
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCF3-substituted sulfonium cations as efficient chalcogen bond donors towards cyanometalates†
Tim-Niclas
Streit
a,
Rosa M.
Gomila
 b,
Robin
Sievers
a,
Antonio
Frontera
*b and
Moritz
Malischewski
*a
b,
Robin
Sievers
a,
Antonio
Frontera
*b and
Moritz
Malischewski
*a
aFreie Universität Berlin, Institut für Anorganische Chemie, Fabeckstr. 34/36, D-14195 Berlin, Germany. E-mail: moritz.malischewski@fu-berlin.de
bDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 11th January 2024
Abstract
S–(CF3)Thianthrenium and S–(CF3)dibenzothiophenium cations form potent chalcogen bonds (ChBs) with [Mo(CO)5CN]−, yielding S2N2 supramolecular motifs. Crystal structures reveal shorter S⋯N contacts opposite the CF3 group compared to the aryl substituents. The energetic features of the ChBs have been studied using DFT calculations demonstrating the structure guiding role of ChBs.
The chalcogen bond (ChB) is an attractive interaction characterized by an electrophilic chalcogen atom (ChB donor) interacting with an electron-rich site (ChB acceptor).1 In the solid state, ChBs have been compared to halogen bonding in terms of directionality and supramolecular synthons.2 Understanding the physical nature of ChB interactions is complex, since it involved electrostatic effects, polarization, dispersion contributions, and n → σ* orbital delocalization. In recent years, ChB has gained prominence in synthesis, catalysis, material design, and coordination chemistry, particularly for crystal engineering.2,3 Supramolecular hierarchies and design strategies have been devised to create molecules with large σ-holes and high electrostatic potentials. Achieving this typically involves incorporating multiple electron-withdrawing substituents.4
Fluorine's significance in pharmaceuticals and agrochemicals has led to extensive research on methodologies for introducing fluorine or fluorinated groups into organic molecules.5 Notably, the incorporation of CF3 groups is of particular interest.6 Umemoto's work in the 1990s introduced dibenzothiophenium cations as effective electrophilic trifluoromethylating agents, with reactivity fine-tuned by varying the chalcogen atom and incorporating substituents in the aryl rings.7 Recently, Ritter8 introduced S-(trifluoromethyl) thianthrenium triflate as a readily available trifluoromethylating agent. Given the pronounced σ-holes in sulfonium cations, we explored whether these cationic S–CF3 reagents could serve as potent ChB donors. As ChB acceptors, we considered cyanometalates due to their high Lewis basicity9 of the negatively polarized nitrogen atoms and their suitability as halogen bond acceptors.10
Surprisingly, this approach remains relatively unexplored in the context of ChB. Only two X-ray structures11,12 have been reported in the CCDC involving sulfonium cations R3S+ and cyanometalate anions, where the S⋯N contacts fall below the sum of van der Waals radii (∑vdW(SN): 3.35 Å).13
