
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous syntheses of anthracene-based mixed-ligand coordination polymers and their structural and optical properties†
Antti
Marttinen
 and
Manu
Lahtinen
*
and
Manu
Lahtinen
*
Department of Chemistry, University of Jyväskylä, Jyväskylä, Finland. E-mail: manu.k.lahtinen@jyu.fi
First published on 28th May 2024
Abstract
Series of mixed-ligand coordination polymers (CP) and metal–organic frameworks (MOFs) based on bulky 9,10-di(1H-imidazol-1-yl)anthracene (dia) were synthesized from aqueous solutions even with very short (<10 min) reaction times. Two different metals (M = Cu, Ni) and three different secondary ligands were used to generate six new structures: [Cu2(dia)2(5-aip)2]·6H2O (1), [Ni2(dia)2(5-aip)2]·5H2O (2), [Cu2(dia)2(2-ata)2]·4H2O (3), [Ni(dia)(2-ata)·Ni(dia)2(2-ata)]·2.5H2O (4), [Cu8(dia)2(5-sip)4(OH)4] (5) and [Ni3(dia)5(5-sip)2]·6DMF·14H2O (6). Structure analyses involving single-crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD) and Fourier-transform infrared spectroscopy (FT-IR) were conducted along with studies determining the thermal stabilities and optical properties of the compounds. The results indicate that the developed fast and green syntheses can produce pure CP and MOF phases that have various thermally stable and visible light absorbing structures which may find applications in photocatalysis.
Introduction
Coordination polymers (CP) are a class of hybrid compounds incorporating inorganic and organic components into coordinate covalently (datively) bonded repeating units which extend, for example, to one-dimensional polymeric chains or three-dimensional networks. Metal–organic frameworks (MOFs) are a subclass of coordination polymers characterized by their porous three-dimensional structures. They have been studied for various applications such as gas storage, CO2 capture or luminescent material design, and their vast selection is steadily growing.1–3 Some MOFs have even reached industrial production scale and are moving towards commercial applications.2 In recent years MOFs have also been studied as light harvesting materials or photocatalysts for example in sensing applications, photocatalytic water splitting or carbon dioxide reduction.4–7 Easy modifiability, vast variety and high specific surface areas of MOF and CP structures are highly desirable properties in creating such materials.Conventionally MOFs have been synthesized by high pressure/temperature (over 100 °C) solvothermal methods in organic media (e.g., dimethylformamide (DMF), diethylformamide (DEF)) with long reaction times (several days). Upscaling of such syntheses is not economically or environmentally reasonable, and so it has been a limiting factor for the application of multiple potential MOFs.8,9 Instead, syntheses of MOFs in low temperatures and aqueous media with fast reaction times are highly desirable synthesis methods to pursue. Furthermore, preparation of MOF compounds from water could indicate their higher stability in the presence of water, promoting their applicability for example as catalysts in aqueous media. Aqueous synthesis of MOFs is usually limited by the poor solubility of the organic linkers, but this problem can be circumvented by using organic salts. Organic salts can have a significantly better water solubility and reactivity than their neutral counterparts, and so the formation kinetics of MOFs can be influenced.10
In this study six new mixed-ligand MOFs/CPs, namely [Cu2(dia)2(5-aip)2]·6H2O (1), [Ni2(dia)2(5-aip)2]·5H2O (2), [Cu2(dia)2(2-ata)2]·4H2O (3), [Ni(dia)(2-ata)·Ni(dia)2(2-ata)]·2.5H2O (4), [Cu8(dia)2(5-sip)4(OH)4] (5) and [Ni3(dia)5(5-sip)2]·6DMF·14H2O (6), were designed and characterized. 9,10-Di(1H-imidazol-1-yl)anthracene (dia) was selected as the primary ligand, of which the chromophoric anthracene core has been incorporated in many light responsive or photocatalytic complexes and MOFs.11–14 Dia ligand was mixed with three different simple functionalized carboxylic acids (Scheme 1) to produce mixed-ligand MOFs/CPs and possibly induce a bathochromic shift to the synthesized compound through the amino or sulfo groups incorporated on the ligands.6 Mixed-ligand MOFs can also benefit from improved charge transfer and widened light absorption range through the use of complementary ligands.15 Copper and nickel were selected as node metals due to their commercial availability, high abundance and beneficial electronic properties. Developing noble-metal free photocatalysts could also lessen the costs and so promote the feasibility of photocatalytic applications.16 Fast aqueous synthesis conditions using salt forms of the chosen organic ligands were examined and optimized. The hydrothermally synthesized MOFs/CPs have been characterized by Fourier-transform infrared spectroscopy (FT-IR), single-crystal and powder X-ray diffraction (SCXRD and PXRD, respectively) methods. Thermal properties of the compounds were analyzed by thermogravimetric analysis, and solid-state UV-vis spectroscopy was used to determine their optical properties.
