6(2)° (vs. Ni1–N–C of 175.1(2)°) and Cu1–N–C–Ni2 torsion angle of 120.2(2)° (vs. 13.8(2)° in 1).
Comparison of the packing mode in 1 and 2vs. Hofmann coordination polymers
In 2013, Hu et al. attributed the formation of the [M(L)(H2O)n][M'(CN)4] family of compounds to the use of longer linear ligands L.21 They proposed that shorter linear diaza ligands, such as pyrazine, 4,4′-bipyridine, 4,4′-dipyridylacetylene, 1,2-bis(4-pyridyl)ethylene, or 1,2-di(4-pyridyl)ethane (bpe), were generally employed to construct Hofmann-type 3-D materials, [ML][M'(CN)4], in similar reactions.21 However, in 2015, Tian and his collaborators obtained the [M(L)(H2O)n][M'(CN)4] family of compounds using a shorter ligand, 4,4′-bipyridine,23 contradicting Hu's proposal. In our syntheses, we also obtained this packing [M(L)(H2O)2][M'(CN)4] using the ligand 1,2-di(4-pyridyl)ethane (bpe). This ligand has been used to construct Hofmann-type polymers with nickel and tetracyanonickelate [Ni(CN)4] by Culp and collaborators18 but, in our work, it has been used to construct the packing mode variant of the Hofmann polymer. Therefore, we can conclude that the size of the ligand is not the only parameter to consider in the formation of this family of compounds.It's also important to note that the synthesis conditions may play a crucial role in the formation of this new packing. When comparing the synthesis method of 1 to that of the Hofmann-type coordination polymer Ni(Bpane)[Ni(CN)4] (bpane = 1,2-di(4-pyridyl)ethane),18 the main difference lies in the reaction conditions. In the synthesis of 1, all reactants are mixed in an aqueous solution, leading to the direct formation of the precipitate, while in the synthesis of Ni(Bpane)[Ni(CN)4] a Hofmann coordination polymer, the starting compound Ni(H2O)2[Ni(CN)4]·3H2O (obtained by slowly mixing 0.1 M aqueous solutions of K2[Ni(CN)4] and Ni(NO3)2·6H2O, followed by refluxing the resulting mixture overnight) is heated to 140 °C under a N2 flux before adding the ligand.18 These differences in reaction conditions may have led to the formation of different products.
Culp et al. synthesized their Hofmann-type coordination polymers by starting with the preparation of Ni(H2O)2[Ni(CN)4]. They then vacuum-dried this compound at 130 °C for 2 to 3 hours to obtain Ni2(CN)4, which they dissolved with the ligand in a mixture of dry toluene and dry acetonitrile, followed by refluxing under nitrogen.38
We used the same method for 2 by forming Cu(H2O)2[Ni(CN)4], drying it, then washing it with water, and finally drying it again. We air-dried our compound at room temperature for 12 hours and then dissolved it in an aqueous ammonia solution, followed by adding 1,2-bis(4-pyridyl)ethane dissolved in ethanol, but this synthesis produced the aqua complex 2 rather than a classic Hofmann polymer. This suggests that the length of the ligand is not a parameter to consider when considering the formation of this family of compounds, but the synthesis method may play a role. For specific reagents, it remains necessary to find a suitable synthetic method.
Characterization of the bulk material
Thermal analysis
Fig. 4 shows the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) obtained for compounds 1 and 2 heated in the temperature range of 10–600 °C and 10–550 °C respectively. | ||
| Fig. 4 TGA and DSC curves of 1 (a) and 2 (b). | ||
A two-stage thermal behavior is observed in 1. In the first stage, compound 1 lost the guest and the coordinated water molecules at 110 °C with theoretical and experimental mass losses of 15.10% and 13.72%, respectively (Fig. 4a). The DSC thermogram confirms that this mass loss is endothermic with a peak at 96 °C. This compound started to decompose at 300 °C with an abrupt mass loss at 595 °C which includes the loss of the organic ligand. During the initial phase of thermal analysis, compound 2 lost its ethanol guest molecule at 102 °C with a large, broad endothermic peak on the DSC curve. The theoretical and experimental mass losses at this first stage are 9.35% and 9.72%, respectively. The network decomposed in a stepwise fashion over a temperature range of 200–500 °C.
