
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural diversity of supramolecular networks formed between polycyanometalates and sulfur-based chalcogen bond donors†
Tim-Niclas
Streit
a,
Jan
Langwald
ab,
Rosa M.
Gomila
 c,
Antonio
Frontera
*c and
Moritz
Malischewski
*a
c,
Antonio
Frontera
*c and
Moritz
Malischewski
*a
aInstitut für Anorganische Chemie, Freie Universität Berlin, Fabeckstr. 34/36, D-14195 Berlin, Germany. E-mail: moritz.malischewski@fu-berlin.de
bInstitut für Anorganische Chemie, Universität zu Köln, Greinstraße 6, 50939 Köln, Germany
cUniversitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, SPAIN. E-mail: toni.frontera@uib.es
First published on 11th June 2024
Abstract
The co-crystallization of S-(CF3)-dibenzothiophenium (“Umemoto” – abbreviated Ume) cations and polycyanometalates with different coordination geometries yields a large structural variety of supramolecular assemblies. In the case of [Ume+]2[Ni(CN)4]2−·CH2Cl2 all four cyanide groups of the square-planar dianion act as chalcogen bond acceptors whereas the sulfur atoms of the Umemoto cations act as biaxial chalcogen bond donors. For tetrahedral [Zn(CN)4]2− a more complex assembly of four Umemoto cations and two dianions is observed. In the case of [Ume+]3[Fe(CN)6]3−·CH2Cl2 one Umemoto cation even acts as a triaxial chalcogen bond donor whereas only 4 out of the 6 cyanide groups act as chalcogen bond acceptors. For [Ume+]3[W(CN)8]3− a polymeric chain structure is observed in which all Umemoto cations act as biaxial chalcogen bond donors. DFT investigations focus on the physical nature of the chalcogen bonds, encompassing energetic assessments, MEP surface plots, QTAIM, and NBO analysis, shedding light on the nature of chalcogen donor–acceptor interactions. The energetic results evidence that total contributions of chalcogen bonds in different synthons range from −7.6 to −16.3 kcal mol−1, surpassing that of hydrogen bonds that range from −4.9 to −11.8 kcal mol−1.
Introduction
The concept of chalcogen bonding (ChB)1 describes an interaction between an electrophilic chalcogen atom and an electron rich Lewis base. Several decades ago, this interaction was first observed in sulfur2 and selenium3 compounds, featuring unusually short intermolecular contacts below the sum of van der Waals radii, sparking unexpected interest.4 Nowadays, extensive knowledge of related non-covalent interactions such as hydrogen-bonding (HB) and halogen-bonding (HaB) helps to categorize and understand the now called chalcogen bond.5 Its relevance is illustrated by applications in materials science,6 molecular sensors,7 host–guest interactions,8 as well as in synthesis9 and catalysis.10 Similar to HaB in many aspects such as polarization, electrostatic effects and charge transfer contributions, a key difference lies in the capability of binding multiple substituents to the chalcogen donor altering the nature of the ChB. The larger resulting σ-hole11 gives way to more bonding options. As these are less directional than halogen bonds this can lead to less predictable supramolecular networks.Suitable ChB donors usually display two to three covalent bonds at the chalcogen atom, as regions of electrostatic potential12 form opposite of the covalent bonds.13 Umemoto type dibenzothiophenium reagents are excellent candidates fulfilling these prerequisites. Initially developed in the 1990s as electrophilic trifluoromethylation reagents,14,15 they have become popular reagents16,17 since they are accessible with various substitution patterns and chalcogen atoms.18 In contrast to its known reactivity, its ChB donor properties have only been reported very recently by us.19 Purely organic ChB is extensively reported in the literature,20,21 whereas reports of inorganic bonding partners are so far underrepresented.9,22 Polycyanometalates are attractive building blocks for complex supramolecular structures as they are highly charged anions with exceptional nucleophilic properties and variable topologies.23 They have successfully been implemented to achieve three dimensional arrays using hydrogen-bonding24 or halogen-bonding,25,26 with different organic electrophiles.
Having recently established sulfur based Umemoto type reagents as efficient chalcogen bond donors towards the cyanide ligand of [Mo(CO)5CN]−,19 we were interested in the potential complex supramolecular assemblies that could be achieved in combination with polycyanometalates with different coordination geometries. For this study, we are focussing on multiple-charged [Ni(CN)4]2−, [Zn(CN)4]2−, [Fe(CN)6]3− and [W(CN)8]3− anions as square planar, tetrahedral, octahedral and square anti-prismatic/triangular dodecahedral building blocks, respectively. For linear [Au(CN)2]− no cocrystallization could be observed, merely starting material was recovered (Scheme 1). In general, [Ume]+[CF3SO3]− was reacted in a salt metathesis in CH2Cl2 with polycyanometalate salts containing large lipophilic cations which led to the corresponding [Ume+]z[M(CN)x]z− adduct. In the solid state, these salts exhibited multiple supramolecular synthons (I–V) where chalcogen bonding (ChB) and hydrogen bonding cooperatively influenced the assembly processes (see Scheme 1, bottom). Specifically, the σ-holes opposite to the aromatic rings and aromatic H-atoms facilitated the supramolecular binding. Notably, for the salt of the octahedral [Fe(CN)6]3− anion, all three sigma-holes at the sulfur atom of the Umemoto cation (Ume+) were involved in forming synthon-III, a phenomenon that, to our knowledge, has not been previously reported for inorganic donors. In the case of [Zn(CN)4]2−, ChB and anion–π interactions cooperatively formed synthon-IV. Additionally, with tungsten (M = W) as the central metal, synthon-V was observed, where the anion connects two sulfonium cations through two distinct cyano ligands. To study these diverse synthons, we utilized DFT energy calculations, supplemented by quantum theory of atoms in molecules (QTAIM) and natural bond orbital (NBO) analyses, which helped elucidate the solid-state architecture of the Ume+ salts. Our primary objective is to elucidate the physical nature and energetic characteristics of the chalcogen bonds (ChBs) in all synthons, rather than analyzing all interactions present in the X-ray structures of the Umemoto salts.