 | ||
| Scheme 1 Structures of the neutral forms of the ligands used in this study. | ||
Experimental
Ligand syntheses
All reagents unless otherwise stated were commercially purchased, and used as received in the syntheses. All water (H2O) used in the syntheses and experiments was deionized. Dia ligand was synthesized according to a reported method17 with some modifications as follows. 9,10-Dibromoanthracene (2.57 g, 7.66 mmol), imidazole (2.08 g, 30.6 mmol) and powdered sodium hydroxide (1.22 g, 30.6 mmol) were transferred with 100 ml of dimethylformamide (DMF) to a 250 ml round flask under magnetic stirring. The mixture was heated under reflux at 120 °C for 24 hours. After that, the mixture was cooled down to room temperature and poured into 250 ml of water. The yellow precipitate was filtered, washed with water (2 × 50 ml), toluene (2 × 50 ml) and hexane (1 × 50 ml) to obtain a pale yellow powdery product which was dried in a desiccator for 24 h. Yield 1.12 g, 47%. 1H NMR (Fig. S1 and S2†): δ 7.83 (d, 2H), 7.52–7.57 (m, 8H), 7.49 (s, 2H), 7.33 (d, 2H).5-Aminoisophtalic acid and 2-aminoterephtalic acid disodium salts (Na2aip·H2O and Na2ata·H2O, respectively) used in the MOF/CP syntheses were prepared according to reported methods18,19 with some modifications as follows. Commercially purchased neutral acid (1.0 g, 5.5 mmol) was weighed into a beaker and 11 ml of 1 M aqueous sodium hydroxide solution (11.0 mmol) was added. The mixture was magnetically stirred and heated at roughly 60 °C for 5 minutes and then left to evaporate almost to dryness. When most of the water had evaporated, 10 ml of ethanol and 50 ml of diethyl ether was added to the solution, which precipitated the salt. After filtering, the obtained pale/light brown powders were dried in a desiccator. Yield (Na2aip·H2O) 1.26 g, 94%. Yield (Na2ata·H2O) 1.28 g, 95%. The presence of monohydrated crystal forms on both salts was confirmed by thermogravimetric analysis (Fig. S3,† experimental values of 1.07·H2O and 0.90·H2O). FT-IR spectra (Fig. S4 and S5†) confirmed the successful syntheses based on the observation of characteristic carboxylate peaks at 1550–1570 cm−1 region.
To improve water solubility and by that the reactivity of the dia ligand, it was treated with HNO3 to form a new nitrate salt (diaH2(NO3)2). 0.84 g (2.71 mmol) of dia ligand was added into a 50 ml round-bottomed flask and 11 ml of 2.5 M HNO3 (27.5 mmol) was added. The flask was closed, and the mixture was briefly mixed before leaving it to react in room temperature. After 24 hours the powdery ligand had changed color from pale yellow to light orange. The powder was filtered and washed quickly with 10 ml of H2O. Light orange powder (yield 0.97 g, 82%) was dried in a desiccator. Characterization of the prepared salt is presented in the ESI.†
Single-crystal crystallizations
Bulk synthesis procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 as in: diaH2(NO3)2 (50 mg, 0.11 mmol), Na2ata·H2O (52 mg, 0.21 mmol) and Ni(OAc)2·4H2O (56 mg, 0.23 mmol), instead of 1:2:1 ratio as in the case of compound 1. The resulting light green/yellow powder was dried in a desiccator (yield 67 mg, 63%).
:2:2 as in: diaH2(NO3)2 (50 mg, 0.11 mmol), Na2ata·H2O (52 mg, 0.21 mmol) and Ni(OAc)2·4H2O (56 mg, 0.23 mmol), instead of 1:2:1 ratio as in the case of compound 1. The resulting light green/yellow powder was dried in a desiccator (yield 67 mg, 63%).
Single-crystal X-ray diffraction (SCXRD)
Single-crystal data was measured using two devices; Rigaku SuperNova dual-source (Cu and Mo micro-sources) X-ray diffractometer with an HyPix-Arc 100° detector or Atlas CCD detector; or Rigaku XtaLAB Synergy-R single source high flux rotating anode X-ray diffractometer equipped with an HyPix-Arc 100° detector. Both diffractometers are equipped with multilayer optics producing monochromatized Cu Kα (1.54187 Å) radiation. The data collection, reduction, and analytical face-index-based absorption correction of the data were carried out using the CrysAlisPRO program (v.42).20 All structures were solved by intrinsic phasing (ShelXT21) and refined on F2 by full-matrix least-squares techniques with the ShelXL22 subprogram in the Olex2 (v.1.5)23 structure solving program that utilizes the SHELXL-2013 module. All C–H hydrogen atoms were calculated to their optimal positions and treated as riding atoms using isotropic displacement parameters of 1.2 (sp2 group) larger than the host atom. Occupancies of the disorder model atoms were first refined freely and then fixed to specific values if settled closer to 1:1 ratio. Structure dependently occupancies of some disordered solvent molecules were fixed to 0.5 or 0.25. In the case of dried crystals which displayed highly disordered solvent molecules, solvents were first identified from electron density maps but could not be modelled satisfactorily and therefore they were removed by Olex2 solvent mask. When analyzing hydrogen bonds, donor–acceptor maximum distance limit of 3.0 Å and minimum angle limit of 120° were used. When analyzing π–π interactions, centroid–centroid maximum limit of 4.0 Å and intercentroid shift maximum limit of 3.0 Å were used. Crystallographic data, selected bond distances and angles along with hydrogen bond distances and angles are listed in Tables S2–S16,† and the asymmetric units of the structures are presented in Fig. S8–S15.†
Powder X-ray diffraction (PXRD)
Powdery products were analyzed using a Panalytical X'Pert PRO MPD diffractometer with Cu Kα radiation (λ = 1.54187 Å; Ni β-filter; 45 kV, 40 mA). Powder samples were attached to a silicon based zero background generating plate with petroleum jelly. Diffraction intensities were recorded by a position-sensitive X'Celerator detector at room temperature with a 2θ range of 3–60°, a step size of 0.017°, and a counting time of 50–280 s per step. Data processing and pattern comparisons were carried out with the program X'pert HighScore Plus (v. 4.9).24Solid-state UV-vis spectroscopy
UV-vis absorption spectra of the synthesized powder products were measured using a Perkin Elmer Lambda 850 spectrophotometer with an integrating sphere. First, a blank reference was determined by measuring the absorbance of two glass slides. Then a powder sample was placed between the two glass slides, and they were set in the center of the integrating sphere with a clip style center mount sample holder. Both the transmittance and reflectance were measured simultaneously in the range of 300–900 nm to determine the total absorbances of the samples.Other methods
