
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The atypical solubility of silver(I) pentafluorobenzoate and its use as a catch-and-release agent†
Jas S.
Ward
 *
*
University of Jyvaskyla, Department of Chemistry, 40014 Jyväskylä, Finland. E-mail: james.s.ward@jyu.fi
First published on 20th May 2024
Abstract
Silver(I) pentafluorobenzoate (1) possesses an atypical solubility for a silver(I) carboxylate, demonstrating solubility in a range of etheric and aromatic solvents. This versatile solubility has been probed crystallographically by single-crystal X-ray diffraction (SCXRD) due to the myriad of solid-state structures that were isolated from a variety of solvents, providing insights into how 1 can be utilised as a reuseable crystallisation aid. This potential as a reuseable ‘catch-and-release’ agent to obtain the solid-state structures of liquid natural products was screened with the racemic dihydrocarvone, achiral eucalyptol, and the chiral R-carvone, and found to be viable. The results of these screenings demonstrated improved crystallographic outcomes than other prior attempts utilising different methodologies (e.g., encapsulation).
Introduction
The chemistry of silver is rich, diverse, and stretches back centuries. The flexibility of silver to accommodate a myriad of coordination numbers (commonly ranging from 2–6),1 its straightforward incorporation into molecules, its ability to cleanly form insoluble halide salts via metathesis, and commercial availability have all contributed to its continued utility in the present.A renewed interest in silver(I) carboxylates has been driven by the recent revival of carbonyl hypoiodite chemistry, for which the analogous silver(I) carboxylates are their synthetic precursors.2–7 The intrinsic heteroleptic nature of stabilised carbonyl hypoiodites has facilitated the closest observation of the elusive iodine(I)-pyridine cation to date,3 a pursuit that might be furthered through the use of strongly electron-withdrawing substituents on the carboxylate being incorporated into the hypoiodite moiety. Known since 1967,8 silver(I) pentafluorobenzoate has already found utility being generated in situ for incorporation into coordination polymers and clusters,9,10 and therefore presented itself as a natural progression for future hypoiodite studies.
Silver(I) carboxylates typically have negligible solubility, necessitating strongly polar solvents like (CH3)2SO (DMSO) to effect dissolution for subsequent solution-based studies, though this solubility is concomitant with decomposition of the complex on a timescale of hours to sometimes minutes. Attempts to generate more soluble silver(I) carboxylates have included the silver(I) derivative of pivalic acid, silver(I) pivalate (AgCO2tBu),5 though even this complex still required strongly polar solvents to dissolve it. The recalcitrant nature of silver(I) carboxylates is aptly explained via the solid-state structure of silver(I) pivalate, which revealed a polymeric bilayer structure of Ag⋯O interactions (Fig. 1).5 However, the effective insolubility of silver(I) carboxylates does contribute to their straightforward synthesis in H2O, given that all starting materials, side-products, and potential impurities can be washed out of the products, nor did it hinder their use as intermediates toward the formation of carbonyl hypoiodites.3–6
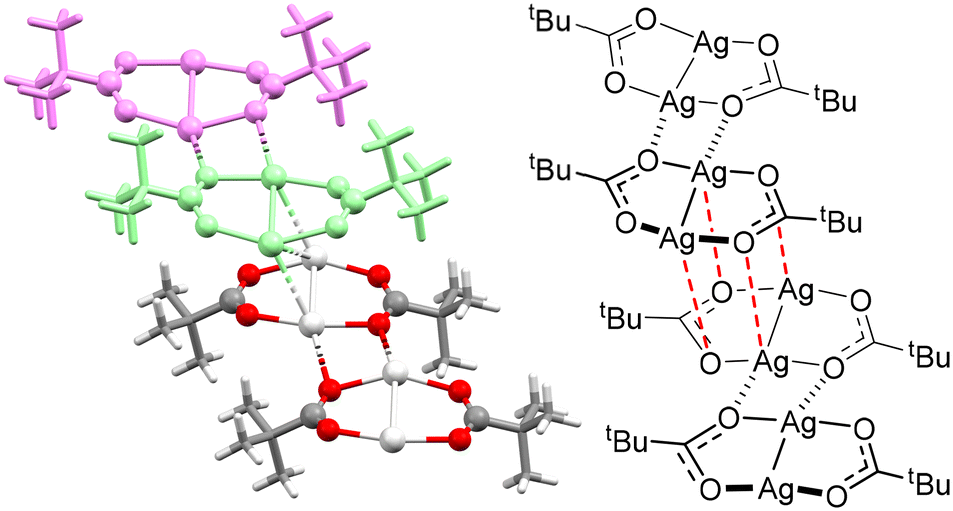 | ||
| Fig. 1 The solid-state (left; dimeric subunits of the upper layer block coloured for clarity) and structural (right; dashed bonds highlighting the connectivity of dimeric subunits along the layer [black] and between the layers [red]) representations of the polymeric bilayer structure previously observed for (AgCO2tBu)n.5 | ||
Results and discussion
Synthesis and solubility
Silver(I) pentafluorobenzoate (1) was synthesised in effectively quantitative yield from pentafluorobenzoic acid via formation of the sodium salt using NaOH, followed by cation exchange of the sodium with silver(I) using AgNO3 (Scheme 1). However, given the atypically soluble nature of 1, the work-up diverges from that of other silver(I) carboxylates by Et2O extraction of the product from the H2O reaction mixture, yielding the product in 99% yield upon evaporation under reduced pressure of the Et2O extract. This method can be completed in its entirety on a gram scale in under an hour, and is therefore preferred over prior methods in the literature.11–13 | ||
| Scheme 1 The one-pot synthesis of complex 1 from pentafluorobenzoic acid via its sodium salt by cation exchange using AgNO3. | ||
Initially intrigued by the unexpected solubility of 1 in both Et2O and H2O,‡ a range of other ethers were explored. Complex 1 was found to be soluble in Et2O, iPr2O, tBuOMe, Ph2O, Bn2O, THF, and dioxane, which with the exception of Bn2O, were all without decomposition. The more polar solvents MeOH, EtOH, iPrOH, MeOAc, EtOAc, acetone, MeCN, pyridine, (CH3)2NC(O)H (DMF), (CH3)2SO were also found to dissolve 1. However, visually-apparent decomposition in solution of the silver(I) to silver(0) was observed (sometimes within seconds) for MeOH, EtOH, MeCN, (CH3)2NC(O)H, and (CH3)2SO. Given the well-established precedence of arene adducts of AgClO4,14,15 the aromatic solvents benzene, toluene, p-xylene, chlorobenzene, and nitrobenzene were also tested, and all were found to dissolve 1. It would perhaps be more expedient to list the common solvents in which 1 was found to have negligible solubility: alkanes, CH2Cl2, and CHCl3. Though it might be tempting to think that the atypical solubility of 1 is a property that would be shared by other analogous strongly electron-withdrawing silver(I) carboxylates, the silver(I) complexes of CF3CO2, 4-BrC6H4CO2, and 4-NO2C6H4CO2 only demonstrate partial solubility in strongly polar solvents such as (CH3)2SO.3 Whilst the good solubility of 1 is unusual for a silver(I) carboxylate, it is reminiscent of silver(I) trifluoromethanesulfonate (AgOTf), for which there are numerous prior examples of its derivatives serendipitously forming adducts via the silver(I) centre with organic solvents (e.g., Et2O, tetrahydrofuran) in the solid state.1,16–19
Solution studies
Despite the lack of hydrogen atoms, NMR spectroscopy was initially thought to be an ideal technique to study the effects of potential adduct formation, however, 13C NMR studies in multiple different solvents failed to observe the adduct or even the C6F5 resonances, suggesting fluxionality on the NMR timescale. Nevertheless, the 19F NMR resonances were clearly observed, though differences originating from the formation of potentially different adducts was difficult to differentiate from simple solvent effects, with differences of ∼1–3 ppm for the various solvents attempted (C6D6, d8-THF, CD3CN, and (CD3)2SO). The solid-state IR of 1 was similarly limited, due to the absence of the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch observed at 1710 cm−1 for pentafluorobenzoic acid, and the unchanged stretch at 1648 cm−1 observed in both the precursor acid and in 1. It should be noted that the desolvating nature of the adducts of 1, though minimal for most of the solvents tested herein, hindered some solid-state studies (e.g., IR, TGA) of these combinations.
