
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHost compounds based on the rigid 9,10-dihydro-9,10-ethanoanthracene framework: selectivity behaviour in mixed isomeric dichlorobenzenes†
Benita
Barton
 *a,
Mino R.
Caira
*b,
Ulrich
Senekal
a and
Eric C.
Hosten
a
*a,
Mino R.
Caira
*b,
Ulrich
Senekal
a and
Eric C.
Hosten
a
aDepartment of Chemistry, Nelson Mandela University, PO Box 77000, Port Elizabeth, 6031, South Africa. E-mail: benita.barton@mandela.ac.za
bDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: mino.caira@uct.ac.za
First published on 4th March 2024
Abstract
The behaviour of host compounds dimethyl trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate (H1), trans-α,α,α′,α′-tetraphenyl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H2) and trans-α,α,α′,α′-tetra(p-chlorophenyl)-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H3), based on the rigid 9,10-dihydro-9,10-ethanoanthracene framework, was investigated in the presence of singular dichlorobenzene (o-DCB, m-DCB and p-DCB) guest isomers as well as various mixtures of these in order to determine whether these guest compounds may be separated/purified by means of host–guest chemistry strategies. H1 failed to complex with these DCBs and so was disregarded for any further investigations, while H2 enclathrated each one; H3, on the other hand, only formed complexes with o-DCB and m-DCB. When presented with mixtures of the DCBs, H2 demonstrated a marginal affinity for o-DCB in most cases, and H3, complementarily, m-DCB. Remarkably, it was revealed that H3 has the ability to separate binary guest DCB mixtures with 17.2% m-DCB/82.8% p-DCB and 49.5% m-DCB/50.5% p-DCB (in favour of m-DCB, K = 24.0 and 14.0, respectively). This result is significant given that m-DCB/p-DCB mixtures, in particular, are extremely arduous to separate in the chemical industry by means of the more conventional distillation/crystallization methods. Of the five novel complexes produced in this work, three were subjected to SCXRD analyses (the remaining two complexes were powders): H2 retained the o-DCB and m-DCB guests in the complex through (host)C–H⋯π(guest), (host)C–H⋯C–Cl(guest) and (guest)C–H⋯C–C(host) contacts (in 3(H2)·m-DCB were also observed additional (host)C–H⋯Cl(guest) interactions). In H3·m-DCB, only one host⋯guest interaction could be identified, namely a (guest)C–Cl⋯π(host) contact measuring 3.864(2) Å (134.2(1)°): this complex therefore approaches that of a true clathrate. Finally, thermoanalytical experiments explained the marginal affinity of H2 for o-DCM (this complex possessed the greater thermal stability of the three inclusion compounds), while this technique was less informative with respect to understanding the preference of H3 for the meta isomer.
1. Introduction
The dichlorobenzenes (DCBs) are a group of isomeric aromatic compounds that play multifaceted roles in various industries. To illustrate, o-, m- and p-dichlorobenzene (o-DCB, m-DCB and p-DCB, Scheme 1) are employed in the preparation of a wide variety of end products, including insecticides, herbicides, fumigants (when released into soil or the air), bacteriostats (found in deodorants), dyes, disinfectants, pharmaceuticals, and even as a moth control agent.1–4 More specifically and as examples, o-DCB serves as a solvent in the synthesis of toluene diisocyanate, while p-DCB is instrumental in the production of poly(phenylene sulfide) resin (PPS).1 This varied array of applications underscores the significance of these DCBs in the various industrial processes and highlights their pivotal role in the synthesis of essential products.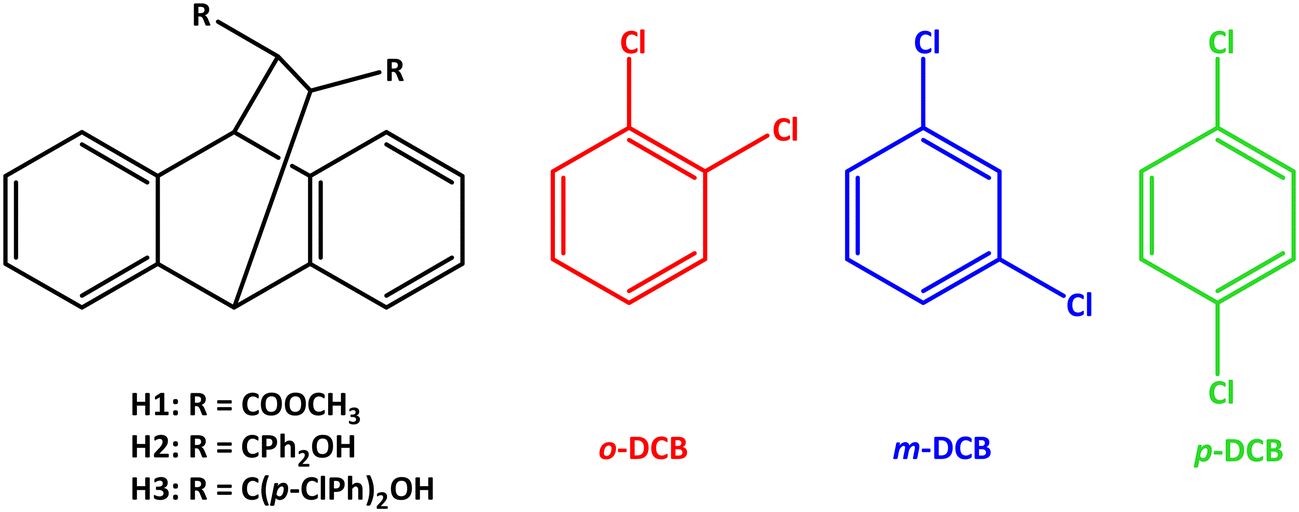 | ||
| Scheme 1 Molecular structures of the host compounds dimethyl trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate (H1), trans-α,α,α′,α′-tetra-phenyl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H2) and trans-α,α,α′,α′-tetra(p-chlorophenyl)-9,10-dihydro-9,10-ethano-anthracene-11,12-dimethanol (H3), and the dichlorobenzene isomers (o-, m- and p-DCB). | ||
The DCBs may be synthesized by means of the Sandmeyer reaction using the appropriate chloro-substituted aniline via diazotization,5–7 or through the chlorination of benzene and mono-chlorobenzene.8 Unfortunately, the latter halogenation processes furnish a mixture of all three isomers. In fact, an elevated temperature alone facilitates the isomerization of these DCBs. Problematically, each DCB isomer possesses similar boiling points, ranging from 173 (m-DCB) to 180 °C (o-DCB); p-DCB boils at 174 °C. Consequently, separating these compounds by means of fractional distillation is extremely challenging. Additionally, these DCBs melt between −25 (m-DCB) and 53 °C (p-DCB) (the melting point of o-DCB is −17 °C),1 but fractional crystallization separation processes would also not be feasible despite the fact that p-DCB has a distinctly different melting point (53 °C) compared with, for example, m-DCB (−25 °C): this particular binary system has a eutectic point when the mixture contains 88% by weight of the meta isomer, which implies that only one of these compounds may ultimately be isolated in pure form through this methodology. Furthermore, since m-DCB melts at such a low temperature, this technique would inevitably have negative consequences with respect to economics.5 Therefore, effecting the isolation of these dichlorobenzene isomers in pure form, if even practicable, necessitates multiple rounds of fractional crystallization and/or distillation processes.1,9
Investigations to facilitate the separation of DCB mixtures using metal organic frameworks (MOFs), membranes, zeolites and liquid and gas chromatography (employing suitable stationary phases) have been reported in detail in the recent literature.10–14 Unfortunately, these purification and separation procedures consume significant time and energy and are, oftentimes, extremely costly to perform. Therefore, an alternative approach for these separations is particularly appealing.