The thermal events described above were observed using hot-stage microscopy (HSM). The crystal of 1 begins to become opaque with heating to 80 °C, which can be attributed to the dehydration of the crystal. It maintains its solid state from room temperature (21 °C) to 300 °C thus confirming that the material decomposes without melting. (Fig. S3†).
As shown in Fig. S4,†2 begins to become opaque and becomes more light blue with heating to 100 °C which is attributed to the loss of the ethanol guest. With gradual decomposition, the compound changes colour and becomes light-yellow at 245 °C, and then progressively changes from light-yellow to yellow at 300 °C, then to brown at 320 °C, and finally to black at 330 °C.
It appears from this hot stage analysis that crystals of 1 and 2 undergo desolvation and then decompose without melting.
Powder X-ray Diffraction (PXRD) analysis
The powder X-ray diffraction (PXRD) patterns of 1 and 2 materials are shown in Fig. 5. The materials are crystalline and that the experimental PXRD patterns of compounds 1 and 2 agree with the simulated patterns generated based on structural data, confirming that 1 and 2 are pure materials. The presence of the first peak observed in the experimental powder XRD pattern of 2 and its absence in the simulated one could be attributed to differences in the size and texture of the sample during the analysis.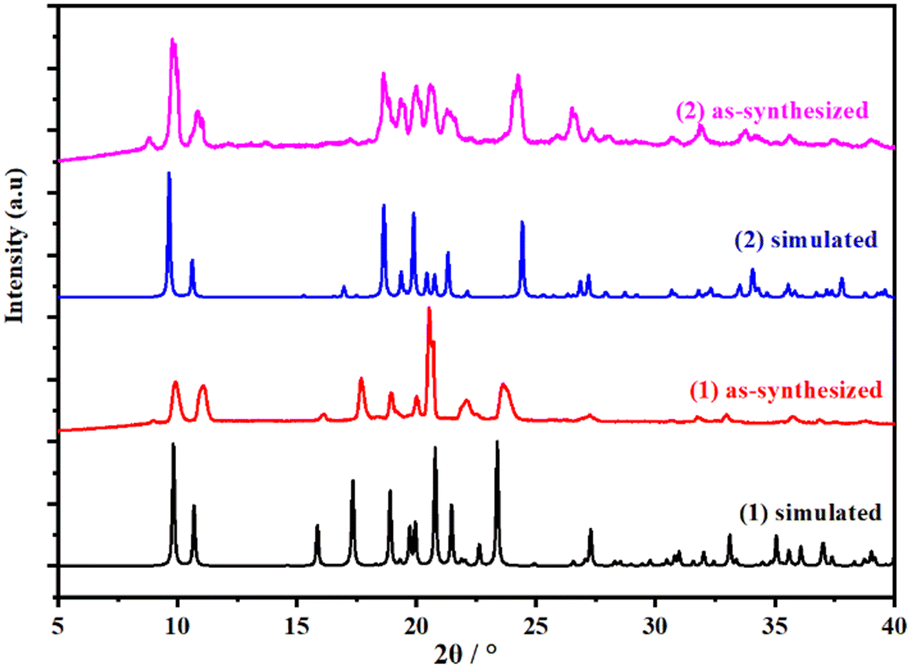 | ||
| Fig. 5 Powder X-ray diffraction patterns of as-synthesized and simulated 1 and 2 from the crystal structure analysis. | ||
Determination of the structural changes occurring during dehydration/rehydration
We followed the structural changes occurring on heating using variable-temperature PXRD (VT-PXRD) (Fig. S12†). Results are shown in full in the ESI† and summarized here in Fig. 6. 1 and 2 show modest shifts in the peak position as the sample is placed under vacuum. A new phase is evident in each case at 50 °C (the midpoint of the solvent loss event on TGA). This phase is retained, and the samples remain crystalline at 350 °C and are unchanged when the samples are cooled to 25 °C.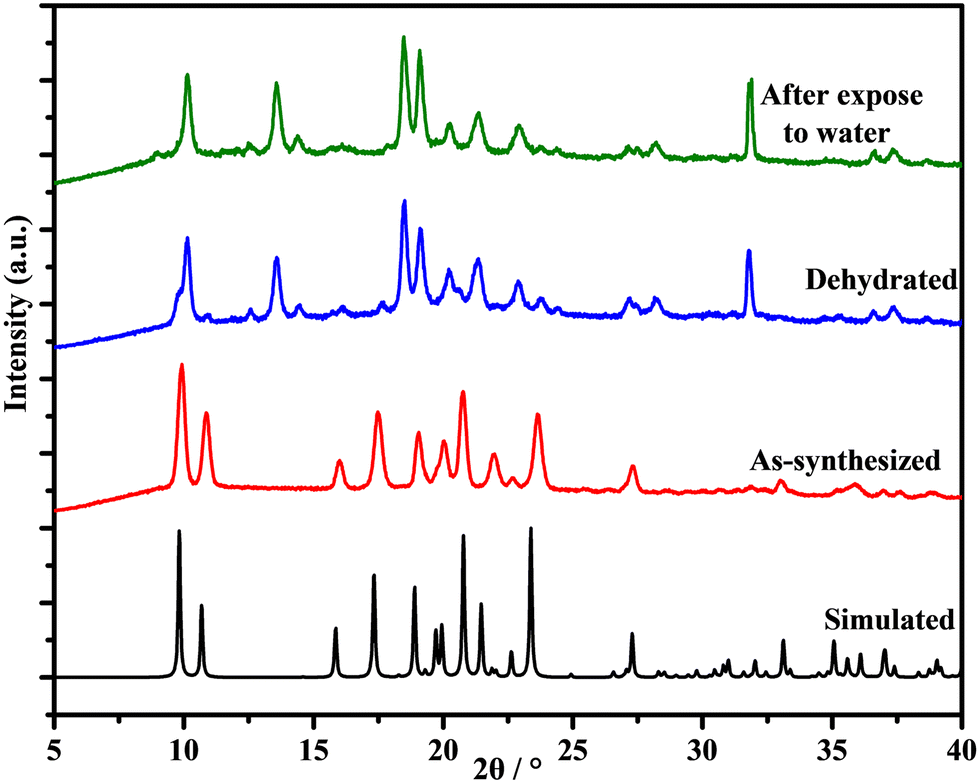 | ||