O stretch observed at 1710 cm−1 for pentafluorobenzoic acid, and the unchanged stretch at 1648 cm−1 observed in both the precursor acid and in 1. It should be noted that the desolvating nature of the adducts of 1, though minimal for most of the solvents tested herein, hindered some solid-state studies (e.g., IR, TGA) of these combinations.
Solid-state studies
Crystal engineering is an important aspect of silver(I) salts,20–23 especially with respect to the formation of silver(I) arene complexes.24,25 Investigating new types of crystalline silver(I) materials by incorporating so called super-weak anions such as [B12F12]2−, is widely pursued toward ultimately preparing significantly more reactive metal centres than with traditional anions (e.g., BF4, PF6, ClO4, etc.).24,26 Owing to the widely soluble nature of 1, a myriad of solid-state structures of solvent adducts of 1 could be prepared by vapour diffusion and determined via single-crystal X-ray crystallography (SCXRD; Fig. 2 and Tables S1 and S2†). This offered a rare opportunity to study a large subset of solvated solid-state structures of the exact same parent complex, from which direct comparisons could be drawn with respect to the differing coordinating groups of the solvents explored. However, whilst a few of the solid-state structures herein can be considered as solvated structures of 1, i.e., with solvent molecules trapped in a cavity or coordinated by very weak interactions, the majority of the solid-state structures are adducts of 1, the advantages of which will be discussed below.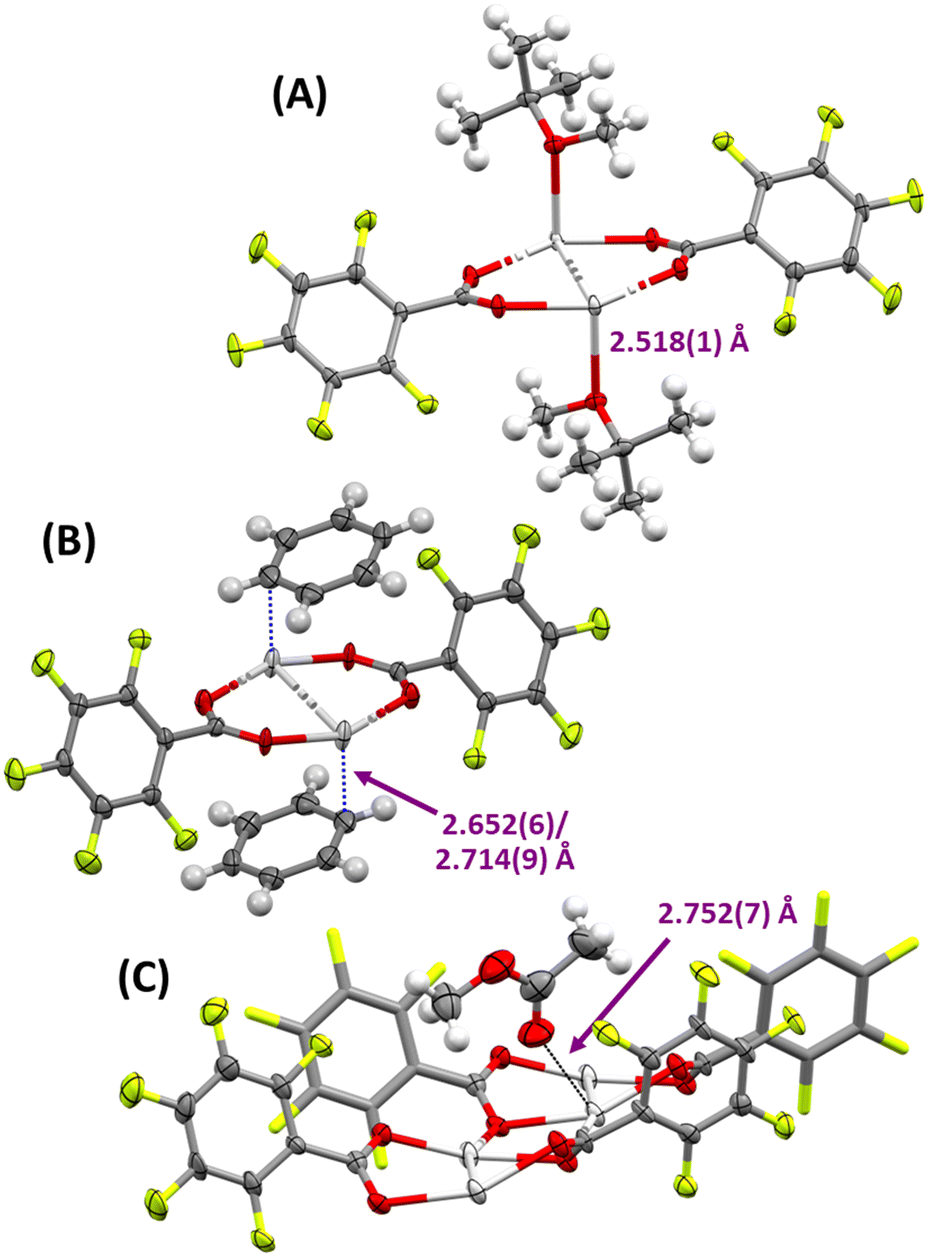 | ||
| Fig. 2 The solid-state structures of A) 1·tBuOMe, B) 1·Benzene, and C) 1·MeOAc) with the silver(I)-adduct interaction distances annotated, as representative examples of the functional group variability (ethers, arenes/alkenes, ketone/esters, respectively) possible for adduct formation with 1 (thermal ellipsoids at 50% probability). | ||
The solid-state structures of 1 could be broadly differentiated into two groups: oxygen-adducts and π-adducts. The ethers Et2O, tBuOMe, THF, and dioxane were found to be straightforward oxygen-coordinated adducts to the silver(I) metal centre. iPr2O was also presumably behaving in an analogous fashion as it was able to dissolve 1, though it could not be confirmed in the solid state. These oxygen-bound adducts featured similar Ag–O bond lengths of 2.476(6) Å (1·Et2O), 2.482(1) Å (1·THF), 2.518(1) Å (1·tBuOMe), and 2.464(2)–2.7747(19) Å (1·Dioxane), as expected for their similar electronic and steric environments. The adducts 1·Et2O, 1·THF, and 1·tBuOMe crystallised in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [1]:[adduct] ratios, and retained the dimeric subunits of 1 in their packing as 1D chains via Ag⋯O interactions to two neighbouring dimeric subunits. However, whilst the bidentate nature of dioxane yielded the same [1]:[adduct] ratio of 1:1 in 1·Dioxane (not including an additional 0.125 molecules of solvated dioxane that were also present), it was observed as 2D sheets in their packing, with dioxane adducts bridging across two different dimeric subunits of 1.