We envisaged the use of the Ritter reagent [TT]+[CF3SO3]− as a soluble ChB donor (Scheme 1). To fine-tune the ChB donor ability, a series of Umemoto reagents with different substituents was also used (Scheme 1). To avoid the formation of polymeric network structures, [NEt4]+[Mo(CO)5CN]− was used as a building block. Salt metathesis of [NEt4]+[Mo(CO)5CN]− and [TT]+[CF3SO3]− in CH2Cl2 led to [TT]+[Mo(CO)5CN]− which crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The sulfur atom carrying the CF3 substituent acts as a biaxial ChB donor whereas the nitrogen of the cyanide ligand acts as a bifurcated ChB acceptor, resulting in the formation of an intermolecular S2N2 ring motive with two different S⋯N distances (Fig. 1). The shorter contact of 2.775(2) Å is situated in elongation of the S–CF3 group, whereas the longer contact of 3.212(1) Å is in the plane of the aryl rings. Both values are below the sum of van der Waals radii of 3.35 Å of sulfur and nitrogen.13 Whereas the C–S⋯N angles are close to linear for the short S⋯N contact (C100–S1–N1: 172.31(22)°) the deviation from linearity is greatly pronounced for the longer S⋯N interaction (C25–S1–N1: 95.95(19)°). The bifurcation of the cyanide ligand leads to C
. The sulfur atom carrying the CF3 substituent acts as a biaxial ChB donor whereas the nitrogen of the cyanide ligand acts as a bifurcated ChB acceptor, resulting in the formation of an intermolecular S2N2 ring motive with two different S⋯N distances (Fig. 1). The shorter contact of 2.775(2) Å is situated in elongation of the S–CF3 group, whereas the longer contact of 3.212(1) Å is in the plane of the aryl rings. Both values are below the sum of van der Waals radii of 3.35 Å of sulfur and nitrogen.13 Whereas the C–S⋯N angles are close to linear for the short S⋯N contact (C100–S1–N1: 172.31(22)°) the deviation from linearity is greatly pronounced for the longer S⋯N interaction (C25–S1–N1: 95.95(19)°). The bifurcation of the cyanide ligand leads to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯S angles of 117.91(37)° and 123.59(39)°. Surprisingly, the second sulfur atom of the S-(trifluoromethyl) thianthrenium cation is also a ChB donor. Here, the S⋯O contact is 3.264(4) (∑vdW(SO): 3.32 Å (ref. 13)) to the negatively polarized oxygen atom of the carbonyl ligand (Fig. S1†). This interaction is furthermore supported by short contacts between the hydrogen atoms in β-position to sulfur to the oxygen atoms of the anions (H–O distances 2.524(1) and 2.588(1) Å).
N⋯S angles of 117.91(37)° and 123.59(39)°. Surprisingly, the second sulfur atom of the S-(trifluoromethyl) thianthrenium cation is also a ChB donor. Here, the S⋯O contact is 3.264(4) (∑vdW(SO): 3.32 Å (ref. 13)) to the negatively polarized oxygen atom of the carbonyl ligand (Fig. S1†). This interaction is furthermore supported by short contacts between the hydrogen atoms in β-position to sulfur to the oxygen atoms of the anions (H–O distances 2.524(1) and 2.588(1) Å).
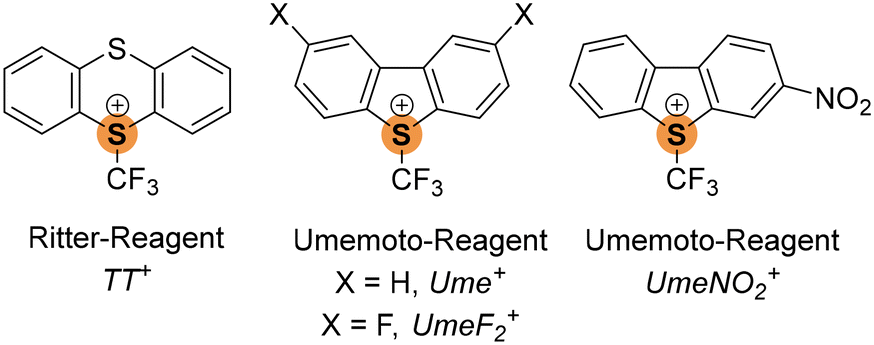 | ||