The 1H NMR spectrum was measured using Bruker Avance III 500 MHz spectrometer with deuterated chloroform (CDCl3) as solvent and the residual CHCl3 solvent signal (7.26 ppm) as internal standard. FT-IR spectra were measured by Bruker Alpha Platinum-ATR FT-IR spectrometer in the range of 400–4000 cm−1 with step of 1.4 cm−1. Thermogravimetric data were collected by Perkin Elmer STA 6000 simultaneous TG/DSC thermal analyzer. Temperature calibration of the analyzer was done using melting points of the indium (156.6 °C), zinc (419.5 °C) and aluminum (660.3 °C) standards. The weight balance was calibrated with standard weight of a 50.00 mg at room temperature. Na2aip·H2O and Na2ata·H2O samples were heated under an air flow (40 ml min−1) in a platinum pan to 175 °C at 10 °C min−1 rate and then held there for 30 minutes. MOF/CP samples were heated under an air flow in a platinum pan at 10 °C min−1 rate in the range of 22–600 °C. Sample weights used in the measurements were in the range of 5–10 mg.Results and discussion
Crystal structure of 1 and 2
Crystal structures of compounds 1 and 2 proved to be isostructural and both compounds crystallize in the monoclinic space group of P21/n. The asymmetric unit (AU; Fig. S8†) consists of a single copper(II) atom (or nickel(II) atom in 2, Fig. S9†) with two halves of dia ligand, one 5-aminoisophtalate ligand and three (2.5 in 2, see Fig. S9†) uncoordinated water molecules. Metal cations of the structures have a distorted octahedral coordination geometry, in which two dia ligands and three 5-aminoisophtalates coordinate to the metal nodes. 5-Aminoisophtalate shows three different coordination motifs with the metal nodes; through its monodentate carboxylate (Cu–O, 1.974(1) Å; Ni–O, 2.007(2) Å), bidentate carboxylate (Cu–O, 2.017(1) and 2.713(2) Å; Ni–O, 2.079(2) and 2.278(2) Å), or via its amino group (Cu–N, 2.300(2) Å; Ni–N, 2.112(3) Å). Dia ligand is coordinated through its terminal imidazole nitrogen atoms (Cu–N, ∼1.981 Å; Ni–N, ∼2.046 Å) and the ligand has a trans-conformation. Imidazole rings are rotated ∼72° with respect to the anthracene plane. Coordination bonds of the nickel-noded compound 2 differ slightly from 1 due to lack of Jahn–Teller elongation, which was observed especially in bidentate carboxylate bonding of 1.In both systems the distorted octahedral metal centers with the bridging ligands form a polymeric 3D MOF-structure. Alternating metal–ligand coordination represent as wavy 1D-chains, which are linked to each other by the carboxylate groups of 5-aip and as consequence 2D sheet structure is formed (Fig. 1a). These 2D sheets then penetrate each other diagonally as can be seen in Fig. 1b. Furthermore, the 2D sheets are linked together by the amino groups that are coordinated to the metal nodes (Fig. 1a and c), eventually forming a self-penetrating 3D framework (Fig. 1c). The framework has small channels running along a-axis containing all the lattice water molecules.
 | ||
| Fig. 1 Partial structure views of 1, a) 2D sheet structure of 1 viewed along (100) plane, b) 2D sheet penetration shown by blue and red colored parts viewed along (110) plane, c) 3D framework of 1 and its channels viewed along the a-axis. Hydrogen atoms have been omitted for clarity. | ||
Crystal structure of 3
Compound 3 crystallizes in the triclinic space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , having two Cu(II) atoms on special positions, one dia ligand, two halves of 2-aminoterephtalates and two water molecules (one uncoordinated and one coordinated) in the AU (Fig. S10†). In this case the dia ligand has adopted a cis-conformation. Amino-groups of the 2-aminoterephtalate ligands are disordered over two or four sites with partial occupation of either 0.5 or 0.25. The two crystallographically independent copper cations display Jahn–Teller elongated octahedral coordination geometries. Cu1 is coordinated to two N-atoms of dia ligands and to two bidentate carboxylates. Cu1–O1 bond distances are 1.961(2) Å but Cu1–O2 distances are significantly longer and weaker by showing contact distance of 2.604(2) Å. Cu2 is also coordinated to two N-atoms of dia ligands, but unlike Cu1, it is coordinated to two monodentate carboxylates (1.951(2) Å) and to two water molecules in the axial positions. Axial bonds to water molecules are elongated significantly with bond distances of 2.535(2) Å. Coordination through the dia ligands creates a 1D-chain as shown in Fig. 2a. Bidentatively coordinated carboxylates join these chains together to form 2D sheets (Fig. 2b). Finally, the monodentate carboxylates join the 2D sheets together diagonally to form a 3D framework somewhat similarly as in 1 and 2. Along the c-axis the structure forms a quadrilateral grid (Fig. 2c), which has the planar anthracene rings of the neighboring sheets overlapping each other slightly.
, having two Cu(II) atoms on special positions, one dia ligand, two halves of 2-aminoterephtalates and two water molecules (one uncoordinated and one coordinated) in the AU (Fig. S10†). In this case the dia ligand has adopted a cis-conformation. Amino-groups of the 2-aminoterephtalate ligands are disordered over two or four sites with partial occupation of either 0.5 or 0.25. The two crystallographically independent copper cations display Jahn–Teller elongated octahedral coordination geometries. Cu1 is coordinated to two N-atoms of dia ligands and to two bidentate carboxylates. Cu1–O1 bond distances are 1.961(2) Å but Cu1–O2 distances are significantly longer and weaker by showing contact distance of 2.604(2) Å. Cu2 is also coordinated to two N-atoms of dia ligands, but unlike Cu1, it is coordinated to two monodentate carboxylates (1.951(2) Å) and to two water molecules in the axial positions. Axial bonds to water molecules are elongated significantly with bond distances of 2.535(2) Å. Coordination through the dia ligands creates a 1D-chain as shown in Fig. 2a. Bidentatively coordinated carboxylates join these chains together to form 2D sheets (Fig. 2b). Finally, the monodentate carboxylates join the 2D sheets together diagonally to form a 3D framework somewhat similarly as in 1 and 2. Along the c-axis the structure forms a quadrilateral grid (Fig. 2c), which has the planar anthracene rings of the neighboring sheets overlapping each other slightly.