| Fig. 6 Powder X-ray diffraction patterns of simulated, synthesized, dehydrated, and hydrated 1. | ||
Fig. 6 shows the PXRD of a sample of 1 after heating at 70 °C for 2 hours. TGA (Fig. S15†) confirms that 1 had been dehydrated in this process. Fig. 6 shows that the compound remained highly crystalline upon removal of crystallized water molecules. Although the structure of the dehydrated phase of 1 has changed, as we can see by small differences in the diffraction peaks, it appears from this result that the removal of water does not result in the collapse of the framework. The dehydration PXRD patterns observed are the same with the VTPXRD patterns when 1 is heated at 50 °C in vacuum (Fig. S12†).
The compound [Zn(L)(H2O)2][M(CN)4]·3H2O (L = N,N′-bis(4-pyridylformamide)-1,4-benzene) synthesized by Hu exhibited reversible dehydration/rehydration of water guest molecules after heating at 150 °C under vacuum for 12 h.21 Thus, we also considered possible rehydration of 1 after heating, and exposed the dried powder to water vapour at 40 °C for 24 hours using a solvent diffusion reaction (Fig. S16†). However, PXRD confirmed that there was no phase change (Fig. 6 top) and the TGA curve of compound 1 in Fig. S17† shows that there is no hydration when the dehydrated phase is exposed to air for 24 hours, so we conclude that the dehydration of compound 1 is irreversible, and the removal of water leads to the modification of the structure of this material. Fig. S19 and S20† show that after water vapor adsorption experiments, the phase of compound 1 remains unchanged, while the crystalline phase of compound 2 shows some small shifts in peak positions.