| Scheme 1 Available sulfonium-type chalcogen bond donors containing S–CF3 moieties. | ||
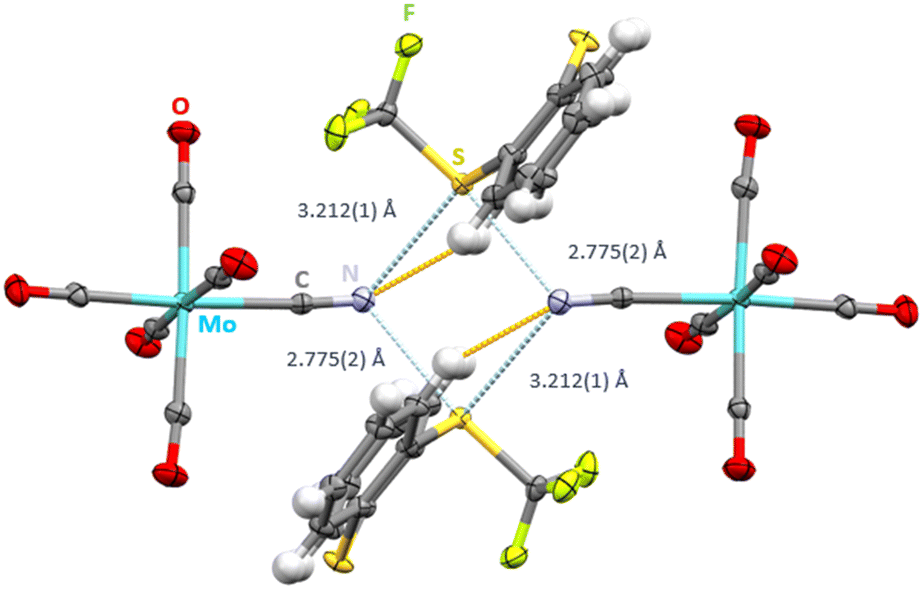 | ||
| Fig. 1 Intermolecular S⋯N contacts (light blue, distances given) and hydrogen bonds (orange) in the crystal structure of [TT]+[Mo(CO)5CN]−. Thermal ellipsoids at the 50% probability level. | ||
Interestingly, when salt metathesis between the unsubstituted [Ume]+[CF3SO3]− and [NEt4]+[Mo(CO)5CN]− was attempted, only single crystals of both starting materials were observed, without any salt metathesis. Consequently, derivatives of the Umemoto cation containing electron-withdrawing substituents were prepared. Salt metathesis of the double fluorinated Umemoto cation [UmeF2]+[B(C6F5)4]− with [NEt4+][Mo(CO)5CN]− in CH2Cl2 gave single crystals of [UmeF2]+[Mo(CO)5CN]−·CH2Cl2 (space group C2/c, Fig. 2). In agreement with the previous structure the contact trans to the CF3 group (2.719(3) Å) is shorter than the contact trans to the aryl rings (3.084(3)). The C–S⋯N ChB is strongly directional with an angle of 176.68(23)°. Bifurcation around the cyanide nitrogen atoms leads to CN⋯S angles in the range of 129.64(38)–141.48(41)°. As in all other structures the S–CF3 bonds are longer (1.884(5)–1.886(7) Å) than the S-aryl bonds (1.776(6)–1.782(6) Å).
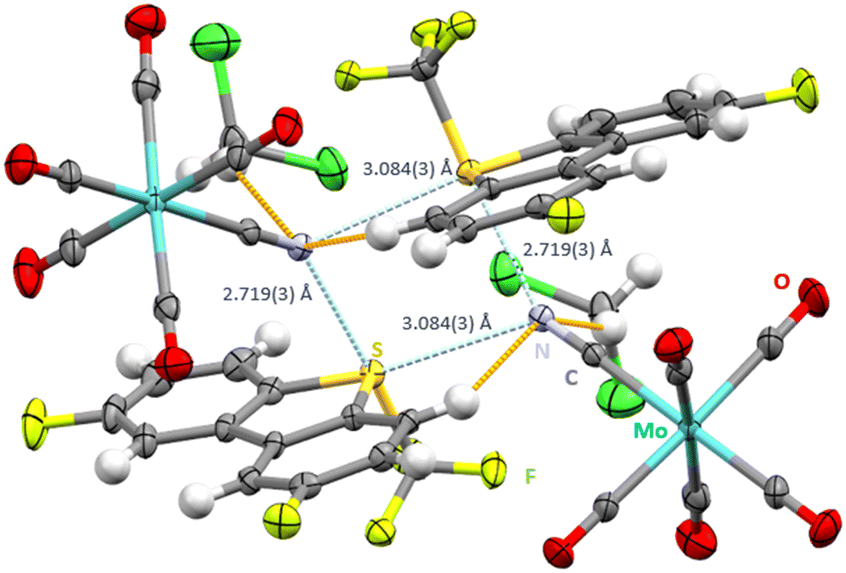 | ||
| Fig. 2 Intermolecular S⋯N contacts (light blue, distances given) and hydrogen bonds (orange) in the crystal structure of [UmeF2]+[Mo(CO)5CN]−·CH2Cl2. Thermal ellipsoids at the 50% probability level, solvent molecules not shown. | ||
Salt metathesis of nitro-substituted [UmeNO2]+[SbF6]− with [NEt4]+[Mo(CO)5CN]− in CH2Cl2 proved challenging due to extensive decomposition reactions. In one case, the desired cocrystal was obtained, but it was later discovered that the old batch of the molybdenum compound used was contaminated with around 18% of the analogous tungsten compound. Experiments with the pure molybdenum compound were unfortunately not successful despite numerous attempts. [UmeNO2]+[Mo0.822W0.178(CO)5CN]−·CH2Cl2 crystallizes in the space group P, displaying the same intermolecular S2N2 motive (Fig. 3). Interestingly, shortening of the S⋯N contacts to 2.722(4) Å (trans to the CF3 group) and 2.953(4) Å (trans to the aryl group) was observed. In contrast to the previous structure, both ChB are more linear (C13–S1–N2 174.36(25)° and C4–S1–N2 172.01(21)°). The bifurcated nitrogen atom of the cyanide group shows C15–N2–S1 angles of 130.92(46)° and 127.24(42)°. Furthermore, the nitrogen atoms display close contacts to two hydrogen atoms of neighboring cations (N2–H9 2.681(7) Å) and solvent molecules (N2–H20b 2.728(7) Å). Additionally, three carbonyl oxygens and both oxygen atoms of the nitro group display O–H contacts in the range of 2.543(7)–2.683(9) Å and 2.678(7) Å, respectively. The oxygen atoms of the nitro group form also intramolecular O⋯H bonds to the hydrogen atoms in ortho-position (2.433(6) Å and 2.455(7) Å).