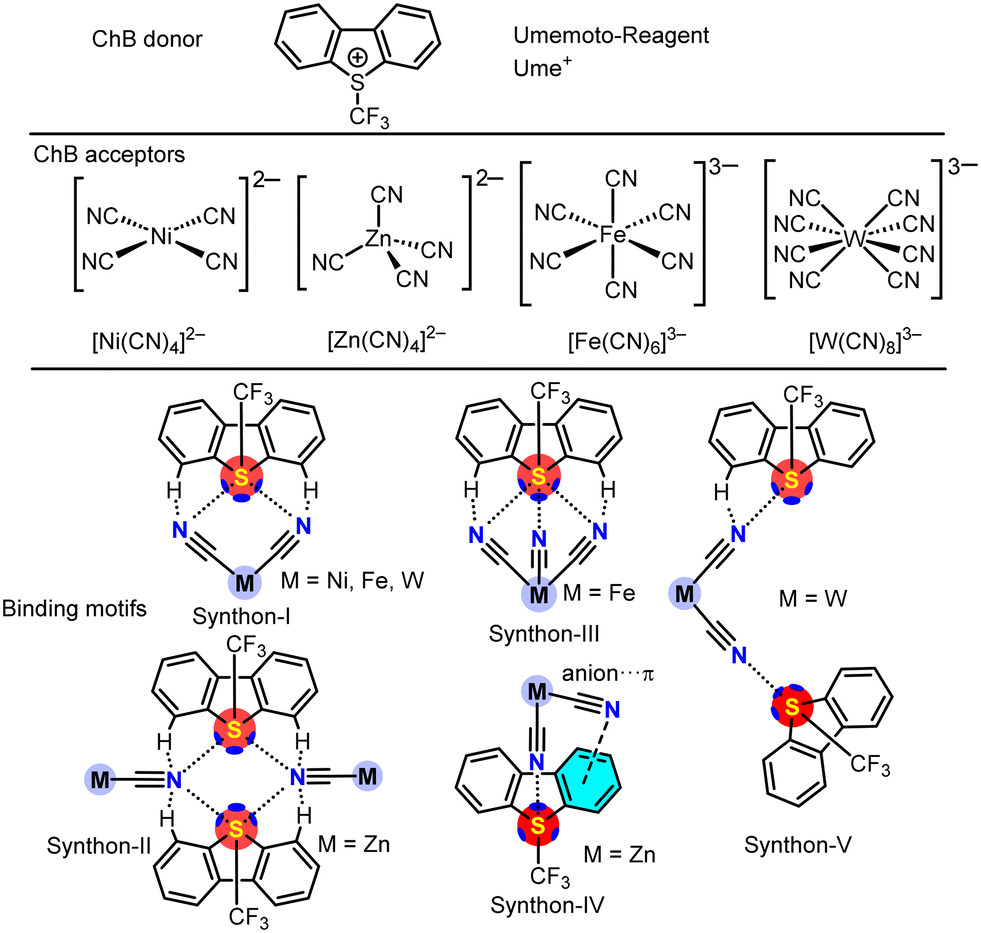 | ||
| Scheme 1 List of chalcogen bond donors, acceptors and binding motifs studied in this work. | ||
Results and discussion
Crystallizing in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) [Ume+]2[Ni(CN)4]2−·CH2Cl2 shows a sulfur atom acting as a chalcogen donor to two neighbouring cyanide groups from the [Ni(CN)4]2− building block (Fig. 1). A set of two different chalcogen bonds between S⋯N can be identified with 3.010(1) Å as the shorter contact and 3.249(2) Å as the longer contact (synthon-I). These values place below the combined van der Waals radii of 3.35 Å of sulfur and nitrogen.4 Bonding solely takes place in the horizontal plane to the aryl ring plain and differs slightly from linearity in both cases (C7–S1–N2: 166.56(75)° and C1–S1–N1: 168.72(80)°). The resulting C
[Ume+]2[Ni(CN)4]2−·CH2Cl2 shows a sulfur atom acting as a chalcogen donor to two neighbouring cyanide groups from the [Ni(CN)4]2− building block (Fig. 1). A set of two different chalcogen bonds between S⋯N can be identified with 3.010(1) Å as the shorter contact and 3.249(2) Å as the longer contact (synthon-I). These values place below the combined van der Waals radii of 3.35 Å of sulfur and nitrogen.4 Bonding solely takes place in the horizontal plane to the aryl ring plain and differs slightly from linearity in both cases (C7–S1–N2: 166.56(75)° and C1–S1–N1: 168.72(80)°). The resulting C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯S angles are close to 90° with slight deviations of 92.920(15)° for the longer contact and 98.42(15)° for the shorter one. Each S⋯N interaction is supported by further hydrogen contacts ranging from 2.494(1) Å and 2.649(1) Å to either dibenzothiophenium moieties or solvent molecules.
N⋯S angles are close to 90° with slight deviations of 92.920(15)° for the longer contact and 98.42(15)° for the shorter one. Each S⋯N interaction is supported by further hydrogen contacts ranging from 2.494(1) Å and 2.649(1) Å to either dibenzothiophenium moieties or solvent molecules.
