
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of solvent molecules on crystallisation of heterotrinuclear MII–TbIII–MII complexes with tripodal nonadentate ligands†
Kazuma
Takahara
a,
Yuki
Horino
a,
Koki
Wada
a,
Hiromu
Sakata
a,
Daichi
Tomita
b,
Yukinari
Sunatsuki
 c,
Hiroshi
Isobe
d,
Masaaki
Kojima
a and
Takayoshi
Suzuki
*ad
c,
Hiroshi
Isobe
d,
Masaaki
Kojima
a and
Takayoshi
Suzuki
*ad
aGraduate School of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan. E-mail: suzuki@okayama-u.ac.jp
bFaculty of Science, Okayama University, Okayama 700-8530, Japan
cAdvanced Science Research Center, Okayama University, Okayama 700-8530, Japan
dResearch Institute for Interdisciplinary Science, Okayama University, Okayama 700-8530, Japan
First published on 10th January 2024
Abstract
The crystal structures and crystallisation behaviours of MII–TbIII–MII heterotrinuclear complexes, [(L)MTbM(L)]NO3 (M = Mn and Zn; L3− stands for a conjugated base of H3L = 1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane), obtained from various organic solvents (MeOH, EtOH, CH2Cl2 and CHCl3) were investigated. The trinuclear complex cation has two asymmetric centres (Δ or Λ) at two MII sites as a result of the twisted tripodal arms of L3−. Single-crystal X-ray diffraction analysis revealed that all the analysed Zn–Tb–Zn complexes had homochiral structures (Δ,Δ- or Λ,Λ-enantiomers) in each single crystal; however, the type of crystallisation behaviour showed clear differences depending on the type of solvent molecule. Specifically, crystallisation from MeOH or CH2Cl2 resulted in the exclusive formation of the Λ-conglomerates with the Λ,Λ-enantiomers—a phenomenon we recently termed ‘absolute spontaneous resolution’. The analogous Mn–Tb–Mn complex crystallised from MeOH also resulted in the same phenomenon as that of Zn–Tb–Zn. In contrast, the meso-type (Δ,Λ) achiral isomer of the Mn–Tb–Mn complex was deposited for the first time in a series of MII–LnIII–MII trinuclear complexes from a CH2Cl2 or EtOH solution. Density functional theory calculations were performed to compare the thermodynamic stability of homochiral (Λ,Λ) and meso-type (Δ,Λ) complex cations of [(L)MnTbMn(L)]+ in MeOH and EtOH. Results were consistent with the molecular structures observed in the crystallographic analysis of the compounds deposited from these solvents.
Introduction
In most cases, a solution containing a racemic mixture of chiral compounds (a racemic solution) provides racemic crystals (racemic compounds) that contain both left- and right-handed enantiomers in a symmetry-related manner. In contrast, it is well known that ca. 5% of chiral compounds exhibit spontaneous resolution, depositing equal amounts of both enantiomorphic crystals (conglomerates) (Fig. 1a).1–6 In previous papers, we reported on an interesting example of spontaneous resolution for a series of zinc(II)-derived 3d–4f–3d heterometallic trinuclear complexes, [(L)ZnIILnIIIZnII(L)]NO3·nMeOH (Ln = Y and all lanthanoids except Pm and Tm; n = 2 or 4; H3L = 1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane), bearing two achiral tripodal nonadentate ligands (L3−).7,8 It was confirmed by means of single-crystal X-ray diffraction (SC-XRD) analysis that the crystal structures of the Zn–Ln–Zn complexes were dependent on the central lanthanoid(III) ions (Fig. 1b). The compounds of [(L)ZnLnZn(L)]NO3·4MeOH with early lanthanoids up to PrIII crystallised in a centrosymmetric space group P21/c and gave racemic crystals, including both Δ,Δ- and Λ,Λ-enantiomers together with four solvent MeOH molecules. In contrast, compounds of [(L)ZnLnZn(L)]NO3·2MeOH with NdIII or heavier lanthanoids crystallised in a nonenantiogenic (Sohncke) space group P212121 and exhibited spontaneous resolution to form conglomerates, which contain one of the enantiomers in a crystal with two MeOH molecules. Solid-state circular dichroism (CD) spectroscopy was also used to clarify the new crystallisation phenomenon, in which all crystals deposited by spontaneous resolution were left-handed enantiomorphic crystals (the Λ-conglomerates with the Λ,Λ-enantiomer) in the case of Ln = Eu (f6), Tb (f8), Ho (f10) and Lu (f14) with an even number of 4f electrons (Fig. 1b).8,9 These results were indicative of the importance of the type of each central lanthanoid(III) ion for the crystallisation behaviour of spontaneously resolved compounds. | ||
| Fig. 1 (a) Definition of total and absolute spontaneous resolution together with normal spontaneous resolution and formation of racemic crystals (racemic compounds) and (b) classification of crystallisation behaviours of a series of Zn–Ln–Zn heterotrinuclear complexes. | ||
In this study, we examined the effect of various solvents (methanol, ethanol, dichloromethane and chloroform) on the crystal structures and crystallisation behaviours of the Zn–Tb–Zn heterotrinuclear complex. In addition, the analogous Mn–Tb–Mn complex was investigated similarly, because the crystal structures of both compounds were very similar to each other in the orthorhombic space group of P212121 with Z = 4,7,8 and because a high-spin MnII ion would give a rapid racemisation in solution, which is necessary for inducement of the above-mentioned novel crystallisation behaviour. All the resulting crystals contained the corresponding solvent molecules, formulated as [(L)MTbM(L)]NO3·nsolvent (M = Zn or Mn, n = 2–4, solvent = MeOH, EtOH, CH2Cl2 and CHCl3), and their crystallisation behaviours were dependent on the solvent. In particular, in the case of crystallisation of the MnII complex from EtOH, a meso-type (Δ,Λ) complex cation was obtained. Density functional theory (DFT) calculations were performed to compare the relative stability between homochiral (Λ,Λ) and meso-type (Δ,Λ) complex cations of [(L)MnTbMn(L)]+ in solution.