Host–guest chemistry, which is a subfield of the broader supramolecular chemistry realm, has been suggested as a very likely proxy for distillation/crystallization separation strategies. This field of science has already been demonstrated to be an efficient separatory protocol, and examples include the separation of the lutidines by Nassimbeni and coworkers15 and the xylene isomers by Barbour and Lusi,16 amongst numerous others.
In our own laboratories, investigations into the employment of host–guest chemistry for the challenging separations of difficult-to-separate mixtures are ongoing. More specifically, the synthesis and selectivity behaviour of host compounds derived from tartaric acid,17 xanthone and thioxanthone,18 and anthracene19 have been analysed in mixtures of isomers with commendable outcomes. To illustrate, the host compound DED (trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid) was assessed for its potential to separate the DCBs with remarkable results: this roof-shaped compound was observed to have a complete selectivity towards p-DCB when crystallized from various mixtures of the DCBs.20 Owing to this pleasing observation, three additional host compounds having the roof-shaped geometry, namely dimethyl trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate (H1), trans-α,α,α′,α′-tetraphenyl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H2) and trans-α,α,α′,α′-tetra(p-chlorophenyl)-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H3) (Scheme 1) were subsequently synthesized and their selectivity behaviour investigated in these mixed DCBs in the continued search for host compounds with greater efficiencies or complementary selectivities. These experiments and the results thereof have not been carried out nor reported on a prior occasion. In this particular work, we describe our findings in detail, and provide also the results obtained from single crystal X-ray diffraction analyses, where possible, and thermal analyses.
2. Experimental
2.1 General
All starting and guest materials were purchased from Merck (South Africa) and were used as obtained.
13C NMR spectroscopy was used to determine the guest![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :guest (G:G) ratios for any mixed complexes arising from the o-DCB/p-DCB and o-DCB/m-DCB/p-DCB crystallization experiments, while the overall host:guest (H:G) ratios were obtained using 1H NMR spectroscopy. These 1H and 13C NMR analyses were carried out by means of a Bruker Ultrashield Plus 400 MHz spectrometer; CDCl3 was the deuterated solvent. Data from these experiments were analysed by means of MNOVA and Topspin software.
:guest (G:G) ratios for any mixed complexes arising from the o-DCB/p-DCB and o-DCB/m-DCB/p-DCB crystallization experiments, while the overall host:guest (H:G) ratios were obtained using 1H NMR spectroscopy. These 1H and 13C NMR analyses were carried out by means of a Bruker Ultrashield Plus 400 MHz spectrometer; CDCl3 was the deuterated solvent. Data from these experiments were analysed by means of MNOVA and Topspin software.
The complexes 3(H2)·m-DCB and H3·m-DCB were of suitable crystal quality for analyses by means of SCXRD experiments. These were analysed using a Bruker Kappa Apex II diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The data were collected using APEXII, whereas cell refinement and data-reduction were achieved by employing SAINT; numerical absorption corrections were carried out with SADABS.21 Twinned data were corrected with TWINABS-2012/1. The structures were solved with SHELXT-2018/222 and refined by means of SHELXL-2018/323 (using least-squares procedures) together with SHELXLE24 as the graphical interface. All non-hydrogen atoms were refined anisotropically, while the carbon- and oxygen-bound hydrogen atoms were inserted in idealized geometrical positions in a riding model. An alternative diffractometer was used for the complex H2·o-DCB. Intensity data were collected on a Bruker D8 VENTURE single crystal X-ray diffractometer using graphite-monochromated MoKα-radiation, with the crystal specimen cooled to 173(2) K with nitrogen vapour from a crysostream (Oxford Cryosystems). Data-collection, performed with ω- and ϕ-scans of width 0.5°, was controlled using APEX3/v2019.1-0 (Bruker) software and refinement of the unit cell and data-reduction were performed with program SAINT v8.40A (Bruker).24 Absorption corrections were applied using the multi-scan method with program SADABS (2014/5).25 The structure was solved by direct methods and refined by full-matrix least-squares (programs in the SHELX suite).26 As a graphical user interface (GUI), version 4.0 of X-Seed (a program for supramolecular crystallography) was employed.27 In the final cycles of refinement, all non-hydrogen atoms were treated anisotropically, while H atoms were added in idealized positions in a riding model following their unequivocal location in successive difference Fourier maps. The crystal structures for H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB were deposited at the Cambridge Crystallographic Data Centre (CCDC) and their CCDC numbers are 2329242, 2330022 and 2330021.
A Young Lin YL6500 gas chromatography instrument coupled to a flame ionization detector (GC-FID) was required in order to obtain the G:G ratios for any mixed complexes emanating from the o-DCB/m-DCB and m-DCB/p-DCB crystallization experiments. An Agilent J&W Cyclosil-B column was used as the stationary phase while both hydrogen gas (30 mL min−1) and air (300 mL min−1) served as the mobile phase. The split ratio was 80:1. The inlet (200 °C) and detector (300 °C) metal plates were maintained at these temperatures throughout. The dissolution solvent was dichloromethane. The method involved an initial column temperature of 50 °C which was held for 1 min, followed by applying a heating rate of 15 °C min−1 until 150 °C was reached. This final temperature was maintained here for 0.3 min. The total flow of the gas mixture was 1.5 mL min−1.
All of the single solvent complexes were analysed by means of thermal analyses in order to determine their relative thermal stabilities. After isolating the solids from the solutions by means of vacuum filtration, washing these with petroleum ether (bp 40–60 °C) and patting them dry in folded filter paper, these were subjected to such experiments by means of a TA SDT Q600 Module system (with the data analysed using TA Universal Analysis 2000 software) or a Perkin Elmer STA6000 Simultaneous Thermal Analyser (with the data analysed by means of Perkin Elmer Pyris 13 Thermal Analysis software). Samples were placed in ceramic pans, and an empty pan served as the reference. The purge gas was high purity nitrogen, and the samples were heated from approximately 40 to 400 °C at a heating rate of 10 °C min−1.
2.2 Preparation of the host compounds dimethyl trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate (H1), trans-α,α,α′,α′-tetraphenyl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H2) and trans-α,α,α′,α′-tetra(p-chlorophenyl)-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol (H3)
Host compounds H1–H3 were synthesized using methods published on a prior occasion.28–302.3 Single solvent crystallization experiments
The single solvent crystallization experiments were conducted in glass vials (which had press-in polyethylene screw lids) to determine whether the host compounds possessed enclathration potential for each of the DCB guest compounds. As such, H1–H3 (0.04–0.05 g) were independently dissolved in an excess of each of the guest solvents (3 mmol for H1 and H2, and 20 mmol for H3). Complete dissolution was facilitated by means of a hot water bath in most instances. Dichloromethane was employed as a cosolvent in the case of p-DCB since this guest is a solid at ambient conditions. Furthermore, an additional filtration step was required for all the experiments involving H3 since some undissolved host compound remained: this filtration was carried out through a glass pipette dropper with an added cotton plug. After dissolution, the vials were left open at ambient pressure and temperature to encourage the formation of crystals by allowing some guest solvent/co-solvent to escape to the gas phase. The so-formed crystals were isolated by means of vacuum filtration, washed with petroleum ether (bp 40–60 °C) and analysed using 1H NMR spectroscopy. A comparison of the integrals of relevant host and guest resonance signals on the resultant 1H NMR spectra of successfully formed complexes provided the H:G ratios.
2.4 Guest competition experiments
In order to determine whether the host compounds in this investigation (H1–H3) possessed selectivity for any particular DCB guest species present in a mixture of DCBs, host crystallization experiments were carried out from such guest mixtures. These experiments were carried out in glass vials with press-in polyethylene screw lids. Two different guest competition experiments were employed: in the first of these, equimolar DCB guest mixtures were prepared using every possible guest combination from the DCB guest series while, in the second, only binary DCB guest mixtures were prepared but in which the molar amounts of each one was varied sequentially. More details for these two methods will now be described separately.:G and G:G ratios.