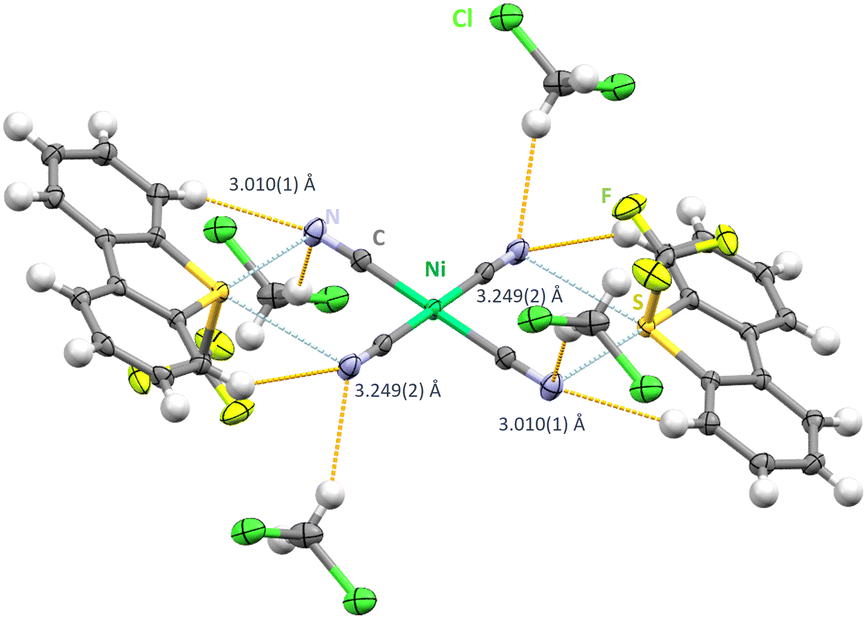 | ||
| Fig. 1 Chalcogen bonding in the crystal structure [Ume+]2[Ni(CN)4]2−·CH2Cl2. Thermal ellipsoids at the 50% probability level. Chalcogen bonds in light blue and hydrogen bonds in orange. | ||
In order to investigate a tetrahedral polycyanometalate, [PPh4+]2[Zn(CN)4]2− was reacted in a salt metathesis with [Ume]+[CF3SO3]−. The reaction product yielded [Ume+]2[Zn(CN)4]2−·H2O·CH2Cl2 as single crystals in the P space group. The crystal structure reveals that two of the four cyanide groups are involved in ChB (Fig. 2). N1 and N2 form exclusively hydrogen bonds with water, (1.986(2) Å and 2.003(5) Å), CH2Cl2 (2.484(3) Å) and backbone hydrogens of dibenzothiophenium moieties (2.553(3) Å see ESI†). Bifurcated cyanide groups display N⋯S contacts of 3.085(2) Å and 3.215(2) Å to the ChB donor atoms, slightly varying in distance to the contacts of the previously discussed square planar structure. A four membered S2N2 ring results which displays bonding angles of S2–N4–S2: 109.16(12)° and N4–S2–N4: 70.84(14)°.
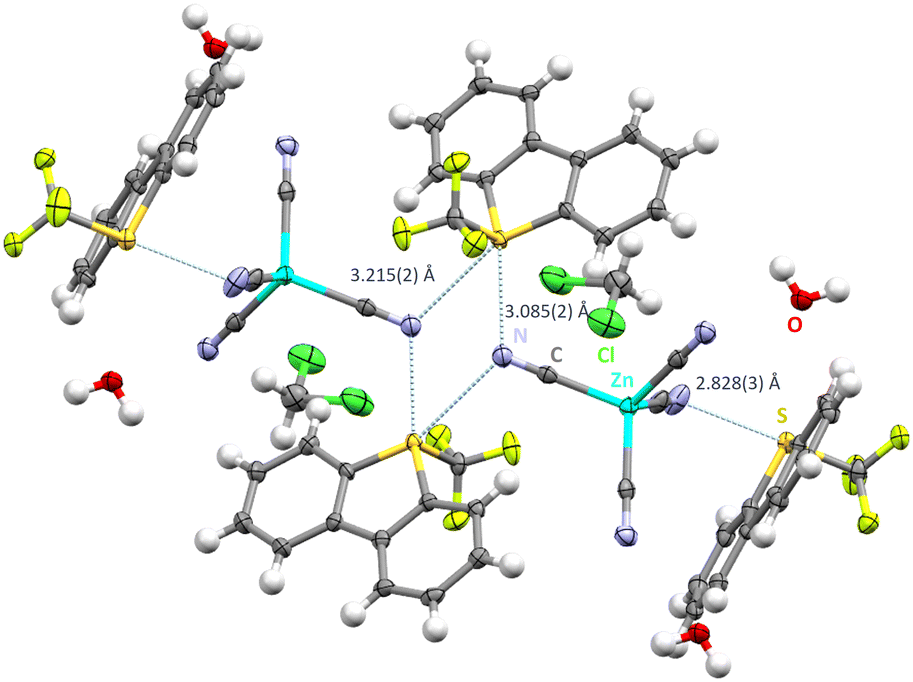 | ||
| Fig. 2 Intermolecular S⋯N contacts in the crystal structure of [Ume+]2·[Zn(CN)4]2−·H2O·CH2Cl2. Thermal ellipsoids at the 50% probability level. Chalcogen bonds in light blue. Hydrogen bonds omitted for clarity. Monomeric unit displaying hydrogen bonds in Fig. S1 in the ESI.† | ||
The formation of supramolecular S2N2 rings (synthon-II) was already observed in our previous study using [Mo(CO)5CN]− as chalcogen bond acceptor.19 Additionally, a significantly shorter N⋯S contact of 2.828(3) Å is also observed, binding in an almost vertical fashion (N3–S1–C5: 175.46(24)°) (synthon-IV).
Interested in higher coordination arrays, [NBu4+]3[Fe(CN)6]3− was reacted in a salt metathesis with [Ume]+[CF3SO3]− in CH2Cl2, to yield crystals of [Ume+]3[Fe(CN)6]3−·CH2Cl2 in the triclinic P1 space group (Fig. 3). Interestingly, two sulfur atoms of the Ume+ cations are monoaxial whereas S3 is a three-axial ChB donor (synthon-III). The shortest N⋯S contact of 2.957(2) Å is observed between S3 and a monofurcated cyanide group (Fig. 3). This bond is horizontal to the S–CF3 group and displays close to linear C–S⋯N angles (C9–S2–N5: 173.90(11)°) which is in line with our previous investigations.19 For the bifurcated cyanide group, two N⋯S contacts of 3.003(2) Å and 3.139(2) are observed in highly directional ChBs (C10–S3–N4: 172.14(12)° and C8–S1–N4: 173.24(11)°). S3 as a three-axial chalcogen bond donor features elongated S⋯N contacts in the range of 3.139(2)–3.212(2) Å. Interestingly, all CN⋯S angles are relatively close to 90° (range of 79.19(29)–86.70(29)°). S⋯N contacts in linear extension of the S–CF3 bond axis are also observed (C10–S3–N4: 172.11(10)°) whereas the other acceptor groups display contacts in the Caryl plain (C35–S3–N1: 172.15(12)° and C37–S3–N2: 166.83(11)°). Additional hydrogen contacts between 2.408(2) Å and 2.710(3) Å support the structural motif.