 | ||
| Fig. 2 Partial structure views of 3, a) 1D-chain structure of 3 viewed along (1−21) plane, b) 2D sheet structure of 3 viewed along (110) plane, c) quadrilateral grid of 3 viewed along the c-axis. Hydrogen atoms have been omitted for clarity. | ||
Crystal structure of 4
Compound 4 crystallizes in the triclinic space group of P, having two crystallographically unique Ni(II) atoms on special positions. Ni1 has two halves of dia ligand and one half of 2-aminoterephtalate coordinated to it (Fig. S11†). Ni2 has one half of dia ligand, one half of 2-aminoterephtalate and one water molecule coordinated. Dia ligands are in trans-conformation as in 1 and 2. The bond lengths between both nickel atoms and the coordinated ligands are very similar with each other with them varying between 2.029(2)–2.111(3) Å. Ni1 forms a 3D framework with the dia and monodentatively coordinated 2-aminoterephtalate ligands (Fig. 3b). Ni2 on the other hand forms 2D sheets (Fig. 3a) since the water molecules terminate the coordination along b-axis. There is also some disordered uncoordinated water between the two structure moieties. Together these form an unusual interpenetrated 2D + 3D framework (Fig. 3c), which is isostructural to a structure retrieved from CSD25 with an identifier of ETAXAZ reported by Lee et al.26 As they proved in their study, the bulkiness and orientation of dia ligand suppresses the interpenetration along the b-axis for the 2D sheets and so prevents formation of a two-fold 3D interpenetrated framework. Contrary to structure ETAXAZ, compound 4 has amino groups (disordered to two sites with fixed occupancies of 0.5) between the 2D sheets and the 3D framework, which could improve the compound's functionality and possibly cause a bathochromic shift for the absorption edge. Nickel coordination in 4 is still octahedral like the cobalt nodes in the case of ETAXAZ structure.
 | ||
| Fig. 3 Partial structure views of 4, a) 2D sheets of 4 viewed along the b-axis, b) 3D framework of 4 viewed along the c-axis, c) 2D (blue) + 3D (red) interpenetrated structure of 4 viewed along the c-axis. Hydrogen atoms have been omitted for clarity. | ||
Crystal structure of 5
Compound 5 crystallizes in the triclinic space group of P with four copper(II) atoms, two 5-sip ligands, two halves of dia ligand and two hydroxyl groups in the AU (Fig. S12†). All the copper atoms have a distorted square pyramidal coordination geometry (geometry index27τ5 = 0.19–0.29) and the dia ligands have a trans-conformation. Again, as in compounds 1 and 3, Jahn–Teller elongation is evident in axial bonds from copper atoms to sulfo-group oxygens (2.295(3)–2.392(2) Å) compared to four equatorial bonds varying between 1.935(2)–2.007(2) Å. Four copper atoms create two crystallographically independent nodes, in which the copper atoms are connected to each other through hydroxyl groups in the center of the node. Cu1 is coordinated to one hydroxyl group, two carboxylate groups, one sulfo group and one imidazole, while Cu2 coordinates to two hydroxyls, two carboxylates and one sulfo group. These together form a unique secondary building unit (SBU) and Cu3 and Cu4 create another SBU with similar bonding but in a different orientation (Fig. 4a and b). The sulfo group that is coordinated to Cu1 and Cu2, is disordered on two rotational orientations with occupancies of 0.9 and 0.1. The SBUs are oriented to each other at roughly 90-degree angle (measured from planes lining up the carboxylates of each SBU). They are connected to each other by the coordination of 5-sip ligand's sulfo groups. Neighboring identical SBUs are connected to each other either through carboxylate coordination or dia ligand coordination. This creates a non-interpenetrated wavy 3D framework (Fig. 4c). The structure is very dense, which is apparent from the fact that not even water molecules are found in the voids of the framework or coordinated to metal nodes.