Results and discussion
Synthesis and characterisation of heterotrinuclear complexes
Syntheses of the ligand precursor, H3L, and the heterotrinuclear complexes of [(L)ZnTbZn(L)]NO3·2MeOH (Zn·2MeOH) and [(L)MnTbMn(L)]NO3 have been reported previously.7,8 However, H3L was now prepared by a modified method using potassium tert-butoxide (t-BuOK) instead of triethylamine to achieve a simpler isolation process, by the formation of a readily separable by-product KCl (Scheme S1†). The manganese(II) complex, [(L)MnTbMn(L)]NO3·3MeOH (Mn·3MeOH), was prepared with the addition of triethylamine by a method similar to that used for Zn·2MeOH in an inert atmosphere (see Experimental section). Recrystallisation of both trinuclear complexes from dry methanol, ethanol, dichloromethane or chloroform afforded single crystals or microcrystals of the corresponding solvated compounds in high purity. The products were identified by elemental analysis, infrared (IR) spectroscopy (Fig. S1†), ultraviolet-visible (UV-vis) absorption spectroscopy (Fig. S2†) and SC-XRD analysis (vide infra).Crystal structures of Zn–Tb–Zn compounds
In addition to the previously reported Zn·2MeOH, the crystal structures of the Zn–Tb–Zn compounds obtained from EtOH, CH2Cl2 and CHCl3 were analysed by SC-XRD. The crystallographic data are given in Table 1. The compound from EtOH was crystallised in the monoclinic crystal system and a centrosymmetric space group P21/n with Z = 4. The asymmetric unit contained a heterotrinuclear complex cation, [(L)ZnTbZn(L)]+, a NO3− anion, and four EtOH molecules, [(L)ZnTbZn(L)]NO3·4EtOH (Zn·4EtOH). The molecular structure of the complex cation in Zn·4EtOH is shown in Fig. 2. Two tripodal ligands were coordinated to each ZnII ion via the N3O3 atoms and to a TbIII ion via the O6 atoms, constructing a 3d–4f–3d trinuclear complex cation—similar to those in all the previously reported trinuclear Fe–Ln–Fe, Co–Ln–Co and Zn–Ln–Zn complexes bearing the H3L ligands.7,8 These complexes have two asymmetric centres (Δ and Λ) at each of the ZnII sites, originating from the twist of the tripodal ligand arms. There are theoretically three possible stereoisomers for the resulting trinuclear complex cation: the homochiral Λ,Λ and Δ,Δ enantiomers, and the meso-type (Δ,Λ) achiral isomer.7,8 The observed crystal structure of Zn·4EtOH was the racemic compounds (racemic crystals) of the homochiral enantiomers (Fig. S3 and S4†), which could be classified as [class A] compounds (Table 2), similar to the previously reported Zn–Ln–Zn (Ln = La, Ce and Pr) complexes containing four MeOH molecules.8| Compound | [(L)ZnTbZn(L)]-NO3·2MeOH (ref. 8) | [(L)ZnTbZn(L)]-NO3·4EtOH | [(L)ZnTbZn(L)]-NO3·2CH2Cl2 | [(L)ZnTbZn(L)]-NO3·4CHCl3 | [(L)MnTbMn(L)]-NO3·3MeOH | [(L)MnTbMn(L)]-NO3·4EtOH | [(L)MnTbMn(L)]-NO3·2CH2Cl2 | [(L)MnTbMn(L)]-NO3·4CH2Cl2 | [(L)MnTbMn(L)]-NO3·4CHCl3 |
|---|---|---|---|---|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | |||||||||
| CCDC no. | 2191540 | 2303369 | 2303370 | 2303371 | 2303373 | 2303378 | 2303374 | 2303375 | 2303376 |
| Chemical formula | C60H68N7O17TbZn2 | C66H84N7O19TbZn2 | C60H64Cl4N7O15TbZn2 | C62H64Cl12N7O15TbZn2 | C61H72Mn2N7O18Tb | C66H84Mn2N7O19Tb | C60H64Cl4Mn2N7O15Tb | C62H68Cl8Mn2N7O15Tb | C62H64Cl12Mn2N7O15Tb |
| Formula weight | 1448.92 | 1569.11 | 1554.70 | 1862.35 | 1460.08 | 1548.23 | 1533.82 | 1703.68 | 1841.46 |
| T/K | 192(2) | 192(2) | 192(2) | 192(2) | 192(2) | 192(2) | 192(2) | 192(2) | 192(2) |
| Crystal colour, shape | Yellow, block | Yellow, block | Yellow, block | Yellow, block | Yellow, block | Yellow, block | Yellow, block | Yellow, block | Yellow, block |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group, Z | P212121, 4 | P21/n, 4 | P212121, 4 | P21, 2 | P212121, 4 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 1 , 1 |
P21/c, 4 | P21/c, 4 | P21, 2 |
| Crystal size/mm3 | 0.70 × 0.70 × 0.60 | 0.80 × 0.80 × 0.60 | 0.80 × 0.50 × 0.50 | 0.45 × 0.30 × 0.30 | 0.70 × 0.65 × 0.45 | 0.60 × 0.55 × 0.55 | 0.50 × 0.40 × 0.30 | 0.75 × 0.35 × 0.30 | 0.30 × 0.30 × 0.10 |
| a/Å | 15.8173(6) | 15.6078(8) | 15.2744(6) | 12.1126(9) | 15.7164(8) | 11.8404(17) | 18.4535(8) | 23.9154(18) | 12.3427(11) |
| b/Å | 15.8158(7) | 19.1265(10) | 15.3918(5) | 18.5566(14) | 15.9773(9) | 11.8606(14) | 14.8720(5) | 17.9324(14) | 18.1743(13) |
| c/Å | 24.3596(9) | 24.1307(8) | 25.2915(9) | 16.3863(11) | 24.5892(12) | 14.328(2) | 25.4136(8) | 16.4008(11) | 16.3681(14) |
| α/° | 90 | 90 | 90 | 90 | 90 | 104.939(4) | 90 | 90 | 90 |
| β/° | 90 | 110.387(2) | 90 | 98.127(2) | 90 | 90.305(4) | 115.697(2) | 98.864(2) | 100.677(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 117.451(4) | 90 | 90 | 90 |
| U/Å3 | 6093.9(4) | 6752.3(5) | 6181.2(4) | 3646.1(5) | 6174.5(6) | 1707.1(4) | 6284.7(4) | 6946.2(9) | 3608.1(5) |
| D calc/g cm−3 | 1.579 | 1.543 | 1.671 | 1.696 | 1.567 | 1.506 | 1.621 | 1.629 | 1.695 |
| μ (Mo Kα)/cm−1 | 20.025 | 18.158 | 21.451 | 21.166 | 16.14 | 14.625 | 17.479 | 17.389 | 18.238 |