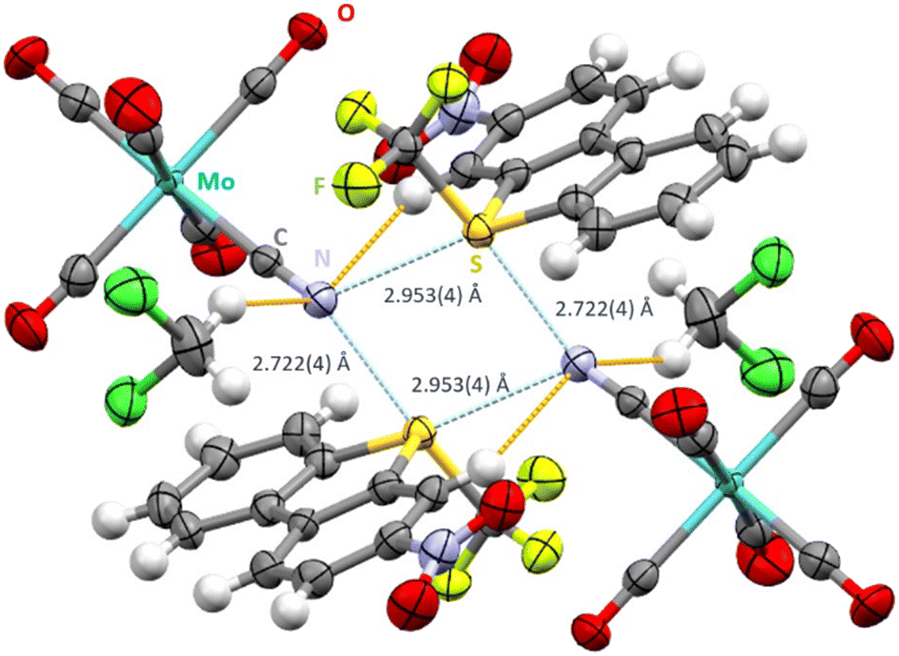 | ||
| Fig. 3 Intermolecular S⋯N contacts (light blue, distances given) and hydrogen bonds (orange) in the crystal structure of [UmeNO2]+[Mo0.822W0.178(CO)5CN]−·CH2Cl2. Thermal ellipsoids at the 50% probability level. | ||
In order to rationalize the strong ChB donor ability of the CF3-substituted sulfonium cations, electrostatic potential maps (MEPs) were calculated, allowing to establish a hierarchy between them. In this context, the influence of electron-withdrawing groups (F, NO2) on the strength of the σ-holes was of particular interest. Sulfonium cations (R3S+) exhibit three σ-holes extending from the three C–S bonds, as previously demonstrated.14 This configuration results in a substantial region of maximum electrostatic potential centered around the sulfur atom. In the [TT]+ cation (MEP surface in Fig. 4a), all three σ-holes extending from the C–S bonds exhibit MEP maxima with almost identical values, ranging from 107 to 109 kcal mol−1. However, in the case of the two Umemoto cations (shown in the lower part of Fig. 4), the maximum MEP values at the σ-holes significantly increase, ranging from 117 to 130 kcal mol−1.