Gas sorption studies on 1 and 2
After dehydration (activation), compounds 1 and 2 were found to show porosity for various gases, as shown in Fig. 7 for nitrogen, hydrogen, and water vapor and Fig. 8 for carbon dioxide. | ||
| Fig. 7 Sorption isotherms of 1 and 2 for (a) nitrogen at 77 K, (b) hydrogen at 77 K, and (c) water vapor at 298 K. | ||
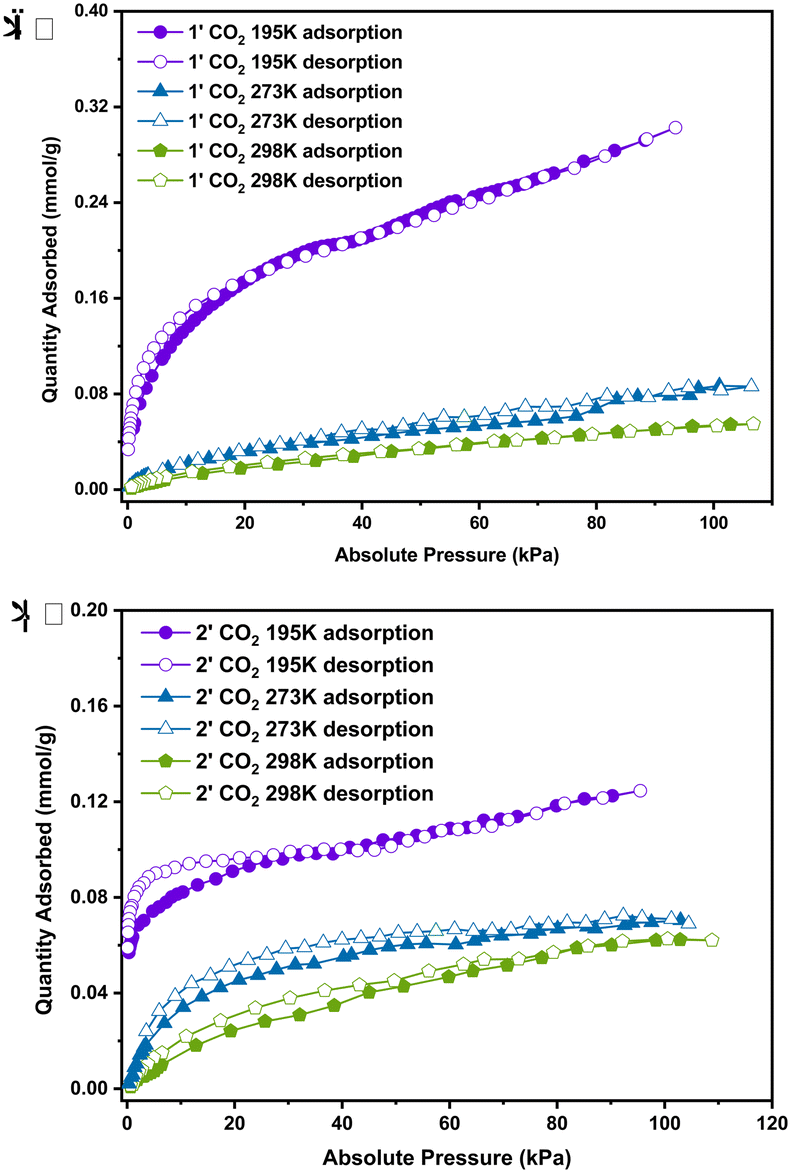 | ||
| Fig. 8 CO2 uptake isotherms at 195 K, 273 K and 298 K of 1 (a) and 2 (b). | ||
Nitrogen sorption isotherms recorded at 77 K (Fig. 7) indicated that 1 and 2 show reversible type-II isotherms.39 These isotherms show a maximum N2 uptake of 2.47 mmol g−1 and 1.17 mmol g−1 for 1 and 2, respectively. 1 has a BET surface area of 28.16 m2 g−1 while 2 has a negligible surface area of 4.74 m2 g−1. These specific surfaces are very low and confirm the flexibility of 1 and 2 which undergo structural deformation after activation as indicated by VT-PXRD.