| R int | 0.0348 | 0.0410 | 0.0292 | 0.0574 | 0.0230 | 0.0253 | 0.0311 | 0.0779 | 0.0498 |
| No. reflns./params. | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 884/794 884/794 |
15445/868 |
14129/810 |
16689/902 |
14086/813 |
7757/454 | 14400/813 |
15782/867 |
16275/900 |
| R 1 [I > 2.0 s(I)]a | 0.0306 | 0.0366 | 0.0273 | 0.0409 | 0.0191 | 0.0238 | 0.0242 | 0.0589 | 0.0436 |
| wR2 [all data]b | 0.0735 | 0.1022 | 0.0632 | 0.1090 | 0.0475 | 0.0729 | 0.0634 | 0.1498 | 0.1128 |
| GoF | 1.052 | 1.036 | 1.028 | 1.062 | 1.035 | 1.147 | 1.044 | 1.065 | 1.010 |
 | ||
| Fig. 2 ORTEP drawing of a homochiral Λ,Λ complex cation in [(L)ZnTbZn(L)]NO3·4EtOH (Zn·4EtOH) (ellipsoids are drawn at the 50% probability level; all hydrogen atoms, counterions and solvent molecules are omitted for clarity). | ||
| Compound | Class | Chirality of complex | C1–C30 (C1–C1′)a,b (Å) | U/Z (Å3) |
|---|---|---|---|---|
| a See Fig. 2 and 5 as well as ref. 8. b For the meso-type complexes this should be C1–C1′. | ||||
| Zn·2MeOH | B (Λ) | Λ,Λ | 16.111 | 1523.5 |
| Zn·2CH 2 Cl 2 | B (Λ) | Λ,Λ | 16.062 | 1545.3 |
| Zn·4CHCl 3 | C (Λ)/(Δ) | Λ,Λ/Δ,Δ | 16.041 | 1823.1 |
| Zn·4EtOH | A (rac) | Λ,Λ + Δ,Δ | 16.355 | 1820.8 |
| Mn·3MeOH | B (Λ) | Λ,Λ | 16.184 | 1543.6 |
| Mn·2CH 2 Cl 2 | D (meso) | Δ,Λ | 16.121 | 1571.2 |
| Mn·4CH 2 Cl 2 | D (meso) | Δ,Λ | 16.121 | 1736.5 |
| Mn·4CHCl 3 | C (Λ)/(Δ) | Λ,Λ/Δ,Δ | 16.170 | 1804.1 |
| Mn·4EtOH | D (meso) | Δ,Λ | 16.167 | 1707.1 |
The crystal structure of the compound obtained from CH2Cl2 was found to be nearly isomorphic to that of Zn·2MeOH; it crystallised in the orthorhombic crystal system and a nonenantiogenic (Sohncke) space group P212121 with Z = 4, [(L)ZnTbZn(L)]NO3·2CH2Cl2 (Zn·2CH2Cl2). The compound exhibits spontaneous resolution and belongs to [class B] conglomerates (Table 2), as defined in a previous paper for the Zn–Ln–Zn (Ln = Nd and heavier lanthanoids) compounds containing two MeOH molecules.8 For Zn·2CH2Cl2, all crystals analysed to date by the SC-XRD method were Λ-conglomerates with the Λ,Λ-enantiomers (confirmed using the Flack parameter), similar to the Fe–Tb–Fe and Mn–Tb–Mn and a series of [class B] Zn–Ln–Zn complexes reported earlier.7,8
In the case of the Zn–Tb–Zn compound deposited from a CHCl3 solution, [(L)ZnTbZn(L)]NO3·4CHCl3 (Zn·4CHCl3), it crystallised in the monoclinic crystal system and a nonenantiogenic space group P21 with Z = 2. The molecular structure of the trinuclear complex cation was very similar to the structures of the above-mentioned complexes; however, the crystal structure and packing pattern of Zn·4CHCl3 differ from that of Zn·2MeOH; thus it is classified as [class C] (Table 2).
In all the Zn–Tb–Zn compounds analysed in our previous studies and the present study, no obvious hydrogen bonds or π–π stacking interactions were recognised between the complex cations and the molecules of solvation or the NO3− anion. However, the molecular size of the trinuclear complex cation, [(L)ZnTbZn(L)]+, estimated by the intramolecular distance between C1 and C30, is larger for the racemic compound [class A] of Zn·4EtOH (16.355 Å) than for the corresponding conglomerates [class B] and [class C] (16.041–16.111 Å, Table 2). When the estimated volume of asymmetric unit (U/Z) was calculated for the [class B] Zn·2MeOH and Zn·2CH2Cl2 compounds (conglomerates by P212121), where U is the volume of a unit cell and Z is the number of asymmetric units in a unit cell, Zn·2CH2Cl2 (1545.3 Å3) and Zn·2MeOH (1523.5 Å3) (Table 2) gave similar values—the small difference between them is due to a larger molecular volume of CH2Cl2 than MeOH. In contrast, the values of U/Z for [class A] Zn·4EtOH (1820.8 Å3) (racemic crystal by P21/n) and [class C] Zn·4CHCl3 (1823.1 Å3) (conglomerates by P21) are larger than those of [class B] compounds as the volume and the number of solvating molecules EtOH or CHCl3 are remarkably increased (see Table 2). According to the powder X-ray diffraction (P-XRD) measurements (see Experimental section and Fig. S5†), the [class B] compounds retain the original crystal structures even after complete drying (i.e., in the fully dried state), while [class A] and [class C] compounds immediately changed into the amorphous phase due to the loss of the solvating molecules in air. It is therefore suggested that the trinuclear [(L)ZnTbZn(L)]+ complex cations and NO3− anions are firmly packed in the crystal structures of [class B] compounds to create voids that can incorporate solvent molecules without any intermolecular interaction. Therefore, a suitable number (two in this case) and size of solvating molecules (MeOH and CH2Cl2 in this case) would be able to co-crystallise with complex cations, forming the [class B] compounds (conglomerates by P212121). EtOH and CHCl3 molecules with larger sizes would exceed the void capacity, hence the resulting crystal structures will change to [class A] or [class C] compounds, which easily lost crystallinity upon removal of the solvent molecules.
