
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethylxanthines for halogen bonded cocrystals with 1,4-diiodotetrafluorobenzene: green synthesis, structure, photophysics and DFT studies†
Mónica
Benito
 *a,
Rosario
Núñez
*a,
Sohini
Sinha‡
a,
Claudio
Roscini
b,
Yoan
Hidalgo-Rosa
c,
Eduardo
Schott
d,
Ximena
Zarate
*e and
Elies
Molins
a
*a,
Rosario
Núñez
*a,
Sohini
Sinha‡
a,
Claudio
Roscini
b,
Yoan
Hidalgo-Rosa
c,
Eduardo
Schott
d,
Ximena
Zarate
*e and
Elies
Molins
a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: mbenito@icmab.es; rosario@icmab.es
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology (BIST), Campus UAB, Bellaterra, Barcelona 08193, Spain
cCentro de Nanotecnología Aplicada, Facultad de Ciencias, Ingeniería y Tecnología, Universidad Mayor, Camino La Pirámide 5750, Huechuraba, Santiago 8580745, Chile
dDepartamento de Química Inorgánica, Facultad de Química y Farmacia, Centro de Energía UC, Centro de Investigación en Nanotecnología y Materiales Avanzados CIEN-UC, Pontificia Universidad Católica de Chile, Avenida Vicuña Mackenna, 4860 Santiago, Chile
eInstituto de Ciencias Aplicadas, Facultad de Ingeniería, Universidad Autónoma de Chile, Av. Pedro de Valdivia 425, Santiago, Chile. E-mail: ximena.zarate@uautonoma.cl
First published on 14th June 2024
Abstract
Four new halogen-bonded cocrystals of biological methylxanthines, named caffeine, theophylline and theobromine, have been prepared with 1,4-diiodotetrafluorobenzene as a halogen bond donor by mechanochemical and solution processes. For theophylline, N⋯I and N⋯O interactions were observed, while for caffeine and theobromine, only N⋯I was detected. The solids were characterized by PXRD, SC-XRD, FTIR and thermal methods (TGA-DSC analyses). In addition, the solid-state photoluminescence properties of the methylxanthines and their respective cocrystals have been studied and quantum chemistry calculations have been performed to rationalise and understand the electronic and optical properties of all compounds. This work provides a triad of natural methylxanthines capable of forming halogen-bonded multicomponent systems that can give rise to a cocrystal-to-crystal transformation with off–on luminescence activation.
Introduction
Mechanochemistry, and by extension reactive extrusion, has been identified by IUPAC among the ten top emerging technologies that will change the world to make it more sustainable.1 Some of the advantages of the mechanochemical process include the avoidance of large amounts of solvent, quantitative reactions, reduction of waste products, generally shorter time-scale compared to other solvent-based approaches, and mild temperatures. All these benefits have allowed the use of grinding processes in many different areas of application that range from metallurgic to organic synthesis, catalysis or crystal engineering.2–5Crystal engineering has been a considerable tool for tuning the physical properties of compounds. Among supramolecular materials, cocrystals are defined as a class of multicomponent solids wherein neutral components bonded by non-covalent interactions, including hydrogen and halogen bonds, among others, are present within a crystalline compound in a stoichiometric ratio. The halogen bond has been defined as “a neat attractive interaction between an electrophilic region (σ-hole) associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”.6 The halogen bond is also present in biological systems such as protein–ligand systems used in drug delivery. A few iodinated thyroid hormones (thyroxines T4 and T3) are known to behave as halogen-bond donors. Many active pharmaceutical ingredients (APIs) have been designed to contain halogenated atoms in their backbone, not only to provide steric hindrance but also to improve their lipophilicity, which helps to penetrate the cellular membranes and tissues.7,8
In the last few years, some of us have studied the propensity of different modified nucleobases for the preparation of multicomponent solids (salts and cocrystals) containing both hydrogen and halogen bonds.9–12 Now we study xanthines, which are indeed purine-based nitrogen compounds with a structure similar to that of DNA bases adenine and guanine and their related nucleotides. This even makes them important scaffolds for the exploration of new potential drugs.13 Moreover, some natural methylxanthines, such as caffeine (CAF), theophylline (TPH), and theobromine (TBR), are compounds extracted from medicinal plants. While caffeine (1,3,7-trimethylxanthine) is a trimethylated derivate, theophylline (1,3-dimethylxanthine) and theobromine (3,7-dimethylxanthine) are dimethylated and structural isomers, the main difference being the second methyl group at the C1 or C7 position, respectively (see Chart 1). All of them are recognized for their pharmaceutical applications and biological effects.14,15
 | ||
| Chart 1 Chemical structures of natural methylxanthines and DITFB used in this work. | ||
The crystal engineering of these methylxanthines based on hydrogen-bond interactions is a thoroughly explored area. However, to the best of our knowledge, the preparation and structural study of new crystalline solid forms from them, through halogen bonding interactions, has not been reported yet. Therefore, it remains unexplored how the halogen bond interactions between the methylxanthine and a coformer can influence the crystal structure and their physico-chemical properties. In this study, the well-known halogen-bond donor and ditopic 1,4-diiodotetrafluorobenzene (DITFB, see Chart 1) has been chosen as a coformer for cocrystal screening.
In the last decades, the development of new materials has opened up exciting opportunities for modulating and enhancing photoluminescence (PL) properties for various applications. Although traditionally PL has been mainly studied in molecular systems that contain both electron-donor and electron-acceptor moieties,16 more recently, the interest has focused on the modulation of luminescence in the solid state by self-assembly through molecular interactions.17 In the particular case of cocrystals, progressive advances have been reported following the preparation of halogen bonded cocrystals.18–26 Concerning the PL properties of methylxanthines, little information has been found in the literature; while some references to phosphorescence in solution have been reported in the past,27–34 solid state studies are very scarce34 and specific for caffeine or theobromine derivatives.35,36 On the other hand, the use of DITFB as a coformer in combination with fluorescent organic compounds or aromatic model systems has afforded new cocrystals with luminescence features (fluorescence and phosphorescence) in the solid state.18,19,21,23–26 However, it is desirable to extend these studies to understand the fundamental mechanisms of the luminescence process and find suitable, cheap and really accessible molecules for solid-state fluorescent materials, as well as new applications.
With this idea in mind, in the present work, new cocrystals from these natural methylxanthines and DITFB have been prepared by liquid-assisted grinding (LAG). Crystal structure analysis indicates that these precursors can form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 or even 4:1 molar ratio cocrystals through halogen bonds. Herein, their physical characterization by thermal analysis and spectroscopic techniques including FTIR, UV-vis and solid-state emission spectroscopy has been described. Time-dependent density functional theory (TD-DFT) studies were used to understand the change in the luminescence properties of the cocrystals with respect to the parent compounds.
:1, 2:1 or even 4:1 molar ratio cocrystals through halogen bonds. Herein, their physical characterization by thermal analysis and spectroscopic techniques including FTIR, UV-vis and solid-state emission spectroscopy has been described. Time-dependent density functional theory (TD-DFT) studies were used to understand the change in the luminescence properties of the cocrystals with respect to the parent compounds.
Experimental section
All reagents were from commercial sources and used without further purification. Analytical grade solvents were used for the crystallization experiments.Liquid-assisted grinding
Mechanochemical synthesis of cocrystals were performed using a Retsch mixer mill MM400 in 10 mL agate grinding jars with two 5 mm agate balls. Variable amounts (stoichiometric ratios 1:1, 2:1 or 4:1) of the selected methylxanthine and the coformer DITFB were ground for 30 min at 30 Hz upon addition of two drops of the chosen solvent. To avoid any confusion it should be pointed out that only the methylxanthine:DITFB molar ratio, when two cocrystals have been obtained, is included in the naming of the new cocrystals.
Details for each multicomponent system are described below.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1).