:80 to 80:20 (GA:GB). The vials were treated in a similar fashion to those in the equimolar competition experiments. The solutions of the respective mixtures and the crystals obtained from these were analysed using 13C NMR spectroscopy and/or GC-FID, as appropriate. Selectivity profiles were then constructed by plotting the molar fraction of the guest in the crystals (Z) against the molar fraction of the same guest in the solution (X). Eqn (1) was then employed to calculate KA:B, the selectivity coefficient,31 where XA and XB represent the amount of GA and GB in the solutions and ZA and ZB indicate the molar fraction of GA and GB in the inclusion complexes. The line of no selectivity, where K = 1, is indicated on each selectivity profile by the straight-line plot; this represents a host compound that possesses no selectivity for either of the two guest compounds present.31| KA:B = (ZA/ZB) × (XB/XA), where XA + XB = 1 | (1) |
2.5 Software
Program Mercury32 was used to analyse three of the crystal structures produced in this work (the remainder of the complexes presented as powders and could not be analysed by this technique). Employing this program, diagrams for the unit cells, the host–guest packing, the host–guest interactions and also the voids in which the guests resided, could all be prepared and analysed (in order to observe these voids, the guest molecules were deleted, and the spaces that so formed were investigated by means of a probe with a 1.2 Å radius).3. Results and discussion
3.1 Single solvent crystallization experiments
The results obtained from 1H NMR spectroscopy after crystallizing each of H1–H3 from each of the DCB guest compounds are provided in Table 1.:G ratio data from 1H NMR spectroscopy after crystallization experiments of H1–H3 from each of the DCBsa,b
Host compound H1 was unable to enclathrate any of the three DCBs and guest-free host compound was obtained in each of these three instances, while H2 was able to form a complex with each one, and H:G ratios were 1:1 (o-DCB) and 3:1 (m-DCB and p-DCB) (Table 1). Similarly, H3 formed 3:2 and 1:1 H:G complexes with o-DCB and m-DCB, respectively, but failed to include p-DCB, this latter experiment also resulting in the isolation of the apohost compound of H3 from the glass vessel.
Due to the inability of H1 to form complexes with any of these dichlorobenzenes, this host compound was not investigated further for its selectivity behaviour in mixed DCB guests; H2 and H3, however, were presented with DCB mixtures in order to ascertain their guest preferences since these two host compounds possessed enclathration ability for these guest types.
3.2 Guest competition experiments
| o-DCB | m-DCB | p-DCB | H2 | H3 | ||
|---|---|---|---|---|---|---|
| G:G (% e.s.d.'s) |
Overall H:G |
G:G (% e.s.d.'s) |
Overall H:G |
|||
|
a GC-FID was used to obtain the G:G ratios in the o-DCB/m-DCB and m-DCB/p-DCB experiments, while 13C NMR spectroscopy was used to determine the G:G ratios in the o-DCB/p-DCB and o-DCB/m-DCB/p-DCB experiments; overall H:G ratios were obtained using 1H NMR spectroscopy.
b All experiments were conducted in duplicate and the % e.s.d.'s are provided in parentheses.
|
||||||
| ✗ | ✗ |
68.8:31.2 (1.3) |
2:1 |
15.8:84.2 (2.0) |
3:2 |
|
| ✗ | ✗ |
64.8:35.2 (0.5) |
3:1 |
43.7:56.3 (0.1) |
3:2 |
|
| ✗ | ✗ | 49.5:50.5 (0.2) |
3:2 |
90.2:9.8 (1.5) |
3:1 |
|
| ✗ | ✗ | ✗ |
59.6:6.4:34.0 (1.1)(0.3)(0.8) |
2:1 |
45.5:45.9:8.6 (0.3)(0.1)(0.2) |
3:2 |
From Table 2, it may be concluded that H2 constantly favoured o-DCB when this guest was present in the guest mixture (68.8% in o-DCB/m-DCB, 64.8% in o-DCB/p-DCB and 59.6% in o-DCB/m-DCB/p-DCB). The absence of o-DCB in the binary mixture (m-DCB/p-DCB) resulted in p-DCB (50.5%) being only moderately preferred by H2 compared with m-DCB (49.5%). The ternary DCB mixture furnished crystals containing modest amounts of o-DCB (59.6%), some p-DCB (34.0%) and a small portion of m-DCB (6.4%).
Host compound H3, in contrast, showed a constant preference for m-DCB in o-DCB/m-DCB (84.2%), m-DCB/p-DCB (90.2%) and o-DCB/m-DCB/p-DCB (45.9%) guest mixtures (Table 2). These results, more especially in the case of the o-DCB/m-DCB and m-DCB/p-DCB mixtures, are significant given the difficulty of separating such mixtures by alternative methods owing to their near-identical boiling points. In the absence of the meta isomer (o-DCB/p-DCB), p-DCB was then moderately preferred (56.3%).
The overall H:G ratios of complexes that resulted from these guest mixtures ranged between 3:1 (o-DCB/p-DCB), 2:1 (o-DCB/m-DCB and o-DCB/m-DCB/p-DCB) and 3:2 (m-DCB/p-DCB) for H2, and this ratio was 3:2 for all of the experiments with H3, with the exception of the m-DCB/p-DCB binary solution: here, the crystals that formed had a 3:1 overall H:G ratio (Table 2).
While the selectivity behaviour was only marginal when employing H2 in these mixed DCBs (50.5–68.8%), H3 demonstrated a much-improved preferential behaviour, more especially in the m-DCB/p-DCB experiment, favouring m-DCB. It is therefore feasible to utilize this host compound for the isolation of the meta isomer from that of the para-substituted analogue, an enormous challenge in the chemical industry when using fractional distillations. Interestingly, the selectivity behaviour of H2 (favouring o-DCB) and H3 (preferring m-DCB) complement those observed for DED, which overwhelmingly preferred the p-DCB.20
 | ||
| Fig. 1 The selectivity profiles for H2 in (a) o-DCB/m-DCB, (b) o-DCB/p-DCB and (c) p-DCB/m-DCB binary guest mixtures. | ||
 | ||
| Fig. 2 The selectivity profiles for H3 in (a) m-DCB/o-DCB, (b) p-DCB/o-DCB and (c) m-DCB/p-DCB binary guest mixtures. | ||
Immediately evident from Fig. 1a (o-DCB/m-DCB) and Fig. 1c (p-DCB/m-DCB) is that H2 was rather unselective for either guest species present. In the former set of experiments, H2 only favoured o-DCB (67.5 and 71.1%, Fig. 1a) when the amount of this guest species present was ≥50%; the K values calculated for the experiments in favour of the ortho isomer were low and ranged between 1.0 and 1.5. As a result, H2 is not a suitable host compound for these separations according to Nassimbeni et al., who reported that K values should be 10 or greater for effective separations.33 In the case of the p-DCB/m-DCB experiments, H2 displayed a very marginal preference for m-DCB (22.2%) when this guest was present in low concentrations (13.0%, Fig. 1c); here, K = 1.9, and this was the highest K value calculated in this set of experiments, and so separations of these mixtures with this host compound are also not practicable. In the o-DCB/p-DCB experiments (Fig. 1b), results were significantly more encouraging: in 18.3% o-DCB/81.7% p-DCB, the crystals that resulted contained 2.0% o-DCB and 98.0% p-DCB, and K = 11.0, alluding to the viability of the effective separation on an industrial scale of these kinds of mixtures. Interestingly, the selectivity of H2 in these experiments depended on the guest ratios and, unfortunately, the remaining o-DCB/p-DCB experiments had K values of 9.3 or less.