Crystallisation behaviour of the dichloromethane and chloroform solvates, Zn·2CH2Cl2 and Zn·4CHCl3
In earlier work, we reported on the novel phenomenon referred to as ‘absolute spontaneous resolution’, which gave certain handed enantiomorphic crystals exclusively in the crystallisation of [(L)ZnTbZn(L)]NO3 from MeOH, affording Zn·2MeOH.8,9 As mentioned earlier, the crystal structures of the Zn–Tb–Zn compounds varied depending on the type of recrystallisation solvents used; and in the cases of dichloromethane and chloroform, it was found that the first crystals analysed by the SC-XRD method were confirmed, using the Flack parameters, to be the Λ-conglomerates (Table 1) as Zn·2MeOH. We further examined the crystallisation behaviours of Zn·2CH2Cl2 and Zn·4CHCl3 by means of SC-XRD analysis and/or solid-state CD spectroscopy, as we described previously.8,9 We also checked the phase purity of the entire bulk crystalline samples of Zn·2CH2Cl2 and Zn·4CHCl3 using the P-XRD technique. For Zn·2CH2Cl2, the observed spectral patterns are in good agreement with the simulated ones from SC-XRD analyses (Fig. S5b and c†), indicating that all bulk samples have the same crystal structures as shown in Table 1. Conversely, the Zn·4CHCl3 gave a slightly broadened spectrum, probably due to a rapid efflorescence.For both Zn·2CH2Cl2 and Zn·4CHCl3, several single crystal samples were selected from a recrystallisation batch and their crystal structures were analysed by SC-XRD. The absolute structure of the single crystal was determined by the Flack method. The results gave the ratio of Λ- and Δ-conglomerates as 8:0 and 7:3 for Zn·2CH2Cl2 and Zn·4CHCl3, respectively (Table 3). These results indicated that, while Zn·4CHCl3 exhibits normal spontaneous resolution yielding both enantiomorphic crystals, Zn·2CH2Cl2 provides only the Λ-conglomerate, similar to Zn·2MeOH.8 In addition, the solid-state CD spectra of the bulk crystalline samples obtained from repeated crystallisation experiments exhibited CD spectra with the same cotton effects as those of Zn·2MeOH (Fig. 3). These results suggest that absolute spontaneous resolution also takes place during the crystallisation of the Zn–Tb–Zn complex from CH2Cl2.
| No. | (a) Zn·2CH2Cl2 | (b) Zn·4CHCl3 | ||
|---|---|---|---|---|
| Chirality | Flack parameter | Chirality | Flack parameter | |
| 1 | Λ | −0.003(3) | Λ | −0.004(4) |
| 2 | Λ | 0.003(3) | Λ | −0.003(3) |
| 3 | Λ | 0.038(15) | Λ | 0.012(6) |
| 4 | Λ | −0.001(3) | Λ | 0.002(4) |
| 5 | Λ | 0.005(3) | Λ | −0.007(3) |
| 6 | Λ | −0.001(3) | Δ | −0.005(4) |
| 7 | Λ | 0.010(4) | Λ | −0.007(3) |
| 8 | Λ | 0.001(3) | Λ | 0.003(4) |
| 9 | — | — | Δ | 0.0294(6) |
| 10 | — | — | Δ | 0.073(17) |
 | ||
| Fig. 3 CD spectra of Zn·2CH2Cl2 prepared from bulk samples from eight repeated crystallisation batches. | ||
Crystal structures of Mn–Tb–Mn compounds
There were significant differences in the crystal structures and crystallisation behaviours of the Zn–Tb–Zn compounds depending on the recrystallisation solvents. We therefore considered it interesting to examine the crystal structures and crystallisation behaviours of the analogous manganese(II) complex, Mn–Tb–Mn, using a variety of different solvents for crystallisation because of the similarity in the crystal structures of both compounds7,8 and a possibility for absolute or total spontaneous resolution resulting from a high-spin MnII state.SC-XRD analysis results for a single crystal of Mn–Tb–Mn compound deposited from MeOH confirmed the formulation of the compound as [(L)MnTbMn(L)]NO3·3MeOH (Mn·3MeOH); it was crystallised in the orthorhombic crystal system and a nonenantiogenic space group P212121 with Z = 4 (Table 1). The crystal structure of the Mn–Tb–Mn complex without any solvent molecule (Mn) was disclosed in a previous paper of ours.7 The crystal packing patterns are quite similar to each other, but the unit cell volumes of two crystals are apparently different (6174.5(6) Å3 for Mn·3MeOHvs. 6127.7(5) Å3 for Mn). At present, we are unable to reproduce the conditions to form Mn (without a MeOH molecule of solvation), but several SC- and P-XRD analyses do indicate that all single crystals obtained in the present study are Mn·3MeOH. The crystal structure of Mn·3MeOH was nearly isomorphic to that of Zn·2MeOH, and the absolute structures of all single crystals analysed by SC-XRD were found to be the Λ-conglomerate. Thus, this compound can be classified as [class B]. The molecular structure of the cationic complex, [(L)MnTbMn(L)]+, was almost identical to those of [(L)ZnTbZn(L)]+ observed in a series of the above-mentioned Zn–Tb–Zn compounds.
The single crystal of the Mn–Tb–Mn complex deposited from CHCl3 was confirmed to crystallise with four CHCl3 molecules in the monoclinic crystal system and a nonenantiogenic space group P21 with Z = 2, [(L)MnTbMn(L)]NO3·4CHCl3 (Mn·4CHCl3) (Table 1). This crystal structure was nearly isomorphic to the corresponding Zn·4CHCl3 and belongs to [class C].
For both Mn–Tb–Mn compounds (Mn·3MeOH and Mn·4CHCl3), obvious hydrogen bonding or π–π stacking interactions were not detected between the complex cations and the solvating molecules or NO3− anions. Comparing the molecular structures of the Zn–Tb–Zn and Mn–Tb–Mn complex cations, the C1–C30 distance of the Mn–Tb–Mn cation was longer than that of the Zn–Tb–Zn analogue, consistent with a larger ion radius of MnII than that of ZnII (Table 2). The estimated volume of asymmetric unit (U/Z) for Mn·3MeOH (1543.6 Å3) is slightly larger than that for Zn·2MeOH (1523.5 Å3) (Table 2). A possible explanation is that the slightly larger size of a complex cation gains a little more space to take one more MeOH molecule in the unit cell in Mn·3MeOH, in comparison with that in Zn·2MeOH. Conversely, the estimated value of U/Z for [class C] (conglomerates by P21) Mn·4CHCl3 compound (1804.1 Å3) is much larger than that of [class B] (conglomerates by P212121) Mn·3MeOH. Taking into account the P-XRD measurements, which gave similar results to those for the analogous Zn–Tb–Zn compounds (see Experimental section and Fig. S5†), the [class B] compounds maintain the crystal structures with trinuclear complex cations [(L)MnTbMn(L)]+ and NO3− anions; there are voids for accommodating three MeOH molecules. Therefore, a suitable number (three in this study) and size of solvating molecules (MeOH in this study) are of importance in providing the [class B] crystal structure (conglomerates by P212121) with quite similar lattice parameters and unit cell volumes to those of zinc(II) analogues. The larger size and number of CHCl3 molecules exceed the void capacity, resulting in the formation of [class C] crystal structures with easy loss of crystallinity upon removal of the solvent molecules.