:1 [1]:[adduct] ratios, and retained the dimeric subunits of 1 in their packing as 1D chains via Ag⋯O interactions to two neighbouring dimeric subunits. However, whilst the bidentate nature of dioxane yielded the same [1]:[adduct] ratio of 1:1 in 1·Dioxane (not including an additional 0.125 molecules of solvated dioxane that were also present), it was observed as 2D sheets in their packing, with dioxane adducts bridging across two different dimeric subunits of 1.
To test if 1 could be used productively to trap previously unknown liquid substrates, the tetrahydropyran derivative isochroman was attempted. This proved successful in an analogous fashion to the other etheric substrates, yielding 1·Isochroman, which is the first time isochroman has been observed in the solid state (Fig. 3). Interestingly, the solid-state structure contained both an adduct and a solvate molecule of isochroman per molecule of 1. However, though both crystallographically independent molecules were observed with minor disorder of the flexible heterocyclic ring, the benefits of adduct formation over simple solvation were demonstrated by the more reliable assignment of the oxygen atom in the adduct due to the presence of the Ag–O coordination bond.
 | ||
| Fig. 3 The solid-state structure of 1·Isochroman, showing the coordinated and solvated molecules of isochroman (minor disordered positions omitted for clarity; thermal ellipsoids at 50% probability). | ||
The arene solvents all featured close η1 Ag⋯C(H) interactions of 2.650(2) Å (1·p-Xylene), 2.652(6) Å (1·Benzene), 2.710(8) Å (1·Toluene), and 2.97(2) Å (1·Chlorobenzene),§ which were both longer and comparable to the η2-silver(I) arene complexes with the OTf anion (cf. 2.387(2)/2.698(4) Å for [Ag(benzene)]OTf or 2.452(3)/2.474(3) Å for [Ag(toluene)]OTf),27 and only slightly longer than the Ag⋯C(H) of 2.563(1)/2.563(1) Å observed for [Ag(benzene)]ClO4.28 The solid-state packing of 1·Benzene and 1·p-Xylene were found to be comparable to their etheric counterparts, with 1D chains comprised of the same dimeric subunits of 1 coordinating via Ag⋯O interactions and [1]:[adduct] ratios of 1:1. However, the structure of 1·Toluene was found adopt a more involved network. The solid-state structure of 1·Toluene comprised of only a single toluene adduct arranged axially along one side of the 1D chain of dimeric subunits of 1, and with a lower [1]:[adduct] ratio of 2:1 compared to the 1:1 ratios of 1·Benzene and 1·p-Xylene that demonstrated alternating axial adducts on each side of the dimeric subunits of 1. However, unlike benzene and p-xylene in 1·Benzene and 1·p-Xylene, respectively, the toluene molecules in 1·Toluene bridged across two 1D chains via an η2 Ag⋯aryl interaction with Ag⋯C(H) distances of 2.776(8)/2.811(8) Å (Fig. 4), which were only slightly longer than the η1 Ag⋯C(H) (cf. 2.710(8) Å). However, the cause of this interesting bridging interaction was not readily apparent, given that the less sterically hindered benzene and more electron-rich arene system of p-xylene do not demonstrate the same behaviour, and instead mimic the solid-state packing of their etheric analogues (cf.1·Et2O, 1·THF, 1·tBuOMe).
 | ||
| Fig. 4 The bridging toluene molecules between 1D chains of dimeric subunits of 1via π-interactions (dashed blue lines) in the solid-state packing of eight molecules of 1·Toluene (C6F5 substituents omitted for clarity; thermal ellipsoids at 50% probability). | ||
The solid-state structure of 1 grown from acetone remained elusive due to rapid desolvation, and that of EtOAc was observed to have heavily disordered channels of overlapping partial-occupancy EtOAc molecules, negating the reliability of its solid-state structure. Nevertheless, the solid-state structure of 1·MeOAc, which straddled two adjacent silver(I) dimers via CO⋯Ag interactions of 2.752(7) and 3.253(7) Å, definitively confirmed that 1 could also be successfully implemented to immobilise ketones and esters. However, the MeOAc in 1·MeOAc could be described as a solvate rather than an adduct given the long Ag⋯O interactions (cf. Ag–O ≈ 2.2–2.4 Å) and minimal MeOAc present per molecule of 1 ([1]:[solvate] = 4:1).
The mixed etheric-aromatic solvents Ph2O and Bn2O were tested to probe the steric limitations and preferences of 1, given their duality in potentially forming both oxygen- and π-adducts with 1. Unfortunately, Bn2O was found to react with 1 yielding 1·AgBenzoate, which was not pursued further. Nevertheless, Ph2O did successfully form an adduct with 1, which was revealed to be an η1-adduct with a Ag⋯C(H) of 2.684(4) Å, comparable to the η1-adducts of 1 observed for benzene, toluene, and p-xylene.
Given the option to potentially form both oxygen- and π-adducts, it is surprising that 1·Ph2O would assume what is considered the weaker of the two interactions in the solid state. However, this can be attributed to the steric bulk of the phenyl rings preventing an oxygen-adduct from forming within the preferred packing of the 1D chain of the dimeric subunits of 1. Another factor is the ability of both phenyl rings of Ph2O to form π-adducts (Fig. 5), even if the second π-adduct is an η2 Ag⋯arene interaction with comparably longer Ag⋯C(H) distances of 3.147(4)/3.255(5) Å compared to the primary η1-adduct of the other phenyl ring (2.684(4) Å), and similar to the two sets of π-adducts observed for each molecule of toluene in 1·Toluene.
 | ||
| Fig. 5 The η1 and η2 π-adducts (dashed blue lines) formed by a single molecule of Ph2O over two dimeric subunits of 1 in 1·Ph2O (second molecule of Ph2O omitted for clarity; thermal ellipsoids at 50% probability). | ||
A general examination of the packing architectures of the solid-state adducts reveal that they are predominantly comprised of argentophilic dimeric subunits (Ag⋯Ag = 2.7370(11)–3.0674(11) Å) that pack as 1D chains via pairs of Ag⋯O interactions to two neighbouring dimeric subunits, with adducts situated axially on the silver(I) atoms of the chain alternating above and below the chain in an overall ratio of 1:1 for [1]:[adduct] (Fig. 6). The exceptions to the aforementioned packing included 1·Dioxane, which formed adducts via both oxygen atoms to give 2D sheets connected via the same Ag⋯O interactions in an 8:9 ratio of [1]:[dioxane], though that includes a single molecule of non-coordinating dioxane (i.e., still a 1:1 ratio for adduct formation with 1). Other exceptions to the 1:1 ratio of [1]:[adduct] were 1·Toluene (2:1), 1·Chlorobenzene (1:0.33), 1·Ph2O (2:1, though this can be considered a 1:1 ratio per coordinating phenyl ring), 1·MeOAc (4:1), and 1·Eucalyptol (3:2; vide infra). Given the wide range of adducts observed for 1 (including arene adducts), and despite it possessing a historically strongly-coordinating carboxylate-based ‘anion’, it might be apt to regard 1 as more akin to the super-weak anions currently under research for crystal engineering applications.