H3 was more decided in its selectivity behaviour and favoured m-DCB across the concentration range in m-DCB/o-DCB (Fig. 2a) and m-DCB/p-DCB (Fig. 2c) mixtures. However, only experiments in m-DCB/p-DCB provided suitably high K values, 24.0 (17.2%/82.8% m-DCB/p-DCB) and 14.0 (49.5%/50.5% m-DCB/p-DCB), in favour of m-DCB, for feasible separation applications. In the m-DCB/o-DCB experiments, the highest K value was 8.9 (in favour of m-DCB,) where the isolated crystals contained 69.0% m-DCB, and these emanated from a solution that had only 20.0% of the meta isomer. Unfortunately, the remaining m-DCB/o-DCB and m-DCB/p-DCB experiments all resulted in complexes with calculated K values less than 10 (K = 6.8–8.9 and 8.1–9.8, respectively). Finally, the selectivity profile obtained from the p-DCB/o-DCB experiments (Fig. 2b) indicated that H3 was distinctly unselective for either guest present as the data points all lie close to the line representing no selectivity.
3.3 Single crystal X-ray diffractometry analyses on complexes H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB
The relevant crystallographic data and refinement parameters for H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB, the only three complexes with suitable quality crystals for SCXRD analyses, are provided in Table 3.| H2·o-DCB | 3(H2)·m-DCB | H3·m-DCB | |
|---|---|---|---|
| Chemical formula | C42H34O2·C6H4Cl2 | 3(C42H34O2)·C6H4Cl2 | C42H3oCl4O2·C6H4Cl2 |
| Formula weight (g mol−1) | 717.68 | 619.68 | 855.45 |
| Crystal system | Monoclinic | Tetragonal | Monoclinic |
| Space group | P21/n |
I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) |
I2/a |
| μ (Mo-Kα)/mm−1 | 0.219 | 0.124 | 0.478 |
| a/Å | 12.4249(18) | 23.2834(9) | 23.236(1) |
| b/Å | 11.7971(18) | 23.2834(9) | 15.6145(8) |
| c/Å | 24.951(4) | 12.4263(6) | 24.0913(10) |
| Alpha/° | 90 | 90 | 90 |
| Beta/° | 95.752(3) | 90 | 115.807(3) |
| Gamma/° | 90 | 90 | 90 |
| V/Å3 | 3638.9(9) | 6736.5(6) | 7869.0(7) |
| Z | 4 | 8 | 8 |
| F(000) | 1504 | 2613 | 3520 |
| Temp./K | 173 | 296 | 200 |
| Restraints | 0 | 108 | 0 |
| Nref | 9059 | 8217 | 9829 |
| Npar | 471 | 460 | 506 |
| R | 0.0577 | 0.0566 | 0.0450 |
| wR2 | 0.1510 | 0.1569 | 0.1082 |
| S | 1.02 | 1.04 | 1.05 |
| θ min–max/° | 1.6, 28.3 | 1.7, 28.3 | 1.6, 28.4 |
| Tot. data | 77693 |
44314 |
9829 |
| Unique data | 9059 | 8217 | 9829 |
| Observed data [I > 2.0sigma(I)] | 6691 | 6147 | 8170 |
| R int | 0.140 | 0.027 | 0.000 |
| Completeness | 0.998 | 0.999 | 1.000 |
| Min. resd. dens./e Å−3 | −0.71 | −0.24 | −0.84 |
| Max. resd. dens./e Å−3 | 0.67 | 0.51 | 0.63 |
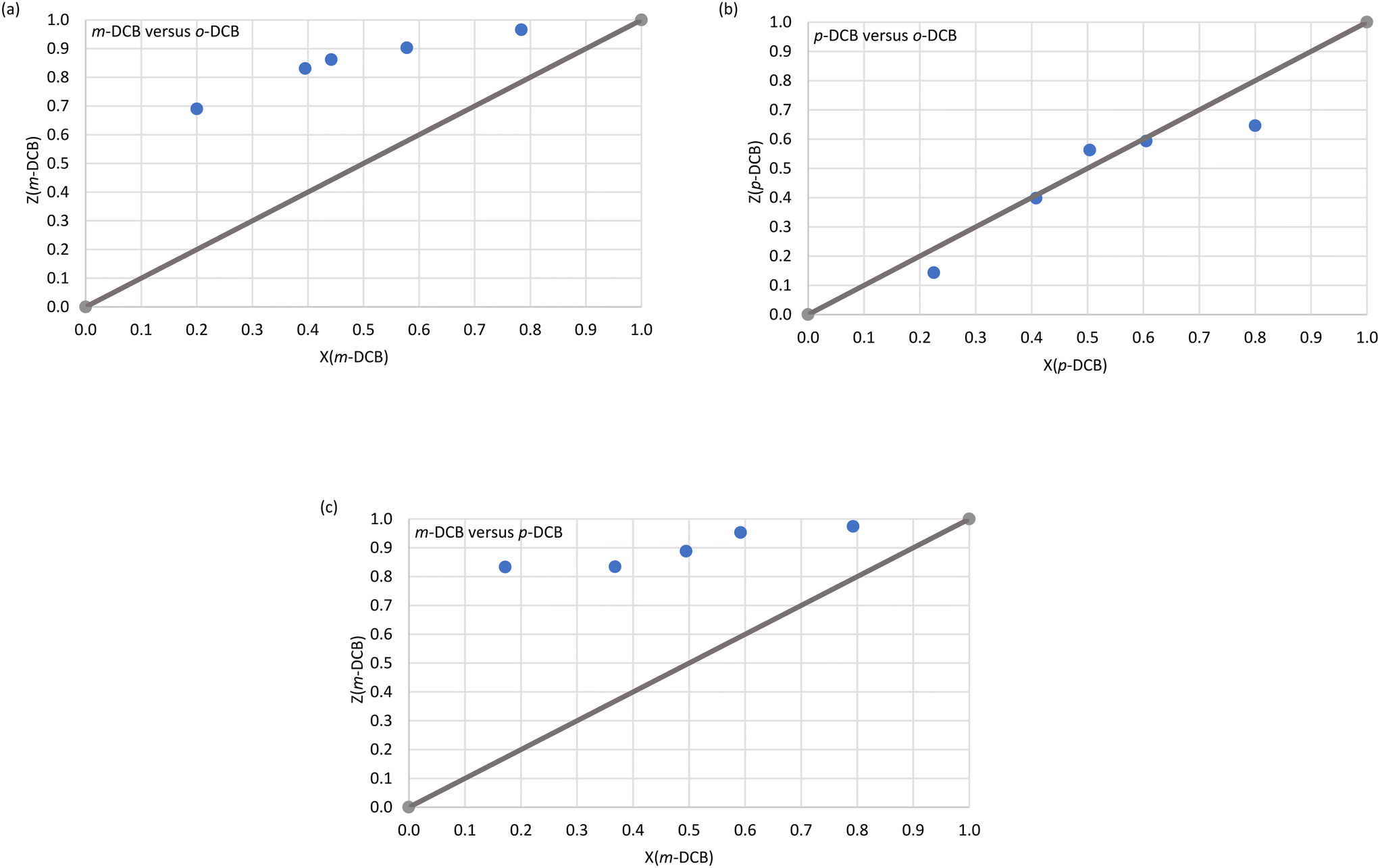
Complexes H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB each formed crystals with distinct space groups (P21/n, I and I2/a, respectively), and H2·o-DCB and H3·m-DCB shared the same monoclinic crystal system, while 3(H2)·m-DCB crystallized in the tetragonal crystal system. The m-DCB guest in 3(H2)·m-DCB was disordered around an inversion centre and two-fold rotation axis, while no guest disorder was observed in H2·o-DCB and H3·m-DCB. Interestingly, the host packing in 3(H2)·m-DCB is isostructural with other complexes recently synthesized in our laboratories, namely H2·0.5(dimethylaniline),34 3(H2)·o-xylene,35 and 2(H2)·toluene and 3(H2)·ethylbenzene.36
The host–guest packing (left) and calculated void (right) diagrams for H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB are provided in Fig. 3a–c.
 | ||
| Fig. 3 The host–guest packing (left) and calculated voids (right) for (a) H2·o-DCB, (b) 3(H2)·m-DCB and (c) H3·m-DCB (views along [100], [001] and [010], correspondingly); host structures are in capped stick and guest molecules in spacefill forms. | ||
From Fig. 3a and c, it is evident that two o-DCB and two m-DCB guest molecules occupied discrete cage-like voids in H2·o-DCB and H3·m-DCB, while the m-DCB guest species in 3(H2)·m-DCB were accommodated in infinite channels along the c-axis (Fig. 3b).