Water sorption isotherms of 1 and 2 are type II isotherms in the IUPAC classification39 (Fig. 7c). At the start of the water sorption process at 298 K on 1, the water molecules are adsorbed quicker than on 2 at low P/P0 and a capacity of 0.15 mmol g−1 is obtained at P/P0 = 0.1. This quantity adsorbed increases gradually from P/P0 = 0.1 to 0.76 from 0.15 mmol g−1 to 0.95 mmol g−1. For 2, the water adsorption is lower and shows gradual sorption with an uptake capacity of 0.20 mmol g−1.
The sorption of carbon dioxide for 1 and 2 was measured at 195 K, 273 K and 298 K. Fig. 8a and b respectively show that 1 and 2 exhibit reversible type-I isotherms at 195 K, 273 K and 298 K. 1 shows a low capacity for CO2 (0.056 mmol g−1 at 298 K, 0.086 mmol g−1 at 273 K and 0.30 mmol g−1 at 195 K) and 2 also adsorbs little CO2 (0.062 mmol g−1 at 298 K, 0.069 mmol g−1 at 273 K and 0.125 mmol g−1 at 195 K). Interestingly, at 195 K, the compound absorbs more than 3 times the amount of carbon dioxide compared to that at 273 K at low absolute pressures, suggesting that the uptake capacity decreases as a function of temperature as expected.
Sorption isotherms measured at temperatures between 273 and 298 K were used to calculate the isosteric heat of adsorption Qst using the Clausius–Clapeyron equation. The Qst values for 1 and 2 (Fig. S18, ESI†) were measured at loading values ranging from 0.0025–0.054 mmol g−1 (STP). For 1, the Qst started higher at 37.83 kJ mol−1 (with 0.0025 mmol g−1) and decreased to 34.87 kJ mol−1 (with 0.0051 mmol g−1) but increased again at 36.06 kJ mol−1 (with 0.0074 mmol g−1) and finally decreased to 15.61 kJ mol−1 with 0.0479 mmol g−1 indicating weak interactions between CO2 and the activated phase of 1. For the activated phase of 2, it started lower at 28.59 kJ mol−1 (with 0.0025 mmol g−1) and increased to 39.37 kJ mol−1 (with 0.0082 mmol g−1) and finally decreased gradually to 18.94 kJ mol−1 with 0.0535 mmol g−1 indicating also weak interactions between CO2 and this material.
Fe(H2O)2(bpy)[Pt(CN)4]·H2O (bpy = 4,4-bipyridine) adsorbs 3 molecules of water per one iron at 298 K and 1 atm with a type I isotherm22 while 1 and 2 adsorb respectively 0.19 mol and 0.04 mol of water vapor per mol of compound at 298 K and 1 atm. Fe(H2O)2(bpy)[Pt(CN)4]·H2O exhibits a 3D accordion-like MOF structure, while 1 and 2 have 2D structures. This dimensional difference could explain the difference in their water adsorption capacities. The 3D structure provides more adsorption sites and cavities, resulting in a higher adsorption capacity, while the 2D structure limits available sites, resulting in a lower adsorption capacity.
