
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallographic, spectroscopic, and computational characterization of a diiodonaphthoquinone diarylethene photochrome: halogen bonding and photocrystallography†
Shea D.
Myers‡
 a,
Zoe Y.
Marr‡
a,
Jade A.
Sency
b,
Travis B.
Mitchell
a,
Jason B.
Benedict
*a and
Dinesh G.
Patel
*b
a,
Zoe Y.
Marr‡
a,
Jade A.
Sency
b,
Travis B.
Mitchell
a,
Jason B.
Benedict
*a and
Dinesh G.
Patel
*b
aDepartment of Chemistry, University at Buffalo, Buffalo, NY 14228, USA. E-mail: jbb6@buffalo.edu
bDepartment of Chemistry, The Pennsylvania State University, Hazleton, PA 1820, USA. E-mail: dgp15@psu.edu
First published on 23rd April 2024
Abstract
We report the synthesis and characterization of 2,3-bis(5-iodo-2-methylthiophen-3-yl)naphthalene-1,4-dione and its ring-closed isomer. The ring-closed isomer crystallizes in the space group P21 with unit cell parameters of a = 11.93119(10), b = 7.4644(6), c = 12.4586(10), β = 117.024(2)°. Photoisomerization in solution is reversible and similar to that of other quinone based diarylethenes. The crystal structure of the ring-closed isomer consists of halogen bonded 2-D sheets. Upon irradiation, single crystals of the ring-closed isomer fracture along well-defined cleavage planes parallel to the sheet layers. Subsequent X-ray diffraction measurements revealed that the light-induced fracturing is due to the formation of the ring-open isomer.
Introduction
Photochromism is defined as the ability of a compound to photochemically switch between two isomeric states with each of these states having its own unique optical, structural, and electrochemical characteristics.1 Among the many classes of photochromic compounds, diarylethenes (DAEs, as an example quinone containing compounds 1 and 2, Scheme 1) have gained attention owing to their fatigue resistance2–4 and potential for use as data storage media,5,6 sensors,7,8 and probes9 including dosimeters.10 Additionally, they have been used in biological applications11,12 and even for catalyst control.13,14 | ||
| Scheme 1 The synthesis and photochromic response of DAE compound 1. The chemical synthesis of closed-form 1b from open-form 1a is also presented. | ||
As newer and more complex applications are envisioned, the desire to find alternatives to the high energy UV light typically required for the photoisomerization reactions has come forward as a major concern with some groups focusing on modifying the aryl portion of diarylethene photochromes.15–17 It has been demonstrated that the use of either a quinone or naphthoquinone ethene bridge allows for visible light to initiate photoisomerization in both the ring-opening and ring-closing directions.18–20 Quinone based headgroups are also electrochemically active21,22 and found in important biological molecules,23,24 thus making them excellent candidates for potential therapeutic agents.25
The rational design of DAE-based crystalline materials remains challenging and is due in part to the conformational flexibility associated with the free rotation of the aryl groups responsible for the photoactivity.26–28 This, combined with the fact that many reported DAEs lack functional groups capable of forming reasonably strong supramolecular interactions (hydrogen bonding, halogen bonding, etc.) limits DAE's potential for use in crystal engineering.2,5,29 The synthesis of DAE building units in the ring-closed form which contain functional groups capable of supramolecular interactions30–33 should facilitate the production of photoactive crystalline materials and lay the foundation for the rational design of photoactive crystalline solids.