 | ||
| Fig. 6 A dimeric subunit of 1·THF (left; thermal ellipsoids at 50% probability) and the alternating axial coordination of the THF adducts in a 1D chain of eight molecules of 1·THF (right) above and below the plane (block coloured green and purple, respectively) of the polymeric chain of dimeric subunits of 1. | ||
Catch-and-release agent
The co-crystallisation of liquid-phase natural products, e.g., essential oils, to obtain their solid-state structures is an established method to circumvent, as Fujita aptly stated, “the intrinsic limitation that the target molecules must be obtained as single crystals”.29 Nevertheless, such methods have historically relied upon the formation of strong intermolecular interactions like hydrogen bonding,30,31 which necessitates the presence of hydrogen-bond capable donor and acceptor functional groups. In the absence of such functional groups, and their strong intermolecular forces directing the solid-state packing of these co-crystallisations, methodologies have instead shifted to trapping the liquid substrates within cavities or inside porous networks.29,32–36 These methodologies are impressive in the scope of their substrates successfully co-crystallised, especially for substrates that lack any readily-exploited functional groups (e.g., alkanes).33 However, the required porosity of such networks can result in large unit cells comprised of primarily light-atom molecules (Z < Si) can act as double-edged swords, given that such systems can necessitate the use of powerful synchrotron radiation sources.37 This in turn results in other common limitations of macro-molecular crystallography, e.g., high R-factors (R[F2 > 2σ(F2)] > 5%) relative to the crystallography of small discrete molecules. It is in this respect that 1 may offer advantages over other prior co-crystallisation methodologies.The adduct formation of 1 with ethers, arenes, and ketones/esters demonstrates the same ability to direct the orderly packing of the adduct co-crystals with 1 as observed for hydrogen-bond directed co-crystals, but without the presence of a strong intermolecular interaction such as hydrogen bonding. As a result of this weak adduct formation, a surprising outcome of co-crystallisations of 1 was the ability to recover it after adduct formation (for solvents where decomposition had not been observed) through simple evaporation. Similar recovery and reuse had been previously reported for the closely-related AgOTf salt when it was utilised as a catalyst.38 Isolation of the liquid substrates could also be envisioned through the same evaporation process, if desired, though was not attempted herein. Subsequent reuse of the recovered 1 with other solvents could then generate new adducts. A test sample was successfully reused five times to form various etheric or aromatic adducts, as confirmed from the crystallographic unit cells by SCXRD, and most of the X-ray datasets reported herein were themselves grown from recovered samples of 1. It was envisioned that this ability would be an attractive feature for organic chemists to crystallise liquid-phase natural products, without the need for low-temperature crystallisation or advanced in situ crystallisation techniques, followed by sample recovery via evaporation. This ‘catch-and-release’39 agent would minimise the amount of substrate consumed to attempt crystallisation, which would be ideal for natural products where the amount of sample is limited (e.g., if they are difficult to synthesise or isolate). In this way, 1 can be favourably likened to Fujita's crystalline sponges,29,35,36 with 1 being easier to produce, and the resulting crystallographic modelling being generally more straightforward. Additional benefits would include the presence of an atom of greater atomic number than Si (i.e., Ag), which would permit the absolute stereochemistry to be assigned for exclusively light-atom molecules even using Mo radiation X-ray diffractometers, which may prove challenging with the crystalline sponge methodology.40,41
To test this potential of 1, four additional liquid substrates were screened: the cyclic ethers 2-methyltetrahydrofuran (2-MeTHF; racemic) and eucalyptol, as well as the ketone- and alkene-containing dihydrocarvone (racemic) and R-carvone. All substrates were able to successfully dissolve a sample of 1, with crystals successfully obtained for all of them (1·2-MeTHF, 1·Eucalyptol, 1·Dihydrocarvone, and 1·R-Carvone) via pentane vapour diffusion. The 2-MeTHF adduct in 1·2-MeTHF was coordinated via the oxygen atom (Ag–O = 2.40(2)/2.42(1) Å) and did not display any narcissistic self-sorting of the R and S enantiomers, with both crystallising together in the solid-state structure. Similarly, dihydrocarvone also crystallised as the racemate in a 1:1 ratio with 1 (1·Dihydrocarvone), coordinating via the oxygen atom of its carbonyl group with a Ag–O bond length of 2.490(2) Å). No solid-state structure of dihydrocarvone previously existed in the literature, highlighting the potential of 1 to elucidate new structures. Whilst 1 had some solubility in neat dihydrocarvone, screening tests revealed that the use of a 1:1 dihydrocarvone:acetone mixture proved best to yield single crystals, with acetone being seen as an ideal co-solvent given its excellent solubility toward 1, coupled with its aforementioned highly-labile nature as an adduct/solvate of 1. This further demonstrates the potential applicability of 1 with substrates in which it has limited solubility through the use of co-solvents like acetone that form highly-labile adducts/solvates and are therefore easily out-competed by other substrates. Additionally, the use of a mixture of solvents can help reduce the amount of substrate used, should they be expensive or difficult to synthesise. The achiral 1·Eucalyptol was also observed as an oxygen adduct (Ag–O = 2.367(3)/2.409(3) Å). The solid-state structure of eucalyptol was previously obtained via in situ crystallisation and co-crystallisation with 1,4-dihydroxyenzene.1,42,43 However, the crystals of 1·Eucalyptol could be prepared overnight without the need for low-temperature crystallisation, and the stronger Ag–O adduct formation provided crystals that did not suffer from the rapid desolvation reported for the 1,4-dihydroxyenzene co-crystal,43 which is beneficial for subsequent handling during SCXRD studies. Lastly, the enantiopure configuration of R-carvone in 1·R-Carvone was successfully determined crystallographically via refinement of the Flack parameter in the non-centrosymmetric C2 space group. The R-carvone was observed to coordinate via both its oxygen and terminal alkene,¶ bridging across two 1D chains of 1 (Fig. 7), reminiscent of the previously discussed coordination of 1·Dioxane.