Conclusion
In this article, the synthesis, spectral and thermal properties and crystal structure of new cyano-bridged coordination polymers with the 1,2-bis(4-pyridyl)ethane (bpe) ligand have been investigated. We attempted to rationalize the synthesis of Hofmann coordination polymers by exploring previously reported synthetic conditions. In our study, we found that the length of the ligand does not control the outcome of the synthesis. Despite using a longer linker, we obtained the aqua-derivatives of Hofmann coordination polymers. These were characterized, and their gas sorption properties were studied. Hot-stage microscopy, XRD, FTIR, BET, and TGA analyses were employed to characterize these materials, {[Ni(bpe)(H2O)2][Ni(CN)4]·2H2O}n (1) and {[Cu(bpe)(H2O)2][Ni(CN)4]·ethanol}n (2). IR spectra confirm the presence of all functional groups expected in 1 and 2. Thermal decomposition of the prepared compounds showed that in the initial phase of thermogravimetric analysis, compound 1 lost its guest water molecule while compound 2 releases an ethanol molecule. Powder X-ray diffraction revealed that the experimental powder XRD patterns of these compounds are in accord with the simulated patterns confirming the purity of the synthesized product as a single phase. Single X-ray diffraction revealed that 1 and 2 crystallize in P. In both compounds, one Ni(II) ion is four-coordinated with four cyanide-carbon atoms in a square planar geometry geometry, and the other Cu(II) or Ni(II) ion adopts a distorted octahedral coordination by two bpe ligands, two bridging cyano groups, and two water molecules. The compounds form a 2D sql network linked by hydrogen bonds to adjacent networks to form a 3D supramolecular network. Compounds 1 and 2 have maximum potential void spaces of 12.6% (64.80 Å3) and 25.4% (131.11 Å3), respectively. The presence of disordered terminal cyanide ligands in 1 is attributed to subtle differences in molecular conformation about the bridging cyanides and a larger space available to the terminal cyanide in 1. The gas adsorption of 1 and 2 was studied and it was found that their activated phases have had no pores and BET surface areas of 28 m2 g−1 and 5 m2 g−1 showed low affinity for carbon dioxide or hydrogen. Both showed better affinity for nitrogen and water vapor. 1′ was capable of sorbing nearly four times as much water as 2′, suggesting that the framework is flexible and able to adapt to the presence of gases and water vapor.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds 1 and 2 have been deposited at the CCDC under deposition numbers 2341512–2341513.Author contributions
VBNE: investigation, analysis, writing – original draft; PKT: conceptualisation, supervision, writing – review & editing; TK: project management, supervision; CG: investigation, analysis; NLS: supervision & editing; SAB: conceptualisation, supervision, writing – review & editing.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was carried out with the support of the Organization for Women in Science for the Developing World (OWSD) and the Swedish International Development Cooperation Agency (SIDA). VBNE is grateful to the University of Cape Town (UCT) in South Africa for hosting her during successive research visits. The authors also thank the Cambridge Crystallographic Data Centre (CCDC) for the promotion of structural studies at the University of Dschang (Cameroon) through the FAIRE programme.References
- P. K. Narayan and S. Narayan, Energy Policy, 2018, 38, 661–666 CrossRef.
- Y. Zheng, R. Shan, W. Xu and Y. Qiu, Commun. Earth Environ., 2024, 5, 1–11 CrossRef.
- D. Balsalobre-L, M. Shahbaz, D. Roubaud and S. Farhani, Energy Policy, 2018, 113, 356–367 CrossRef.
- Y. Gao, X. Gao and X. Zhang, Engineering, 2017, 3, 272–278 CrossRef.
- A. Aftab, A. Shariff, S. Garg, B. Lal, M. Shaikh and N. Faiqa, Chem. Eng. Res. Des., 2018, 131, 385–392 CrossRef CAS.
- S. Kasahara, E. Kamio, A. R. Shaikh, T. Matsuki and H. Matsuyama, J. Membr. Sci., 2016, 503, 148–157 CrossRef CAS.
- M. Babar, M. A. Bustam, A. Ali, S. A. Maulud, U. Shafiq, A. Mukhtar, N. S. Shah, K. Maqsood, N. Mellon and A. M. Shariff, Cryogenics, 2019, 102, 85–104 CrossRef CAS.
- T. Ghanbari, F. Abnisa and W. M. A. W. Daud, Sci. Total Environ., 2019, 707, 135090 CrossRef PubMed.
- J. A. Delgrado, M. A. Uguina, J. Sotelo, B. Ruiz and J. Gomez, Adsorption, 2006, 12, 5–18 CrossRef.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- B. F. Abrahams, R. Babarao, A. D. Dharma, J. L. Holmes, T. A. Hudson, H. E. Maynard-Casely, F. McGain, R. Robson and K. F. White, Inorg. Chem., 2020, 59, 8871–8881 CrossRef CAS PubMed.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- J. Yong, J. Chen, Y. Chen, Y. Cai and J. Gao, Microporous Mesoporous Mater., 2022, 344, 112233 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- W. Wong-Ng, J. T. Culp, Y.-S. Chen, P. Zavalij, L. Espinal, D. W. Siderius, A. J. Allen, S. Scheins and C. Matranga, CrystEngComm, 2013, 15, 4684–4693 RSC.