Benzoquinone and naphthoquinone based diarylethenes are attractive candidates for these materials given the long lifetimes of the ring closed isomer and the ability of these molecules to undergo acid-promoted ring-closing reactions12,18,34 thus allowing for the chromatographic isolation of pure ring-closed forms in good yields. A solution of the ring-closed isomer of 2,3-bis(2,5-dimethylthiophen-3-yl)-5,6-dimethyl-1,4-benzoquinone exhibited no ring-opening isomerization when left for 14 days in the dark at room temperature34 indicating its thermal stability. In solution photochemical, ring closure of dithienyl quinone and naphthoquinone containing DAEs is known to proceed with efficiencies less18,19,34 than that of the better known and structurally related perfluorocyclopentene containing DAEs.35 However, chemically induced ring closure can be achieved in high yield when three equivalents of AlCl3 in methylene chloride are added to the quinone and naphthoquinone containing ring-open isomers.18
Given that DAEs can provide a means for optical control of material properties, they have been synthesized with both carboxylic acid20,36 and pyridyl17,37 end groups that can allow for supramolecular assemblies through hydrogen bonding and metal–ligand interactions. Another important class of interaction in directing self-assembly, halogen bonding, has risen in popularity over the past 20 years.38–41 However, the utilization of halogen bonding in the preparation of DAE-based crystalline solids remains relatively underexplored compared to materials based upon hydrogen bonding36,42,43 or metal–ligand interactions.44–48 Many of the properties that are unique to halogen bonding, such as tunability and variable size of the donor atom,38 may offer specific advantages for materials designed and synthesized using these interactions.
Herein we report the synthesis, photochemistry, and structural characterization of 2,3-bis(5-iodo-2-methylthiophen-3-yl)naphthalene-1,4-dione (1, Scheme 1), a naphthoquinone dithienylethene containing iodine functional groups. The ring closed isomer 1b was produced in good yield and subsequently crystallized. Analysis of the structure revealed the presence of halogen bonding interactions between the iodine and carbonyl functional groups. Photocrystallography measurements on single crystals of the ring closed isomer revealed that photo-induced cleavage of the crystals was driven by the formation of the ring-open isomer upon irradiation with visible light.
Materials and methods
Materials
All chemicals were obtained from commercial sources and used as received unless otherwise noted. Acetonitrile was distilled from calcium hydride and stored over 4 Å molecular sieves prior to use. Flash column chromatography was accomplished using silica gel purchased from Fisher Scientific (230–400 mesh, grade 60, catalog number S825-212). Polyester backed TLC silica plates (grade 60 with UV indicator) were obtained from EMD Millipore (catalog number 1.05735.0001).Synthesis of compound 1
In a 75 mL glass pressure vessel was combined iodine (2.315 g, 9.12 mmol), previously reported compound 2 (0.500 g, 1.14 mmol),20 and anhydrous K3PO4 (0.487 g, 2.29 mmol). Anhydrous acetonitrile (10 mL) was then added in a single portion. This reaction sequence is outlined in Scheme 1. Upon sealing the vessel, the reaction was heated to 100 °C for 24 hours. After cooling to room temperature, the resulting dark solution was diluted with methylene chloride (100 mL) and washed with aqueous sodium thiosulfate (15 wt% aqueous, 2 × 50 mL) followed by one portion of saturated, aqueous sodium bicarbonate (50 mL). The remaining organic layer was dried over magnesium sulfate, filtered, and solvent removed under reduced pressure to afford a dark orange-brown solid. The solid was adsorbed onto silica, irradiated with 430 nm light, and chromatographed on silica eluting with 100% hexanes to 10% ethyl acetate in hexanes. The purple-colored closed form 1b eluted first followed by a yellow-orange band of open form 1a. The pure fractions of closed form were allowed to slowly evaporate yielding X-ray quality, dark purple crystals. The open form fractions were combined and solvent removed to give a dark solid (0.680 g, 99%). Spectral characterization matched that previously reported in the literature.18Preparation of 1b
In a 20 mL glass vial was placed compound 1 (0.100 g, 0.17 mmol), methylene chloride (10 mL), and AlCl3 (0.066 g, granular) as depicted in Scheme 1. The dark solution was capped and stirred, turning from brown to dark green over approximately 1–2 minutes, for 1 hour and then diluted with water (20 mL). The organic phase was collected, washed with brine, and the crude product quickly adsorbed onto silica followed by chromatography on silica eluting with 10% ethyl acetate in hexanes. The first eluted compound was collected affording a dark purple/black solid upon removal of solvent (0.043 g, 43%). X-ray quality crystals can be obtained by slow evaporation of the chromatographic fractions.Spectroscopic characterization