Crystallisation behaviour of Mn–Tb–Mn compounds
Because both Mn·3MeOH and Mn·4CHCl3 exhibit spontaneous resolution, similar to the corresponding Zn compounds, the crystallisation behaviour of these compounds should be carefully examined. To this end, use was made of SC-XRD and solid-state CD techniques, as described in a previous section. SC-XRD analysis of Mn·4CHCl3 indicated Λ- and Δ-conglomerate formation in the same crystallisation batch which is indicative of the exhibition of normal spontaneous resolution (Table S3†). In the case of Mn·3MeOH, the solid-state CD spectra for nine single crystals selected from a recrystallisation batch were measured; the results are shown in Fig. 4a. All of the resulting CD spectra showed the same positive cotton effect around 400 nm. This suggested that all crystals measured were exclusively the Λ-conglomerates, as in the case of the analogous Zn·2MeOH. Having confirmed the phase purity of the entire bulk crystalline samples of Mn·3MeOH by the P-XRD technique (Fig. S5d†), we then examined solid-state CD measurements for bulk crystalline samples from repeated crystallisation batches of Mn·3MeOH (Fig. 4b). The resulting CD spectra were very similar to those of single crystal samples; therefore, it is indicative of the possibility of absolute spontaneous resolution.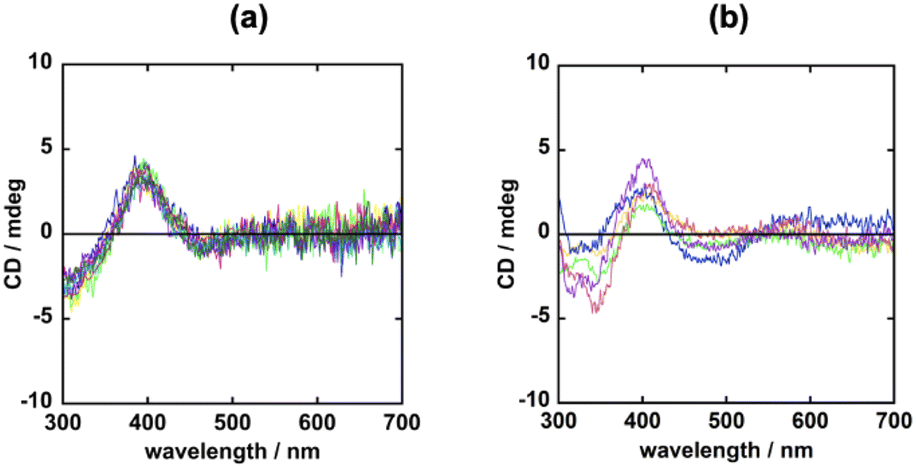 | ||
| Fig. 4 Solid-state CD spectra of Mn·3MeOH samples prepared from (a) nine single crystals resulting from a crystallisation batch and (b) bulk samples from five repeated crystallisation batches. | ||
To summarise, the TbIII complexes in the [class B] compounds of Zn·2MeOH, Zn·2CH2Cl2 and Mn·3MeOH, which possess similar lattice parameters, exhibited absolute spontaneous resolution, giving the Λ-conglomerates exclusively.8 The crucial reason behind the appearance of this novel phenomenon is still unclear. It is nonetheless evident that, at least for the crystallisation of a series of Zn–Tb–Zn and Mn–Tb–Mn complexes, a suitable solvent for crystallisation is necessary to exhibit absolute spontaneous resolution.
Crystal structures of Mn–Tb–Mn compounds obtained from EtOH or CH2Cl2
A single crystal of the Mn–Tb–Mn compound deposited from an EtOH solution was analysed by SC-XRD. Results confirmed that the compound was crystallised in the triclinic crystal system and a centrosymmetric space group P with Z = 1. The asymmetric unit contains half of a trinuclear [(L)MnTbMn(L)]+ complex cation, in which a crystallographic inversion centre is located at the terbium(III) ion, a disordered NO3− anion with 0.5 occupancy, and two EtOH molecules; thus, the crystals are formulated as [(L)MnTbMn(L)]NO3·4EtOH (Mn·4EtOH) (Table 1). Due to the inversion centre at the terbium(III) ion, this crystal consists of not the homochiral Λ,Λ and Δ,Δ enantiomers but the meso-type Δ,Λ achiral complex cation (Fig. 5). It is notable that such a meso-type complex cation had not been observed in any of the H3L-derived trinuclear M–Ln–M complexes reported previously.7,8
 | ||
| Fig. 5 ORTEP drawing of the meso-type Δ,Λ complex cation in [(L)MnTbMn(L)]NO3·4EtOH (Mn·4EtOH) (ellipsoids are drawn at the 50% probability level and all hydrogen atoms are omitted for clarity). | ||
Recrystallisation of the Mn–Tb–Mn compound from CH2Cl2 afforded two kinds of single crystals with similar crystal shapes but different compositions in a certain crystallisation batch: [(L)MnTbMn(L)]NO3·2CH2Cl2 (Mn·2CH2Cl2) and [(L)MnTbMn(L)]NO3·4CH2Cl2 (Mn·4CH2Cl2). Both compounds crystallised in the monoclinic crystal system and a centrosymmetric space group P21/c with Z = 4 (Table 1). For each crystal, the asymmetric unit contains two halves of trinuclear [(L)MnTbMn(L)]+ complex cations, where both the TbIII centres are located on the crystallographic inversion centres, a counterion NO3−, and two or four CH2Cl2 molecules. The phase purity of the entire bulk crystalline sample was not clearly determined due to high efflorescence. It could nonetheless at least be concluded that both crystals contained meso-type Δ,Λ achiral complex cations due to the existence of a crystallographic inversion centre at the terbium(III) ion in the trinuclear complex cation, similar to the molecular structures of Mn·4EtOH. Thus, the crystal structures of Mn·2CH2Cl2 and Mn·4CH2Cl2 as well as Mn·4EtOH can be classified as a new class [class D].
It is similarly expected from the amorphous phase transition, as confirmed by P-XRD measurements, that the larger values of U/Z for [class D] compounds, Mn·4EtOH (1707.1 Å3), Mn·2CH2Cl2 (1571.2 Å3), and Mn·4CH2Cl2 (1736.5 Å3) (see Table 2), compared with those of [class B] Mn·3MeOH resulted from the greater volumes and numbers of solvating molecules for the voids expected for corresponding [class B] compounds (see Experimental section and Fig. S5†).
Regarding the coordination sphere around a central terbium(III) ion of all classes of compounds, no significant differences were observed in the coordination bond lengths or bond angles (Tables S6 and S7†). However, the first to third smallest bond angles Φ1–Φ3 defined by Omethoxy–TbIII–Omethoxy angles, where two methoxy oxygen atoms are located at each of the facing tripodal ligands (Fig. 6a and Table S8†), are 61.08–62.68° for [class D] compounds; these values are higher than 57.32–61.53° for [class A], [class B] and [class C] compounds. The torsion angles θ1–θ6 between two lines defined by two N⋯N and O⋯O atoms around the 3d metal centres (Fig. 6b and Table S9†) are 13.64–17.72°; these values are smaller than 11.89–28.60° for [class A], [class B] and [class C] compounds (θ = 60° for an ideal octahedron; 0° for an ideal trigonal prism). Taking these values into account, it is assumed that the steric hindrance between the tripodal ligand arms in the meso-type (Δ,Λ) complex cations is slightly larger than that of the homochiral (Δ,Δ or Λ,Λ) cations, and the distance between adjacent oxygen atoms increases around the central terbium(III) centre. Indications are therefore that the larger distortion of the six-coordinated polyhedron around the 3d metal centre from an ideal octahedron (defined by the values of θ) for meso-type cations in [class D] compounds than for homochiral cations in [class A] to [class C] is caused by the more sterically hindered tripodal arms' positions (defined by the values of Φ).
 | ||
| Fig. 6 Definition of (a) the first to third closest bond angles Φ1–Φ3 defined by Omethoxy–TbIII–Omethoxy and (b) the torsion angles θ1–θ6 between two lines defined by N⋯N and O⋯O atoms around 3d metal centers in a series of M–Tb–M complexes. | ||
Theoretical evaluation of the structural stability of molecular cation in solution phase
To explore the structural preference of either a homochiral enantiomeric cation (Λ,Λ and Δ,Δ) or a meso-type Δ,Λ cation of [(L)MnTbMn(L)]+ in MeOH and EtOH solutions, we performed DFT calculations using the dispersion-corrected B3LYP-D3(BJ) functional to assess their relative stability and potential contributing factors.10–14 Our primary goal was to address the question of whether meso-type isomers predominantly exist in EtOH solution without forming a racemic mixture.We first calculated the energies of gas-phase homochiral Λ,Λ- and meso-type Δ,Λ-[(L)MnTbMn(L)]+ cations in their ground state. The resulting energy gap, defined as ΔE = E(Λ,Λ) − E(Δ,Λ), is negative (−1.1135 kcal mol−1) (Fig. 7). This negative value indicates that the free homochiral Λ,Λ cation is thermodynamically more stable than the meso-type Δ,Λ cation, which then suggests that the [(L)MnTbMn(L)]+ cation is likely to predominantly form a racemic mixture in the gas phase because the stability of both enantiomers (Λ,Λ and Δ,Δ) is exactly the same to each other.
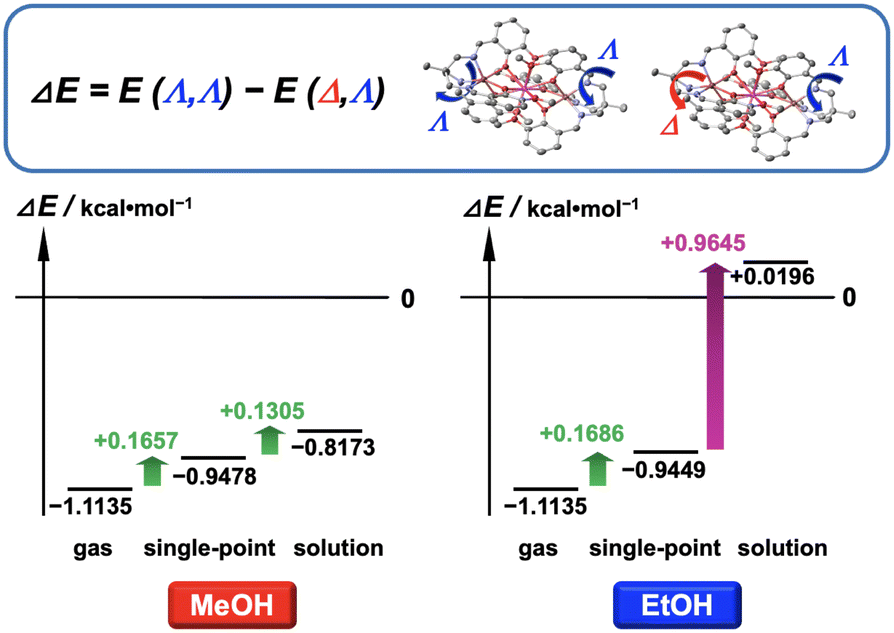 | ||
| Fig. 7 Theoretical energy gap between a homochiral Λ,Λ and a meso-type Δ,Λ configuration of the [(L)MnTbMn(L)]+ cation, defined as ΔE = E(Λ,Λ)-E(Δ,Λ), in the gas phase, single-point fixed phase and solution phase. | ||
Subsequently, we performed single-point calculations using an implicit solvation model, focusing exclusively on the electrostatic interaction between the complex cation and solvent molecules. In this approach, the complex cation is immersed in a dielectric field with a permittivity value of MeOH, ε = 32.613, or EtOH, ε = 24.852, while retaining its gas-phase geometry.12 This assumption implies that the cation is situated within a hypothetical MeOH or EtOH solution with negligible molecular volume. The ground-state energy gap ΔE was calculated to be −0.9478 and −0.9449 kcal mol−1 in MeOH and EtOH, respectively (Fig. 7). These values are not so different from those in the gas phase, indicating that the stable and dominant cationic configuration is still the homochiral enantiomers.
Finally, we incorporated the structural relaxation of the complex cation, accounting for how it responds to the electrostatic interaction with the dielectric continuum of either MeOH or EtOH. This approach provides a more accurate representation of realistic conditions in solution phases. The results gave ΔE values of −0.8173 and +0.0196 kcal mol−1 in MeOH and EtOH, respectively (Fig. 7). Here, the ΔE value in EtOH solution dramatically increases and shifts slightly towards the positive side, while the ΔE value in MeOH exhibits only a marginal change. One could infer from the permittivity values of the MeOH and EtOH dielectric fields that the higher polarity of MeOH molecules might promote the formation of homochiral cations with a higher dipole moment in a free solution state. Conversely, the lower polarity of EtOH molecules might not provide sufficient stabilisation for homochiral cations, allowing only achiral meso-type cations with no dipole moment to be present in an EtOH solution.
The results suggest that in an EtOH solution the meso-type cationic structure (Λ,Λ) is more stable and prevalent than the homochiral (Λ,Λ and Δ,Δ) cations, while in a MeOH solution a racemic mixture with homochiral cations is preferably formed. These results also provide reasonable support for experimental observations, such as the formation of Λ-conglomerates with homochiral Λ,Λ-enantiomers deposited in MeOH and the growth of centrosymmetric crystals with achiral meso-type Δ,Λ-cations deposited in EtOH. The reason behind the completely different crystallisation behaviours of the Zn–Tb–Zn and Mn–Tb–Mn complexes in CH2Cl2 and CHCl3 solutions remains unclear due to computational challenges. However, the difficulty might be associated with significant contributions from higher order terms in the multipole expansion of the electrostatic potential, which are essential for achieving precise computational predictions.
Conclusion
In this study, the crystallisation behaviours of M–Tb–M heterotrinuclear complexes, [(L)MTbM(L)]NO3 (M = Mn or Zn), were examined in various solvents: MeOH, EtOH, CH2Cl2 and CHCl3. Results of SC-XRD analysis revealed that all Zn–Tb–Zn complexes analysed in our previous and present studies have homochiral structures (Δ,Δ- or Λ,Λ-enantiomer) in the solid state. The types of crystallisation behaviours of these complexes were dependent on the type of solvent, and their crystal structures were classified into [class A] (racemic compounds), [class B] (conglomerates by P212121) and [class C] (those by P21).The analogous Mn–Tb–Mn complexes resulting from MeOH and CHCl3 exhibited the same behaviour as the Zn–Tb–Zn complexes. In particular, the Zn–Tb–Zn complex deposited from a CH2Cl2 solution and the Mn–Tb–Mn complex deposited from a MeOH solution exclusively gave the Λ-conglomerate in all crystallisation experiments—termed ‘absolute spontaneous resolution’—as in the case of the Zn–Tb–Zn complex from a MeOH solution.8
In addition, the [class D] meso-type (Δ,Λ) achiral isomer was obtained for Mn–Tb–Mn complexes from a CH2Cl2 or EtOH solution. The meso-type diastereoisomer was first observed in the series of the H3L-derived trinuclear M–Ln–M complexes. DFT calculations were performed to compare the structural stability of the homochiral (Λ,Λ) and meso-type (Δ,Λ) complex cations of [(L)MnTbMn(L)]+ in MeOH and EtOH solutions. The findings reasonably supported the preferable molecular configuration of trinuclear complex cations observed in the resulting crystal structures from each solution. These results give some insight into differences in the crystal structures and the crystallisation behaviours of M–Tb–M heterotrinuclear complexes by recrystallisation solvents.
Experimental section
All chemicals and solvents were reagent grade and used as received. The tripodal ligand precursor, 1,1,1-tris(aminomethyl)-ethane (tame), was prepared using a previously reported method.15Preparation of tripodal ligand: 1,1,1-tris[(3-methoxysalicylidene-amino)methyl]ethane (H3L)
Pottasium t-butoxide (1.68 g, 15.0 mmol) was slowly and completely dissolved in MeOH (100 mL), followed by the addition of tame·3HCl (1.14 g, 5.0 mmol). The colourless solution gradually turned to pale yellow and a white suspension of KCl formed. After stirring for several minutes, o-vanillin (2.28 g, 15.0 mmol) was slowly added and the solution colour immediately changed to deep yellow. The reaction mixture was stirred for 3 h at room temperature. It was then poured into a round-bottomed flask and the solvents were removed under reduced pressure (evaporated to dryness). The product comprised a white KCl powder and a deep yellow coloured oil as the main product. Dichloromethane (20 mL) was added to the residue to extract the oily product from KCl. The solution was then evaporated to dryness, affording a yellow crystalline powder of H3L. Yield: 2.02 g (78.4%). IR (KBr): ν(O–H) 3454, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) 1629 cm−1. 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 3H, –CH3), 3.66 (s, 6H, N–CH2), 3.93 (s, 9H, O–CH3), 6.80–6.96 (m, 9H, aryl H), 8.39 (s, 3H, NCH), 13.95 (s, 3H, –OH).
N) 1629 cm−1. 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 3H, –CH3), 3.66 (s, 6H, N–CH2), 3.93 (s, 9H, O–CH3), 6.80–6.96 (m, 9H, aryl H), 8.39 (s, 3H, NCH), 13.95 (s, 3H, –OH).
Preparation of heterotrinuclear complexes
The synthetic procedure for the zinc(II) complex, [(L)ZnTbZn(L)]NO3·2MeOH (Zn·2MeOH), has been reported previously.8 The analogous manganese(II) complex, [(L)MnTbMn(L)]NO3·3MeOH (Mn·3MeOH), was prepared by a modified method (reported by Yamaguchi et al.7) using triethylamine as a base.N) 1619, ν(NO) 1474 and ν(NO3) 1384 cm−1.
Elemental analysis and physical measurements
Elemental analysis was performed at the Advanced Science Research Center, Okayama University. The crystalline samples were allowed to stand in air to complete any possible efflorescence and/or hygroscopicity prior to measurements. IR spectra were recorded on a Jasco FT/IR FT-001 spectrophotometer. Samples were prepared as KBr disks. UV-vis absorption spectra and CD spectra of the complexes in methanol were obtained on a Jasco V-550 spectrophotometer and Jasco J-1500 spectropolarimeter, respectively, at room temperature. Proton nuclear magnetic resonance (1H NMR) measurements were carried out at room temperature using a Varian Mercury 400 spectrometer.Crystallography
P-XRD data were collected on a Rigaku RINT-TTR III diffractometer at room temperature. For the [class B] compounds (Zn·2CH2Cl2 and Mn·3MeOH), the crystalline samples were completely dried in vacuo and ground in an agate mortar before mounting for the measurements. Conversely, for the other classes of compounds (Zn·4EtOH, Zn·4CHCl3, Mn·4EtOH and Mn·4CHCl3), the wet crystalline samples were directly taken from the experimental sample tubes for measurements. This was done in an effort to prevent quick amorphous phase transition by efflorescence (and moisture adsorption) in air. The mixture of Mn·2CH2Cl2 and Mn·4CH2Cl2 compounds could not show any promising signals under wet conditions due to the rapid amorphous phase transition by efflorescence (and moisture adsorption), while the other compounds gave well-resolved diffraction patterns corresponding to the simulated spectra from SC-XRD analysis (Fig. S5†). The SC-XRD data were collected on a Rigaku R-AXIS RAPID diffractometer equipped with an imaging plate area detector with a Mo Kα radiation source (λ = 0.71069 Å), using a graphite monochromator. A suitable single crystal was mounted with a cryoloop from paraffin oil and flash cooled in a stream of cold nitrogen gas. The diffraction data were processed using the Process-Auto software package,16 and numerical absorption corrections17 were applied. The structures were solved using a direct method, employing the SIR2008, SIR2011 or SIR2014 (ref. 18) software packages, and refined on F2 (with all independent reflections) using the SHELXL2014 (ref. 19) software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced at the theoretical positions and refined using riding models. Absolute configurations of the crystals used for the SC-XRD analysis were established using anomalous scattering effects in diffraction measurements (using the Flack parameter). All calculations were carried out using the CrystalStructure software package.20Solid-state CD measurement
The solid-state CD spectra were measured on a JASCO J-720 or J-1500 spectropolarimeter using KBr disks of the samples, in transmittance mode. A disk was prepared from a single piece of crystal or an entire batch of microcrystals of the compound with crystalline KBr. The following procedure was followed. A mixture of sample and KBr was well ground in an agate mortar and then vacuum pressed to form a uniform disk (Fig. S11†). The disk was placed in a sample holder and then positioned in the cell holder of the spectropolarimeter. For each disk, the CD signals were measured from both sides, with a disk rotation of 180° (a total of four times), then the average was taken. A blank disk was used to measure background spectra.8 The data were corrected accordingly.DFT calculations
Computational models for DFT calculations were constructed from the SC-XRD models of Mn·3MeOH (for a homochiral Λ,Λ cation) and Mn·4EtOH (for a meso-type Δ,Λ cation), with all solvent molecules and counterions excluded. All calculations were performed using the B3LYP functional10 augmented with the D3 version of Grimme's empirical dispersion correction and the Becke–Johnson amping function,11 as implemented in Gaussian 16.12 The Stuttgart/Dresden SDD basis set/potential13a was used to represent MnII, while two tripodal nonadentate ligands (L3−) were described using the 6-31G(d) basis set. Given that 4f electrons do not engage in bonding with the ligands, the inner shell of TbIII including all 4f electrons ([Kr]4d104f8) was replaced by the large-core relativistic effective core potential (MWB54), along with the optimised basis set for 5s, 5p, 6s, 5d and 6p valence electrons.13b Geometry optimisations were carried out with a spin multiplicity of 11. The effect of solvation was considered through an implicit solvation model using the integral equation formalism (IEFPCM),14 with a dielectric constant of 32.613 (MeOH) or 24.852 (EtOH).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST through the establishment of University Fellowships toward the Creation of Science Technology Innovation, Grant Number JPMJFS2128 (for K. T.), and by JSPS KAKENHI, Grant Number 21K05084 (for T. S.).Notes and references
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, John Wiley & Sons, Inc., USA, 1981 Search PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 371 CrossRef CAS PubMed.
- I. Katsuki, Y. Motoda, Y. Sunatsuki, N. Matsumoto, T. Nagashima and M. Kojima, J. Am. Chem. Soc., 2002, 124, 629 CrossRef CAS PubMed.
- (a) R. E. R. Steendam, J. B. T. van Benthem, M. E. E. Huijs, H. Meekes, J. P. W. van Enckevort, J. Raap, P. J. T. F. Rutjes and E. Vlieg, Cryst. Growth Des., 2015, 15, 3917 CrossRef CAS; (b) R. E. Steendam and H. J. ter Horst, Cryst. Growth Des., 2017, 17, 4428 CrossRef CAS.
- C. L. Yadav, G. Rajput, K. K. Bisht, M. G. B. Drew and N. Singh, Inorg. Chem., 2019, 58, 14449 CrossRef CAS PubMed.
- A. Cucinotta, C. Kahlfuss, A. Minoia, S. Eyley, K. Zwaenepoel, G. Velpula, W. Thielemans, R. Lazzaroni, V. Bulach, M. W. Hosseini, K. S. Mali and S. D. Feyter, J. Am. Chem. Soc., 2023, 145, 1194 CrossRef CAS PubMed.
- T. Yamaguchi, J.-P. Costes, Y. Kishima, M. Kojima, Y. Sunatsuki, N. Bréfuel, J.-P. Tuchagues, L. Vendier and W. Wernsdorfer, Inorg. Chem., 2010, 49, 9125 CrossRef CAS PubMed.
- K. Takahara, Y. Horino, K. Wada, H. Sakata, Y. Sunatsuki, M. Kojima and T. Suzuki, Cryst. Growth Des., 2023, 23, 5244 CrossRef CAS.
- M. Matsushima, K. Wada, Y. Horino, K. Takahara, Y. Sunatsuki and T. Suzuki, CrystEngComm, 2020, 22, 458 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS; (b) M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- E. B. Fleischer, A. E. Gebala, A. Levey and P. A. Tasker, J. Org. Chem., 1971, 36, 3042 CrossRef CAS.
- Rigaku Co. Ltd., Process-Auto, Akishima, Tokyo, 1998 Search PubMed.
- Rigaku Co. Ltd., NUMABS, Akishima, Tokyo, 1999 Search PubMed.
- (a) M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, J. Appl. Crystallogr., 2007, 40, 609 CrossRef CAS; (b) M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357 CrossRef CAS; (c) M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- Rigaku Co. Ltd., CrystalStructure, Akishima, Tokyo, 2000–2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic scheme of H3L; IR and UV-vis absorption spectra of Mn·3MeOH; molecular structures of both enantiomers of [(L)ZnTbZn(L)]+; estimated and observed P-XRD patterns of all compounds (except for Mn·2CH2Cl2 and Mn·4CH2Cl2); CD spectrum of a methanol solution of Mn·3MeOH; packing diagrams of all compounds; tables of crystallographic data and selected structural parameters of all crystals measured in this study. CCDC 2303369–2303378. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01192e |
| This journal is © The Royal Society of Chemistry 2024 |