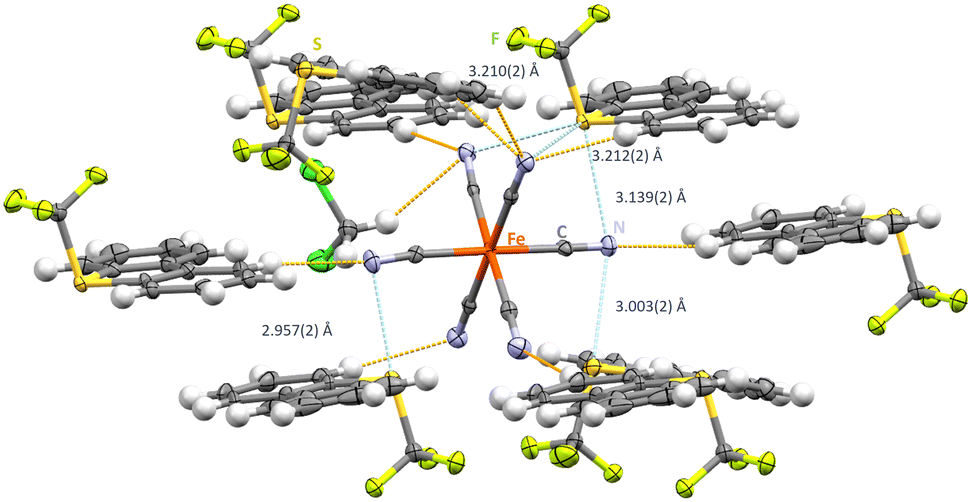 | ||
| Fig. 3 Intermolecular S⋯N contacts in the crystal structure of [Ume+]3[Fe(CN)6]3−·CH2Cl2. Thermal ellipsoids at the 50% probability level. Chalcogen bonds in light blue and hydrogen bonds in orange. | ||
Salt metathesis of [Ume]+[CF3SO3]− and [NBu4+]3[W(CN)8]3− in CH2Cl2 resulted in single crystals of [Ume+]3[W(CN)8]3− (space group C2/c). Interestingly, only 6 out of 8 cyanide ligands are chalcogen bond acceptors, all of which are monofurcated (Fig. 4). Nonetheless each cyanide group is involved in hydrogen contacts ranging from 2.408(2)–2.744(8) Å. In contrast to the previous compounds with discrete structural entities, a polymeric chain structure with approximately right angles is observed since the Umemoto cation featuring S1 acts as a biaxial chalcogen bond donor towards cyanide groups of two different [W(CN)8]3− moieties (N1–S1–N4: 91.836(93)°). The Umemoto cations containing S2 and S3 act also as biaxial chalcogen bond donors but here to two neighbouring cyanide ligands from the same [W(CN)8]3− anion. These N⋯S contacts display distances from 2.986(3)–3.034(3) Å and are all arranged to be in the plain of the Caryl backbone of their respective [Ume]+ cation (N5–S2–C35 – 167.62(12)° and N8–S3–C36 – 165.24(12)° (Fig. 4)). For S1, S⋯N contacts shorten to 2.836(3) Å and 2.859(3) Å, respectively. In contrast to the previous structures a very broad range of CN⋯S angles is observed. Four angles are in the range of 103.92(23)–105.59(23)° whereas also angles of 142.72(27)° (C4–N4–S1) and 165.74(30)° (C1–N1–S1) can be found.
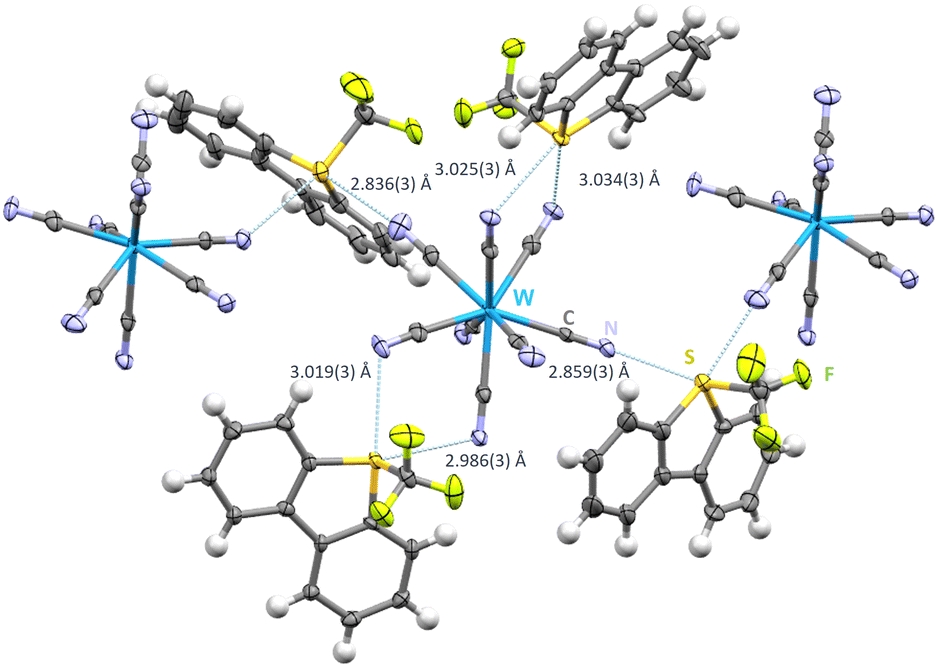 | ||
| Fig. 4 Intermolecular S⋯N contacts in the crystal structure of [Ume+]3[W(CN)8]3−. Thermal ellipsoids at the 50% probability level. Chalcogen bonds in light blue. Hydrogen bonds omitted for clarity. The monomeric unit with hydrogen bonds can be found in Fig. S2 in the ESI.† | ||
To elucidate the strong chalcogen bond (ChB) donor capabilities of the Ume+ cation, the molecular electrostatic potential (MEP) surface was calculated. It was found that the sulfonium cation displays three σ-holes associated with the three C–S bonds, consistent with previous findings. The highest MEP values, located at the σ-holes opposite the aromatic C-atoms (118.2 kcal mol−1), are likely enhanced by the adjacent H-atoms of the aromatic rings. The σ-hole adjacent to the CF3 group exhibits a slightly lower value (111.1 kcal mol−1), which aligns with prior observations and is rationalized by hyperconjugation effects. Additionally, the MEP surface shows another electrophilic region (109.8 kcal mol−1) at the carbon atom of the CF3 group, opposite one C–F bond, indicating the presence of a σ-hole at this carbon atom (Fig. 5).