A mixture of theobromine (refcode: SEDNAQ39) (75.01 mg, 0.416 mmol) and DITFB (167.24 mg, 0.416 mmol) was placed in the grinding jar with two drops of methanol. The mixture was milled for 30 min at 30 Hz yielding a crystalline solid.
:1).
A mixture of TBR (100.11 mg, 0.555 mmol) and DITFB (111.72 mg, 0.278 mmol) was placed in the grinding jar with two drops of nitromethane. The mixture was milled for 30 min at 30 Hz and yielded a crystalline solid. The solid obtained using the same amounts of TBR and DITFB in methanol and the same conditions was used for growing single crystals by heating in a mixture of methanol–dimethylsulfoxide (DMSO) and ethanol (1:1:10). The solution was filtered using a syringe filter (nylon, 0.2 μm) and allowed to evaporate slowly at rt. Needle-type crystals were obtained after 4 weeks.
:1).
A mixture of theobromine (refcode: SEDNAQ39) (75.01 mg, 0.416 mmol) and DITFB (167.24 mg, 0.416 mmol) was placed in the grinding jar with two drops of methanol. The mixture was milled for 30 min at 30 Hz yielding a crystalline solid.
:1).
A mixture of TBR (100.11 mg, 0.555 mmol) and DITFB (111.72 mg, 0.278 mmol) was placed in the grinding jar with two drops of nitromethane. The mixture was milled for 30 min at 30 Hz and yielded a crystalline solid. The solid obtained using the same amounts of TBR and DITFB in methanol and the same conditions was used for growing single crystals by heating in a mixture of methanol–dimethylsulfoxide (DMSO) and ethanol (1:1:10). The solution was filtered using a syringe filter (nylon, 0.2 μm) and allowed to evaporate slowly at rt. Needle-type crystals were obtained after 4 weeks.
Powder X-ray diffraction (PXRD)
PXRD data were collected using a Siemens D5000 powder X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å), with 40 kV and 39 mA voltage and current applied. An amount of powder was gently pressed on a glass slide to afford a flat surface and then analyzed. The samples were scanned in the 2θ range of 2–50° using a step size of 0.02° and a scan rate of 1 s per step.Single crystal X-ray diffraction (SC-XRD)
Single crystals of compounds CAF-DITFB, TPH-DITFB and TBR-DITFB (2:1) were selected and mounted for X-ray single crystal diffraction experiments. Crystallographic data were collected on a Bruker APEX-II CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Crystallographic data were collected at 294(2) K. Data reduction was performed using SAINT V6.45A, SORTAV,40 and SADABS41 in the diffractometer package. The structural resolution procedure was made using SHELXT42 and the refinement by least squares on F2 was performed using SHELXL2014/7 inside the WinGX program environment.43,44 For CAF-DITFB, although several crystallizations were performed and after checking different crystals, the best dataset for CAF-DITFB was selected for the X-ray study. A major component was integrated, although other minor components were observed in the frame images. The full set of data was used in the refinement (even the high Rint value), which afforded the best results with a reasonable quality of the final crystal structure. Non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Hydrogen atoms were introduced in calculated positions and refined riding on their parent atoms. Selected crystal and data collection parameters are reported in the corresponding Table 1.
:1)
| Compound | CAF-DITFB | TPH-DITFB |
TBR-DITFB (2:1) |
|---|---|---|---|
| Empirical formula | C19 H20 F2 I N8 O4 | C13 H8 F4 I2 N4 O2 | C20 H16 F4 I2 N8 O4 |
| Formula weight | 589.33 | 582.03 | 762.21 |
| Temperature (K) | 294(2) | 294(2) | 294(2) |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions | a = 21.904(18) Å | a = 19.924(8) Å | a = 4.0269(18) Å |
| b = 4.044 (4) Å | b = 4.4267(18) Å | b = 7.451(3) Å | |
| c = 25.87(2) Å | c = 20.419(8) Å | c = 20.660(9) Å | |
| α = 90° | α = 90° | α = 82.022(8)° | |
| β = 110.90(2)° | β = 113.293(9)° | β = 88.669(8)° | |
| γ = 90° | γ = 90° | γ = 80.671(9)° | |
| Volume (Å3) | 2140(3) | 1654.1(11) | 605.8(5) |
| Z | 4 | 4 | 1 |
| Density calc. (mg m−3) | 1.829 | 2.337 | 2.089 |
| Absorption coefficient (mm−1) | 1.561 | 3.860 | 2.673 |
| F(000) | 1172 | 1088 | 366 |
| Crystal size (mm3) | 0.32 × 0.07 × 0.06 | 0.28 × 0.05 × 0.02 | 0.12 × 0.08 × 0.07 |
| Theta range for data collection (°) | 1.623 to 28.944 | 1.837 to 28.019 | 1.991 to 28.506° |
| Index ranges | −29 <= h < 29, −5 <= k <= 5, −34 <= l <= 35 | −26 <= h <= 26, −5 <= k <= 5, −26 <= l <= 26 | −5 <= h <= 5, −9 <= k <= 9, −27 <= l <= 27 |
| Reflections collected | 58277 |
32965 |
12902 |
| Independent reflections | 5576 [R(int) = 0.3691] | 3979[R(int) = 0.0509] | 3074 [R(int) = 0.0698] |
| Completeness to θ max (%) | 100.0% | 100.0% | 100% |
| Max. and min. transmission | 1 and 0.54 | 1 and 0.695 | 1 and 0.618 |
| Refinement method | Full-matrix least-squares on F2 | ||
| Data/restraints/parameters | 5576/0/313 | 3979/0/228 | 3074/0/174 |
| Goodness-of-fit on F2 | 0.970 | 1.021 | 1.008 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0715, wR2 = 0.1398 | R 1 = 0.0306, wR2 = 0.0676 | R 1 = 0.0529, wR2 = 0.1238 |
| R indices (all data) | R 1 = 0.1566, wR2 = 0.1686 | R 1 = 0.0453, wR2 = 0.0748 | R 1 = 0.0850, wR2 = 0.1410 |
| Largest diff. peak and hole (e Å−3) | 1.095 and −0.936 | 0.568 and −0.798 | 1.199 and −1.623 |
| CCDC no. | 2304678 | 2304676 | 2304677 |
Mercury 2021.1.0 software was also used to calculate the PXRD powder patterns of the new cocrystals based on the single-crystal X-ray structures and to predict the Bravais, Friedel, Donnay and Harker (BFDH) morphology using the CSD Materials module.45
Complete crystallographic data for the structural analysis have been deposited to the Cambridge Crystallographic Data Centre, CCDC no. 2304676–2304678.
Thermogravimetric analysis – differential scanning calorimetry (TGA-DSC)
A simultaneous thermogravimetric analysis (TGA)-differential scanning calorimetry/differential thermal analysis (heat flow DSC/DTA) system NETZSCH-STA 449 F1 Jupiter was used to study the thermal properties of the solids. Samples (3–8 mg) were placed in an open alumina pan and measured at a scan speed of 10 °C min−1 from ambient temperature to 300 °C under a N2 atmosphere as protective and purge gas (their respective flow velocities were 20 and 40 mL min−1).Spectroscopic measurements
A Jasco 4700LE spectrophotometer with an attenuated total reflectance accessory was used to record the FT-IR spectra of the starting products and the new cocrystals in the range from 4000 to 400 cm−1 and at a resolution of 4.0 cm−1 and 32 scans. Fluorescence images were taken using an Olympus BX51 microscope with a DP20 camera and a mercury lamp U-RFL-T accessory for the sample excitation in the UV-visible region. Diffuse reflectance analyses were performed using a Jasco UV-vis-NIR V-780 spectrophotometer with a diffuse reflectance sphere accessory in the scan range of 200–800 nm. The diffusion reflectance was converted in the F(R) function through the Kubelka–Munk law. Steady-state luminescence spectra and fluorescence quantum yields for all the compounds in the solid state were measured on a Hamamatsu absolute PL quantum yield spectrometer C9920-02G.Theoretical calculations
:1, 2:1, or 4:1 for TPH-DITFB, TBR-DITFB (2:1), and CAF-DITFB, respectively. The initial configuration was derived from the optimized crystal structure of each of these cocrystals. (see Fig. S1a†). In the case of free methylxanthine, the structural model for the simulations was created using the available crystallographic data for caffeine, theophylline, and theobromine (see Fig. S1b†).