 | ||
| Fig. 7 The solid-state structure of 1·R-Carvone illustrating the R-carvone molecule bridging across two 1D chains of 1via both its terminal alkene (dashed blue lines) and oxygen atom (minor disordered position omitted for clarity; thermal ellipsoids at 50% probability). | ||
The solid-state packing of these natural-product co-crystals of 1, as observed for the other simpler adducts described previously, retained their dimeric subunits of 1 upon adduct formation, with Ag⋯Ag distances of 2.8598(4)/2.8835(4) Å for 1·Eucalyptol, 2.8451(4) Å for 1·Dihydrocarvone, and 2.795(1)/2.982(1) Å for 1·R-Carvone. The structures were also analogously observed as 1D chains in their packing via Ag⋯O interactions between neighbouring dimeric subunits of 1, with Ag⋯O distances of 2.461(3)–2.541(2) Å (1·Eucalyptol), 2.496(2) Å (1·Dihydrocarvone), and 2.384(3)–2.522(3) Å (1·R-Carvone). Finally, it should be noted that the natural-product crystals of eucalyptol or R-carvone with 1, despite crystallographic disorder being present in both, gave excellent SCXRD structures with R1/wR2 values of 3.1/8.0% and 2.2/5.3%, respectively, which have lower R1 values for both substrates in comparison to all prior literature structures.1 The structure of dihydrocarvone (1·Dihydrocarvone) was without any apparent disorder present and was also of excellent quality (R1/wR2 = 2.7/6.0%), though there were no literature examples for comparison. The 1·Eucalyptol structure (3.1%) is comparable to the R1 values of the several hydrogen-bonded co-crystals of eucalyptol (ranging from 4.6 to 8.3%)43,44 and the low-temperature isolated structure (4.7%),42 and a notable improvement over encapsulation within a porous organic cage (11.3%).45 Similarly, 1·R-Carvone (2.2%) is comparable to the R1 values of hydrogen-bonded (4.2%)46 or organic co-crystals (4.3 and 5.1%)33 of R-carvone, as well as the low-temperature isolated structure (3.0%),47 whilst again being an improvement over co-crystallisation within porous organic cages (ranging from 7.1 to 15.3%).45,48,49
Conclusions
In conclusion, the easy to synthesise complex silver(I) pentafluorobenzoate (1) was found to be soluble in a myriad of organic solvents, which was atypical when compared to other silver(I) carboxylates, even those incorporating similarly strong electron-withdrawing substituents on the carboxylates. Solid-state adducts of 1 could be straightforwardly crystallised for liquid substrates coordinated via a range of functional groups including ethers, ketones, esters, alkenes, and arenes. The ease and low cost of synthesising 1 in bulk also makes it ideal for the inexpensive, large-scale screening of substrates (including determination of their absolute stereochemistry), such as liquid-phase natural products that are of interest to researchers. The possibility of using substrate:acetone solvent mixtures for crystallisation also present an economic pathway should the natural products be expensive or difficult to synthesise. After SCXRD studies, 1 could often be recovered from these substrates by evaporation and subsequently reused; a favourable trait toward the pursuit of greener chemistry. The adducts of 1 with isochroman and dihydrocarvone also served to highlight the potential of 1 to yield previously unreported solid-state structures of liquid substrates. The solid-state structures of the natural-product adducts of 1 tested herein demonstrated excellent crystallographic metrics, which were found to have improved outcomes compared to those from other methodologies like encapsulation. Just as mechanochemical techniques minimise environmental impact by circumventing the use of toxic solvents like CH2Cl2,50–52 the recovery and reuse of crystallisation materials, as demonstrated with 1 herein, could offer an environmentally-friendly alternative when attempting to prepare suitable crystals for SCXRD.
Experimental
General considerations
All reagents and solvents were obtained from commercial suppliers and used without further purification. For structural NMR assignments, 19F NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer to an external KF standard, with chemical shifts reported on the δ scale in ppm to a CFCl3 standard calculated from the following conversion: [chemical shift referenced to CFCl3] = (−125.3 ppm + [chemical shift referenced to KF]). Multiplicities are denoted as: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). Infrared (IR) spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer.The single crystal X-ray diffraction (SCXRD) data were collected at 120 K using mirror-monochromated Cu-Kα (λ = 1.54184 Å) radiation on a Rigaku XtaLAB Synergy-R diffractometer with a HyPix-Arc 100 detector. All structures were solved by intrinsic phasing (SHELXT)53 and refined by full-matrix least squares on F2 using Olex2,54 utilising the SHELXL module.55 Anisotropic displacement parameters were assigned to non-H atoms and isotropic displacement parameters for all H atoms were constrained to multiples of the equivalent displacement parameters of their parent atoms with Uiso(H) = 1.2 Ueq(CH) or 1.5 Ueq(CH2, CH3, OH) of their respective parent atoms. The X-ray single crystal data and CCDC numbers (2310151–2310162, 2311134) of all new structures are included below, excluding the connectivity model for 1·Chlorobenzene.
Synthesis and characterisation
SCXRD
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 5.4941(2) Å, b = 9.8998(3) Å, c = 12.7267(5) Å, α = 82.824(3)°, β = 80.970(3)°, γ = 75.189(3)°, V = 658.31(4) Å3, Z = 2, Dcalc = 1.983 gcm−3, F(000) = 384, μ = 12.94 mm−1, T = 120.0(1) K, θmax = 74.5°, 2554 independent reflections, 2411 with Io > 2σ(Io), Rint = 0.071, 2554 data, 216 parameters, 89 restraints, GooF = 1.07, 1.50 > Δρ > −0.82 e Å−3, R[F2 > 2σ(F2)] = 0.045, wR(F2) = 0.131.
(no. 2), a = 5.59808(5) Å, b = 10.23969(10) Å, c = 12.79989(10) Å, α = 97.6393(7)°, β = 96.3916(7)°, γ = 102.3278(7)°, V = 703.01(1) Å3, Z = 2, Dcalc = 1.923 gcm−3, F(000) = 400, μ = 12.15 mm−1, T = 120.0(1) K, θmax = 74.5°, 2849 independent reflections, 2791 with Io > 2σ(Io), Rint = 0.027, 2849 data, 194 parameters, no restraints, GooF = 1.06, 0.39 > Δρ > −0.37 e Å−3, R[F2 > 2σ(F2)] = 0.014, wR(F2) = 0.033.
(no. 2), a = 5.5278(1) Å, b = 8.6031(1) Å, c = 8.6031(1) Å, α = 103.225(1)°, β = 94.823(2)°, γ = 100.843(2)°, V = 593.50(2) Å3, Z = 2, Dcalc = 2.188 gcm−3, F(000) = 380, μ = 14.35 mm−1, T = 120.0(1) K, θmax = 74.5°, 2418 independent reflections, 2357 with Io > 2σ(Io), Rint = 0.028, 2418 data, 181 parameters, no restraints, GooF = 1.12, 0.39 > Δρ > −0.46 e Å−3, R[F2 > 2σ(F2)] = 0.016, wR(F2) = 0.042.
(no. 2), a = 7.2534(3) Å, b = 11.0121(5) Å, c = 13.8225(6) Å, α = 85.841(4)°, β = 85.713(3)°, γ = 84.263(3)°, V = 1093.11(8) Å3, Z = 1, Dcalc = 1.784 gcm−3, F(000) = 588, μ = 8.08 mm−1, T = 120.0(1) K, θmax = 74.5°, 4439 independent reflections, 3829 with Io > 2σ(Io), Rint = 0.052, 4439 data, 363 parameters, 38 restraints, GooF = 1.05, 1.53 > Δρ > −1.43 e Å−3, R[F2 > 2σ(F2)] = 0.036, wR(F2) = 0.079.