Solution UV-vis spectra were obtained using a Shimadzu UV-1800 spectrometer using a home-built light source20 for irradiation experiments. Quartz cuvettes (1 cm pathlength) were used for all solution absorption measurements. The UV-vis spectrum of solid 1b was obtained by gently grinding approximately 20 single crystals between two quartz plates (Starna Type 20C) to produce a microcrystalline film (thickness 5.5 μm). The plates were mounted in a cell holder (Starna CH-2049) and placed in multi-port cuvette holder (Thorlabs, CVH100) with fiber optics. The UV-vis spectrum was obtained using a broadband UV-vis light source (Ocean Insight, DH-2000-BAL) and spectrometer (Ocean Insight, HR4Pro). 1H-NMR characterization was performed on a Varian Inova 400 with a broadband probe operating at 400 MHz and all spectra are referenced to the residual chloroform-d solvent peak at 7.26 ppm.Computational details
All density functional theory (DFT) calculations were performed using the computational software suite Gaussian 09 Rev. D.01 with the def2-SVP basis set.49 The M06-2x functional was selected due to its ability to accurately model noncovalent halogen bonding interactions.50 Gas phase geometry optimizations were performed on 1a, 1b, and a halogen bonded dimer consisting of two molecules of 1b. Vibrational frequency calculations were performed for each optimized geometry to ensure the energetic minimum was obtained.Time-dependent DFT (TD-DFT) was used to simulate the theoretical UV-vis spectra for both the open- and closed-ring isomer and the halogen bonded dimer (XB-dimer). Twenty of the lowest energy excited states were computed for each geometry. The spectra were then generated using a Gaussian broadening function with a half-width of 4000 cm−1 for each transition.
X-Ray diffraction
A single crystal of 1b suitable for X-ray diffraction was mounted on the tip of a glass fiber with oil and placed on a Bruker SMART APEX II CCD diffractometer installed with a rotating anode source (Mo-Kα radiation, λ = 0.71073 Å) with a detector distance of 40.00 mm from the crystal and a 2θ-angle of −30°. During the data collection, the crystal was cooled to 90(1) K using an Oxford cryostream nitrogen gas-flow apparatus. A total of 1800 frames were collected using five 180° ω-scans (0.5° scan width) at different φ-angles (φ = 0° to 288° in 72° increments), nominally covering complete reciprocal space.Following irradiation with broadband white light from an inverted microscope (Olympus IMT-2), a fragment of a photoirradiated slice of 1b suitable for X-ray diffraction was mounted on the tip of a glass fiber with oil and placed on a Bruker SMART APEX II CCD diffractometer installed at a rotating anode source (Mo-Kα radiation, λ = 0.71073 Å) with a detector distance of 40.00 mm from the crystal and a 2θ-angle of −20°. During the data collection, the crystal was cooled to 90(1) K using an Oxford cryostream nitrogen gas-flow apparatus. A total of 1550 frames (25 second exposure) were collected using five 155° ω-scans (0.5° scan width) at different φ-angles (φ = 0° to 288° in 72° increments).
Data reduction was completed using SAINT version 8.40A, and a multi-scan absorption correction was applied using SADABS version 2016 included in the Bruker APEX3 software suite.51 Space-group determination was performed using the XPREP utility included in the SHELXTL software package.52 Using Olex2,53 the structure was solved with ShelXT54 using intrinsic phasing and refined with ShelXL55 using least squares minimization (full-matrix least-squares on F2). For the irradiated 1b, conversion percentages in the lattice were determined from the refinement of the disordered sulfur atoms which arise from the superposition of the ring open and ring closed structures within the lattice. The occupancies of an open/closed pair of sulfur atoms were constrained to sum to 1.0. The resulting occupancies were taken to be the conversion percentage within the lattice where 100% is a crystal consisting entirely of ring closed molecules. Numerous attempts were made to model the disordered thiophene corresponding to the ring-open isomer. For example, refinement using the thiophene fragment from the database in Olex2 combined with EADP, DFIX, and RIGU commands added over 150 restraints, 20 parameters and increased R1 by approximately 0.3 relative to the model 1b-irradiated described herein. This suggests that the geometry of the thiophene groups corresponding to the ring-open isomer may not be appropriately and reliably captured through discrete modelling of these groups.