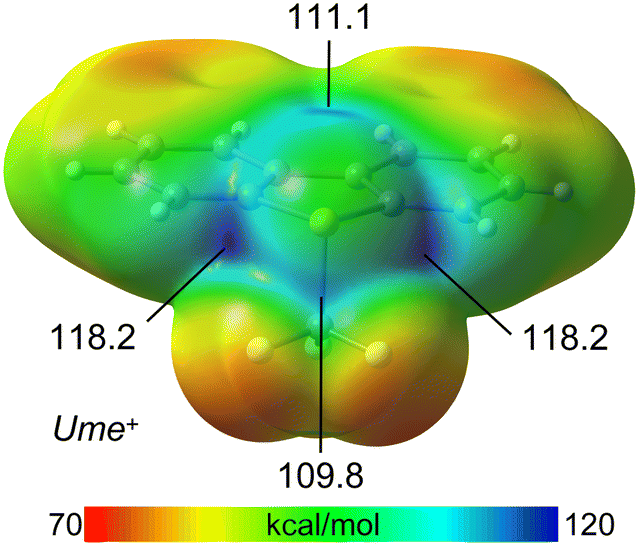 | ||
| Fig. 5 MEP surface of Ume+. The values at the σ-holes are indicated in kcal mol−1. Isovalue 0.001 a.u. | ||
Chalcogen bonds (ChBs) and hydrogen bonds (HBs) in synthons I–IV were investigated using DFT calculations and the quantum theory of atoms-in-molecules (QTAIM). Fig. 6 presents the QTAIM analysis of the [Ume+]2[Ni(CN)4]2− trimer, highlighting the formation of four ChBs and four HBs, each characterized by bond critical points (BCPs) and bond paths depicted as orange lines. BCPs for HBs are shown as blue spheres, while those for ChBs are red, facilitating clear differentiation. The assembly exhibits a substantial formation energy of −237.4 kcal mol−1, attributed to the strong coulombic attraction between one dianion and two cations, corresponding to two instances of synthon-I. To evaluate the strengths of ChB and HB independently of coulombic forces, interaction energies were calculated using QTAIM parameters at the BCPs, as outlined in the Theoretical Methods section (see ESI†). This analysis confirmed that ChBs dominate, contributing −7.6 kcal mol−1, compared to −5.7 kcal mol−1 from HBs. Additionally, it is notable that the positioning of the CN groups in the assembly aligns with the locations of the maximum electrostatic potential (σ-holes) on the MEP surface of Ume+.
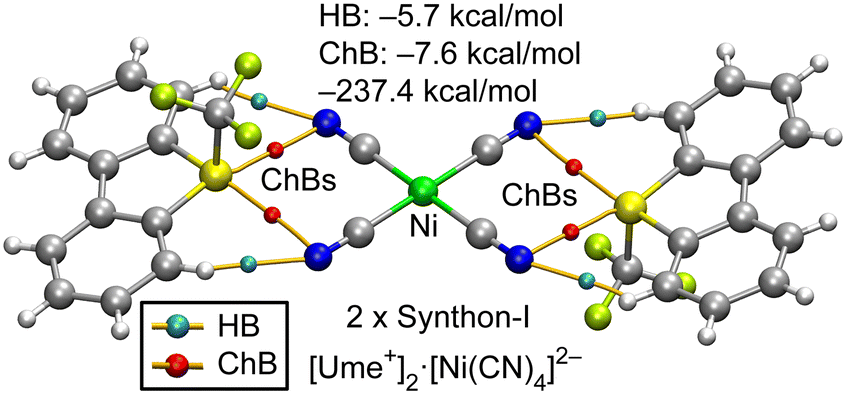 | ||
| Fig. 6 QTAIM analysis for the trimeric assembly of the [Ume+]2[Ni(CN)4]2− salt. Only intermolecular HB and ChBs are represented for clarity. The energies of the assemblies are also indicated. The contributions of HB and ChB interactions derived from the QTAIM parameters are indicated. | ||
The analysis of the [Ume+]2·[Zn(CN)4]2− structure, as depicted in Fig. 7, highlights the formation of synthon-II (S2N2 supramolecular ring, depicted in the middle of Fig. 7) and two symmetrically equivalent instances of synthon-IV. Additionally, Fig. 7 includes BCPs that characterize HBs (blue spheres), ChBs (red spheres) and several BCPs (magenta spheres) linking the anion to the aromatic rings, verifying the presence of anion–π (A–π) interactions.
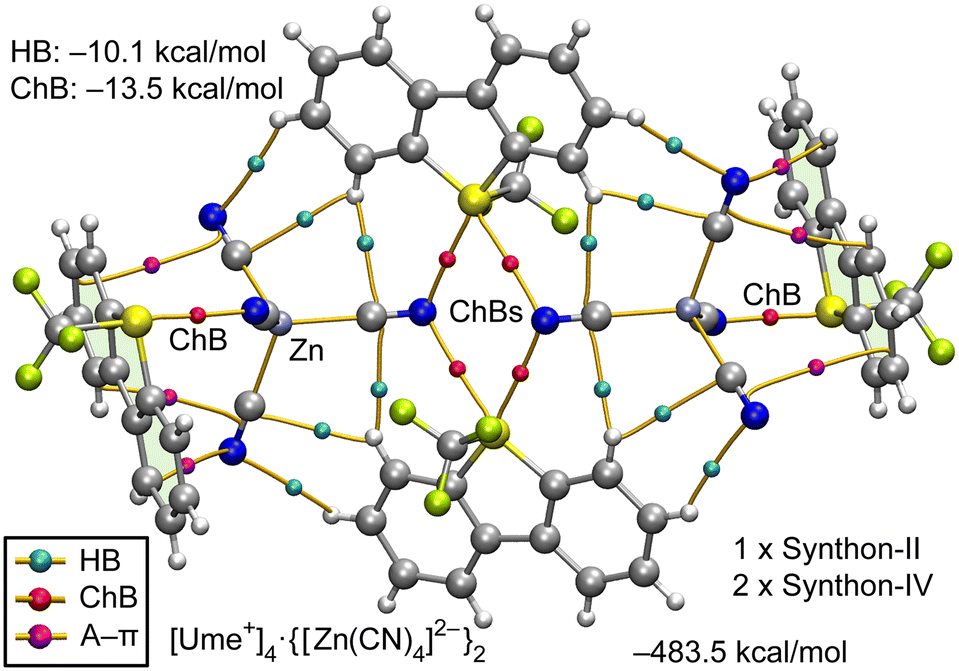 | ||
| Fig. 7 QTAIM analysis for the hexameric assembly of the [Ume+]2[Zn(CN)4]2− salt. Only intermolecular HB, ChBs and A–πs are represented for clarity. The energies of the assemblies are also indicated. The contributions of HB and ChB interactions derived from the QTAIM parameters are indicated. | ||
The hexameric assembly exhibits a substantial stabilization energy of −483.5 kcal mol−1, attributable to its ion-pair nature. The QTAIM energy prediction shows that ChBs exert a more significant influence, with an energy of −13.5 kcal mol−1, compared to the HBs, which have an energy of −10.1 kcal mol−1.