607 independent reflections, 9585 with Io > 2σ(Io), Rint = 0.025, 10607 data, 815 parameters, 36 restraints, GooF = 1.08, 0.39 > Δρ > −0.64 e Å−3, R[F2 > 2σ(F2)] = 0.020, wR(F2) = 0.054.
(no. 2), a = 5.69188(13) Å, b = 12.5817(3) Å, c = 17.9282(6) Å, α = 105.107(3)°, β = 90.214(2)°, γ = 90.163(2)°, V = 1239.51(6) Å3, Z = 2, Dcalc = 2.128 gcm−3, F(000) = 768, μ = 13.71 mm−1, T = 120.0(1) K, θmax = 74.5°, 5044 independent reflections, 4271 with Io > 2σ(Io), Rint = 0.040, 5044 data, 379 parameters, no restraints, GooF = 1.15, 1.91 > Δρ > −1.39 e Å−3, R[F2 > 2σ(F2)] = 0.044, wR(F2) = 0.109.
(no. 2), a = 5.5552(2) Å, b = 16.0610(7) Å, c = 20.9338(6) Å, α = 74.645(3)°, β = 89.103(3)°, γ = 80.350(3)°, V = 1774.79(12) Å3, Z = 2, Dcalc = 2.526 gcm−3, F(000) = 1280, μ = 19.00 mm−1, T = 120.0(1) K, θmax = 74.5°, 7191 independent reflections, 4740 with Io > 2σ(Io), Rint = 0.090, 7191 data, 588 parameters, no restraints, GooF = 1.01, 0.80 > Δρ > −1.07 e Å−3, R[F2 > 2σ(F2)] = 0.047, wR(F2) = 0.092.
:1 mixture of benzoate:pentafluorobenzoate. Crystals suitable for single crystal X-ray diffraction were obtained from a Bn2O solution of 1 vapour diffused with pentane at ambient temperature. Crystal data for 1·AgBenzoate: CCDC-2310160, C14H5Ag2F5O4, M = 547.92, colourless needle, 0.01 × 0.01 × 0.17 mm, monoclinic, space group P21/n, a = 6.9141(3) Å, b = 5.5610(2) Å, c = 37.1068(11) Å, β = 93.837(3)°, V = 1423.53(9) Å3, Z = 4, Dcalc = 2.557 gcm−3, F(000) = 1040, μ = 22.86 mm−1, T = 120.0(1) K, θmax = 74.5°, 2894 independent reflections, 2336 with Io > 2σ(Io), Rint = 0.051, 2894 data, 226 parameters, no restraints, GooF = 1.04, 1.26 > Δρ > −1.14 e Å−3, R[F2 > 2σ(F2)] = 0.047, wR(F2) = 0.120.
(no. 2), a = 5.51822(13) Å, b = 9.8945(5) Å, c = 13.0489(3) Å, α = 75.275(3)°, β = 85.880(2)°, γ = 79.845(3)°, V = 678.00(4) Å3, Z = 2, Dcalc = 1.984 gcm−3, F(000) = 396, μ = 12.59 mm−1, T = 120.0(1) K, θmax = 74.5°, 2742 independent reflections, 2470 with Io > 2σ(Io), Rint = 0.045, 2742 data, 241 parameters, 132 restraints, GooF = 1.09, 0.98 > Δρ > −0.70 e Å−3, R[F2 > 2σ(F2)] = 0.037, wR(F2) = 0.106.
:acetone (1:1) solution of 1 vapour diffused with pentane at ambient temperature. Crystal data for 1·Dihydrocarvone: CCDC-2352000, C17H16AgF5O3, M = 471.17, colourless plate, 0.01 × 0.04 × 0.22 mm, monoclinic, space group P21/n, a = 13.9355(2) Å, b = 5.5125(1) Å, c = 22.5133(2) Å, β = 93.604(1)°, V = 1726.04(4) Å3, Z = 4, Dcalc = 1.813 gcm−3, F(000) = 936, μ = 10.00 mm−1, T = 120.0(1) K, θmax = 74.5°, 3513 independent reflections, 3104 with Io > 2σ(Io), Rint = 0.045, 3513 data, 237 parameters, no restraints, GooF = 1.05, 0.55 > Δρ > −0.53 e Å−3, R[F2 > 2σ(F2)] = 0.027, wR(F2) = 0.060.
(no. 2), a = 12.4062(3) Å, b = 13.45738(19) Å, c = 13.9666(3) Å, α = 85.3721(16)°, β = 70.664(2)°, γ = 86.9586(16)°, V = 2192.24(8) Å3, Z = 2, Dcalc = 1.917 gcm−3, F(000) = 1244, μ = 11.70 mm−1, T = 120.0(1) K, θmax = 74.5°, 8953 independent reflections, 8507 with Io > 2σ(Io), Rint = 0.029, 8953 data, 925 parameters, 705 restraints, GooF = 1.03, 2.54 > Δρ > −0.70 e Å−3, R[F2 > 2σ(F2)] = 0.031, wR(F2) = 0.080.
(no. 2), a = 5.4941(2) Å, b = 9.8998(3) Å, c = 12.7267(5) Å, α = 82.824(3)°, β = 80.970(3)°, γ = 75.189(3)°, V = 658.31(4) Å3, Z = 2, Dcalc = 1.983 gcm−3, F(000) = 384, μ = 12.94 mm−1, T = 120.0(1) K, θmax = 74.5°, 2554 independent reflections, 2411 with Io > 2σ(Io), Rint = 0.071, 2554 data, 216 parameters, 89 restraints, GooF = 1.07, 1.50 > Δρ > −0.82 e Å−3, R[F2 > 2σ(F2)] = 0.045, wR(F2) = 0.131.
(no. 2), a = 5.59808(5) Å, b = 10.23969(10) Å, c = 12.79989(10) Å, α = 97.6393(7)°, β = 96.3916(7)°, γ = 102.3278(7)°, V = 703.01(1) Å3, Z = 2, Dcalc = 1.923 gcm−3, F(000) = 400, μ = 12.15 mm−1, T = 120.0(1) K, θmax = 74.5°, 2849 independent reflections, 2791 with Io > 2σ(Io), Rint = 0.027, 2849 data, 194 parameters, no restraints, GooF = 1.06, 0.39 > Δρ > −0.37 e Å−3, R[F2 > 2σ(F2)] = 0.014, wR(F2) = 0.033.
(no. 2), a = 5.5278(1) Å, b = 8.6031(1) Å, c = 8.6031(1) Å, α = 103.225(1)°, β = 94.823(2)°, γ = 100.843(2)°, V = 593.50(2) Å3, Z = 2, Dcalc = 2.188 gcm−3, F(000) = 380, μ = 14.35 mm−1, T = 120.0(1) K, θmax = 74.5°, 2418 independent reflections, 2357 with Io > 2σ(Io), Rint = 0.028, 2418 data, 181 parameters, no restraints, GooF = 1.12, 0.39 > Δρ > −0.46 e Å−3, R[F2 > 2σ(F2)] = 0.016, wR(F2) = 0.042.