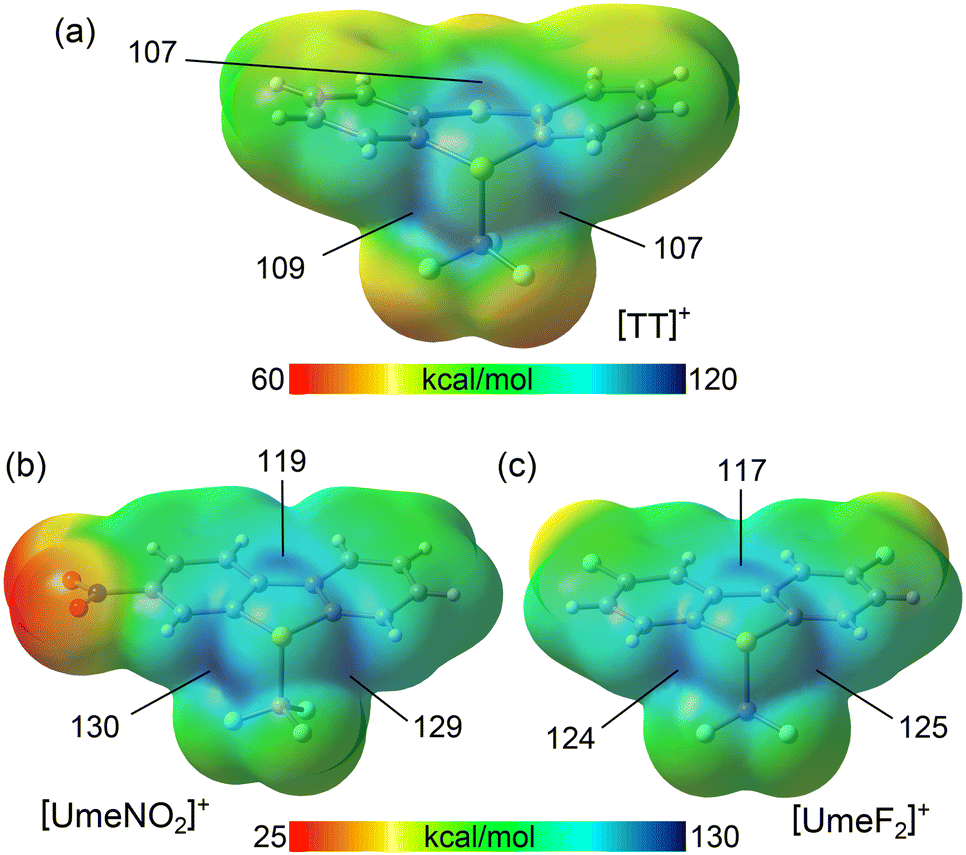 | ||
| Fig. 4 MEP surfaces (isovalue 0.001 a.u.) of the Ritter (a) and Umemoto (b and c) reactants. The values at the σ-holes are indicated in kcal mol−1. | ||
The σ-holes opposite the S–CAr bonds are more positive than those opposite the S–CF3 bond due to the influence of the adjacent hydrogen atoms. Notably, in the [UmeNO2]+ cation (Fig. 4b), the most positively charged σ-hole is opposite to the unsubstituted aromatic ring, driven by the enhanced acidity of the H-atom adjacent to the nitro group. The σ-hole opposite the S–CF3 bond is less positive due to π(arene) → σ*(S–C) charge transfer through hyperconjugation, reducing the MEP.
ChBs were further analyzed using DFT calculations and quantum theory of atoms-in-molecules (QTAIM), coupled with noncovalent interaction plot (NCIPlot) for real-space visualization. For [UmeNO2]+[Mo0.822W0.178(CO)5CN]−·assembly, only the major component [Mo(CO)5CN]− was considered in the calculations. Fig. 5 displays the results for the three tetrameric assemblies. Within these analyses, we observe four bond critical points (BCPs, red spheres) and bond paths (solid orange lines) connecting S and N atoms, confirming the existence of S2N2 supramolecular rings (pink in Fig. 5) in all three assemblies. These ChBs are characterized by green/blue reduced density gradient (RDG) isosurfaces coinciding with the BCP locations. The colour of these RDG isosurfaces indicates stronger N⋯S–CF3 interactions than N⋯S–CAr interactions, consistent with the experimental distances but in disagreement with the MEP surface analysis (the σ-hole opposite the S–CF3 bond is the least positive). This can be attributed to a shortening of the ChB distance due to the concurrent formation of anion–π interaction (see ESI,† Fig. S3). Additionally, charge transfer effects play a more prominent role in N⋯S–CF3 interactions than in N⋯S–CAr interactions (vide infra). The QTAIM analysis also reveals two ancillary hydrogen bonds (HBs) between N-atoms of both anions and two aromatic C–H bonds, each characterized by a BCP, bond path, and green RDG isosurface.
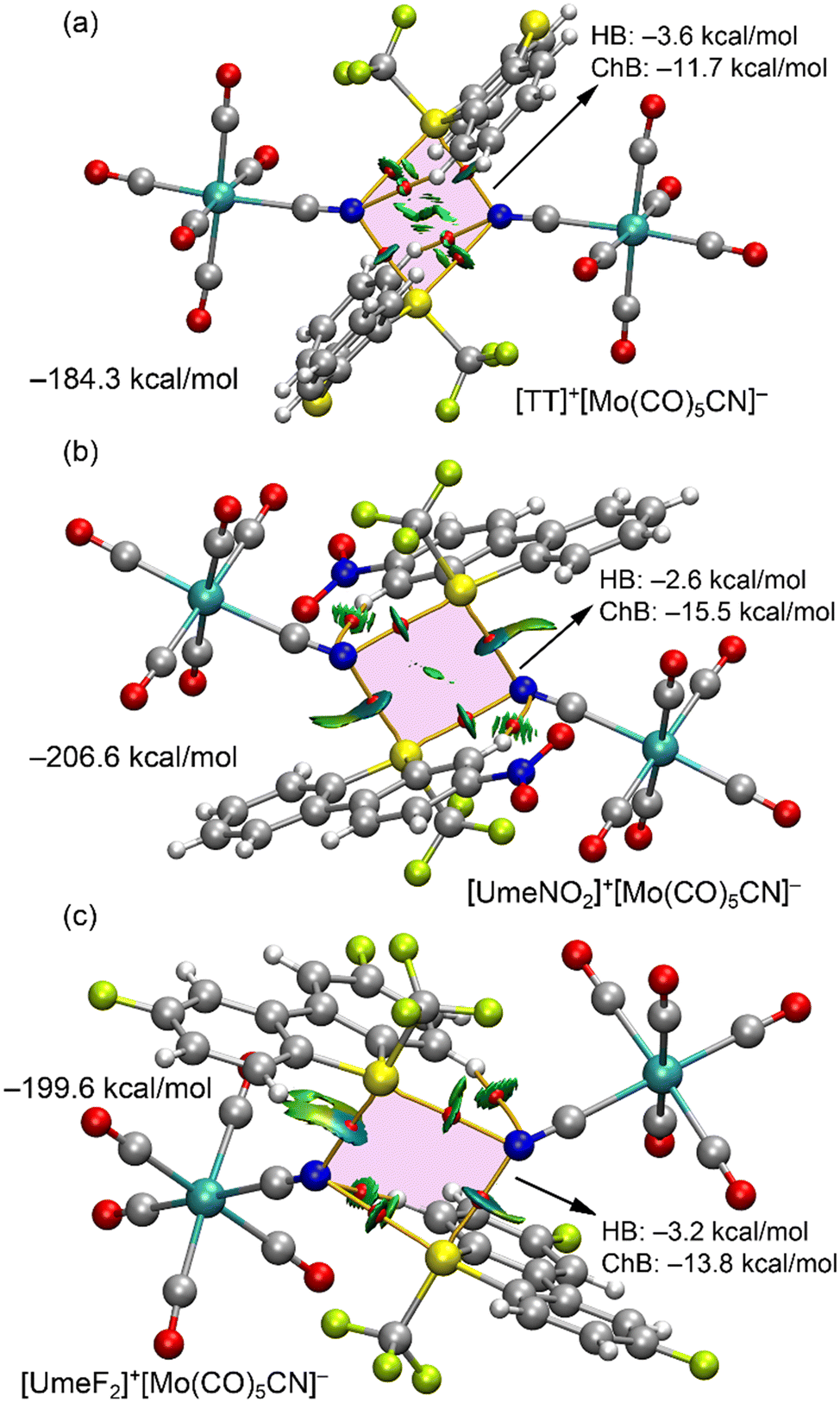 | ||
Fig. 5 QTAIM/NCIplot analyses for the tetrameric assemblies of [TT]+[Mo(CO)5CN]− (a), [UmeNO2]+[Mo(CO)5CN]− (b) and [UmeF2]+[Mo(CO)5CN]− (c). Only intermolecular HB and ChBs are represented for clarity. The energies of the assemblies are also indicated. The contributions of HB and ChB interactions derived from the QTAIM parameters are indicated. NCIplot settings: RDG = 0.5, ρ cut-off = 0.04 a.u.; colour range −0.035 ≤ sign![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) λ2ρ ≤ 0.035 a.u. λ2ρ ≤ 0.035 a.u. | ||
The assemblies' formation energies in Fig. 5 are large (ranging from −184 to −207 kcal mol−1) due to strong coulombic attraction between two anions and two cations. The strongest interaction corresponds to the [UmeNO2]+[Mo(CO)5CN]− assembly, consistent with MEP analysis. To assess ChB and HB strength independently of coulombic forces, interaction energies based on QTAIM parameters at the BCPs were calculated (see Theoretical methods, ESI†). The results in Fig. 5 emphasize the dominance of ChBs. For the [TT]+[Mo(CO)5CN]− assembly, the ChB contribution is −11.7 kcal mol−1, with a HB contribution of −3.6 kcal mol−1. In the case of the [UmeF2]+[Mo(CO)5CN]− assembly (Fig. 5c), HB contribution is almost equivalent to that of [TT]+[Mo(CO)5CN]− assembly, while ChB contribution increases to −13.9 kcal mol−1. The [UmeNO2]+[Mo(CO)5CN]− assembly presents the most substantial ChB contribution of −15.5 kcal mol−1, in agreement with experimental distances and MEP surface analysis.
Finally, we analyzed orbital charge transfer effects in the [TT]+[Mo(CO)5CN]− and [UmeNO2]+[Mo(CO)5CN]− assemblies using the natural bond orbital (NBO) method. Fig. 6 illustrates the relevant orbitals in both assemblies and their associated stabilization energies. It is important to note that, for clarity, we depict only one anion in Fig. 6, though our calculations considered the entire tetrameric assembly. Consequently, only one set of ChB interactions is shown. In both assemblies, we observe electron donation from the LP located at the sp-hybridized N-atom of the anion to the antibonding S–C bonds of the cations. The corresponding E(2) stabilization energies from the LP(N) → σ*(S–C) charge transfer are shown in Fig. 6. In line with interaction energies and experimental distances, the [UmeNO2]+[Mo(CO)5CN]− assembly displays higher stabilization energies than [TT]+[Mo(CO)5CN]−. Furthermore, we note that second-order stabilization energies E(2) for LP(N) → σ*(S–CF3) electron donation surpass those for LP(N) → σ*(S–CAr). This heightened electron transfer aligns with shorter equilibrium distances and stronger N⋯S–CF3 ChBs compared to N⋯S–CAr interactions which would not be expected if just ESP are taken into account.