Results and discussion
Synthesis and solution spectroscopic characterization of compound 1
Compound 1 was obtained using the procedure of Larrosa56 starting with dicarboxylic acid DAE 2, the synthesis of which has been previously reported.20 Briefly, 2 is deprotonated using potassium phosphate and then iodinated using molecular iodine in acetonitrile in a one-pot reaction (Scheme 1). Critical to the reaction's success is the temperature, which must be kept at 100 °C; consequently, the reaction was run in a pressure vessel to achieve this temperature. Irradiation of 1, which has been adsorbed onto silica, with 430 nm light allows photoisomerization of 1a to 1b such that separation of open form 1a and closed form 1b can be achieved by column chromatography eluting with 100% hexanes to 10% ethyl acetate in hexanes. Under these conditions, 1b elutes slightly faster than 1a. Alternatively, pure 1b can be obtained by treatment of 1 with AlCl3.Worthy of note, compound 1 has been previously synthesized by Deng and Liebeskind18 using a Stille coupling route; iodination was achieved at the unsubstituted thiophene 5-position using iodic acid and iodine in a mixture of acetic acid and carbon tetrachloride. In this study, compound 1 is obtained from compound 2 (Scheme 1), which is conveniently obtained through Suzuki coupling methodology.20
Compound 1a is readily soluble in chlorinated solvents and presents a faint yellow color in dilute solutions with λmax values at 311 nm and 434 nm in methylene chloride. Treatment with aluminum chloride leads to ring closure and the formation 1b with the appearance of new absorption bands at 406 nm and 504 nm and a shoulder near 600 nm (Fig. 1 and S1†). There is a slight increase in intensity of the 434 nm band. Isosbestic points are observed at 319 nm and 353 nm confirming the irradiated solutions contain only two chemical species, namely the ring-open (1a) and ring-closed (1b) isomers. Fig. 1 illustrates 1a, 1b, and the photostationary state obtained after irradiation of 1a with 430 nm light for 10 seconds. The time resolved spectra of the conversion is of 1a to 1b is presented in Fig. S2.† The original spectrum is regenerated upon irradiation with 623 nm light for 100 seconds (Fig. S3†).
 | ||
| Fig. 1 The UV-vis spectra of 1a, 1b, and the photostationary state obtained after irradiation of 1a with 430 nm light for 10 seconds. Concentrations are 9.6 × 10−5 M in methylene chloride. | ||
Structure and spectroscopic characterization of crystalline 1b
Compound 1b crystallizes in the monoclinic space group, P21, with a single molecule in the asymmetric unit. Crystals of 1b grow as large (001) tablets bounded by (010), (20−1), and (22−1) as shown in Fig. 2. | ||
| Fig. 2 Top: The asymmetric unit of 1b with atom labels. Bottom: Idealized habit of a single crystal of 1b illustrating the observed crystallographic faces.57 Atom colors: carbon (grey), oxygen (red), hydrogen (white), sulfur (yellow), iodine (purple). | ||
Closed-form 1b is highly planar except for the two methyl groups which protrude from the molecular plane. The root-mean-square deviation (RMSD) of all non-H atoms from the mean plane (excluding the methyl carbons) is 0.146 Å. The distance of 1.515 Å between C17 and C19 confirms that the diarylethene molecule is the ring-closed isomer.
Molecules of 1b form 2-D sheets parallel to (010) in which the molecular planes are also parallel to (010). Two crystallographically unique short oxygen-iodine contacts indicate the presence of halogen bonding interactions responsible for the sheet structure with intermolecular I⋯O distances (dI⋯O) of 2.924 Å and 3.338 Å and corresponding C–I⋯O angles (∠C–I⋯O) of 171.704° and 163.007°, respectively. The sum of the van der Waals radii for oxygen and iodine is 3.50 Å.58
The 2-D sheets stack along (010) in repeating and staggered ABAB layers in which a B layer is rotated by 180° around [010]. The layers are staggered such that the protruding methyl groups of two similar sheets (e.g. AA) are directed towards the void of a B layer (Fig. 3).