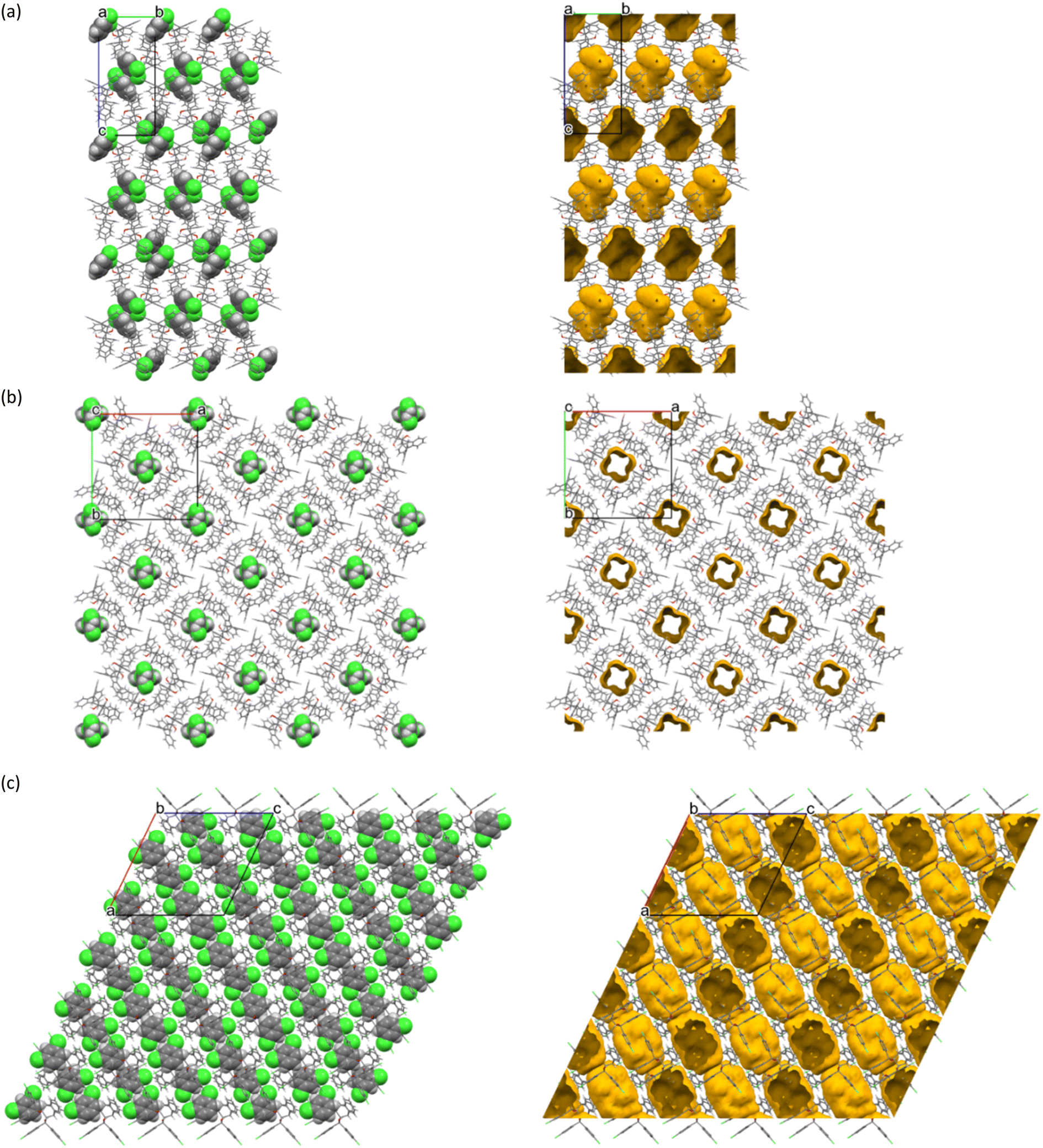
Each of the cages occupied by the two guests in H2·o-DCB was created by four H2 host molecules (Fig. 4). The host interacted with these guests by means of a (host)C–H⋯π(guest) contact (measuring 2.91 Å with an associated angle of 145°) involving the hydrogen atom of the anthracene unit of the host and the aromatic ring of the guest (each of the centrosymmetrically related guests experienced such an interaction, Fig. 5, a stereoview). The guests were also further secured in the crystal with the aid of (host)C–H⋯C–Cl(guest) and (guest)C–H⋯C–C(host) interactions measuring 2.85 Å (146°) and 2.74 Å (133°), respectively (Fig. 6, also a stereoview), which may even be regarded as C–H⋯π interactions (or even π⋯π interactions in the edge-to-face or edge-to-edge orientation).
 | ||
| Fig. 4 The four host molecules surrounding the two o-DCB guests in H2·o-DCB; host molecules are displayed in capped stick configuration and the guests are shown in spacefill form. | ||
 | ||
| Fig. 5 Stereoview showing the (host)C–H⋯π(guest) interactions in H2·o-DCB. | ||
 | ||
| Fig. 6 Stereoview depicting the (host)C–H⋯C–Cl(guest) (blue dashed lines) and (guest)C–H⋯C–C(host) (green dashed lines) interactions in H2·o-DCB. | ||
The crystal packing in this complex was stabilized via (host)C–H⋯π(host) (2.89 Å, 136°) and (host)π⋯π(host) (3.733(1) Å, slippage 1.118 Å) interactions (Fig. 7a). The π⋯π interaction involved the phenyl ring of the anthracenyl moiety and its centrosymmetric counterpart in a second host molecule. The complex did not experience any classical hydrogen bonding interactions between the host molecules. Additionally, the geometry of each host molecule was stabilized by means of one intramolecular π⋯π (3.511(12) Å, slippage 1.647 Å) and two O–H⋯π (2.582 (161°) and 2.672 (153°) Å) contacts as depicted in Fig. 7b (the reason for the ALERT level B in the checkcif report is that the two hydroxyl groups in H2 do not form H-bonds with single acceptor atoms, but instead engage in these intramolecular O–H⋯π interactions with the phenyl groups of the anthracene unit). Since no strong host intramolecular hydrogen bonding interactions were evident in this complex, this host molecular geometry is referred to as the “inactive” form of H2, as defined by Csöregh et al.37–39
 | ||
| Fig. 7 The (a) intermolecular host⋯host C–H⋯π (green dashed lines) and π⋯π (orange dashed line) interactions and (b) intramolecular host π⋯π and O–H⋯π contacts (green dashed lines) in H2·o-DCB. | ||
As was the case in 3(H2)·o-Xy from a previous investigation,35 the m-DCB guest species in 3(H2)·m-DCB were surrounded by four molecules of H2 creating host–guest motifs that stretched out along the c-axis, forming tunnel voids in which the guests were housed. Fig. 8a is an illustration of these, while the intermolecular host⋯host interactions of the C–H⋯π type (2.79 Å and 2.93 Å, each with an associated angle of 137°) reinforced the crystal structure geometry, as observed in Fig. 8b.