The electronic structure for all the electronic states (S0, S1 and T1) were computed using the Perdew–Burke–Ernzerhof (PBE) function52 and the basis set def2-TZVPP.53,54 The ma-def2-SVP basis set with the Def2-ECP pseudopotentials was used to treat iodine atoms. The RIJ approach for the Coulomb term with the “chain of spheres” COSX approximation and their related auxiliary basis sets were used to speed up the SCF computation.55,56 A time-dependent DFT (TD-DFT) method51 was used to simulate the optical properties (absorption) of all compounds. The PBE057 hybrid exchange correlation and the def2-TZVPP53 basis set were used to compute the absorption spectra for all systems.
Results and discussion
Synthesis of cocrystals and powder X-ray diffraction
Liquid-assisted grinding was used as a fast and reliable method for solid phase screening. To confirm the phase purity of the bulk solid obtained by LAG, powder X-ray diffraction analysis was performed for all the samples. The results are shown in Fig. 1 and 2. New phases were obtained for CAF or TPH when co-crystallized with the donor coformer in the presence of methanol, in 4:1 (CAF-DITFB) and 1:1 (TPH-DITFB) molar ratios, respectively. Further, the peak positions were correlated with the corresponding simulated powder patterns from the crystal structures for these two compounds confirming the great agreement for all of them (Fig. 1a and b, respectively).
 | ||
| Fig. 1 PXRD patterns of former compounds and the new cocrystals (experimental and calculated from crystal structures) for a) CAF-DITFB and b) TPH-DITFB systems. | ||
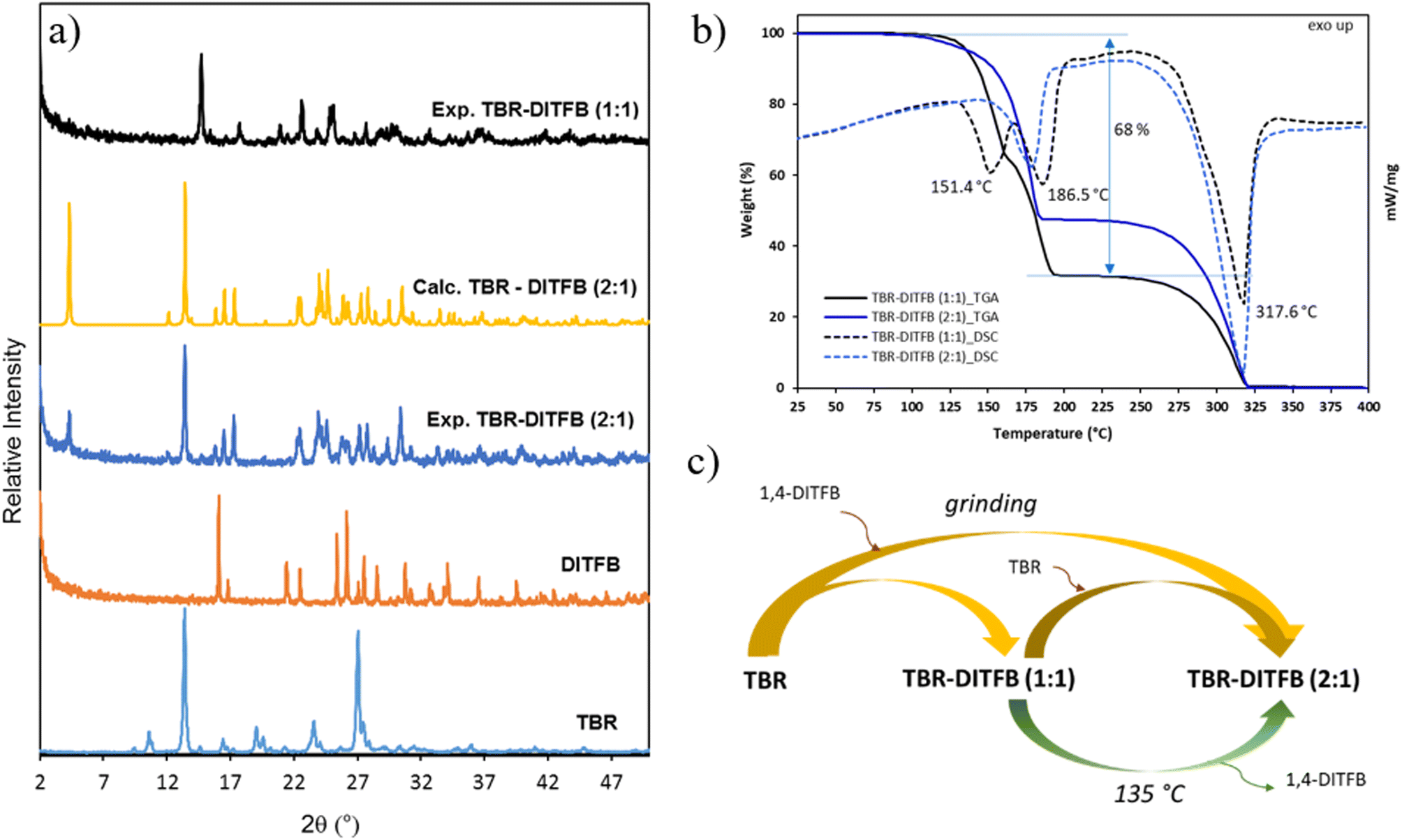 | ||
| Fig. 2 a) PXRD patterns of starting materials (TBR and DITFB) and the new cocrystals (experimental and calculated from crystal structures); b) TGA (solid lines)-DSC (dashed lines) traces of cocrystals TBR-DITFB (1:1) and TBR-DITFB (2:1) and c) phase transformations among TBR and its cocrystals. | ||
In the case of the TBR-DITFB system, the grinding process at a 1:1 ratio, with a few drops of methanol, afforded the cocrystal TBR-DITFB (1:1), as suggested by a powder pattern, which was different from the pristine compounds. On the other hand, grinding TBR-DITFB at a 2:1 ratio, the process did not proceed effectively as the powder pattern of the bulk solid contained a mixture of the same phase observed previously for the TBR-DITFB (1:1) system, along with the phase corresponding to the starting TBR.
Surprisingly, when the process was carried out in nitromethane, chloroform, or dichloromethane as catalytic solvents, the process was quantitative and a new phase, corresponding to the cocrystal TBR-DITFB (2:1), was yielded, as shown in Fig. 2a.
Finally, from a mixture of methanol, EtOH and DMSO, it was possible to isolate single crystals of the corresponding new TBR-DITFB (2:1) cocrystal.
Unfortunately, despite the efforts performed to isolate single crystals of the TBR-DITFB (1:1) cocrystal, they could not be obtained. TBR showed very low solubility in many common solvents and in all cocrystal syntheses a residual part of non-cocrystallized TBR was always found even when working in an excess of DITFB. This was ascribed to the different solubility between TBR and DITFB and the thermal stability of the cocrystals, as it will be commented later in the thermal analysis section.