 | ||
| Fig. 3 Crystal packing diagrams of 1b viewed down [010] (top) and down [100] (middle) and 1b-irradiated viewed down [100] (bottom, disordered sulfur atoms colored green). Blue dashed lines indicate halogen bonding interactions. | ||
Viewed under a microscope, single crystals of 1b appear very dark purple/black and are essentially opaque. Given the strong absorbance, attempts to measure the visible absorption spectrum of a single crystal of 1b were unsuccessful. Instead, approximately 20 single crystals were gently ground between quartz plates to produce a microcrystalline film (thickness 5.5 μm). The absorption spectrum of the microcrystalline film of 1b features a maximum at approximately 350 nm and broad absorbance similar to that in solution (Fig. S4†).
Electronic structure
DFT methods were used to determine the geometry optimized structures and electronic transitions for 1a and 1b. Calculations were also performed on a halogen bonded dimer (XB-dimer) derived from the crystal structure to assess the impact of the halogen bonding interaction on the spectroscopic properties.Above 400 nm, 1a exhibits two strong electronic transitions at 405.81 nm (f = 0.0302) and 427.55 nm (f = 0.0155) as shown in Fig. 4. The maxima at 410 nm in the calculated spectrum is blue shifted by approximately 24 nm relative to 1a in solution. The largest orbital contributions for the 405 nm and 427 nm transitions are HOMO to LUMO (40.7% and 50.5%) and HOMO−8 to LUMO (27.3% and 30.7%), respectively. The HOMO to LUMO transition may be described as a dithienylethene to napthoquinone transition, whereas the HOMO−8 to LUMO is largely a napthoquinone-based π to π* transition (Fig. 5).
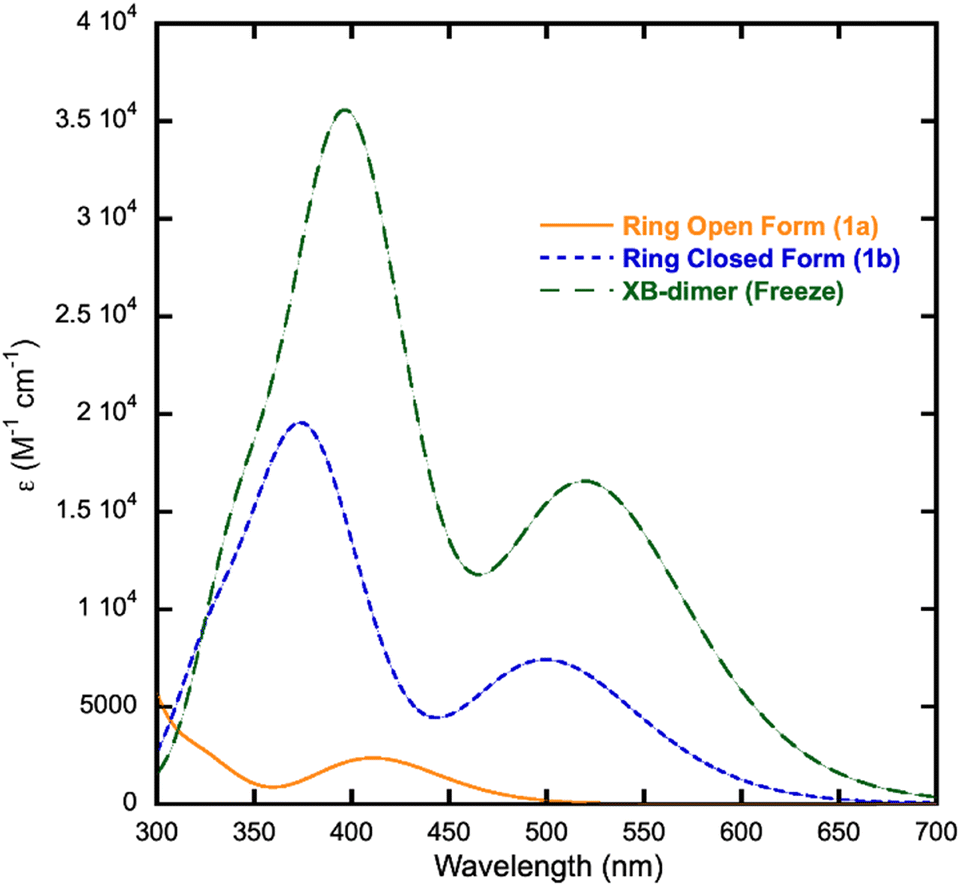 | ||
| Fig. 4 Calculated UV-vis spectra for 1a, 1b, and the XB-dimer. | ||
 | ||
| Fig. 5 Images illustrating the molecular orbitals associated with the HOMO to LUMO transition (top) and the HOMO−8 to LUMO transition (bottom) in 1a. | ||
Isomer 1b exhibits two strong electronic transitions at 382.28 nm (f = 0.2685) and 500.02 nm (f = 0.1413) with two additional weaker transitions at 360.91 nm and 327.7 nm (Fig. 4). It should be noted that the stronger calculated oscillator strengths of 1b relative to 1a are consistent with the larger extinction coefficients measured for 1b relative to 1a (Fig. S1†). The orbital contribution of the transition at 500 nm is 97.2% HOMO to LUMO whereas the contribution of the 382.27 nm transition is 88.1% HOMO to LUMO+1. The HOMO of 1b consists largely of π orbitals residing on the dithienylethene and quinone portion of the molecule (Fig. 6). The LUMO and LUMO+1 consist of π* orbitals spanning the entire molecule. Thus, both the 382 nm and 500 nm excitations may be described as π to π* transitions.
 | ||
| Fig. 6 Images illustrating the molecular orbitals associated with the HOMO to LUMO transition (top) and HOMO to LUMO+1 transition (bottom) in 1b. | ||