As mentioned earlier, the formation of synthon-III, where all three σ-holes concurrently participate in the interaction of Ume+ with the [Fe(CN)6]3− anion, is depicted in Fig. 8. This figure illustrates a single anion engaging with three adjacent cations through the synthon-III binding mode. It is observed that the sulfur atom (S) of Ume+ is linked to three different cyanide ligands via three BCPs (red spheres) and corresponding bond paths. In this assembly, ChBs are predominant, with an interaction energy of −16.3 kcal mol−1, overshadowing the role of HBs, which contribute only −4.9 kcal mol−1. The formation energy of this pentameric assembly totals −472.6 kcal mol−1, suggesting that each synthon-III contributes approximately −157.5 kcal mol−1 (−472.6 divided by 3). This is in comparison to the energy associated with synthon-I, deduced from the assembly of the Ni salt shown in Fig. 6, which is −118.7 kcal mol−1 (−237.4 divided by 2). Thus, synthon-III is more energetically favourable, likely due to the additional chalcogen bond opposite to the S–CF3 bond.
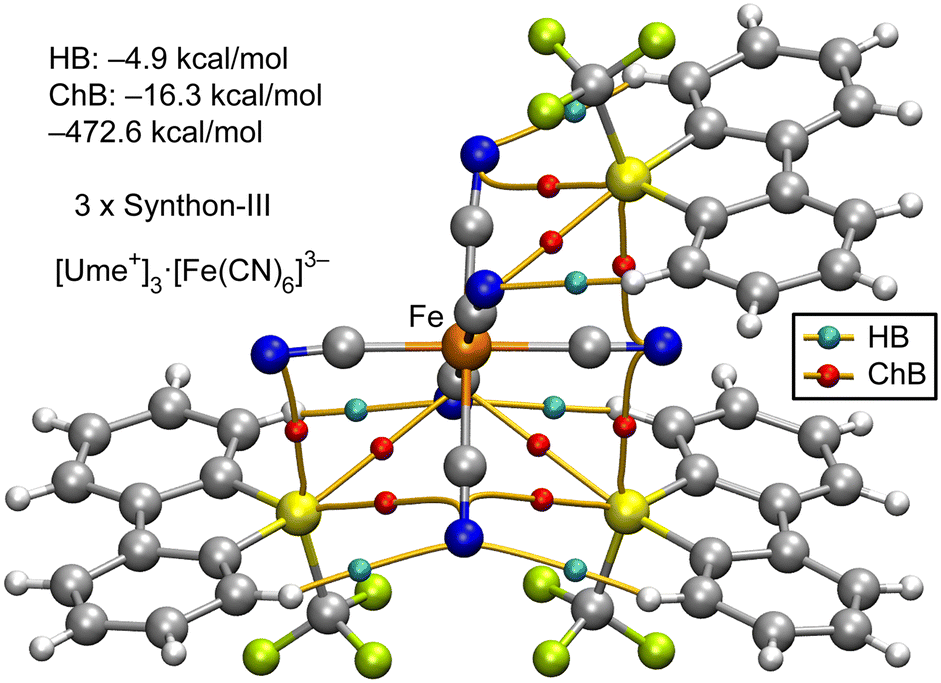 | ||
| Fig. 8 QTAIM analysis for the tetrameric assembly of the [Ume+]3[Fe(CN)6]3− salt. Only intermolecular HB and ChBs are represented for clarity. The energies of the assemblies are also indicated. The contributions of HB and ChB interactions derived from the QTAIM parameters are indicated. | ||
Fig. 9 presents the QTAIM and energetic analysis of a pentameric assembly centered around a [W(CN)8]3− unit with four surrounding Ume+ cations. Two of these cations interact with the anion via synthon-I, demonstrating its predominance throughout the series of salts examined in this study. The remaining two cations engage with the anion through synthon-V, in which each Ume+ unit establishes a single chalcogen bond (ChB); one utilizes the σ-hole facing the C–CF3 bond, and the other utilizes the σ-hole opposite the S–CAr bond. Additionally, the QTAIM analysis reveals extra CH⋯NC interactions with cyano groups not involved in ChBs, which provide further stabilization to synthon-V (marked in Fig. 9 as magenta asterisks). The formation energy of this assembly is −467.1 kcal mol−1, which is comparable to the energies observed in the hexameric and pentameric assemblies of the Zn and Fe salts, respectively. Consistent with previous observations, the ChB interactions play a more significant role than the HBs in stabilizing the assembly.
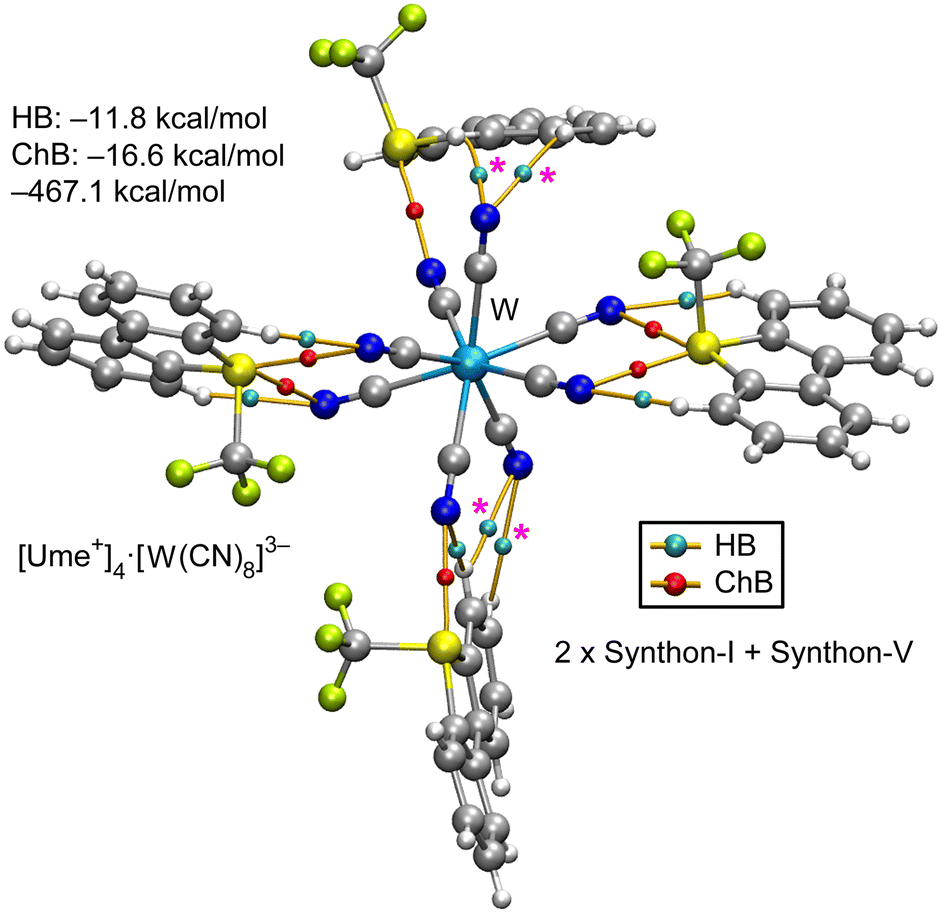 | ||
| Fig. 9 QTAIM analysis for the pentameric assembly of [W(CN)8]3− surrounded by four [Ume+] cations as a structural model for the polymeric structure of [Ume+]3[W(CN)8]3−. Only intermolecular HB and ChBs are represented for clarity. The energies of the assemblies are also indicated. The contributions of HB and ChB interactions derived from the QTAIM parameters are indicated. CH⋯NC interactions with cyano groups not involved in ChBs, are marked using magenta asterisks. | ||
We conducted an analysis of orbital charge transfer effects across representative assemblies of all synthons using the natural bond orbital (NBO) method. Fig. 10 depicts the relevant orbitals in these assemblies along with their associated stabilization energies. To illustrate charge transfer effects in synthon-I, we examined the [Ume+]2[Ni(CN)4]2− salt, identifying two key donor–acceptor interactions: from the lone pair (LP) on the N-atom of the cyanide group to the antibonding σ*(S–C) orbital, and from one of the π-orbitals of the CN triple bond to the same antibonding σ*(S–C) bond. For synthon-I, the π(CN) → σ*(S–C) charge transfer is more prominent; these orbitals are depicted in Fig. 10a. The corresponding second-order stabilization energies, indicated in Fig. 10a, are 1.05 kcal mol−1 for LP(N) → σ*(S–C) and 1.67 kcal mol−1 for π(CN) → σ*(S–C). These values, when compared to the total ChB contribution of −7.6 kcal mol−1 (see Fig. 6), suggest that orbital effects are substantial. Fig. 10b illustrates the NBOs involved in the assembly of [Ume+]2[Zn(CN)4]2− salt, which represent elements of both synthon-II and synthon-IV. Here, the stabilization energies for the LP(N) → σ*(S–C) charge transfer exceed those for the π(CN) → σ*(S–C) charge transfer, marking a distinction from synthon-I. Notably, the stabilization energies are higher for synthon-IV, correlating with the experimentally observed shorter S⋯N distances, indicative of better orbital overlap.
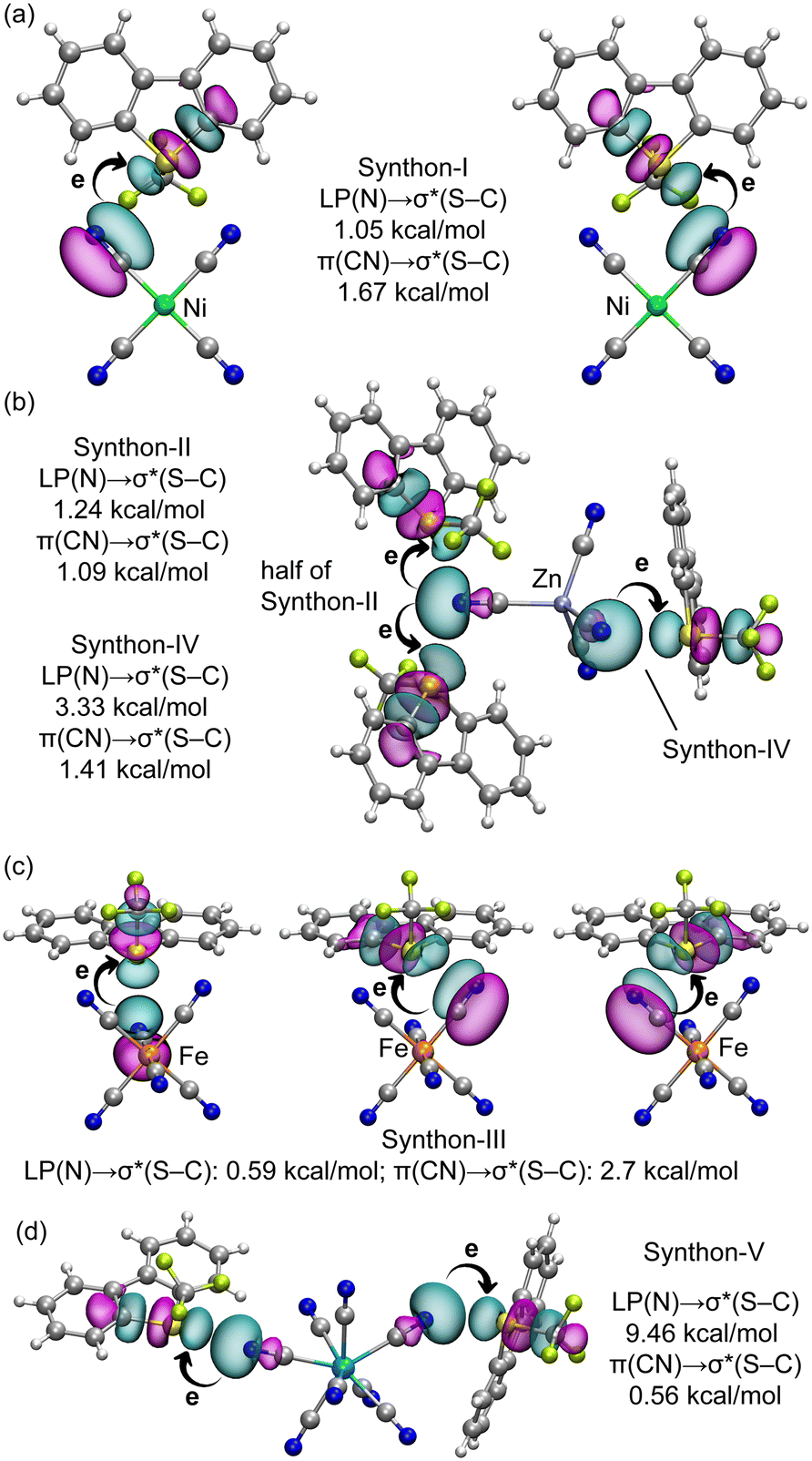 | ||
| Fig. 10 Plot of the NBOs involved in the LP(N) → σ*(S–C) and π(CN) → σ*(S–C) for synthon-I (a), synthon II and IV (b), synthon-III (c) and synthon-V (d) reported in this work. The second order perturbation energies are given in kcal mol−1. The charge transfer is illustrated using curved arrows. | ||
The NBO analysis for synthon-III, shown in Fig. 10c, reveals that the π(CN) → σ*(S–C) charge transfer (2.70 kcal mol−1) predominates over the LP(N) → σ*(S–C) transfer (0.59 kcal mol−1), aligning with the trends observed in synthon-I. Finally, for synthon-V, the LP(N) → σ*(S–C) transfer is notably significant, contributing 9.46 kcal mol−1, which clearly predominates over the π(CN) → σ*(S–C) transfer, measured at 0.56 kcal mol−1. The magnitude of charge transfers across synthons I to III ranges from 0.5 to 1.67 kcal mol−1. For synthon-IV and V, the LP(N) → σ*(S–C) charge transfer proves more critical, likely influenced by shorter the S⋯N distance, directionality and the participation of the σ-hole opposite to the S–CF3 group in both synthons.
Conclusions
In conclusion, this study has provided detailed insights into the structural diversity and stability of supramolecular networks formed through the interaction of sulfur-based Umemoto cations with various polycyanometalates. Our findings underscore the pivotal role of chalcogen bonds (ChBs) in driving the assembly of these complexes, often surpassing the influence of hydrogen bonds in terms of energetic contribution and structural stability. Notably, the synthon-III structure, where the three σ-holes in sulfur concurrently participate in ChBs with the [Fe(CN)6]3− anion, is a significant highlight. The integration of QTAIM and NBO analyses further elucidated the electronic factors contributing to the stability and specificity of the interactions, highlighting the cooperativity of chalcogen and hydrogen bonds and also significant charge transfer effects that play a crucial role in the stabilization of these assemblies. The ability of these assemblies to form diverse structural motifs with significant energetic favorability opens potential pathways for designing novel materials with tailored properties. As we advance our understanding of these complex interactions, it becomes increasingly clear that the subtle interplay of molecular forces and electronic effects can be harnessed to create sophisticated molecular architectures. This study not only expands the repertoire of known chalcogen bond donors and acceptors but also sets the stage for further exploration of these interactions in other chemical environments.Data availability statement
Experimental and computational details as well as a summary of the most important crystallographic parameters have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by DFG (project number MA 7817/3-1). The authors acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. AF and RGM acknowledge the financial support by the MICIU/AEI of Spain (project PID2020-115637GB-I00, FEDER funds).References
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- R. E. Rosenfield, R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860–4862 CrossRef CAS.
- R. Laitinen, R. Steudel and R. Weiss, J. Chem. Soc., Dalton Trans., 1986, 1095–1100 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- P. C. Ho, J. Z. Wang, F. Meloni and I. Vargas-Baca, Coord. Chem. Rev., 2020, 422, 213464 CrossRef CAS.
- R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098–7125 RSC.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed.
- K. T. Mahmudov, A. V. Gurbanov, V. A. Aliyeva, M. F. C. Da Guedes Silva, G. Resnati and A. J. Pombeiro, Coord. Chem. Rev., 2022, 464, 214556 CrossRef CAS.
- J. Bamberger, F. Ostler and O. G. Mancheño, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- P. Politzer and J. Murray, Crystals, 2017, 7, 212 CrossRef.
- B. Galmés, A. Juan-Bals, A. Frontera and G. Resnati, Chem. – Eur. J., 2020, 26, 4599–4606 CrossRef PubMed.
- T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS.
- N. Shibata, A. Matsnev and D. Cahard, J. Org. Chem., 2010, 6, 65 Search PubMed.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS PubMed.
- T. Koike and M. Akita, J. Fluorine Chem., 2014, 167, 30–36 CrossRef CAS.
- T. Umemoto, K. Adachi and S. Ishihara, J. Org. Chem., 2007, 72, 6905–6917 CrossRef CAS PubMed.
- T.-N. Streit, R. M. Gomila, R. Sievers, A. Frontera and M. Malischewski, CrystEngComm, 2024, 26, 594–598 RSC.
- R. Zeng, Z. Gong and Q. Yan, J. Org. Chem., 2020, 85, 8397–8404 CrossRef CAS PubMed.
- V. D. P. N. Nziko and S. Scheiner, J. Org. Chem., 2015, 80, 2356–2363 CrossRef CAS PubMed.
- R. M. Gomila, A. Bauzá and A. Frontera, Dalton Trans., 2022, 51, 5977–5982 RSC.
- E. V. Alexandrov, A. V. Virovets, V. A. Blatov and E. V. Peresypkina, Chem. Rev., 2015, 115, 12286 CrossRef CAS PubMed.
- I. Cvrtila and V. Stilinović, Cryst. Growth Des., 2017, 17, 6793–6800 CrossRef CAS.
- M. Sellin, S. M. Rupf and M. Malischewski, Cryst. Growth Des., 2021, 21, 5515–5520 CrossRef CAS.
- M. Sellin, S. M. Rupf, Y. Zhang and M. Malischewski, Cryst. Growth Des., 2020, 20, 7104–7110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2352918–2352921. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00454j |
| This journal is © The Royal Society of Chemistry 2024 |