Structural description of single crystals
Cocrystal CAF-DITFB crystallizes in the monoclinic space group P21/c with two different molecules of caffeine and half a molecule of DITFB in the asymmetric unit (Fig. 3a). At first sight, self-assembled caffeine molecules are linked through hydrogen bonds established between the carbonyl groups and C–H groups as no N–H moieties are available in this trimethylated xanthine. This yields zigzag chains, which are also bridged by halogen bonds with the coformer through N⋯I interactions (Fig. 3c). Additionally, zig-zag chains of caffeine molecules are also assembled like in DNA. All these interactions are the responsible for the final 4:1 molar ratio observed in the CAF-DITFB cocrystal, showing a needle habit in agreement with the BFDH predicted morphology (Fig. 3b and d). Although not very common, recently we reported another cocrystal containing the same molar ratio with 5-fluorouracil and DITFB.12
 | ||
| Fig. 3 a) ORTEP with atom numbering. Note only half of the DITFB molecule contains atom labelling, indicating the equivalence of the other half, and thus, giving a 4:1 molar ratio, b) BFDH predicted morphology, c) hydrogen and halogen bonding interactions in the packing (view along the b axis). Additionally, self-assembly of other caffeine molecules giving a zigzag chain, and d) perspective view along the a axis of cocrystal CAF-DITFB. | ||
Compound TPH-DITFB crystallizes from a methanol solution as needles, in agreement with the BFDH predicted morphology (Fig. 4b), in the monoclinic space group P21/n, with one molecule of TPH and one molecule of the ditopic coformer (Fig. 4a). TPH molecules are self-assembled by hydrogen bonds between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(6)⋯H(7)–N(7) atoms. Each TPH molecule in the dimer is bridged to two molecules of DITFB through halogen bonds by the same carbonyl group (CO(6)⋯I(2)) as well as by N(9)⋯I(1) interactions, leading to twisted tapes (Fig. 4c). These tapes are interconnected through the C(1)–H(1A)⋯O(2) and C(1)–H(1B)⋯O(2) hydrogen bonds and C(3)–H(3A)⋯I(1) and C(3)–H(3B)⋯I(2) halogen bond interactions which afford the final tridimensional packing. Distances and angles for the halogen bond interactions observed for these two cocrystals, CAF-DITFB and TPH-DITFB, are shown in Table 2, while H-bond interactions are summarized in Table S1 in the ESI.†
O(6)⋯H(7)–N(7) atoms. Each TPH molecule in the dimer is bridged to two molecules of DITFB through halogen bonds by the same carbonyl group (CO(6)⋯I(2)) as well as by N(9)⋯I(1) interactions, leading to twisted tapes (Fig. 4c). These tapes are interconnected through the C(1)–H(1A)⋯O(2) and C(1)–H(1B)⋯O(2) hydrogen bonds and C(3)–H(3A)⋯I(1) and C(3)–H(3B)⋯I(2) halogen bond interactions which afford the final tridimensional packing. Distances and angles for the halogen bond interactions observed for these two cocrystals, CAF-DITFB and TPH-DITFB, are shown in Table 2, while H-bond interactions are summarized in Table S1 in the ESI.†
 | ||
| Fig. 4 a) ORTEP with atom numbering, b) BFDH predicted morphology, c) hydrogen and halogen bonds in tapes along the b axis and d) tapes (perspective view along the c axis) of cocrystal TPH-DITFB. | ||
Cocrystal TBR-DITFB (2:1) also crystallized as needles from a methanol solution in agreement with the BFDH predicted morphology (see Fig. 5a). It belongs to the triclinic space group P, with one molecule of TBR and half a molecule of the halogen bond donor coformer in the asymmetric unit, Fig. 5b. In this case, self-assembly between two TBR molecules is also observed through the hydrogen bond interactions N(1)–H(1)⋯O(6), C(7)–H(7A)⋯O(2) and C(7)–H(7C)⋯O(6), which yielded zigzag tapes of TBR molecules. These are bridged by only one type of halogen bond between N(9) and I(1) atoms through the N(9)⋯I(1) interaction (see Table 2 for distances and angles), forming first a folded structure which resembles a ladder where the TBR molecules represent its steps (see Fig. 5c). Finally, the tridimensional packing is promoted by additional F⋯F interactions between DITFB molecules from different chains, F⋯H–C interactions with the methyl groups of TBR, and π–π stacking between coformer rings or xanthine molecules (Fig. 5d).
 | ||
| Fig. 5 a) ORTEP with numbering. Note only half of the DITFB molecule contains atom labelling, indicating the equivalence of the other half, and thus, giving a 2:1 molar ratio, b) BFDH predicted morphology, c) hydrogen and halogen bonds in tapes and d) perspective view of the ladders of cocrystal TBR-DITFB (2:1). | ||
Thermal analysis
The thermal behavior of the new cocrystals and the starting materials was studied by simultaneous TGA-DSC. No mass loss was observed before melting, confirming that the four cocrystals are unsolvated and with the component ratio confirmed by SC-XRD. In Table 3 the melting points (Mp) of precursors and cocrystals are collected. The DSC trace for cocrystal CAF-DITFB shows two endothermic peaks at Tpeak = 165.9 and 236.6 °C. The loss weight of 31% observed up to approximately 170 °C was associated with the removal of the donor coformer (theoretical amount of weight 34.1%) of the 4:1 CAF-DITFB cocrystal. A second step of 67% weight loss, corresponding to the endo peak of the DSC trace at 236 °C, was ascribed to the melting-decomposition of caffeine (Mp = 236 °C, from the literature) (Fig. S2†).58
| Compound | Mp (°C) | Cocrystal | T peak (°C) |
|---|---|---|---|
| DITFB | 108–110 | ||
| CAF | 236 | CAF-DITFB | 165.9 and 236.6 |
| TPH | 273 | TPH-DITFB | 148.0, 164.8 and 271.5 |
| TBR | 357 |
TBR-DITFB (1:1) |
151.4, 186.5 and 317.6 |
|
TBR-DITFB (2:1) |
178.1 and 317.5 |
For cocrystal TPH-DITFB, in the DSC, two sharp endothermic peaks were observed, at a Tpeak of 148.0 °C, with a wide shoulder at 164.8 °C, and 271.5 °C (Fig. S3†). The weight loss measured (68.9%) in the TGA analysis in agreement with the first event at 148.0 °C, matches well with the removal of a molecule of DITFB (theoretical weight amount of 69%). The latter endothermic peak corresponds to the melting decomposition of TPH (Mp = 273 °C).54
In the DSC of the compound TBR-DITFB (1:1), two endothermic peaks at around 151.4 and 186.5 °C were observed and the TGA trace suggests a two-step weight loss (68% in total) ascribed to the removal of the halogenated coformer (Fig. 2b). The last endothermic signal (at 317.6 °C) was attributed to the decomposition of TBR.59
Finally, the TGA-DSC for compound TBR-DITFB (2:1) showed a single endothermic peak at 178.1 °C with the expected weight loss of 52.4%, corresponding to the weight of the DITFB coformer.
Phase transformations
Phase transformation between cocrystals with different stoichiometric ratios are scarcely studied although a few examples have been described in the literature.60–62 Interestingly, these multicomponent solids are expected to show different physical properties depending on the phase, which extends the plethora of the properties of the solid forms available.In view of our results, we aimed to study the relationship between TBR and its cocrystals (TBR-DITFB (1:1) and (2:1)) and their interconversion through grinding or sublimation, as shown in Fig. 2c. When the two precursors (TBR and DITFB) were ground together using the appropriate molar ratio (1:1 or 2:1) and solvent (methanol or chloroform, respectively), two different cocrystals were obtained. Also, solid phase transformation from the cocrystal TBR-DITFB (1:1) to the cocrystal TBR-DITFB (2:1) was possible upon the extra addition of TBR while grinding in chloroform. In methanol, a mixture of both cocrystals resulted from the extra addition of TBR. On the other hand, by heating the TBR-DITFB (1:1) cocrystal at 135 °C, the formation of long needles at the top of the vial was observed, which through single-crystal X-ray diffraction were assigned to the halogenated coformer DITFB.
In a similar way, by heating compounds CAF-DITFB and TPH-DITFB at 165 °C and 130 °C, respectively, long needles were noticed at the top of the vial, which were ascribed to the DITFB compound (Fig. S4†). The residual solid was further analyzed by PXRD and the observed phases confirmed the presence of the precursors β-CAF along with some remaining cocrystal or TPH form II. This result confirmed that for TPH the TPH-DITFB (2:1) cocrystal could not be obtained (Fig. S3b†).
FT-IR spectroscopic analysis of cocrystals
FT-IR was performed to i) corroborate the presence of both components, i.e. methylxanthines and DIFTB and to ii) ensure the presence of hydrogen and halogen bonds in the cocrystals, taking advantage of the variation of their more characteristic vibrational modes. The vibrational modes for the free coformer DIFTB appeared at 1456 (νC–C stretching aromatic ring), 937 (νC–F stretching) and 755 (νC–I asymmetric stretching) cm−1. Some of these stretching modes appeared slightly shifted in the FT-IR spectra of the new cocrystals (see Fig. S5 and Table S2†) suggesting the existence of N⋯I and O⋯I interactions already observed by SC-XRD.For the methylxanthines, the changes of CO, CN and N–H modes were examined.
By careful analysis of the single-crystal structure of the starting β-CAF, no H-bonds could be detected, while π–π stacking and other short interactions were present. In the cocrystal CAF-DITFB, higher wavenumbers for the CO and CN modes were recorded, suggesting that some kind of new interaction for these functional groups has taken place. SC-XRD showed that self-assembly occurred through the carbonyl and C–H groups. The halogen bonds between the only available nitrogen, N(27), and the ditopic coformer were responsible for these shifts (Fig. S5a†).
TPH form II (refcode BAPLOT06) self-assembles through N–H⋯N synthons. In a similar way, in the cocrystal TPH-DITFB, the self-assembly also occurred but through N–H⋯OC interactions. This is reflected by a shift to lower frequencies of the vibrational modes corresponding to CO and CN stretchings, which are also involved in the halogen bond interactions (CO(6)⋯I(2) and N(9)⋯I(1)) (as shown in Fig. 4c).
Finally, for TBR (refcode SEDNAQ), self-assembly occurred through N(1)–H⋯O(2)C interactions. Despite the crystal structure not being obtained, in the FT-IR spectrum of cocrystal TBR-DITFB (1:1), an increase of wavenumbers corresponding to the C=O stretching was observed, suggesting that this group was also involved in the new network formed by hydrogen and halogen bonds. Finally, for the TBR-DITFB (2:1) cocrystal, a shift to higher wavenumbers for the CO and imine bands was observed, due to the establishment of hydrogen and halogen bonds, as described in the previous SC-XRD section. For all these compounds, in the region 3100–2900 cm−1, corresponding to N–H modes, some peak shifts were also observed and ascribed to these interactions.
Absorption and luminescence properties
In order to study the effects of cocrystallization and the molecular packing on the optical features, the absorption and emission properties of the solid compounds were studied.Diffuse reflectance (and corresponding absorption) (DR-UV-vis) and fluorescence emission (FE) spectra of the pristine methylxanthines and their corresponding cocrystals were measured in the solid state (Fig. 6 and S6†). Both cocrystals and methylxanthines showed absorption in the UV region. In all cases, the bands of the cocrystals slightly broadened with respect to the solid pure xanthine forming a tail up to 350 nm, suggesting a variation of the intermolecular interactions in the cocystals.
 | ||
| Fig. 6 Absorption and fluorescence emission spectra of CAF and CAF-DITFB cocrystals (a and b) and TBR and its cocrystals TBR-DITFB (1:1) and TBR-DITFB (2:1) (c and d). | ||
The three methylxanthines exhibited blue emission (λmax = 402, 450, and 455 nm) with different absolute PL quantum yield values (ΦPL = 54.7, 12.4, and 53.6%), whereas the emission considerably dropped after the formation of the corresponding cocrystals (Table 4). CAF showed a maximum emission band at 402 nm when excited at 350 nm, while in the cocrystal CAF-DITFB the emission band was slightly red-shifted to 406 nm upon excitation at 345 nm (Fig. 6a). It is important to stress the significant decrease of the ΦPL after the introduction of DITFB to form the cocrystal with respect to the pristine CAF (54.7% versus 2.4%). In the case of TPH, excitation at 330 nm afforded an emission band at 450 nm with a ΦPL of 12.4%. The formation of the cocrystal TPH-DITFB produced a significant decrease of the luminescence and negligible ΦPL (Table 4). Remarkably, TBR exhibited blue emission with a maximum at 455 nm upon excitation at 330 nm and a high ΦPL value of 53.6%. Both cocrystals TBR-DITFB (1:1) and TBR-DITFB (2:1) exhibited a blue-shift of the emission bands (at around 404–406 and 449–451 nm, respectively) when compared to the pristine TBR and, as for the previous cocrystals, a large decrease of the ΦPL values (Table 4 and Fig. 6d). Notably, TPH showed much lower luminescence quantum efficiency than its analogues CAF and TBR, which in turn presented similar quantum efficiency in the solid state. The large variation of the fluorescence intensity between the pristine compounds and the corresponding crystals could be easily observed even through fluorescence microscopy images, under UV irradiation (330–385 nm range), as shown in Fig. 6 (insets).
| λ ex (nm) | λ em (nm) | Φ (%) | |
|---|---|---|---|
| CAF (β polymorph) | 350 | 402 | 54.7 |
| CAF-DITFB | 345 | 406 | 2.4 |
| TPH (form II) | 330 | 450 | 12.4 |
| TPH-DITFB | 350 | 429 | <0.2 |
| TBR | 330 | 405, 455 | 53.6 |
| TBR-DITFB (1:1) |
346 | 404, 451 | 3.7 |
| TBR-DITFB (2:1) |
346 | 406, 449 | 8.2 |
In general, the important quenching of luminescence that occurs after the formation of the cocrystals, compared to the corresponding methylxanthines, evidences that a) the interactions between the coformer DITFB and the different methylxanthines are established and b) the incorporation of DITFB molecules in the network does not improve the luminescence properties of the methylxanthines in the solid state, probably due to the overall dilution of the intermolecular interactions among methylxanthines (vide infra). Furthermore, as described in the thermal analysis section, since these cocrystals display cocrystal-to-crystal transformations by heating (Fig. S4†), a new system with thermally-induced off–on luminescence modulation was achieved, which might be of interest for the optical detection of the starting natural methylxanthines in optoelectronics or biological applications.
Computational results
:1) the cell parameters are α = 81.99°, β = 80.63°, γ = 88.66° and a = 4.10 Å, b = 7.42 Å and c = 20.59 Å. These results are in excellent agreement with the crystallographic data obtained for each of all three cocrystals, see Table S3.† In all three systems, the computed halogen bond interactions between methylxanthines and DITFB through N⋯I bonds are in the range from 2.80 Å to 2.96 Å, being the experimental values for the cocrystal, between 2.86 Å and 3.03 Å. Besides, the calculations reveal similar results regarding the C–I⋯A bond angles, which are between 174.4° and 176.6°. These values agree with the experimental crystallographic data, which are in a range from 175.4° to 177.1°, see Table S4.†
Optical properties of free methylxanthines and cocrystals
To have a benchmark for our results, the energies of the states of the isolated methylxanthines were calculated, using as references the previously reported experimental data. The UV-vis absorption spectra were calculated using the input data, the optimized structures of free methylxanthines, i.e., CAF, TPH and TBR. From these results, we found that the main calculated absorption bands are centered at 259, 255 and 251 nm for CAF, TPH and TBR, respectively. The oscillator strength (f) for these transitions agrees between the simulated UV-vis absorption spectra of CAF, TPH and TBR and the previously reported experimental results (λmax = 299, 256 and 291 nm, for CAF, TPH and TBR, respectively).63,64 The observed theoretical value variations from the experimental results of around 40 nm are within the TD-DFT error range that is commonly reported.64–66 In addition, the mentioned electronic transitions are characterized by π-type transitions, as indicated by the active molecular orbitals (see Table S5†). Thus, these three methylxanthines are characterized by π–π* transitions, which involve charge transfer (CT) between π-type molecular orbitals (MOs) with an important contribution from the nitrogen lone pairs.Based on previous studies, we have shown how crucial it is to consider the first excited electronic states (S1 or T1) to accurately estimate the luminescence properties in molecular systems66–68 and materials like MOFs.69,70 Fig. S7† illustrates the most probable emission pathway for the free methylxanthines considering the energy of their ground and first excited electronic states. The frontier molecular orbital (FMO) analysis revealed that the MOs involved in the emissive states of the free CAF, TBR and TPH are located on the core of methylxanthine, i.e., MOs located along the rings, both pyrimidine and imidazole, of the same monomer. Fig. S8–S10† display the FMOs of CAF, TBR and TPH, respectively. It should be noted that the characteristic emissive state is observed for both S1 and T1 electronic states.
The theoretical analysis of the optical properties of the herein studied cocrystals was performed using a finite fragment. The UV-vis absorption spectrum for CAF-DITFB showed two intense absorption bands, very close in energy, which appeared localized at 256 and 257 nm. The calculations showed that these bands are π-type electronic transitions. Also, it was found that the active MOs in these electronic transitions are localized on the CAF unit (Table S5†). However, when the FMOs of this system were analyzed, it appeared that the electron density of the lowest unoccupied molecular orbital (LUMO) is distributed on the DITFB unit (see Fig. S11a†). The simulated UV-vis absorption spectra of TPH-DITFB and TBR-DITFB showed their main absorption bands between 200 and 300 nm, which agree with the experimental data (see Table 5). In this sense, for both cocrystals the calculations showed that the MOs involved in this transition are localized on the xanthine moieties i.e., THP and TBR, respectively, see Table S5.† The FMOs of these two cocrystals, similar to that of the CAF-DIFTB system, also revealed that the HOMO is located on the methylxanthine unit, i.e., TPH and TBR, whereas the LUMO is a MO distributed on the DITFB unit (Fig. S11b and c†).
| System | E HL(eV) | E(eV) | λ(nm) | f | Assignment | Transition | Weight |
|---|---|---|---|---|---|---|---|
| Excitation wavelength (λ/nm), energy (E/eV), oscillator strength (f) and the corresponding molecular orbitals (MOs) involved in the electronic transitions, as also the band assignment. The HOMO–LUMO energy differences (EHL/eV) are also included. | |||||||
| CAF-DITFB | 3.79 | 4.83 | 257 | 0.32 | π → π* | H-1 → L + 6 | (44%) |
| H → L + 7 | (43%) | ||||||
| 4.86 | 255 | 0.43 | π → π* | H-3 → L + 4 | (44%) | ||
| H-2 → L + 5 | (44%) | ||||||
| CAF | 5.29 | 4.79 | 259 | 0.52 | π → π* | H-1 → L | (90%) |
| TPH-DITFB | 5.08 | 4.75 | 249 | 0.29 | π → π* | H → L + 1 | (84%) |
| TPH | 5.40 | 4.86 | 255 | 0.23 | π → π* | H → L + 1 | (83%) |
| 4.94 | 251 | 0.18 | π → π* | H-1 → L | (82%) | ||
| TBR-DITFB | 5.39 | 4.85 | 255 | 0.67 | π → π* | H-1 → L | (30%) |
| π → π* | H → L + 1 | (32%) | |||||
| 5.18 | 239 | 0.23 | π → π* | H-1 → L + 1 | (70%) | ||
| TBR | 4.87 | 4.93 | 251 | 0.14 | π → π* | H-2 → L | (90%) |
| 247 | 0.28 | π → π* | H → L + 2 | (83%) | |||
As shown for the free methylxanthines, we have also considered the most probable emission pathway for each cocrystal taking into account the electronic configuration of their S0, S1 and T1, (Fig. S12†). According to the experimental emission spectra, it seemed that the incorporation of DITFB in the cocrystal network significantly contributed to a change in the photoluminescence of the cocrystals with respect to the free methylxanthines. Thus, for example, the quantum yield for the CAF-DITFB cocrystal exhibited a noticeable reduction in comparison to the free CAF. Therefore, we expected to see a significant contribution of DITFB to the CAF-DITFB cocrystal's luminescence properties. With this aim, the electronic configuration of the S1 and T1 electronic states of CAF-DITFB were investigated to fully understand the deactivation mechanism. The FMOs analysis of the S1 and T1 electronic states of the cocrystal is summarized in Fig. S12.† The single occupied molecular orbital (SOMO) is an orbital where the electron density is located on the DITFB unit. However, the FMO analysis showed that this excited electron relaxes to a MO distributed mostly along the DITFB unit and partially on the CAF units in both configurations (S1 and T1). It should be noted that the T1 electronic configuration for CAF-DITFB is not populated under the experimental conditions. According to Kasha's rule, the lowest excited electronic state of a given multiplicity leads to the radiative deactivation of the excited state. Therefore, in the CAF-DITFB system the emission drops due to a CT process, which is faster than the radiative deactivation of the excited state (implying a CT channel from the CAF to DITFB orbital). The electron relaxes after the photoexcitation process and enters the first excited electronic state, with a significant contribution from DITFB, which shows a weak fluorescence.21
Following the same procedure, the first excited states S1 and T1 were also investigated for the TPH-DITFB and TBR-DITFB systems. The S1 and T1 electronic states of these cocrystals exhibited a MO with mix distribution on DITFB as well as the TPH and TBR molecules, as observed before for CAF-DITFB, see Fig. S14 and S15.†
Based on these results, as well as the Franck–Condon principle, Kasha's rule, and selection rules, we propose that the MO composition of the first excited electronic state involves MOs distributed mostly on the DITFB unit and with a minor component on the methylxanthines in the emissive state. This phenomenon contributes to a luminescence quenching effect, highlighting the significant impact of DITFB on the modulation of the emission in the studied cocrystals. These findings shed light on the interaction of molecular components and provide valuable insights for further understanding of the factors influencing luminescence in this system. These results lead us to the conclusion that incorporation of DITFB does not favor the emission process in these cocrystals, producing luminescence quenching.
Conclusions
Over the past few years, we have paid attention to the preparation of cocrystals and salts of active pharmaceutical ingredients or structural synthons mostly based on hydrogen-bonds, with interest in the pharmaceutical industry. Herein, we have reported the synthesis and characterization of new halogen bonded cocrystals based on three natural methylxanthines, caffeine, theophylline and theobromine, and the well-known 1,4-diiodotetrafluorobenzene. The several stoichiometric ratios obtained for these cocrystals (1:1 for TPH-DITFB, 1:1 and 2:1 for TBR-DITFB and 4:1 for CAF-DITFB) remark the importance of continuing exploration of these types of multicomponent solid forms and their physicochemical properties. Although the fluorescence of the prepared new cocrystals is greatly diminished probably as a consequence of the molecular packing and interactions with the coformer, the isolated methylxanthines have interestingly revealed excellent luminescence properties with high PL quantum yields in the solid state. This suggests that methylxanthines can be considered suitable candidates for future luminescent materials as they are industrially produced and readily available. Moreover, our cocrystals displayed cocrystal-to-crystal transformations by heating and represent a new thermally-induced off–on luminescent system, allowing for solid thermofluorescent switches and the identification of these precursors. Furthermore, by means of TD-DFT calculations, the decrease of the photoemission observed with respect to the free methylxanthines and the herein proposed cocrystals seems to indicate that this effect was due to the generation of a molecular orbital in the excited electronic state configuration with a mixed composition of DITFB, as the major component, and the corresponding methylxanthine. Therefore, the introduction of DITFB does not enhance the emission process within these cocrystals, due to the possible CT process between the methylxanthine and the coformer.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MCIU (PID2021-1245720B-C32 and PID2022-136892NB-I00), MICINN (PID2019-106832RB-I00/AEI/10.13039/501100011033). M. B., R. N., S. S. and E. M. acknowledge financial support from the State Investigation Agency, through the Severo Ochoa Programme for Centres of Excellence (CEX2019-00917-S project, CEX2023-001263-S). The ICN2 is supported by the Severo Ochoa Centres of Excellence programme, Grant CEX2021-001214-S, funded by MCIN/AEI/10.13039.501100011033. R. N. and S. S. thank Generalitat de Catalunya (AGAUR 2021-SGR-00442 project). This work was supported by grant TED2021-131709B-I00 funded by MCIN/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. We are also thankful for the financial support from ANID/Chile under Projects ANID Postdoctoral 3230141, FONDECYT 1201880, FONDECYT 1231194, ANID/FONDAP/1523A0006; Millennium Science Initiative Program – NCN2021_090, and the Anillos de Ciencia y Tecnología ACT210057. S. S. acknowledges financial support from DOC-FAM, European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 754397. The authors thank the X-ray diffraction, thermal analysis and spectroscopic services from the ICMAB.References
- X. Fernando Gomollón-Bel, Ten Chemical Innovations That Will Change Our World, Chemistry International, 2019, 41(2), 12–17, DOI:10.1515/ci-2019-0203.
- J. L. Howard, Q. Cao and D. L. Brown, Mechanochemistry as an emerging tool for molecular synthesis: what can it offer?, Chem. Sci., 2018, 9(12), 3080–3094, 10.1039/C7SC05371A.
- S. Hwang, S. Grätz and L. Borchardt, A guide to direct mechanocatalysis, Chem. Commun., 2022, 58, 1661–1671, 10.1039/D1CC05697B.
- D. Braga, L. Maini and F. Grepioni, Mechanochemical preparation of co-crystals, Chem. Soc. Rev., 2013, 42, 7638–7648, 10.1039/C3CS60014A.
- G. Bolla and A. Nangia, Pharmaceutical cocrystals: walking the talk, Chem. Commun., 2016, 52, 8342–8360, 10.1039/C6CC02943D.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Definition of the halogen bond (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1711–1713, DOI:10.1351/PAC-REC-12-05-10.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Halogen bonds in biological molecules, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794, DOI:10.1073/pnas.0407607101.
- V. Govindaraj, H. Ungati, S. Jakka, S. Bose and G. Mugesh, Directing traffic: halogen-bond-mediated membrane transport, Chem. – Eur. J., 2019, 25, 11180–11192, DOI:10.1002/chem.201902243.
- Y. Roselló, N. Bagués, N. Martínez, A. Moradell, M. Benito, I. Mata, J. Galcerà, M. Barceló-Oliver, A. Frontera and E. Molins, 9-Ethyladenine: Mechanochemical Synthesis, Characterization, and DFT Calculations of Novel Cocrystals and Salts, Cryst. Growth Des., 2020, 20, 2985–2997, DOI:10.1021/acs.cgd.9b01628.
- Y. Roselló, M. Benito, E. Molins, M. Barceló-Oliver and A. Frontera, Adenine as a Halogen Bond Acceptor: A Combined Experimental and DFT Study, Crystals, 2019, 9, 224, DOI:10.3390/cryst9040224.
- Y. Rosselló, M. Benito, M. Barceló-Oliver, A. Frontera and E. Molins, 1-Ethyluracil, a New Scaffold for Preparing Multicomponent Forms. Synthesis, Characterization and Computational Studies, Cryst. Growth Des., 2021, 21, 4857–4870, DOI:10.1021/acs.cgd.1c00175.
- M. Benito, Y. Rosselló, M. Barceló-Oliver, A. Frontera and E. Molins, Uracil Derivatives for Halogen-bonded Cocrystals, Int. J. Mol. Sci., 2021, 22, 10663, DOI:10.3390/ijms221910663.
- N. Singh, A. K. Shreshtha, M. S. Thakur and S. Patra, Xanthine scaffold: scope and potential in drug development, Heliyon, 2018, 4, e00829, DOI:10.1016/j.heliyon.2018.e00829.
- A. M. Comer, C. M. Perry and D. P. Figgitt, Caffeine citrate. A review of its use in apnoea of prematurity, Paediatr. Drugs, 2001, 3(1), 61–79, DOI:10.2165/00128072-200103010-00005.
- W. Martindale, The Extra Pharmacopoeia, 30th edn, 1993, pp. 1318–1319 Search PubMed.
- S. Sinha, Z. Kelemen, E. Hümpfner, I. Ratera, J.-P. Malval, J. P. Jurado, C. Viñas, F. Teixidor and R. Núñez, O-Carboran-based fluorophores as efiicient luminescent systems both as solids and as water-dispersible nanoparticles, Chem. Commun., 2022, 58, 4016–4019, 10.1039/D1CC07211K.
- J. R. Otaegui, A. Carracull-Marín, D. Ruiz-Molina, J. Hernando and C. Roscini, Multimodal fluorescence switching materials: one dye to have them all, Adv. Opt. Mater., 2022, 10, 2200083, DOI:10.1002/adom.202200083.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, A cocrystal strategy to tune the luminescent properties of stilbene-type organic solid-state materials, Angew. Chem., Int. Ed., 2011, 50, 12483–12486, DOI:10.1002/anie.201106391.
- J. D. Wuest, Co-crystals give light a tune-up, Nat. Chem., 2012, 4, 74–75, DOI:10.1038/nchem.1256.
- D. Yan and D. G. Evans, Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids, Mater. Horiz., 2014, 1, 46–57, 10.1039/C3MH00023K.
- W. Zhu, R. Zheng, Y. Zhen, Z. Yu, H. Dong, H. Fu, Q. Shi and W. Hu, Rational design of charge-transfer interactions in halogen-bonded co-crystals toward versatile solid-state optoelectronics, J. Am. Chem. Soc., 2015, 137(34), 11038–11046, DOI:10.1021/jacs.5b05586.
- Y. Huang, Q. Gong, J. Ge, P. Tang, F. Yu, L. Xiao, Z. Wang, H. Sun, J. Yu, D.-S. Li, Q. Xiong and Q. Zhang, Green grinding-coassembly engineering toward intrinsically luminescent tetracene in cocrystals, ACS Nano, 2020, 14, 15962–15972, DOI:10.1021/acsnano.0c07416.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Recent advances of cocrystals with room temperature phosphorescence, Adv. Opt. Mater., 2021, 9, 2002197, DOI:10.1002/adom.202002197.
- L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, Creating organic functional materials beyond chemical bond synthesis by organic cocrystal engineering, J. Am. Chem. Soc., 2021, 143, 19243–19256, DOI:10.1021/jacs.1c07678.
- S. D'Agostino, F. Spineeli, P. Taddei, B. Ventura and F. Grepioni, Ultralong organic phosphorescence in the solid state: the case study of triphenylene cocrystals with halo- and dihalo-penta/tetrafluorobenzene, Cryst. Growth Des., 2019, 19, 336–346, DOI:10.1021/acs.cgd.8b01443.
- A. Azzali, S. d'Agostino, M. Capacci, F. Spinelli, B. Ventura and F. Grepioni, Assembling photoactive materials from polycyclic aromatic hydrocarbons (PAHs): room temperature phosphorescence and excimer-emission in co-crystals with 1,4-diiodotetrafluorobenzene, CrystEngComm, 2022, 24, 5748–5756, 10.1039/D2CE00720G.
- M. M. Andino, C. G. de Lima and J. D. Winefordner, Luminescence characteristics of caffeine and theophylline, Spectrochim. Acta, Part A, 1987, 43, 427–437, DOI:10.1016/0584-8539(87)80129-0.
- M. D. Gaye and J. J. Aaron, The effect of pH on the room-temperature phosphorescence properties of several purine and pyrimidine derivatives, Talanta, 1989, 36, 445–449, DOI:10.1016/0039-9140(89)80226-7.
- D. H. Murgida and R. Erra-Balsells, Low-temperature luminescence of purine derivatives: salt effects, J. Lumin., 1999, 85, 129–136, DOI:10.1016/S0022-2313(99)00059-9.
- D. Chuan, W. Yan-Li and S. Shao-Min, Study on the paper substrate room temperature phosphorescence of theobromine, caffeine and theophylline and analytical application, Spectrochim. Acta, Part A, 2003, 59, 1469–1476, DOI:10.1016/s1386-1425(02)00359-1.
- Y.-L. Wei, C. Dong, S.-M. Shuan and D.-S. Liu, Study for luminescence performance of three methyl xanthine derivatives, Spectrochim. Acta, Part A, 2005, 61, 2584–2589, DOI:10.1016/j.saa.2004.09.024.
- P. Changenet-Barret, L. Kovács, D. Markovitsi and T. Gustavsson, Xanthines studied via femtosecond fluorescence spectroscopy, Molecules, 2016, 21, 1668, DOI:10.3390/molecules21121668.
- A. Camiruaga, I. Usabiaga, V. C. D'mello, G. A. García, S. Watergaonkar and J. A. Fernández, Revisiting the spectroscopy of xanthine derivatives: theobromine and theophylline, Phys. Chem. Chem. Phys., 2019, 21, 26430, 10.1039/C9CP05068J.
- J. Tan, R. Li, Z.-T. Jiang, S.-H. Tang and Y. Wang, Rapid and non-destructive prediction of methylxanthine and cocoa solid contents in dark chocolate by synchronous front-face fluorescence spectroscopy and PLSR, J. Food Compos. Anal., 2019, 77, 20–27, DOI:10.1016/j.jfca.2019.01.001.
- Y. Huang, Y. Liu, P. J. W. Sommerville, W. Kaminsky, D. S. Ginger and C. K. Luscombe, Theobromine and direct arylation: a sustainable and scalable solution to minimize aggregation caused quenching, Green Chem., 2019, 21, 6600, 10.1039/C9GC03391B.
- Y. Huang, T. A. Cohen and C. K. Luscombe, Green syntheses of stable and efficient organic dyes for organic hybrid light-emitting diodes, J. Mater. Chem. C, 2021, 135, 1085, 10.1039/D1TC01567B.
- C. W. Lehmann and F. Stowasser, The crystal structure of anhydrous β-caffeine as determined from X-ray powder diffraction data, Chem. – Eur. J., 2007, 13, 2908–2911, DOI:10.1002/chem.200600973.
- Y. Ebisuzaki, P. D. Boyle and J. A. Smith, Methylxanthines. I. Anhydrous Theophylline, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 775–777, DOI:10.1107/S0108270197000516.
- K. A. Ford, Y. Ebisuzaki and P. D. Boyle, Methylxanthines. II. Anhydrous Theobromine, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1980–1983 CrossRef.
- R. H. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38, DOI:10.1107/S0108767394005726.
- SAINT, Bruker A.X.S. Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal structure refinement with Shelxl, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/s2053229914024218.
- L. J. Farrugia, WinGX and ORTEP for Windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854, DOI:10.1107/S0021889812029111.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0-new features for the visualization and investigation of crystal structures, J. Appl. Crystallogr., 2008, 41, 466–470, DOI:10.1107/S0021889807067908.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036, DOI:10.1063/1.478401.
- A. Dal Corso, Pseudopotentials periodic table: From H to Pu, Comput. Mater. Sci., 2014, 95, 337–350, DOI:10.1016/j.commatsci.2014.07.043.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car and C. Cavazzoni, et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, QUANTUM ESPRESSO 2, 2009 Search PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli and M. Calandra, et al. Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901, DOI:10.1088/1361-648X/aa8f79.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78, DOI:10.1002/wcms.81.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/b508541a.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065, 10.1039/b515623h.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree-Fock exchange, Chem. Phys., 2009, 356, 98–109, DOI:10.1016/j.chemphys.2008.10.036.
- G. L. Stoychev, A. A. Auer and F. Neese, Automatic Generation of Auxiliary Basis Sets, J. Chem. Theory Comput., 2017, 13, 554–562, DOI:10.1021/acs.jctc.6b01041.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170, DOI:10.1063/1.478522.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2016 Search PubMed.
- M. Gołdyn, D. Larowska, W. Nowak and E. Bartoszak-Admska, Synthon hierarchy in theobromine cocrystals with hydroxybenzoic acids as coformers, CrystEngComm, 2019, 21, 7373–7388, 10.1039/C9CE01195A.
- B. Saikia, D. Pathak and B. Sarma, Variable stoichiometry cocrystals: occurrence and significance, CrystEngComm, 2021, 23, 4583–4606, 10.1039/D1CE00451D.
- C. Zhao, W. Li, Z. Li, W. Hu, S. Zhang and S. Wu, Preparation and solid-state characterization of dapsone pharmaceutical cocrystals through supramolecular synthon strategy, CrystEngComm, 2021, 23, 6690–6702, 10.1039/D1CE00945A.
- H. Abourahma, D. D. Shah, J. Melendez, E. J. Johnson and K. T. Holman, A Tale of Two Stoichiometrically Diverse Cocrystals, Cryst. Growth Des., 2015, 15, 3101–3104, DOI:10.1021/acs.cgd.5b00357.
- G. V. S. Mota and A. M. J. C. Neto, Theoretical UV spectroscopy proprieties of methylxanthines, J. Comput. Theor. Nanosci., 2010, 7, 205–208, DOI:10.1166/jctn.2010.1346.
- S. Gómez, T. Giovannini and C. Cappelli, Absorption spectra of xanthines in aqueous solution: A computational study, Phys. Chem. Chem. Phys., 2020, 22, 5929–5941, 10.1039/c9cp05420k.
- R. Ramakrishnan, M. Hartmann, E. Tapavicza and O. A. Von Lilienfeld, Electronic Spectra from TDDFT and Machine Learning in Chemical Space, J. Chem. Phys., 2015, 143, 084111, DOI:10.1063/1.4928757.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, X. Zarate and D. Páez-Hernández, Understanding the Selective-Sensing Mechanism of Al 3+ Cation by a Chemical Sensor Based on Schiff Base: A Theoretical Approach, J. Phys. Chem. A, 2019, 123, 6970–6977, DOI:10.1021/acs.jpca.9b03366.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, X. Zarate and D. Páez-Hernández, Radiative decay channel assessment to understand the sensing mechanism of a fluorescent turn-on Al3+ chemosensor, Int. J. Quantum Chem., 2020, 120, 26083, DOI:10.1002/qua.26083.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, D. Páez-Hernández and X. Zarate, Fluorescence turn-on and turn-off mechanisms of a dual-selective chemosensor of Bi3+ and pH changes: Insights from a theoretical perspective, Dyes Pigm., 2021, 185, 108934, DOI:10.1016/j.dyepig.2020.108934.
- Y. Hidalgo-Rosa, M. A. Treto-Suárez, E. Schott, X. Zarate and D. Páez-Hernández, Sensing mechanism elucidation of a chemosensor based on a metal-organic framework selective to explosive aromatic compounds, Int. J. Quantum Chem., 2020, 120, DOI:10.1002/qua.26404.
- Y. Hidalgo-Rosa, K. Mena-Ulecia, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Insights into the selective sensing mechanism of a luminescent Cd(II)-based MOF chemosensor toward NACs: roles of the host–guest interactions and PET processes, J. Mater. Sci., 2021, 56, 13684–13704, DOI:10.1007/s10853-021-06196-3.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information concerning hydrogen-bond interactions of the new compounds, FT-IR spectra, TGA-DSC thermograms, a structural model used in the calculations, computed values of selected bond lengths (Å) for cocrystals, frontier molecular orbitals of methylxanthines (caffeine, theophylline and theobromine) and their cocrystals, molecular orbitals located on the analyte that is involved in the electronic transitions of caffeine, theobromine and theophylline-DITFB cocrystals are included. CCDC numbers 2304676–2304678. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00138a |
| ‡ Present address: Catalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and her email: sohini.sinha@icn2.cat |
| This journal is © The Royal Society of Chemistry 2024 |