Calculations were also performed on a halogen bonded dimer (XB-dimer) using the geometry observed in the crystal structure to assess the impact of this interaction upon the electronic properties of the molecule. The calculated orbitals and contributions for the transitions of XB-dimer are very similar to those of 1b (Tables S1 and S2†). The two lowest energy and strongest transitions were calculated to be 400.77 nm (f = 0.472) and 520.66 nm (f = 0.3058), a red shift of approximately 20 nm relative to the 1b monomer. The molecular orbitals of XB-dimer associated with the strongest electronic transitions are similar to those for the analogous transitions in 1b (Fig. S5†). Likewise, the spectrum of XB-dimer strongly resembles that of 1b, although the maxima are red-shifted. Thus, the presence of halogen bonding interactions is likely responsible for the observed low energy visible absorption.
Photocrystallography
Upon irradiation with visible light, crystals of 1b exhibit the formation of striations approximately parallel to [010] as shown in Fig. 7. With continued exposure, the striations separate thus transforming the original single crystal into a collection of smaller crystal fragments described as thin plates or needles. Although these fragments are extremely fragile, one thin plate was mechanically separated, isolated, and analysed by XRD. | ||
| Fig. 7 Images of a single crystal of 1b viewed normal to (001) before (left) and after (right) irradiation with visible light. Photoinduced cleavage along (010) is indicated with a dashed white line. | ||
Initial refinement of the structure resembled 1b; however, several q-peaks corresponding to regions of unassigned electron density were observed near the sulphur atoms of the thiophenes. The positions of these q-peaks were consistent with the positions of sulphur atoms of the ring-open isomer, 1a, that would be formed because of the irradiation. The structure was refined using a sulphur disorder model to assess the relative fractions of 1b and 1a within the lattice. The resulting occupancies calculated from this model indicated that the lattice contained approximately 7.5(9)% ring-open isomer (1a).
Examination of the crystal structure of 1b reveals that the cleavage is consistent with separation of halogen-bonded planes. At the molecular level the separation is likely driven by the strain induced from the formation of the ring-open isomer. Upon ring opening the methyl groups exert a force upon a neighbouring layer which is consistent with the displacement of the disordered sulfur atoms away from the molecular plane (Fig. 3). That (010) serves as a photoinduced cleavage plane is likely a consequence of the relatively weak van der Waals interactions between neighbouring (010) sheets when compared to the intermolecular halogen-bonding interactions found within these layers (Fig. 5).