 | ||
| Fig. 8 The (a) host–guest motifs along the c-axis and (b) intermolecular (host)C–H⋯π(host) interactions that reinforced the crystal structure geometry in 3(H2)·m-DCB; in the latter instance, the guest molecules are not shown. | ||
As in H2·o-DCB, in the 3(H2)·m-DCB complex was also identified an intramolecular host⋯host π⋯π contact (3.572(2) Å, slippage 1.537 Å) and two intramolecular O–H⋯π interactions (2.554 Å (154°) and 2.575 Å (165°), Fig. 9), this being, once more, the “inactive” form of H2, since no strong intramolecular host O–H⋯O interactions could be identified in this complex.37–39
 | ||
| Fig. 9 The intramolecular π⋯π and O–H⋯π interactions involved in reinforcing the geometry of each H2 molecule in the complex with m-DCB. | ||
Guest retention was accomplished through host⋯guest C–H⋯Cl (2.75 Å (123 and 155°)), C–H⋯C–C (2.84 Å, 134°) and C–H⋯C–Cl (2.61 Å, 154°) interactions. Fig. 10 illustrates the C–H⋯C–Cl and two C–H⋯Cl contacts in 3(H2)·m-DCB (only one of the disorder guest components is shown here).
 | ||
| Fig. 10 The C–H⋯C–Cl (orange dashed line) and two C–H⋯Cl (green dashed lines) interactions that are present between the hydrogen atoms of the free aromatic rings of the host molecule and the carbon and chlorine atoms of the guest species in 3(H2)·m-DCB (only one disorder guest component is shown). | ||
The host molecule in H3·m-DCB also assumed the “inactive” form37–39 (as in H2·o-DCB and 3(H2)·m-DCB) and, therefore, in this complex were identified intramolecular host π⋯π (3.561(2) Å, slippage 1.396 Å) and two O–H⋯π interactions (2.538 Å (155°) and 2.528 Å (166°), Fig. 11). The host molecules also participated in intermolecular C–Cl⋯π interactions (3.418(1)–3.644(1) Å, 93.8(1)–170.1(1)°, Fig. 12), which resulted in the formation of continuous sheets of host molecules along the [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] plane (Fig. 13 (left); note also the Miller-index representation for this case on the right40), with neighbouring sheets being connected by C–H⋯O–C (2.65 Å, 144°) interactions.
] plane (Fig. 13 (left); note also the Miller-index representation for this case on the right40), with neighbouring sheets being connected by C–H⋯O–C (2.65 Å, 144°) interactions.
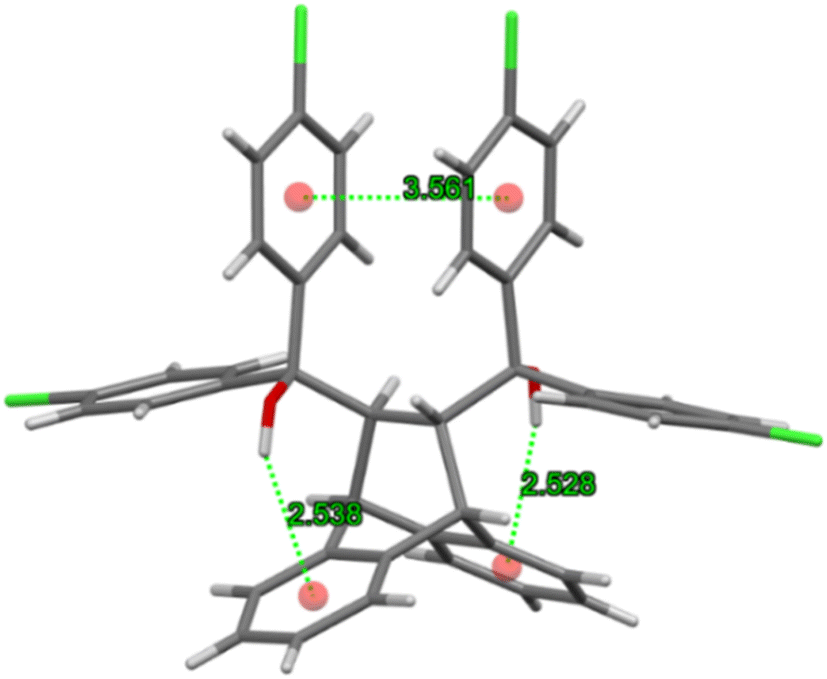 | ||
| Fig. 11 Intramolecular host π⋯π and O–H⋯π interactions in the “inactive” form of H3 in H3·m-DCB. | ||
 | ||
| Fig. 12 The intermolecular C–Cl⋯π interactions (each of the four interactions is indicated in purple, orange, blue and pink) in H3·m-DCB; view along [010]. | ||
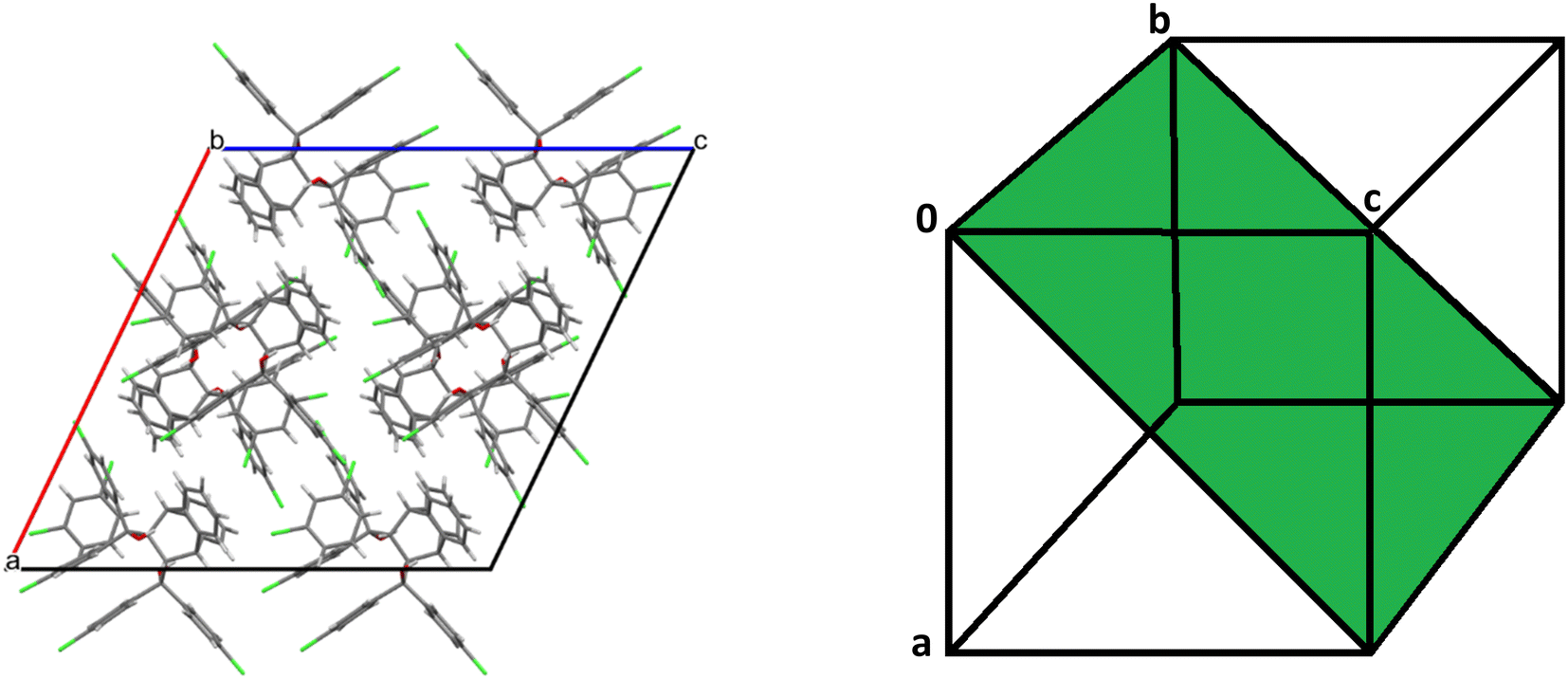 | ||
| Fig. 13 Continuous sheets of host molecules formed along the [10] plane (left) (view along [010]) and the Miller-index representation of the [10] plane (green, right).40 | ||
Guest retention was accomplished through a single C–Cl⋯π interaction (3.864(2) Å, 134.2(1)°) between the chlorine atom of the guest molecule and the anthracene aromatic ring (Fig. 14). This complex may thus be considered as approximating that of a true clathrate owing to the scarcity, in the unit cell, of interactions between the host and guest species.
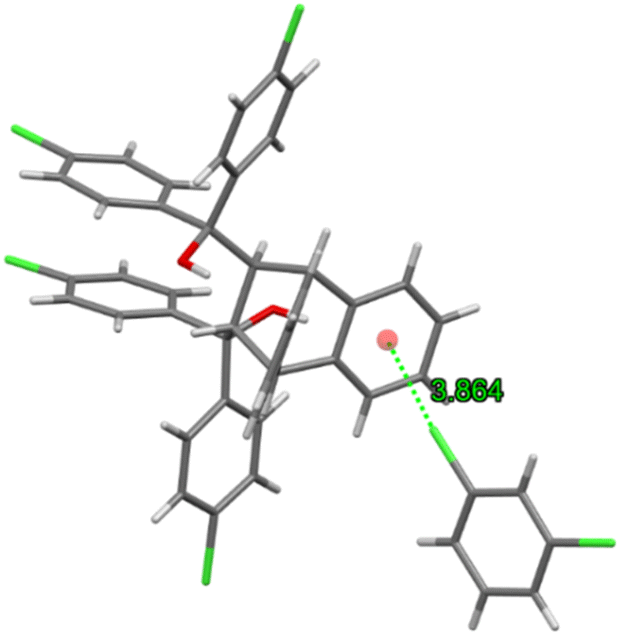 | ||
| Fig. 14 The only guest⋯host interaction, (guest)C–Cl⋯π(host), between H3 and m-DCB in H3·m-DCB. | ||
Noteworthy, H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB each experienced six intramolecular host⋯host C–H⋯O interactions measuring 2.29–2.55 Å (102–112° angles), 2.30–2.48 Å (102–111°) and 2.29–2.44 Å (102–111°), respectively.
We subsequently conducted thermal experiments on each of the five novel complexes produced in this work in order to determine the relative thermal stabilities of each crystalline complex.
3.4 Thermal analyses
The thermal data in Table 4 were obtained from the TG, DSC and/or DTG traces (Fig. 15a–e) obtained after heating each complex from 40 to 400 °C at 10 °C min−1.| Complex | T on /°C | T p /°C | Measured mass loss/% | Expected mass loss/% |
|---|---|---|---|---|
| a T on is the guest release onset temperature. b The temperature at which the guest release is most rapid is indicated by Tp. c T on was estimated from the TG trace after the initial surface solvent had been released. d The Tp values were obtained using the DTG trace. | ||||
| H2·o-DCB | 153.7c | 75.3, 95.6, 180.8d | 20.3c | 20.5 |
| 3(H2)·m-DCB | 57.9 | 165.5, 220.0 | 7.1 | 7.9 |
| 3(H2)·p-DCB | 79.8 | 98.4 | 9.3 | 7.9 |
| 3(H3)·2(o-DCB) | 161.1 | 259.4 | 12.4 | 12.2 |
| H3·m-DCB | 158.9 | 203.1 | 16.1 | 17.2 |
 | ||
| Fig. 15 The TG (red) and DTG (green) traces for (a) H2·o-DCB, and with an added DSC (blue) trace for (b) 3(H2)·m-DCB, (c) 3(H2)·p-DCB, (d) 3(H3)·2(o-DCB) and (e) H3·m-DCB. | ||
Initially evident from Table 4 and Fig. 15a (H2·o-DCB) is that the measured mass loss (33.3%) was much greater than that expected (20.5%). However, it is plausible that the initial mass loss of 13.1% may be attributed to surface solvent (methanol, which was used as a cosolvent during the experiment) as o-DCB has a flash point of 68 °C and methanol a boiling point of 64.7 °C. Therefore, in each of the H2·o-DCB, 3(H2)·p-DCB and H3·m-DCB complexes, the guest species was released in a single step, while this process was more convoluted in 3(H2)·m-DCB and 3(H3)·2(o-DCB) (Fig. 15a–e). Interestingly, the complexes in which the guests occupied discrete cavities (H2·o-DCB and H3·m-DCB) released their guests in a single step (Fig. 15a and e), while m-DCB in 3(H2)·m-DCB, in which guests occupied channels, was released in a more convoluted fashion (Fig. 15b).
The measured mass losses for all the complexes are in close correlation with what was expected (Table 4). The onset temperature for the release of o-DCB from H2·o-DCB, o-DCB being a favoured guest of H2, was 153.7 °C (Fig. 15a, estimated from the TG trace after the initial release of surface solvent occurred), while the complexes of H2 with p-DCB and m-DCB had Ton 79.8 and 57.9 °C (Fig. 15c and b). These results indicate that the 3(H2)·o-DCB complex is significantly more stable than 3(H2)·p-DCB and 3(H2)·m-DCB (153.7 vs. 79.8 and 57.9 °C) which, in turn, explains the selectivity of H2 during the mixed binary and ternary solvent and ratio-dependent competition experiments.
p-DCB, a less preferred guest of H3, failed to form a single solvent complex with this host species and so thermal analysis was not possible in this particular case. However, H3·m-DCB (with the preferred guest species) and 3(H3)·2(o-DCB) (less favoured) had high and comparable Ton values (158.9 and 161.1 °C, respectively, Table 4) and so these data do not fully explain the high affinity of H3 for the meta isomer, as observed in the competition experiments.
4. Conclusions
In this work, H1–H3 were crystallized from each of the DCB solvents, and also binary and ternary DCB mixtures. From the single solvent experiments, it was noted that H1 was unable to include any of the three DCB guests and the resultant solids were merely guest-free apohost H1. H2, on the other hand, was able to enclathrate all three, and H:G ratios were 1:1 (o-DCB) and 3:1 (m-DCB and p-DCB). H3 only formed complexes with o-DCB (3:2) and m-DCB (1:1). During the guest competition experiments, H2 was consistently selective towards o-DCB (with preferences ranging between 59.6 and 68.8%) while H3 preferred m-DCB (the highest selectivity was 90.2%, in the m-DCB/p-DCB binary mixture). It was revealed that H2 has the ability to effectively separate mixtures with approximately 18.3% o-DCB and 81.7% p-DCB (K = 11.0, in favour of the latter guest species). Remarkably, H3 demonstrated the ability to separate binary guest mixtures containing 17.2% m-DCB and 82.8% p-DCB, and 49.5% m-DCB and 50.5% p-DCB (in favour of m-DCB, K values were 24.0 and 14.0, respectively), m-DCB/p-DCB being the most challenging mixtures to separate in the chemical industry. The SCXRD analyses revealed that the o-DCB and m-DCB guests were held in the crystals of their complexes with H2 by means of (host)C–H⋯π(guest), (host)C–H⋯C–Cl(guest) and (guest)C–H⋯C–C(host) contacts (additional (host)C–H⋯Cl(guest) interactions were observed in 3(H2)·m-DCB). The complex of H3 and m-DCB displayed a single (guest)C–Cl⋯π(host) interaction (3.864(2) Å, 134.2(1)°) and may be considered as approaching that of a true clathrate due to the absence of any significant host⋯guest interactions. Thermal analyses explained the selectivity of H2 for o-DCB: the complex with this guest compound possessed the greatest thermal stability of the three complexes. However, the inclusion compound of H3 with m-DCB (the preferred guest compound) had a comparable and high thermal stability as the o-DCM-containing complex (Ton 158.9 and 161.1 °C, respectively).
Author contributions
Benita Barton: conceptualization; funding acquisition; methodology; project administration; resources; supervision; visualization; writing – original draft. Mino R. Caira: resources; visualization; formal analysis. Ulrich Senekal: investigation; methodology; validation. Eric C. Hosten: data curation; formal analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support is acknowledged from the Nelson Mandela University and the National Research Foundation. MRC thanks the University of Cape Town for access to research facilities.References
- D. C. Ayres and D. G. Hellier, Dictionary of Environmentally Important Chemicals, CRC Press, Boca Raton, 1997 Search PubMed.