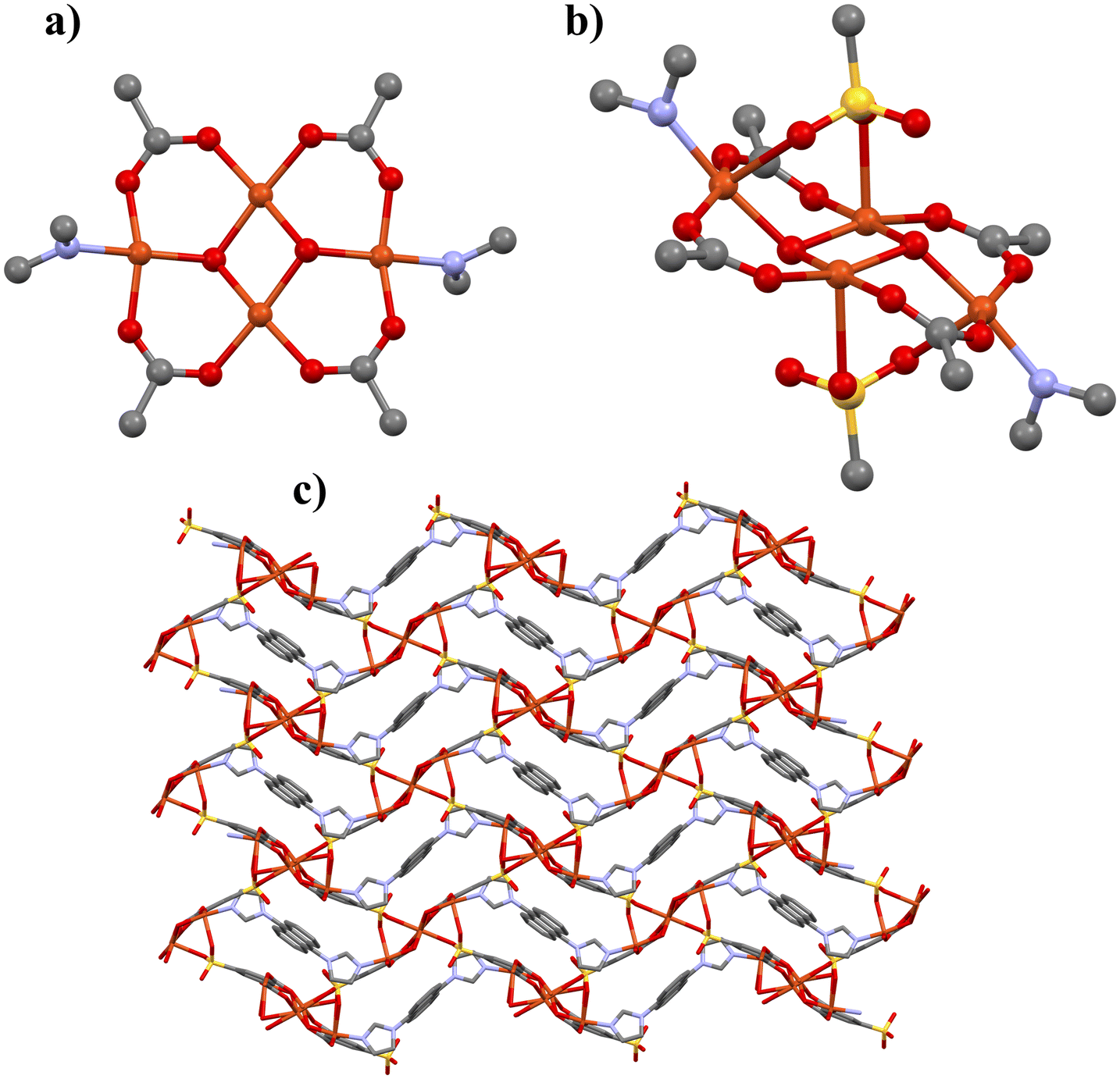 | ||
| Fig. 4 Partial structure views of 5, a) representation of compound 5 SBU (coordination to sulfo groups not shown), b) SBU viewed along (212) plane, c) 3D framework structure of 5 viewed along b-axis. Hydrogen atoms have been omitted for clarity. | ||
Crystal structure of 6
Compound 6 crystallizes in the monoclinic space group of P21/n showing two nickel atoms (one of them in a special position), two and a half dia ligands, one 5-sip ligand, six uncoordinated water molecules, one coordinated water molecule and three dimethylformamide molecules in the AU (Fig. S13†). One of the uncoordinated water molecules is disordered over two positions with occupancies of 0.2 and 0.8. Both nickel atoms are coordinated octahedrally and the dia ligands have a trans-conformation. Ni1 is coordinated to four dia ligand imidazoles (Ni–N 2.078(2)–2.091(2) Å) and two 5-sip ligand sulfo-groups (Ni–O 2.128(1) Å). Second Ni cation is coordinated to three dia ligand imidazoles (Ni–N 2.079(2)–2.097(2) Å), two 5-sip carboxylates (Ni–O 2.058(1)–2.062(2) Å) and one water molecule (Ni–O 2.081(1) Å). As a result, four coordinated nickel atoms form square-shaped openings (Fig. 5a) which are coordinated together forming 2D layers with quadrangular channels (Fig. 5b). These layers are then extended along a-axis by the dia or carboxylate coordination to form a 3D framework (Fig. 5c). The quadrangular channels of the framework contain all the uncoordinated solvent molecules. | ||
| Fig. 5 Partial structure views of 6, a) square-shaped pore opening, b) 2D sheets with DMF and water molecules (in spacefill style) inside the channels viewed along the a-axis, c) 3D framework with DMF and water molecules (in spacefill style) inside the channels viewed along the c-axis. Hydrogen atoms have been omitted for clarity. | ||
Another structure (6B) was measured from the dried crystals of the bulk synthesis of 6. In this case Olex2 solvent masking was used to omit highly disordered uncoordinated solvent molecules. Solvent accessible volume of 2164 Å3 contained 612 electrons which corresponds to 8 DMF and 28 H2O molecules (600 e−) per unit cell or 2 DMF and 7 H2O per asymmetric unit. The structural features and the space group of the MOF remained unchanged (Fig. S14†), but the unit cell dimensions and the void volume reduced significantly, caused by the replacement of four DMF molecules with four water molecules (per unit cell) and the subsequent contraction of the pore structure. To study this behavior further, SCXRD data of 6B was collected at 0 °C (Table S2 and Fig. S15†) alongside with low-temperature data collection at −153 °C. Structure determination revealed that at 0 °C, the unit cell dimensions and the void volume increased significantly (∼160 Å3) due to structural rearrangement albeit the space group remained unchanged and four water molecules (per unit cell) evaporated from the pores (solvent accessible volume of 2336 Å3 contained 560 electrons which corresponds to 8 DMF and 24 H2O molecules (560 e−) per unit cell or 2 DMF and 6 H2O per asymmetric unit). This structural rearrangement, stimulated either by solvent or temperature change, seems to derive from the flexible twisting of the 5-sip ligand and the rotation of the dia ligand. Thus, the flexible crystal structure of 6 differs significantly from 1–5 and makes it more unique and versatile among CP/MOF materials.
Structural comparison and analysis of weak interactions
Along with the strong coordination bonds between the metals and ligands, all structures also show some weaker inter- and intramolecular interactions. In structures 1, 2 and 3 there is a similar hydrogen bond network between the coordinated or uncoordinated water molecules, the carboxylate groups and the amino groups. Fig. 6a shows an example of this in the case of structure 1 where the hydrogen bond network forms an arc shape around the 5-aip ligand. Structure 3 differs from this slightly since the amino group is uncoordinated and on the ortho-position. The amino group forms an intramolecular bond to the carboxylate (similarly as in 4; Fig. 6b) rather than to the water molecules as in 1 and 2. All the bonds in 1–3 are moderately strong with bond lengths (H–A = hydrogen–acceptor) varying between 1.86–2.19 Å (Tables S11–S13†).28 In compound 4 there are intramolecular hydrogen bonds between the amino and carboxylate groups, but more importantly there are intermolecular hydrogen bonds between the interpenetrated 2D and 3D structures (Fig. 6b; Table S14†), which stabilize the whole structure. The dense structure of 5 contains just intramolecular interactions in which the hydroxyl groups in the nodes are hydrogen bonded to the uncoordinated sulfo group oxygens (Fig. 6c; Table S15†). Furthermore, there are weak C–H⋯O bonds between the imidazole and sulfo groups with H–A bond length of 2.37 Å (Fig. 6c). Compound 6 has somewhat similar hydrogen bond network as in 1, 2, 3 where the coordinated or uncoordinated water molecules form hydrogen bonds with each other and the carboxylate groups, but in this case also the DMF molecules and the sulfo groups are involved in the network which covers the whole solvent filled pore from side to side (Fig. 6d; Table S16†). | ||
| Fig. 6 a) Hydrogen bonding around the 5-aip ligand in structure 1. b) Hydrogen bonding between the 2D and 3D networks in structure 4. c) Hydrogen bonding around the SBU in structure 5. d) Hydrogen bond network in the pore of structure 6. Hydrogen bonds presented as light blue lines. | ||
In addition to the hydrogen bonding, structures 1, 2 and 3 have also similar π–π interactions where the anthracene rings form weak parallel offset stacking, as presented in Fig. S16.† Due to steric hindrance, the stacking is possible only by the outermost part of the anthracene rings which also makes the interactions weak with centroid–centroid distances varying between 3.85–3.99 Å. Stacking continues indefinitely along the a-direction in 1 and 2, but in 3 the stacking continues along the c-direction as discrete dual stacking motifs (Fig. 2c). Structures 4, 5 and 6 on the other hand do not contain any conventional π–π interactions or they are even weaker with centroid–centroid distances over 4 Å.23,29,30
When comparing all the presented structures between each other it can be concluded that different metals in 1 and 2, seem to have little effect to the structural assembly as the structures are isostructural. On the other hand, 3 and 4 formed completely different types of structures which could be due to the more restrictive coordination geometry of the 2-ata ligand. Differences between 3 and 4 are most likely caused by the more flexible coordination geometry of copper (due to Jahn–Teller elongation) and the cis-conformation of dia ligand in compound 3. The 5-sip ligand in 5 and 6 has the same coordination geometry as 5-aip in 1 and 2, but due to the differences in the functional group size, shape and coordination preferences, very different coordination motifs could be formed. Lastly, the great contrast between the dense structure of 5 and the porous structure of 6 could be due to the formation of the unique SBU in the case of 5, which may only be possible using the more flexibly coordinative copper metal and aqueous solutions to produce the hydroxyl groups needed for the SBU.
Bulk product syntheses and characterization
Single-crystal formation aimed synthesis methods were also optimized and up-scaled to produce CPs as bulk powders. For compounds 1, 2, 4 and 5, diaH2(NO3)2 was used in syntheses, which yielded pure phases in aqueous media, and with very fast 5–10 min reaction times. For comparison, similar syntheses with the same ligand to metal ratios, solvent volumes and reaction times were attempted using neutral ligands, but these produced only impure mixtures (Fig. S17–S20†). On the other hand, in the case of single-crystal crystallizations, using the neutral dia ligand proved to form better quality crystals than when the nitrate salt was used. Based on these observations, when using the salt forms of the ligands, the compounds seem to self-assemble almost instantly by precipitating out of the solution readily after all reactants are mixed together and heated slightly. But possibly due to the poorer solubility and so slower nucleation, using the neutral dia ligand can produce better quality single-crystals. In the case of compound 3, similar fast synthesis conditions with diaH2(NO3)2 afforded impure powdery products but with slight modification of the synthesis, thus using neutral dia ligand and refluxing conditions instead, phase pure product was afforded. The nitrate salt or the sodium salt of the 2-ata ligand possibly might have caused some side product formation along with the product. Similar synthesis conditions were also examined for compound 6 using diaH2(NO3)2 but the products were amorphous/poorly crystalline, so a scaled up solvothermal crystallization was used to obtain the phase pure product in larger scale. Therefore, longer reaction times are most likely needed to form and crystallize the porous solvent filled structure of 6.The phase purity of the powdery products was studied using powder X-ray diffraction (PXRD). The measured PXRD patterns were compared to the simulated patterns based on the resulted SCXRD data of this study. As can be seen from Fig. S21–S25† the measured PXRD patterns of 1–5 match well with the simulated ones, indicating good phase purities of the powdery products. The patterns of 1, 2, 4 and 5 (Fig. S21, S22, S24 and S25,† respectively) show no sign of neutral ligands or by-products as in the impure samples prepared with neutral ligands (Fig. S17–S20†). This demonstrates that the use of organic ligand salts can be more favorable in MOF/CP-syntheses in terms of reaction time and product purity. In the case of compound 6, the characteristic peaks in the pattern also matched the simulated ones but were significantly shifted to lower 2θ angles (Fig. 7) which was deduced to be due to the flexible structural changes of the network structure, as shown by the 6B structure determination. By comparing the PXRD pattern of 6 to the simulated patterns of 6B collected at 0 °C and at −153 °C, the pattern matches much better to the 0 °C simulated data, as can be seen in Fig. 7 by comparing peak positions e.g., at ∼11°, 12.5°, 19.5° and 22.2° 2θ. Therefore, it can be deduced that when compound 6 is taken out of the reaction solution the major solvent content in the pores starts to evaporate or exchange moisture from the air and the structure adapts to this change. These observations further confirm the dynamic behavior and unit cell differences of structure 6 and 6B.
 | ||
| Fig. 7 a) PXRD-pattern comparison of the synthesized 6 and the simulated patterns of 6B measured in 0 °C or −153 °C. b) Zoomed 2θ range of the same PXRD pattern comparison. | ||
FT-IR spectra of the compounds were also measured to further confirm their chemical composition. As can be seen from the Fig. S26–S31,† the spectra of the compounds containing the same ligands are very similar to each other. Characteristic bands of the amino group (3320–3490 cm−1 region) are present in spectra of 1–4, while bands of the sulfo group (1150–1250 cm−1 region) are present in spectra of 5 and 6. All spectra also contain bands of the coordinating carboxylate group (1550–1610 cm−1 region), the aromatic C–H stretches (3000–3200 cm−1 region) and the broad overlapping bands of coordinated or lattice water molecules (3000–3600 cm−1 region). In the spectrum of compound 6 (Fig. S31†) the bands at ∼1660 cm−1 and 2800–3000 cm−1 are due to the v(CO) and v(CH) stretching of DMF inside the pores of the framework.
Thermal stabilities and optical properties
Thermal analyses were conducted to determine the thermal stabilities and solvent removal of the compounds. In the case of 1–5 the adsorbed, coordinated, or free lattice water molecules escape the structures in the range of 50–200 °C (Fig. 8). In compound 6, water molecules evaporate first in the range of 50–140 °C after which the DMF molecules volatilize at higher temperature range of 140–360 °C. Solvent losses in compound 6 (H2O loss ∼8.13%, DMF loss ∼10.55%) match better with the values based on the solvent mask of 6B collected at 0 °C (calcd: H2O 9.14%, DMF 10.59%) rather than the values of the solvent mask of 6B collected at −153 °C (calcd: H2O 10.31%, DMF 10.46%) or the values from the SCXRD data of 6 (calcd: H2O 8.68%, DMF 15.10%). This confirms the results from the PXRD and the solvent mask calculations. As can be seen from Fig. 8, all the compounds are stable up to 250 °C. The lowest decomposition temperatures were observed for 1 (290 °C) and 3 (284 °C), whereas for 2 and 4 similar onset temperatures at higher temperature range were determined (378 °C and 372 °C, respectively). Thus, the decomposition temperatures of the nickel compounds 2 and 4 are significantly higher (88 °C) than the copper analogues 1 and 3. This is also apparent by comparing Cu-based 5 and Ni-based 6, as the latter has clearly higher thermal decomposition onset temperature (313 and 388 °C, respectively). It is worth mentioning that due to the sudden exothermic decomposition of the compounds, sample temperatures rise momentarily and afterwards cool down which is seen as the curves turning slightly backwards in temperature. | ||
| Fig. 8 TGA curves of compounds 1–6 measured under air flow with heating rate of 10 °C min−1. | ||
Solid state UV-vis absorption spectra of compounds 1–6 reveal that they have very similar light absorption characteristics (Fig. 9). All of the compounds have their absorption edge bathochromically shifted relative to dia ligand, and their total light absorption is relatively much higher. In addition, all compounds have some absorption in the 600–900 nm region, with 1 having the highest band peak at roughly 700 nm. These bands are possibly due to the d–d transitions of Cu2+ and Ni2+ ions.31 Optical band gaps of the compounds were estimated based on their Tauc plots (Fig. S32†). Tauc-analysis has been proven to produce more accurately corresponding results to density functional theory (DFT) calculations in the case of similar MOFs/CPs.32 Assuming direct transitions, the band gaps of compounds 1–6 range from 2.83 to 2.93 eV, which indicates their capability to utilize visible light. The estimated band gaps are also smaller than for example the common TiO2 photocatalyst (∼3.2 eV) and they are comparable to graphitic carbon nitride (g-C3N4) based photocatalysts (∼2.8–3.0 eV).33 It is also worth noting that since compounds 1–5 were synthesized with short reaction times and in aqueous media, their particle sizes are much smaller than in the case of solvothermally synthesized 6. Therefore, more crystalline phases of 1–5 could have significantly reduced band gaps, as crystal size and morphology differences have been proven to affect the absorption edge and so the band gap.34,35
 | ||
| Fig. 9 Solid state UV-vis spectra of compounds 1–6 and dia ligand. | ||
Conclusions
Six new mixed-ligand CPs based on the bulky dia ligand were synthesized and characterized. All compounds exhibit a 3D framework, except 4, which has an unusual interpenetrated 2D + 3D structure. Compound 6 differs from the rest with its flexible structure which is responsive towards temperature or pore guest exchange. Fast aqueous syntheses of 1, 2, 4 and 5 show that the use of organic salts, instead of their neutral ligand form, is highly beneficial in terms of greener CP/MOF syntheses and it introduces new paths for the successful scale-up potential for CP/MOF-chemistry. In addition, for the presented CPs and MOFs, high water-stability is anticipated as the products were afforded from heated aqueous media. Thermal analysis results also indicate that all six compounds are thermally stable and the optical band gap estimates indicate good UV-/visible light absorbing abilities of 1–6 and suggest their potential in photocatalytic applications. Further studies exploring the photocatalytic applicability of 1–6 and developing new MOFs based on these results are under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Magnus Ehrnrooth Foundation and the University of Jyväskylä. The authors would also like to thank Riikka Kuosmanen and Esa Haapaniemi for assistance on NMR measurements and Pasi Myllyperkiö for assistance on solid state UV-vis spectroscopy.References
- C. G. Piscopo and S. Loebbecke, Strategies to Enhance Carbon Dioxide Capture in Metal-Organic Frameworks, ChemPlusChem, 2020, 85, 538–547 CrossRef CAS PubMed.
- P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Multifunctional metal–organic frameworks: from academia to industrial applications, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- H.-Y. Li, Y.-L. Wei, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Novel Tb-MOF Embedded with Viologen Species for Multi-Photofunctionality: Photochromism, Photomodulated Fluorescence, and Luminescent pH Sensing, Chem. Mater., 2015, 27, 1327–1331 CrossRef CAS.
- L. Liu, S. Du, X. Guo, Y. Xiao, Z. Yin, N. Yang, Y. Bao, X. Zhu, S. Jin, Z. Feng and F. Zhang, Water-Stable Nickel Metal–Organic Framework Nanobelts for Cocatalyst-Free Photocatalytic Water Splitting to Produce Hydrogen, J. Am. Chem. Soc., 2022, 144, 2747–2754 CrossRef CAS PubMed.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Photocatalytic CO2 reduction by a mixed metal (Zr/Ti), mixed ligand metal–organic framework under visible light irradiation, Chem. Commun., 2015, 51, 5735–5738 RSC.
- X. Guo, L. Liu, Y. Xiao, Y. Qi, C. Duan and F. Zhang, Band gap engineering of metal-organic frameworks for solar fuel productions, Coord. Chem. Rev., 2021, 435, 213785 CrossRef CAS.
- X. Zhang, W. Wang, Z. Hu, G. Wang and K. Uvdal, Coordination polymers for energy transfer: Preparations, properties, sensing applications, and perspectives, Coord. Chem. Rev., 2015, 284, 206–235 CrossRef CAS.
- P. A. Julien, C. Mottillo and T. Friši, Metal-organic frameworks meet scalable and sustainable synthesis, Green Chem., 2017, 19, 2729–2747 RSC.
- N. Stock and S. Biswas, Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. Sánchez-Sánchez, N. Getachew, K. Díaz, M. Díaz-García, Y. Chebude and I. Díaz, Synthesis of metal–organic frameworks in water at room temperature: salts as linker sources, Green Chem., 2015, 17, 1500–1509 RSC.
- D. Chen, H. Xing, C. Wang and Z. Su, Highly efficient visible-light-driven CO2 reduction to formate by a new anthracene-based zirconium MOF via dual catalytic routes, J. Mater. Chem. A, 2016, 4, 2657–2662 RSC.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, Organic Photocatalyst for Polymerization Reactions: 9,10-Bis[(triisopropylsilyl)ethynyl]anthracene, ACS Macro Lett., 2012, 1, 198–203 CrossRef CAS PubMed.
- D.-M. Chen, C.-X. Sun, C.-S. Liu and M. Du, Stable Layered Semiconductive Cu(I)–Organic Framework for Efficient Visible-Light-Driven Cr(VI) Reduction and H2 Evolution, Inorg. Chem., 2018, 57, 7975–7981 CrossRef CAS PubMed.
- F. Dumur, Recent advances on anthracene-based photoinitiators of polymerization, Eur. Polym. J., 2022, 169, 111139 CrossRef CAS.
- M. Kim, J. S. Oh, B. H. Kim, A. Y. Kim, K. C. Park, J. Mun, G. Gupta and C. Y. Lee, Enhanced Photocatalytic Performance of Nanosized Mixed-Ligand Metal–Organic Frameworks through Sequential Energy and Electron Transfer Process, Inorg. Chem., 2020, 59, 12947–12953 CrossRef CAS PubMed.
- K. Zhang, H. Hu, L. Shi, B. Jia, H. Huang, X. Han, X. Sun and T. Ma, Strategies for Optimizing the Photocatalytic Water-Splitting Performance of Metal–Organic Framework-Based Materials, Small Sci., 2021, 1, 2100060 CrossRef CAS.
- S. I. Vasylevskyi, K. Regeta, A. Ruggi, S. Petoud, C. Piguet and K. M. Fromm, cis- and trans-9,10-di(1H-imidazol-1-yl)-anthracene based coordination polymers of ZnII and CdII: synthesis, crystal structures and luminescence properties, Dalton Trans., 2018, 47, 596–607 RSC.
- I. Pakamorė, J. Rousseau, C. Rousseau, E. Monflier and P. Á. Szilágyi, An ambient-temperature aqueous synthesis of zirconium-based metal–organic frameworks, Green Chem., 2018, 20, 5292–5298 RSC.
- Y. Luo, G. Calvez, S. Freslon, K. Bernot, C. Daiguebonne and O. Guillou, Lanthanide Aminoisophthalate Coordination Polymers: A Promising System for Tunable Luminescent Properties, Eur. J. Inorg. Chem., 2011, 2011, 3705–3716 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro, version 171.42.49, Rigaku Corporation, Wroclaw, Poland, 2022 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, The HighScore suite, Powder Diffr., 2014, 29, S13–S18 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- H.-J. Lee, P.-Y. Cheng, C.-Y. Chen, J.-S. Shen, D. Nandi and H. M. Lee, Controlled assembly of an unprecedented 2D + 3D interpenetrated array of (4,4)-connected and pcu topologies, CrystEngComm, 2011, 13, 4814–4816 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- T. Steiner, The Hydrogen Bond in the Solid State, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- R. Kruszynski and T. Sierański, Can Stacking Interactions Exist Beyond the Commonly Accepted Limits?, Cryst. Growth Des., 2016, 16, 587–595 CrossRef CAS.
- R. R. Choudhury and R. Chitra, Stacking interaction between homostacks of simple aromatics and the factors influencing these interactions, CrystEngComm, 2010, 12, 2113–2121 RSC.
- P. Karthik, A. R. M. Shaheer, A. Vinu and B. Neppolian, Amine Functionalized Metal–Organic Framework Coordinated with Transition Metal Ions: d–d Transition Enhanced Optical Absorption and Role of Transition Metal Sites on Solar Light Driven H2 Production, Small, 2020, 16, 1902990 CrossRef CAS PubMed.
- K. Fabrizio, K. N. Le, A. B. Andreeva, C. H. Hendon and C. K. Brozek, Determining Optical Band Gaps of MOFs, ACS Mater. Lett., 2022, 4, 457–463 CrossRef CAS.
- Q. Wang and K. Domen, Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. Hu, M. Liu, K. Li, Y. Zuo, A. Zhang, C. Song, G. Zhang and X. Guo, Solvothermal synthesis of NH2-MIL-125(Ti) from circular plate to octahedron, CrystEngComm, 2014, 16, 9645–9650 RSC.
- M. Singh, M. Goyal and K. Devlal, Size and shape effects on the band gap of semiconductor compound nanomaterials, J. Taibah Univ. Sci., 2018, 12, 470–475 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental 1H NMR and FTIR spectra, TGA curves, PXRD pattern comparisons, band gap estimates, structure analysis and crystallographic data. CCDC 2309435–2309443. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00399c |
| This journal is © The Royal Society of Chemistry 2024 |