Conclusions
The ability to produce photoactive crystalline materials using ring-closed isomers is an important advance for diarylethene-based crystal engineering. The use of ring-closed isomers effectively eliminates the conformational flexibility present in ring-open isomers that can lead to crystals that contain exclusively photoinactive geometries. This work demonstrates that chemical methods, as opposed to light irradiation, are suitable for producing quantities of ring-closed quinone-based diarylethenes for subsequent crystallization. Future work will assess the potential advantages of chemical ring-closing, namely the suppression of fatigue products that are generated during prolonged irradiation.The crystal structures confirm that the iodine functional groups of 1b form intermolecular halogen bonds with oxygen atoms of the quinone moiety. While crystals of 1b degrade upon exposure to visible light via cleavage planes parallel to the 2-D sheets, this observation confirms that the molecules within the lattice are photoactive and potentially capable of transforming light into mechanical work.
Author contributions
SDM, ZYM, JBB and DGP were responsible for conceptualization. SDM, ZYM, TBM, JAS, and DGP were responsible for the investigation. SDM, ZYM, TBM, DGP, and JBB were responsible for the formal analysis. JBB and DGP were responsible for funding acquisition. All authors participated in writing, reviewing, and editing.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Funding was provided by the National Science Foundation, Directorate for Mathematical and Physical Sciences (award no. DMR-2003932) and a Research Development Grant from the Pennsylvania State University at Hazleton.Notes and references
- Organic Photochromic and Thermochromic Compounds, ed. J. C. Crano and R. J. Guglielmetti, Plenum Press, New York, 1999 Search PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- G. Pariani, M. Quintavalla, L. Colella, L. Oggioni, R. Castagna, F. Ortica, C. Bertarelli and A. Bianco, J. Phys. Chem. C, 2017, 121, 23592–23598 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Y.-Y. Tang, Y.-L. Zeng and R.-G. Xiong, J. Am. Chem. Soc., 2022, 144, 8633–8640 CrossRef CAS PubMed.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- R. Wang, L. Diao, Q. Ren, G. Liu and S. Pu, ACS Omega, 2019, 4, 309–319 CrossRef CAS.
- Y. Zou, T. Yi, S. Xiao, F. Li, C. Li, X. Gao, J. Wu, M. Yu and C. Huang, J. Am. Chem. Soc., 2008, 130, 15750–15751 CrossRef CAS PubMed.
- V. Lemieux and N. R. Branda, Org. Lett., 2005, 7, 2969–2972 CrossRef CAS PubMed.
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966–15967 CrossRef CAS PubMed.
- N. A. Simeth, A. C. Kneuttinger, R. Sterner and B. König, Chem. Sci., 2017, 8, 6474–6483 RSC.
- B. M. Neilson and C. W. Bielawski, Organometallics, 2013, 32, 3121–3128 CrossRef CAS.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- T. Sumi, T. Kaburagi, M. Morimoto, K. Une, H. Sotome, S. Ito, H. Miyasaka and M. Irie, Org. Lett., 2015, 17, 4802–4805 CrossRef CAS PubMed.
- J. Xu, H. Volfova, R. J. Mulder, L. Goerigk, G. Bryant, E. Riedle and C. Ritchie, J. Am. Chem. Soc., 2018, 140, 10482–10487 CrossRef CAS PubMed.
- X. Deng and L. S. Liebeskind, J. Am. Chem. Soc., 2001, 123, 7703–7704 CrossRef CAS PubMed.
- D. G. Patel, M. Boggio-Pasqua, T. B. Mitchell, I. M. Walton, W. R. Quigley and F. A. Novak, Molecules, 2020, 25, 2630 CrossRef CAS PubMed.
- D. G. Patel, T. B. Mitchell, S. D. Myers, D. A. Carter and F. A. Novak, J. Org. Chem., 2020, 85, 2646–2653 CrossRef CAS PubMed.
- M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed.