- P. Schmittinger, Chlorine: Principles and Industrial Practice, Wiley-VCH, Germany, 2000 Search PubMed.
- K. Othmer, Kirk-Othmer Concise Encyclopedia of Chemical Technology 2, Wiley-Interscience, New Jersey, 2007, vol. Set 5th Search PubMed.
- R. Stringer and P. Johnston, Chlorine and the Environment: An Overview of the Chlorine Industry, Kluwer Academic Publishers, Netherlands, 2001 Search PubMed.
- T. Sandmeyer, Ueber die Ersetzung der Amidgruppe durch Chlor in den Aromatischen Substanzen, Ber. Dtsch. Chem. Ges., 1884, 17, 1633–1635 CrossRef.
- T. Sandmeyer, Ueber die Ersetzung der Amid-gruppe durch Chlor. Brom und Cyan in den aromatischen Substanzen, Ber. Dtsch. Chem. Ges., 1884, 17, 2650–2653 CrossRef.
- L. Gattermann, Untersuchungen über Diazoverbindungen, Ber. Dtsch. Chem. Ges., 1890, 23, 1218–1228 CrossRef CAS.
- U. Beck and E. Löser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co., Germany, 2012 Search PubMed.
- G. Erdem, M. Leckebusch, G. Olf, K.-J. Rinck and G. Zuhlke, Process for the Separation of Mixtures Containing m- and p-Dichlorobenzene, US7311807B2, 2007.
- J. J. McKetta, Chemical Processing Handbook, Marcel Dekker, New York, 1993 Search PubMed.
- Q.-P. He, Y.-Y. Wang, P.-F. Wang and X.-M. Dou, Preparation of Modified MFI-Type/PDMS Composite Membranes for the Separation of Dichlorobenzene Isomers via Pervaporation, RSC Adv., 2022, 12, 16131–16140 RSC.
- Q.-P. He, Y. Zou, P.-F. Wang and X.-M. Dou, MFI-Type Zeolite Membranes for Pervaporation Separation of Dichlorobenzene Isomers, ACS Omega, 2021, 6, 8456–8462 CrossRef CAS PubMed.
- C.-X. Yang, S.-S. Liu, H.-F. Wang, S.-W. Wang and P. Y. Yan, High-Performance Liquid Chromatography Separation of Position Isomers using Metal-Organic Framework MIL-53(Al) as the Stationary Phase, Analyst, 2012, 137, 133–139 RSC.
- X. Zhang, H. Ji, X. Zhang, Z. Wang and D. Xiao, Capillary Column Coated with Graphene Quantum Dots for Gas Chromatographic Separation of Alkanes and Aromatic Isomers, Anal. Methods, 2015, 7, 3229–3237 RSC.
- J. S. Bouanga Boudiombo, H. Su, S. A. Bourne, E. Weber and L. R. Nassimbeni, Separation of Lutidine Isomers by Selective Enclathration, Cryst. Growth Des., 2018, 18, 2620–2627 CrossRef CAS.
- M. Lusi and L. J. Barbour, Solid-vapor Sorption of Xylenes: Prioritized Selectivity as a Means of Separating All Three Isomers Using a Single Substrate, Angew. Chem., Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed.
- B. Barton, P. L. Pohl and E. C. Hosten, An Investigation of the Selectivity Behaviour of Host Compound (R,R)-(−)-2,3-Dimethoxy-1,1,4,4-tetraphenylbutane-1,4-diol in Mixed Cresols, CrystEngComm, 2023, 25, 1731–1739 RSC.
- B. Barton, L. de Jager and E. C. Hosten, Comparing the Host Behaviour of N,N'-Bis(9-phenyl-9-thioxanthenyl)ethylenediamine and N,N'-Bis(9-phenyl-9-xanthenyl)ethylenediamine in the Presence of Various Alkylated Aromatic and Aniline Guests: Crystal Engineering Considerations, CrystEngComm, 2019, 21, 4387–4400 RSC.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Complementary Host Behaviour of Three Anthracenyl-derived Roof-shaped Compounds in Mixed Pyridines, CrystEngComm, 2023, 25, 1740–1754 RSC.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, trans-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic Acid: Complete Host Selectivity for Guest Compound para-Dichlorobenzene During Crystal Growth from Mixed Isomeric Dichlorobenzenes, Cryst. Growth Des., 2022, 22, 3385–3394 CrossRef CAS.
- A. Bruker, APEX2, SADABS and SAINT, Bruker AXS Inc., Madison (WI), USA, 2010 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt Graphical User Interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- U. Bruker, APEX3 v2019.1-0, SAINT V8.40A, Bruker AXS Inc., Madison (WI), USA, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of Silver and Molybdenum Microfocus X-ray Sources for Single-crystal Structure Determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, A Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, X-Seed – A Software Tool for Supramolecular Crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- W. E. Bachmann and L. B. Scott, The Reaction of Anthracene with Maleic and Fumaric Acid and Their Derivatives and with Citraconic Anhydride and Mesaconic Acid, J. Am. Chem. Soc., 1948, 70, 1458–1461 CrossRef CAS PubMed.
- P. Yates and P. Eaton, Acceleration of the Diels-Alder Reaction by Aluminium Chloride, J. Am. Chem. Soc., 1960, 82, 4436–4437 CrossRef CAS.
- L. Y. Izotova, D. M. Ashurov, B. T. Ibragimov, E. Weber, M. Perren and S. A. Talipov, Features of Clathrate Formation of trans-9,10-Dihydro-9,10-ethanoanthracene-11,12-dicarboxylic Acid, J. Supramol. Chem., 2005, 46, S103–S108 CAS.
- A. M. Pivovar, K. T. Holman and M. D. Ward, Shape-Selective Separation of Molecular Isomers with Tunable Hydrogen-Bonded Host Frameworks, Chem. Mater., 2011, 13, 3018–3031 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McGabe, E. Pidlock, L. Rodriguez-Mongu, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 – New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- N. M. Sykes, H. Su, E. Weber, S. A. Bourne and L. R. Nassimbeni, Selective Enclathration of Methyl- and Dimethylpiperidines by Fluorenol Hosts, Cryst. Growth Des., 2017, 17, 819–826 CrossRef CAS.
- Unpublished work.
- B. Barton, U. Senekal and E. C. Hosten, trans-α,α,α’,α’-Tetraphenyl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol and its Tetra(p-chlorophenyl) Derivative: Roof-shaped Host Compounds for the Purification of Aromatic C8H10 Isomeric Guest Mixtures, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 77–87 CrossRef CAS.
- Unpublished work.
- E. Weber, T. Hens, O. Gallardo and I. Csöregh, Roof-shaped Hydroxy Hosts: Synthesis, Complex Formation and X-ray Crystal Structures of Inclusion Compounds with EtOH, Nitroethane and Benzene, J. Chem. Soc., Perkin Trans. 2, 1996, 4, 737–745 RSC.
- I. Csöregh, E. Weber and T. Hens, Molecular Recognition Based on Roof-shaped Diol Hosts. X-ray Crystal Structures of Inclusion Compounds with Methanol, Pyridine and Toluene, J. Inclusion Phenom. Macrocyclic Chem., 2000, 38, 397–412 CrossRef.
- I. Csöregh and E. Weber, Molecular Recognition in Solid Inclusion Compounds of Novel Roof-Shaped Diol Hosts, ed. A. W. Coleman, Netherlands, 1998 Search PubMed.
- A. Marikani, Material Science, PHI Learning, Dehli, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The crystal structures for H2·o-DCB, 3(H2)·m-DCB and H3·m-DCB were deposited at the Cambridge Crystallographic Data Centre (CCDC) and their CCDC numbers are 2329242, 2330022 and 2330021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00113c |
| This journal is © The Royal Society of Chemistry 2024 |