- X. Jiang, B. Tao, X. Yu, Y. Wang and H. Xia, RSC Adv., 2015, 5, 19034–19040 RSC.
- M. Shivanna, K.-i. Otake, J.-J. Zheng, S. Sakaki and S. Kitagawa, Chem. Commun., 2020, 56, 9632–9635 RSC.
- J. T. Culp, M. R. Smith, E. Bittner and B. Bockrath, J. Am. Chem. Soc., 2008, 130, 12427–12434 CrossRef CAS PubMed.
- D. Karaağaç, G. S. Kürkçüoğlu, O. Z. Yeşilel and T. Hökelek, Spectrochim. Acta, Part A, 2014, 121, 196–204 CrossRef PubMed.
- L. Piñeiro-López, F. J. Valverde-Muñoz, M. Seredyuk and K. Znovjyak, IUCrData, 2017, 2, x171413 CrossRef.
- A. Hu, X. Chen, H. Zhou, Y. Chen and A. Yuan, J. Coord. Chem., 2013, 66, 3241–3248 CrossRef CAS.
- T. Haraguchi, K. Otsubo, O. Sakata, S. Kawaguchi, A. Fujiwara and H. Kitagawa, Chem. Commun., 2016, 52, 6017–6020 RSC.
- X. Y. Tian, A. Y. Hu, A. H. Yuan, Q. Chen, D. Yang and F. L. Yang, Z. Anorg. Allg. Chem., 2015, 641, 2340–2343 CrossRef CAS.
- D. Tazaki, T. Akitsu, P. S. P. Silva and V. H. N. Rodrigues, Polyhedron, 2015, 102, 297–307 CrossRef CAS.
- B. A. Maynard, P. A. Smith, L. Ladner, A. Jaleel, N. Beedoe, C. Crawford, Z. Assefa and R. E. Sykora, Inorg. Chem., 2009, 48, 6425–6435 CrossRef CAS PubMed.
- B. A. Maynard, P. A. Smith, A. Jaleel, L. Ladner and R. E. Sykora, J. Chem. Crystallogr., 2010, 40, 616–623 CrossRef CAS.
- M. Stojanovic, N. J. Robinson, X. Chen and R. E. Sykora, Inorg. Chim. Acta, 2011, 370, 513–518 CrossRef CAS.
- N. Vannerberg, Acta Chem. Scand., 1964, 18, 2385 CrossRef CAS.
- E. Sayın, G. S. Kürkçüoğlu, O. Z. Yeşilel and M. Taş, J. Coord. Chem., 2016, 69, 1226–1235 CrossRef.
- F. Çetinkaya, G. S. Kürkçüoğlu, O. Z. Yeşilel, T. Hökelek and H. Dal, Polyhedron, 2012, 47, 126–133 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens and M. Towler, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- SAINT-Plus, Madison, Wisconsin, USA, 2004 Search PubMed.
- XPREP, Madison, Wisconsin, USA, 2003 Search PubMed.
- G. M. Sheldrick, SADABS, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- J. T. Culp, C. Madden, K. Kauffman, F. Shi and C. Matranga, Inorg. Chem., 2013, 52, 4205–4216 CrossRef CAS PubMed.
- IUPAC, International Union of Pure and Applied Chemistry, Pure Appl. Chem., 1985, 57, 603–619 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure for K2[Ni(CN)4]·H2O, spectroscopic and crystallographic characterisation, thermal analysis, and details of gas sorption analysis. Crystal structures 1 and 2 data have been deposited at the Cambridge Crystallographic Data Centre with CCDC deposition numbers 2341512–2341513, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00459k |
| This journal is © The Royal Society of Chemistry 2024 |