(no. 2), a = 7.2534(3) Å, b = 11.0121(5) Å, c = 13.8225(6) Å, α = 85.841(4)°, β = 85.713(3)°, γ = 84.263(3)°, V = 1093.11(8) Å3, Z = 1, Dcalc = 1.784 gcm−3, F(000) = 588, μ = 8.08 mm−1, T = 120.0(1) K, θmax = 74.5°, 4439 independent reflections, 3829 with Io > 2σ(Io), Rint = 0.052, 4439 data, 363 parameters, 38 restraints, GooF = 1.05, 1.53 > Δρ > −1.43 e Å−3, R[F2 > 2σ(F2)] = 0.036, wR(F2) = 0.079.
607 independent reflections, 9585 with Io > 2σ(Io), Rint = 0.025, 10607 data, 815 parameters, 36 restraints, GooF = 1.08, 0.39 > Δρ > −0.64 e Å−3, R[F2 > 2σ(F2)] = 0.020, wR(F2) = 0.054.
(no. 2), a = 5.69188(13) Å, b = 12.5817(3) Å, c = 17.9282(6) Å, α = 105.107(3)°, β = 90.214(2)°, γ = 90.163(2)°, V = 1239.51(6) Å3, Z = 2, Dcalc = 2.128 gcm−3, F(000) = 768, μ = 13.71 mm−1, T = 120.0(1) K, θmax = 74.5°, 5044 independent reflections, 4271 with Io > 2σ(Io), Rint = 0.040, 5044 data, 379 parameters, no restraints, GooF = 1.15, 1.91 > Δρ > −1.39 e Å−3, R[F2 > 2σ(F2)] = 0.044, wR(F2) = 0.109.
(no. 2), a = 5.5552(2) Å, b = 16.0610(7) Å, c = 20.9338(6) Å, α = 74.645(3)°, β = 89.103(3)°, γ = 80.350(3)°, V = 1774.79(12) Å3, Z = 2, Dcalc = 2.526 gcm−3, F(000) = 1280, μ = 19.00 mm−1, T = 120.0(1) K, θmax = 74.5°, 7191 independent reflections, 4740 with Io > 2σ(Io), Rint = 0.090, 7191 data, 588 parameters, no restraints, GooF = 1.01, 0.80 > Δρ > −1.07 e Å−3, R[F2 > 2σ(F2)] = 0.047, wR(F2) = 0.092.
:1 mixture of benzoate:pentafluorobenzoate. Crystals suitable for single crystal X-ray diffraction were obtained from a Bn2O solution of 1 vapour diffused with pentane at ambient temperature. Crystal data for 1·AgBenzoate: CCDC-2310160, C14H5Ag2F5O4, M = 547.92, colourless needle, 0.01 × 0.01 × 0.17 mm, monoclinic, space group P21/n, a = 6.9141(3) Å, b = 5.5610(2) Å, c = 37.1068(11) Å, β = 93.837(3)°, V = 1423.53(9) Å3, Z = 4, Dcalc = 2.557 gcm−3, F(000) = 1040, μ = 22.86 mm−1, T = 120.0(1) K, θmax = 74.5°, 2894 independent reflections, 2336 with Io > 2σ(Io), Rint = 0.051, 2894 data, 226 parameters, no restraints, GooF = 1.04, 1.26 > Δρ > −1.14 e Å−3, R[F2 > 2σ(F2)] = 0.047, wR(F2) = 0.120.
(no. 2), a = 5.51822(13) Å, b = 9.8945(5) Å, c = 13.0489(3) Å, α = 75.275(3)°, β = 85.880(2)°, γ = 79.845(3)°, V = 678.00(4) Å3, Z = 2, Dcalc = 1.984 gcm−3, F(000) = 396, μ = 12.59 mm−1, T = 120.0(1) K, θmax = 74.5°, 2742 independent reflections, 2470 with Io > 2σ(Io), Rint = 0.045, 2742 data, 241 parameters, 132 restraints, GooF = 1.09, 0.98 > Δρ > −0.70 e Å−3, R[F2 > 2σ(F2)] = 0.037, wR(F2) = 0.106.
:acetone (1:1) solution of 1 vapour diffused with pentane at ambient temperature. Crystal data for 1·Dihydrocarvone: CCDC-2352000, C17H16AgF5O3, M = 471.17, colourless plate, 0.01 × 0.04 × 0.22 mm, monoclinic, space group P21/n, a = 13.9355(2) Å, b = 5.5125(1) Å, c = 22.5133(2) Å, β = 93.604(1)°, V = 1726.04(4) Å3, Z = 4, Dcalc = 1.813 gcm−3, F(000) = 936, μ = 10.00 mm−1, T = 120.0(1) K, θmax = 74.5°, 3513 independent reflections, 3104 with Io > 2σ(Io), Rint = 0.045, 3513 data, 237 parameters, no restraints, GooF = 1.05, 0.55 > Δρ > −0.53 e Å−3, R[F2 > 2σ(F2)] = 0.027, wR(F2) = 0.060.
(no. 2), a = 12.4062(3) Å, b = 13.45738(19) Å, c = 13.9666(3) Å, α = 85.3721(16)°, β = 70.664(2)°, γ = 86.9586(16)°, V = 2192.24(8) Å3, Z = 2, Dcalc = 1.917 gcm−3, F(000) = 1244, μ = 11.70 mm−1, T = 120.0(1) K, θmax = 74.5°, 8953 independent reflections, 8507 with Io > 2σ(Io), Rint = 0.029, 8953 data, 925 parameters, 705 restraints, GooF = 1.03, 2.54 > Δρ > −0.70 e Å−3, R[F2 > 2σ(F2)] = 0.031, wR(F2) = 0.080.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank M.Sc. Václav Kolařík and Prof. Kari Rissanen for useful discussions, and gratefully acknowledges the Research Council of Finland (grant number: 356187) for funding.Notes and references
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- H. Hartl and M. Hedrich, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1981, 36b, 922–928 CrossRef CAS.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed.
- E. Kramer, S. Yu, J. S. Ward and K. Rissanen, Dalton Trans., 2021, 50, 14990–14993 RSC.
- J. S. Ward, J. Martõnova, L. M. E. Wilson, E. Kramer, R. Aav and K. Rissanen, Dalton Trans., 2022, 51, 14646–14653 RSC.
- L. M. E. Wilson, K. Rissanen and J. S. Ward, New J. Chem., 2023, 47, 2978–2982 RSC.
- V. Kolařík, K. Rissanen and J. S. Ward, Chem. – Asian J., 2024, e202400349 CrossRef PubMed.
- P. Sartori and M. Weidenbruch, Chem. Ber., 1967, 100, 3016–3023 CrossRef CAS.
- F. Lang, D. C. N. G. Singh, A. B. Rao, C. Romer, J. S. Wright, R. Smith, H. Adams and L. Brammer, Chem. – Eur. J., 2022, 28, e202201408 CrossRef CAS PubMed.
- L.-L. Hou, H.-J. Li, D.-N. Yu, X. Cheng, Z.-X. Yao, K.-G. Liu and X.-W. Yan, CrystEngComm, 2021, 23, 7202–7205 RSC.
- J. Vicente, M. D. Bermúdez, M. T. Chicote and M. J. Sanchez-Santano, J. Organomet. Chem., 1989, 371, 129–135 CrossRef CAS.
- E. Szłyk, I. Łakomska and A. Grodzicki, Thermochim. Acta, 1993, 223, 207–212 CrossRef.
- I. O. Koshevoy, P. Lahuerta, M. Sanaú, M. A. Ubeda and A. Doménech, Dalton Trans., 2006, 5536–5541 RSC.
- R. M. Keefer and L. J. Andrews, J. Am. Chem. Soc., 1952, 74, 640–643 CrossRef CAS.
- H. G. Smith and R. E. Rundle, J. Am. Chem. Soc., 1958, 80, 5075–5080 CrossRef CAS.
- D. Gudat, M. Schrott, V. Bajorat, M. Nieger, S. Kotila, R. Fleischer and D. Stalke, Chem. Ber., 1996, 129, 337–345 CrossRef CAS.
- E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. A. Rodríguez, O. Crespo, M. C. Gimeno, A. Laguna and P. G. Jones, Chem. – Eur. J., 2000, 6, 636–644 CrossRef.
- X.-K. Huo, G. Su and G.-X. Jin, Chem. – Eur. J., 2010, 16, 12017–12027 CrossRef CAS PubMed.
- H. Ruffin, S. A. Baudron, D. Salazar-Mendoza and M. W. Hosseini, Chem. – Eur. J., 2014, 20, 2449–2453 CrossRef CAS PubMed.
- M. Munakata, G. L. Ning, Y. Suenaga, T. Kuroda-Sowa, M. Maekawa and T. Ohta, Angew. Chem., Int. Ed., 2000, 39, 4555–4557 CrossRef CAS.
- E. Bosch and C. L. Barnes, Inorg. Chem., 2002, 41, 2543–2547 CrossRef CAS PubMed.
- M. Munakata, L. P. Wu, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga, T. Ohta and H. Konaka, Inorg. Chem., 2003, 42, 2553–2558 CrossRef CAS PubMed.
- S. Q. Liu, T. Kuroda-Sowa, H. Konaka, Y. Suenaga, M. Maekawa, T. Mizutani, G. L. Ning and M. Munakata, Inorg. Chem., 2005, 44, 1031–1036 CrossRef CAS PubMed.
- S. V. Ivanov, S. M. Miller, O. P. Anderson and S. H. Strauss, Cryst. Growth Des., 2004, 4, 249–254 CrossRef CAS.
- Y. Kobayashi, A. A. Popov, S. M. Miller, O. P. Anderson and S. H. Strauss, Inorg. Chem., 2007, 46, 8505–8507 CrossRef CAS PubMed.
- D. V. Peryshkov and S. H. Strauss, Inorg. Chem., 2017, 56, 4072–4083 CrossRef CAS PubMed.
- H. Wadepohl and H. Pritzkow, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 383–384 CrossRef CAS PubMed.
- R. K. McMullan, T. F. Koetzle and C. J. Fritchie Jnr, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 645–653 CrossRef.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- R. Custelcean and M. D. Ward, Cryst. Growth Des., 2005, 5, 2277–2287 CrossRef CAS.
- P. P. Mazzeo, C. Carraro, A. Monica, D. Capucci, P. Pelagatti, F. Bianchi, S. Agazzi, M. Careri, A. Raio, M. Carta, F. Menicucci, M. Belli, M. Michelozzi and A. Bacchi, ACS Sustainable Chem. Eng., 2019, 7, 17929–17940 CrossRef CAS.
- M. Ceborska, K. Szwed, M. Asztemborska, M. Wszelaka-Rylik, E. Kicińska and K. Suwińska, Chem. Phys. Lett., 2015, 641, 44–50 CrossRef CAS.
- F. Krupp, W. Frey and C. Richert, Angew. Chem., Int. Ed., 2020, 59, 15875–15879 CrossRef CAS PubMed.
- H. Li, J. Jiao, W. Xie, Y. Zhao, C. Lin, J. Jiang and L. Wang, ACS Mater. Lett., 2023, 5, 2673–2682 CrossRef CAS.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246–252 CrossRef CAS PubMed.
- N. Zigon, V. Duplan, N. Wada and M. Fujita, Angew. Chem., Int. Ed., 2021, 60, 25204–25222 CrossRef CAS PubMed.
- E. Christoforides, K. Fourtaka, A. Andreou and K. Bethanis, J. Mol. Struct., 2020, 1202, 127350 CrossRef CAS.
- D. K. Roy, K. J. Tamuli and M. Bordoloi, J. Heterocyclic Chem., 2019, 56, 3313–3323 CrossRef CAS.
- A. Peuronen, A. Valkonen, M. Kortelainen, K. Rissanen and M. Lahtinen, Cryst. Growth Des., 2012, 12, 4157–4169 CrossRef CAS.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 501, 262 CrossRef CAS.
- K. Yan, R. Dubey, T. Arai, Y. Inokuma and M. Fujita, J. Am. Chem. Soc., 2017, 139, 11341–11344 CrossRef CAS PubMed.
- A. D. Bond and J. E. Davies, Aust. J. Chem., 2002, 54, 683–684 CrossRef.
- J. Barnes, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 118–120 CrossRef.
- J. C. Barnes, H. A. Sampson and T. J. R. Weakley, J. Chem. Soc., Dalton Trans., 1980, 949–953 RSC.
- Y. Zhou, P. Luo, L.-J. Xu, W. Xu and R.-W. Jiang, Org. Chem. Front., 2023, 10, 1119–1127 RSC.
- S.-Q. Qin, W. Xu, W.-C. Ye and R.-W. Jiang, CrystEngComm, 2022, 24, 8060–8069 RSC.
- J. Sane, J. Ruis, T. Calvet and M. A. Cuevas-Diarte, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 702–707 CrossRef.
- T. Gruber, C. Fischer, W. Seichter, P. Bombicz and E. Weber, CrystEngComm, 2011, 13, 1422–1431 RSC.
- W. de Poel, P. Tinnemans, A. L. L. Duchateau, M. Honing, F. P. J. T. Rutjes, E. Vlieg and R. de Gelder, Chem. – Eur. J., 2019, 25, 14999–15003 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- S. L. James and T. Friščić, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- C. Schumacher, K.-N. Truong, J. S. Ward, R. Puttreddy, A. Rajala, E. Lassila, C. Bolm and K. Rissanen, Org. Chem. Front., 2024, 11, 781–795 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- H. Weigand, W. Tyrra and D. Naumann, Z. Anorg. Allg. Chem., 2008, 634, 2125–2126 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, IR, and SCXRD (CCDC 2310151–2310162, 2311134, 2334781, 2352000). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00441h |
| ‡ 1 as the monohydrate is known (QOJDAV).56 |
| § The solid-state structures of 1·Chlorobenzene featured a symmetry-disordered and partial-occupancy chlorobenzene molecule, which by necessity had to be modelled isotropically. Therefore, the reliability of the value of its Ag⋯C(H) interaction is diminished in comparison to the other 1·[arene] structures. |
| ¶ There was disorder in the position of the silver(I) atoms in 1·R-Carvone, which created ambiguity between the distances of the Ag⋯O and Ag⋯C(H2 = C) interactions of the R-carvone with 1. |
| This journal is © The Royal Society of Chemistry 2024 |