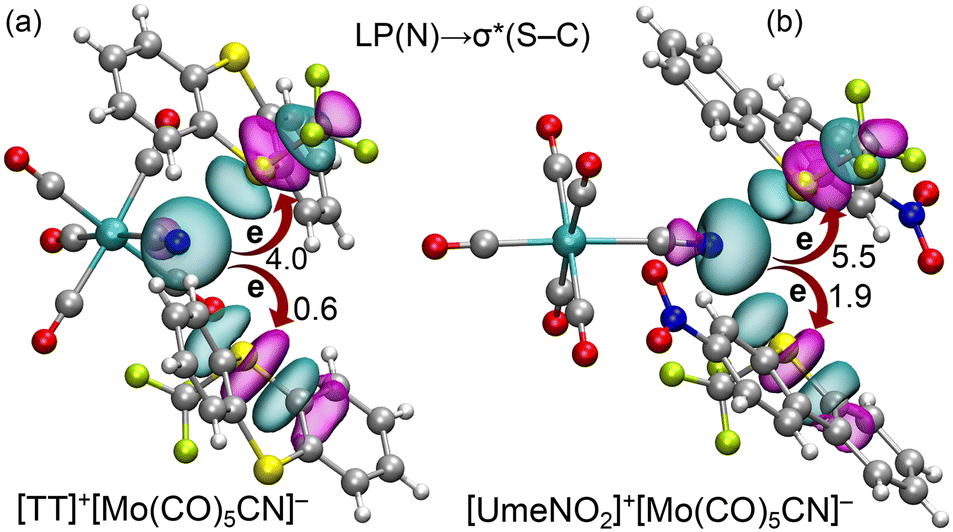 | ||
| Fig. 6 Plot of the NBOs involved in the LP(N) → σ*(S–C) for the tetrameric assemblies of [TT]+[Mo(CO)5CN]− (a) and [UmeNO2]+[Mo(CO)5CN]− (b). One of the anions has been omitted for clarity. The second order perturbation energies are given in kcal mol−1 close to the curved arrows. | ||
In conclusion, our study reveals strong ChB donors in electrophilic trifluoromethylating agents with S–CF3 bonds. These compounds offer tunable σ-hole/ESP maxima through aryl ring substitutions on sulfur. We have demonstrated the relevance of supramolecular S2N2 rings in three crystal structures through X-ray analysis and DFT calculations. These assemblies rely on ChB interactions, with nucleophilic cyanide groups of [Mo(CO)5CN]− acting as bifurcated acceptors. Additionally, the QTAIM analysis uncovers weaker H-bonds within these assemblies compared to the chalcogen bonds.
This work was funded by DFG (project number MA 7817/3-1). Computing time was made available by High-Performance Computing at ZEDAT/FU Berlin. The authors acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. AF and RGM acknowledge the financial support by the MICIU/AEI of Spain (project PID2020-115637GB-I00, FEDER funds). AF thanks the Alexander von Humboldt foundation for the J. C. Mutis research award.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- (a) P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed; (b) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- (a) K. T. Mahmudov, M. N. Kopylovich, M. F. C. G. da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC; (b) K. T. Mahmudov, A. V. Gurbanov, V. A. Aliyeva, M. F. C. Guedes da Silva, G. Resnati and A. J. L. Pombeiro, Coord. Chem. Rev., 2022, 464, 214556 CrossRef CAS; (c) P. C. Ho, J. Z. Wang, F. Meloni and I. Vargas-Baca, Coord. Chem. Rev., 2020, 422, 213464 CrossRef CAS; (d) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- (a) C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed; (b) V. V. Panikkattu, A. S. Sinha and C. B. Aakeröy, CrystEngComm, 2022, 24, 738–742 RSC.
- A. M. Thayer, Chem. Eng. News, 2006, 84, 15–24 Search PubMed.
- (a) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed; (b) N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 Search PubMed; (c) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed.
- (a) T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579–3582 CrossRef CAS; (b) T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- E. V. Alexandrov, A. V. Virovets, V. A. Blatov and E. V. Peresypkina, Chem. Rev., 2015, 115, 12286–12319 CrossRef CAS PubMed.
- (a) J. E. Ormond-Prout, P. Smart and L. Brammer, Cryst. Growth Des., 2012, 12, 205–216 CrossRef CAS; (b) M. Sellin, S. M. Rupf, Y. Zhang and M. Malischewski, Cryst. Growth Des., 2020, 20, 7104–7110 CrossRef CAS; (c) M. Sellin, S. M. Rupf and M. Malischewski, Cryst. Growth Des., 2021, 21, 5515–5520 CrossRef CAS; (d) T. Ohno, K. Nakabayashi, K. Imoto, M. Komine, S. Chorazy and S. I. Ohkoshi, CrystEngComm, 2018, 20, 7236–7241 RSC.
- T. Kitazawa, S. Nishikiori and T. Iwamoto, Mater. Sci. Forum, 1992, 91-93, 257–264 CAS.
- S. S. Basson, J. G. Leipoldt, A. Roodt and W. Purcell, Transition Met. Chem., 1987, 12, 82–84 CrossRef CAS.
- A. Bondi, J. Phys. Chem. A, 1964, 68, 441–451 CAS.
- B. Galmés, A. Juan-Bals, A. Frontera and G. Resnati, Chem. – Eur. J., 2020, 26, 4599–4606 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298717–2298719. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01155k |
| This journal is © The Royal Society of Chemistry 2024 |