- L. F. Fieser and M. Fieser, J. Am. Chem. Soc., 1935, 57, 491–494 CrossRef CAS.
- Y. Kumagai, Y. Shinkai, T. Miura and A. K. Cho, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 221–247 CrossRef CAS PubMed.
- N. El-Najjar, H. Gali-Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, Phytochem. Rev., 2011, 10, 353–370 CrossRef CAS.
- H.-Y. Qiu, P.-F. Wang, H.-Y. Lin, C.-Y. Tang, H.-L. Zhu and Y.-H. Yang, Chem. Biol. Drug Des., 2018, 91, 681–690 CrossRef CAS PubMed.
- M. Walko and B. L. Feringa, Chem. Commun., 2007, 1745–1747, 10.1039/B702264F.
- I. M. Walton, J. M. Cox, C. A. Benson, D. G. Patel, Y.-S. Chen and J. B. Benedict, New J. Chem., 2016, 40, 101–106 RSC.
- J. M. Cox, I. M. Walton and J. B. Benedict, J. Mater. Chem. C, 2016, 4, 4028–4033 RSC.
- M. Irie, Photochem. Photobiol. Sci., 2010, 9, 1535–1542 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC.
- S. Cherukuvada, R. Kaur and T. N. Guru Row, CrystEngComm, 2016, 18, 8528–8555 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed., 1995, 34, 2311–2327 CrossRef CAS.
- E. Saito, T. Ako, Y. Kobori and A. Tsuda, RSC Adv., 2017, 7, 2403–2406 RSC.
- S. Kobatake, T. Yamada, K. Uchida, N. Kato and M. Irie, J. Am. Chem. Soc., 1999, 121, 2380–2386 CrossRef CAS.
- S. Yamamoto, K. Matsuda and M. Irie, Chem. – Eur. J., 2003, 9, 4878–4886 CrossRef CAS PubMed.
- G. Liu, M. Liu, S. Pu, C. Fan and S. Cui, Dyes Pigm., 2012, 95, 553–562 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- V. Nemec, K. Lisac, N. Bedeković, L. Fotović, V. Stilinović and D. Cinčić, CrystEngComm, 2021, 23, 3063–3083 RSC.
- T. Ichikawa, M. Morimoto and M. Irie, Photochem. Photobiol. Sci., 2014, 13, 199–204 CrossRef CAS PubMed.
- T. Fukushima, K. Tamaki, A. Isobe, T. Hirose, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi, M. J. Hollamby and S. Yagai, J. Am. Chem. Soc., 2021, 143, 5845–5854 CrossRef CAS PubMed.
- F. Luo, C. B. Fan, M. B. Luo, X. L. Wu, Y. Zhu, S. Z. Pu, W.-Y. Xu and G.-C. Guo, Angew. Chem., Int. Ed., 2014, 53, 9298–9301 CrossRef CAS PubMed.
- V. I. Nikolayenko, S. A. Herbert and L. J. Barbour, Chem. Commun., 2017, 53, 11142–11145 RSC.
- D. G. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Chem. Commun., 2014, 50, 2653–2656 RSC.
- I. M. Walton, J. M. Cox, T. B. Mitchell, N. P. Bizier and J. B. Benedict, CrystEngComm, 2016, 18, 7972–7977 RSC.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS PubMed.
- Gaussian 09, Revision D.01, Wallingford, CT, 2009 Search PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Theory Comput., 2016, 12, 4303–4325 CrossRef CAS PubMed.
- SMART and SAINTPLUS - Area Detector Control and Integration Software, version 6.01, Bruker AXS, Madison, WI, 1999 Search PubMed.
- XPREP version 2014/2, Bruker AXS Inc, Madison, Wisconsin, USA, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- G. J. P. Perry, J. M. Quibell, A. Panigrahi and I. Larrosa, J. Am. Chem. Soc., 2017, 139, 11527–11536 CrossRef CAS PubMed.
- W. Kaminsky, J. Appl. Crystallogr., 2005, 38, 566–567 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and Tables S1 and S2. CCDC 2329844 and 2330092. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00112e |
| ‡ Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |