
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-organized formation of seven-rod bundle morphology for lanthanum Prussian blue analog microcrystals via a precipitation process†
Fumiyuki
Shiba
 *,
Ren
Nagata
and
Yusuke
Okawa
*,
Ren
Nagata
and
Yusuke
Okawa
Department of Materials Science, Chiba University, 1-33 Yayoicho, Inageku, Chiba 263-8522, Japan. E-mail: shiba@faculty.chiba-u.jp
First published on 11th December 2023
Abstract
This article reports potassium lanthanum hexacyanidoferrate(II) microcrystals with a unique shape consisting of seven hexagonal rods. In an aqueous solution at 55 °C, [Fe(CN)6]3− ions were reduced by ascorbic acid to form [Fe(CN)6]4− ions, which reacted with the surrounding K+ and La3+ ions to generate K0.82La1.11[Fe(CN)6]·4H2O microcrystals. The morphology was spontaneously constructed without growth modifiers via the instantaneous precipitation of the Prussian blue analog with a hexagonal crystal structure under a specific range of reaction conditions.
Introduction
The morphology of crystalline substances, for both natural minerals and synthesized materials, has attracted our interest for a long time. Single crystalline solids have been the center of interest because they tend to show polygonal crystal habits reflecting the crystal structures. For example, on microcrystals synthesized in the aqueous phase, substances with a cubic crystal lattice, such as AgCl,1 are likely to have a cubic shape. Prussian blue, Fe4[Fe(CN)6]3·nH2O, and some of its analogs (PBAs) also tend to be cubic, reflecting their cubic crystal structures.2 Hexagonal prism shapes are often observed for the hexagonal structure materials such as rhabdophane-type YPO4 microcrystals.3,4The crystal habit of microcrystals is usually determined thermodynamically or kinetically based on the crystal structure as a fundamental factor.5,6 In the former cases, called equilibrium forms, the lattice planes with low specific surface energies preferentially appear as the facets of the polyhedral crystal to minimize its overall surface energy, as formulated by Wulff's theorem. In the latter cases, called growth forms, on the other hand, the difference in the growth rate among the surfaces determines the morphology; slow growth rate surfaces remain as the facets by vanishing faster growing ones to become the edges or the corners. The growth forms would be more significant on morphology control since the growth rate constants are expected to be changed more efficiently by changing the reaction conditions, using growth modifiers, or introducing screw dislocations or twins.
Reaction conditions usually determine the growth rates of crystal planes. On AgBr microcrystals (NaCl-type structure), for example, the growth rates for the {100} and {111} faces depend on pBr (≡ −log[Br−]) but have different dependence on pBr.7 Although the growth rates for both faces show a minimum around pBr 3.3 corresponding to the solubility minimum, the growth rate ratio of the {100} face to the {111} face is monotonously increased with a decrease of pBr (i.e., increase of free Br− ion concentration); the ratio is almost unity around pBr 3. As a result, the morphology of AgBr changes from cubic at around pBr 4 to octahedral at around pBr 2. At the intermediate region of around pBr 3, cuboctahedral microcrystals are formed.
The morphology can also be attributed to the reaction temperature. For example, a temperature-dependent height/width ratio has been reported for rhabdophane-type YPO4 microcrystals with a hexagonal prism shape synthesized in a hydrothermal system with sodium citrate.4 The activation energies for width- and height-direction growths are estimated as 97 kJ mol−1 and 112 kJ mol−1, corresponding to the larger width/height ratio at a lower reaction temperature.
Using growth modifiers, whether constituent ions or additives, often enables us to control the morphology more effectively. Hamada et al. reported the synthesis of monodisperse star-like Cu2O microcrystals that consist of six square pyramids orthogonally aligned with each other in an aqueous system containing 1,2-ethylenediamine and hexamethylenetetramine.8 Chen et al. hydrothermally synthesized Cu2O microcrystals under various reaction conditions, including the kind of additives, to form a variety of morphologies, including regular and truncated polyhedra, hopper crystals, multi-pod branching structures, etc.9 Sodium acetate contributes to making the width narrower for wedge-like α-GaOOH microcrystals synthesized via the hydrolysis process.10 Maskasky prepared seven different kinds of polyhedra for AgBr microcrystals with a cubic crystal lattice by using appropriate organic substances that specifically inhibit the growth of the respective surface.11
In this article, we describe the self-organized formation of uniquely shaped microcrystals of potassium lanthanum hexacyanidoferrate(II) (KLa-HCF), a kind of PBA.
Experimental
Materials
All chemicals used in the present study were purchased from FUJIFILM Wako Pure Chemical Co., Japan, except for xylenol orange (XO) from Dojindo Laboratories, Japan. The chemicals were used without additional purifications.Synthesis of KLa-HCF microcrystals
In a glass reaction vessel, 0.04 mol L−1 K3[Fe(CN)6] aqueous solution (5 mL) was mixed with 0.02 mol L−1 La(NO3)3 aqueous solution (10 mL) under magnetic stirring at 55 °C kept in a water bath. Then, 0.2 mol L−1 ascorbic acid (AscA) aqueous solution (5 mL) was introduced into it using a micropipette (time for addition ca. 1 s). Thus, the reactant concentrations after mixing for La(NO3)3, K3[Fe(CN)6], and AscA were CLa = 0.01 mol L−1, CHCF = 0.01 mol L−1, and CAscA = 0.05 mol L−1, respectively. After 30 min, the precipitate was separated from the aqueous phase by suction filtration, and the precipitate was freeze-dried.Characterization of the microcrystal morphology and structure
The microcrystals were observed with an FE-SEM (JEOL JSM-6700F) operated at 15 kV. To prepare the specimen for the FE-SEM observation, the freeze-dried powder of the microcrystals was put on electrically conductive double-sided carbon tape on a brass specimen stub. The specimen was coated with Pt by sputtering before observation. To evaluate the crystal structure of the microcrystals, the freeze-dried powder was subjected to powder XRD measurement using a Bruker D8 Advance with Cu Kα radiation (λ = 1.5418 Å: 40 kV × 40 mA = 1600 W). Selected area electron diffraction (SAED) patterns were obtained with a TEM (Hitachi H-7650) operated at 100 kV.Evaluation for the elemental composition of the microcrystals
The elemental composition of the microcrystal was evaluated by atomic absorption spectrometry (AAS) for K+ and Fe (of [Fe(CN)6]4−) and by colorimetry for La3+. The accurately weighed KLa-HCF powder (25 mg) was dissolved in 2.5 mL of 1 mol L−1 HNO3 and then diluted with distilled water to appropriate concentrations of analyses. The AAS determination was conducted using a Varian SpectrAA 55 with the acetylene/air flame. The absorption lines employed were λ = 404.4 nm and 248.3 nm for K and Fe, respectively. On the colorimetry for La3+, xylenol orange (XO) was used as the coloring agent in the presence of cetylpyridinium chloride (CPC) at pH 8.65 (ref. 12) (see the ESI† for details).The number of hydrated water molecules in the KLa-HCF lattice space was estimated by TG–DTA analysis with a Seiko Instruments TG/DTA6300 at 10 °C min−1 (the DTA reference: Al2O3).
Results and discussion
Fig. 1 shows FE-SEM images for the KLa-HCF microcrystals in the present procedure. Many of the formed microcrystals are in a morphology that resembles a rod bundle shape, as shown in the broad range view in Fig. 1(a). The close-up view, Fig. 1(b)–(d), indicates that each microcrystal is constructed with seven rods; the surrounding six outer rods are connected to the center rod. Each outer rod consists of two stems growing from a bulge at the midpoint of the rod, as shown in Fig. 1(c). The stems become wider toward the rod tips. The top-down view image, Fig. 1(d), indicates that the cross-section of each stem is hexagonal, and the diagonal lines of the hexagons are aligned in the same direction among the seven rods. | ||
| Fig. 1 FE-SEM images of the potassium lanthanum hexacyanidoferrate microcrystals precipitated from the mixed solution of La(NO3)3 and K3[Fe(CN)6] via the reduction reaction by ascorbic acid. (a) A broad range view and close-up views from (b) tilted, (c) side, and (d) top-down angles. | ||
The XRD pattern for the present microcrystals, Fig. 2, is matched with that for KLa[Fe(CN)6]·4H2O (ICDD PDF 00-038-0710; hexagonal lattice; space group P63/m). The lattice parameters are estimated as a = 7.41 Å and c = 14.06 Å from the d-spacings for the 102 and 110 reflections in Fig. 2. The lattice parameter values are in good agreement with those in the literature (a = 7.408 Å and c = 13.934 Å in the PDF data; a = 7.3829 Å and c = 13.871 Å in ref. 13). The hexagonal crystal structure accords with the hexagonal shape of the rod's cross-sections shown in Fig. 1(d), suggesting that the lateral and height directions of the rod correspond to the a- and c-axis directions of the hexagonal lattice, respectively.
 | ||
| Fig. 2 XRD pattern for the KLa-HCF microcrystals shown in Fig. 1 (Cu Kα radiation; λ = 1.5418 Å). The peaks are matched with the reference data for KLa[Fe(CN)6]·4H2O (ICDD PDF 00-038-0710). | ||
The orientation is also confirmed by the SAED patterns shown in Fig. 3; the insets are the microcrystals subjected to the SAED measurements. In Fig. 3(a), Laue spots align in a rectangle arrangement; the distances between the adjacent spots along the height direction correspond to d =14.2 Å of the lattice spacing, which is almost the same as the lattice parameter of c (= 14.06 Å) estimated from the XRD pattern. In Fig. 3(b), on the other hand, the Laue spots suggest a 6-fold symmetry of the crystal structure in the cross-sectional hexagonal planes. Also, from the estimated lattice spacings, the {1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 10} planes stack towards the hexagon's corner directions. Thus, the morphological feature concurs with the ion arrangement in the microcrystals, although the splitting Laue spots by double diffraction in Fig. 3(a) and (b) imply that the microcrystals may have some imperfections.
10} planes stack towards the hexagon's corner directions. Thus, the morphological feature concurs with the ion arrangement in the microcrystals, although the splitting Laue spots by double diffraction in Fig. 3(a) and (b) imply that the microcrystals may have some imperfections.
 | ||
Fig. 3 Selected area electron diffraction patterns for the single KLa-HCF microcrystals indicated in the insets. The patterns correspond to those for the (a) [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] and (b) [00] incident directions of the electron beam. 0] and (b) [00] incident directions of the electron beam. | ||
Even with the unique morphology, the facets would be related to the lattice planes of the crystal. Although the angle of the side face of the rod to the c-axis is different at the position along the height direction, it is about 23°, close to 28° for the {101} planes, around the concaves that divide the rod into the bulge and the stems (Fig. S1 in the ESI†). It is reasonable that the facet is the {101} plane, as the 101 reflection is observed in the XRD pattern in Fig. 2. The angle decrease around the stem tips could be due to the appearance of other indexed faces. In addition, the fringes on the stems shown in Fig. 1(b) would suggest the formation of lamellar twins on the {0001} faces, also contributing to the decrease of the angle.
Since PBAs possibly have compositions deviating from the stoichiometric formulae,14 the elemental composition of the present microcrystals was evaluated by dissolving the freeze-dried powder in HNO3 to apply the AAS (K+ and [Fe(CN)6]4−) and colorimetry (La3+) measurements. The estimated composition formula is K0.82La1.11[Fe(CN)6], which seems to be in the reasonable range of experimental estimation for the ions, as the total cation valence (+4.17) is close to the anion valence (−4).
The number of hydrated water molecules, n, was determined by TG-DTA measurement as shown in Fig. 4. According to the literature, the endothermic weight decrease up to 260 °C is associated with the dehydration of the hydrated water; the decrease above 300 °C is related to the decomposition of CN.13,15 For K0.82La1.11[Fe(CN)6], n = 3.96 is obtained from the weight loss in the former process, 15.2%. The n value agrees with the literature, n = 4. Therefore, the compositional formula of the present microcrystals may be denoted as K0.82La1.11[Fe(CN)6]·4H2O.
 | ||
| Fig. 4 TG–DTA result for the KLa-HCF microcrystals with a seven-rod bundle morphology. The 15.2% weight loss up to 260 °C is due to the dehydration process of the hydrated water. | ||
Interestingly, this unique morphology of KLa-HCF microcrystals was spontaneously formed under a simple precipitation process described in the experimental section. The microcrystal precipitation was triggered by the AscA addition into the mixed solution of La(NO3)3 and K3[Fe(CN)6]: La3+ ions were unreactive with [Fe(CN)6]3− ions, at least under the present conditions; thus the mixed solution was stable until AscA was added. Introducing the AscA solution, the transparent yellowish color of [FeIII(CN)6]3− ions vanished immediately by reduction to [FeII(CN)6]4− ions as:
| 2[FeIII(CN)6]3− + C6H8O6 → 2[FeII(CN)6]4− + C6H6O6 + 2H+, | (1) |
The turbidity continued to increase for about 10 s, suggesting that the crystallization process was completed at least within a few minutes. In fact, the microcrystals sampled at 1 min seem to be the same as those at 30 min in their morphology and size, as shown by the FE-SEM images in Fig. S2 in the ESI.† Owing to the H+ ion generation by the reduction process, the pH after the reaction (pH 2.04; after 30 min from the AscA addition) was lower than that even for the AscA solution (pH 2.36).
Although the present procedure using K3[Fe(CN)6] with AscA has practical advantages for effectively forming the seven-rod bundle morphology of the microcrystals, the low pH seems significant in generating them, rather than the reduction reaction by AscA. Using K4[Fe(CN)6] instead of K3[Fe(CN)6] enables us to precipitate KLa-HCF microcrystals without using AscA, as shown in Fig. 5 (CLa = CHCF = 0.01 mol L−1, 55 °C). Removing the ascorbic acid resulted in the higher reaction pH at pH 6.47, and microcrystals with a pteridophyte leaf-like shape were generated (Fig. 5(a)). Reacting at pH 2.19 by preliminarily adding HNO3 in the La(NO3)3 solution, on the other hand, the precipitate contained rod-bundle-shaped microcrystals (Fig. 5(b)). However, their percentage was smaller than the present K3[Fe(CN)6] system. Thus, it is suggested that the unique morphology of the present KLa-HCF microcrystals originates from their intrinsic nature of dendritic growth without the assistance of specific growth modifiers.
 | ||
| Fig. 5 FE-SEM images of KLa-HCF microcrystals prepared with La(NO3)3 and K4[Fe(CN)6] aqueous solutions at (a) pH 6.47 without adding acid for pH adjusting and (b) pH 2.19 by adding HNO3 to La(NO3)3 aqueous solution before mixing (reaction temperature 55 °C). | ||
To evaluate the effect of reaction temperature on the morphology, the KLa-HCF microcrystals have been prepared at different temperatures using K3[Fe(CN)6] and AscA under the conditions at CLa = CHCF = 0.01 mol L−1 and CAscA = 0.05 mol L−1 (reaction time 30 min). The FE-SEM images are shown in Fig. 6, where the pH values were almost the same among the temperatures: pH 2.06, 2.09, 2.09, and 2.16 at 25, 40, 55, and 70 °C, respectively. At 25 °C, Fig. 6(a), the bundle-morphology microcrystals are not observed; many nubby microcrystals are found instead. Increasing the reaction temperature increases the percentage of the bundle morphology. At 70 °C, almost all the microcrystals are in the bundle morphology, with a smaller size.
 | ||
| Fig. 6 FE-SEM images of the KLa-HCF microcrystals prepared at different temperatures (CLa = CHCF = 0.01 mol L−1, CAscA = 0.05 mol L−1). (a) 25 °C, (b) 40 °C, (c) 55 °C (control), and (d) 70 °C. The inset in (d) is the close-up view. | ||
When AscA was introduced, the turbidity increased more slowly at the lower temperatures, while the yellow color of [Fe(CN)6]3− vanished instantaneously even at 25 °C. This means that the temperature mainly affects the precipitation process rather than the reduction process. As the microcrystal size is determined by the number of microcrystals formed based on the mass-balance relationship, the small size at higher temperatures implies that the nucleation rate increases more significantly than the growth rate with temperature. In other words, at a lower temperature, a smaller number of microcrystals grow relatively slowly, resulting in a decreased consumption rate of the reactant. The highly supersaturated conditions maintained for a relatively long time could cause stacking faults to generate the nubby microcrystals. Hence, the conditions of the fast reaction seem to be preferred for forming the bundle morphology.
Fig. 7 shows the KLa-HCF microcrystals prepared at lower La(NO3)3 and K3[Fe(CN)6] concentrations at CAscA = 0.05 mol L−1 (55 °C, 30 min). The size decrease, corresponding to the decreased reactant amount, suggests that the number of microcrystals is similar among the reactant concentrations. At CLa = CHCF = 0.001 mol L−1 (1/10 of the control condition), the precipitate mainly consists of hexagonal disk microcrystals with relatively thin thickness (pH 2.51). Some bundle-morphology microcrystals are found at CLa = CHCF = 0.003 mol L−1, but with lower height and not-separated rods (pH 2.47). Here, the somewhat higher pH values are due to the decrease of H+ ions generated from AscA by the reduction process.
 | ||
| Fig. 7 FE-SEM images of KLa-HCF microcrystals prepared at 55 °C at lower reactant concentrations of CLa = CHCF = (a) 0.003 mol L−1 and (b) 0.001 mol L−1. CAscA = 0.05 mol L−1. The insets are the close-up views. | ||
Compared to the control condition, the turbidity increases are also slower, requiring a longer time to reach the turbidity maxima under these conditions. As the formation of [Fe(CN)6]4− by AscA reduction is instantaneous, the lower reactant concentration would give a lower supersaturation condition for the KLa-HCF formation. This results in a slower reaction rate, which might be unpreferred to form the bundle morphology but preferred to avoid stacking faults. These results may lead to the tendency for the bundle morphology to be preferably formed at a fast growth rate with a high supersaturation level.
Unfortunately, the formation mechanism for the seven-rod bundle morphology is not yet clear. However, the FE-SEM images in Fig. 1 may give speculations on the self-organized formation of the microcrystals. The bulges of the six outer rods are bound to the center rod at the corner positions of the cross-sectional hexagons, as shown in Fig. 1(b)–(d). The consistently directed hexagons mean the same crystal orientation among the rods. Such morphological features seem similar to snow crystals of a sector-plane type formed under high supersaturation conditions at specific temperature regions, apart from the planar structure of the snow crystals.16,17
Inferring from the analogy of the snowflakes, the rod-bundle structure formation could be associated with the Berg effect, which gives a faster growth rate at the corner than the edges of a hexagonal plate by the supersaturation difference.5,18,19 Compared to the edges, the corners protrude to the diffusion layer of a higher supersaturation region so that the growth around the corners could be selectively promoted to form new hexagons via dendritic growth, becoming the bulges of the outer rods. The following vertical growth from the outer bulges and the center hexagon constructs the rod bundles. The Berg effect possibly contributed also to the rod's shape; faster growth at the outer parts of the rods in a higher supersaturation resulted in the broader width with the shifted centers of cross-sectional hexagons and their tilted-up tips.
The crystal structure of KLa-HCF would be another factor for the dendritic growth that causes the seven-rod bundle morphology. The arrangement of La3+ and [Fe(CN)6]4− ions in the KLa[Fe(CN)6]·4H2O crystals is illustrated in Fig. 8, where the coordination data are from the literature;13 K+ ions and H2O molecules, located in the interstitial space, are omitted in Fig. 8 for simplification. Note that each illustration is an overlapped view of some parallel lattice planes having different ion positions. Reflecting the large ionic radius of La3+, KLa[Fe(CN)6]·4H2O exhibits the inverse NiAs arrangement of the constituent ions (hexagonal, P63/m), in which [Fe(CN)6]4− ions fill all the octahedral holes of the hcp configuration of La3+ ions (on the present microcrystals, some [Fe(CN)6]4− positions would be vacant due to the estimated composition, K0.82La1.11[Fe(CN)6]·4H2O).
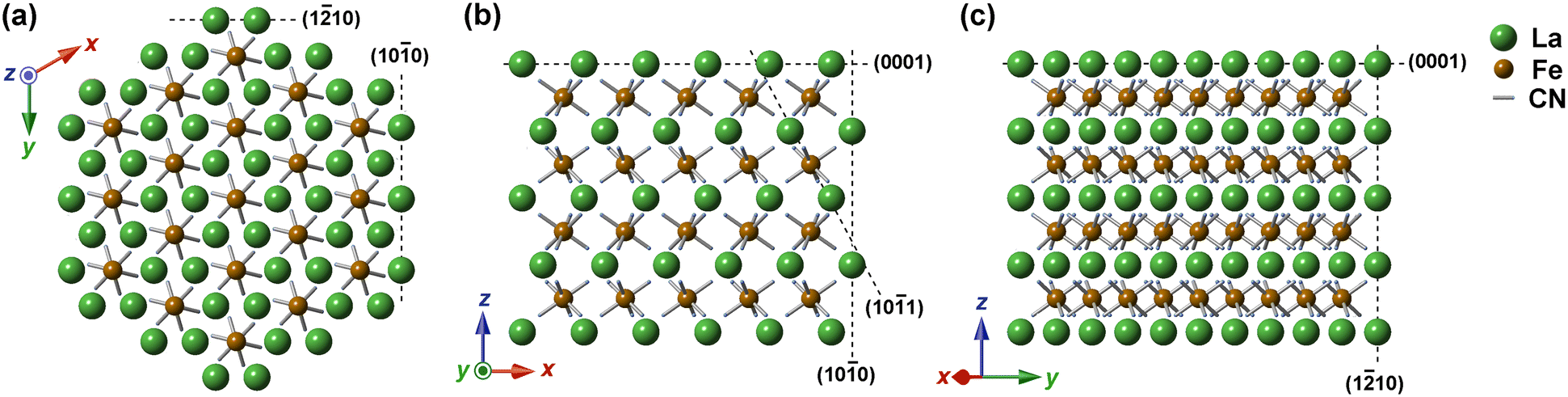 | ||
| Fig. 8 The hexagonal crystal structure for KLa[Fe(CN)6]·4H2O viewed from the (a) [001] (top), (b) [010] (corner), and (c) [0] (edge) directions, where K+ ions and hydrated H2O molecules located in the interstitial space are omitted for simplification. The dotted lines indicate the orientations for the indexed lattice planes vertical to the illustration. Note that the ionic radius ratio of La3+ to Fe2+ is not illustrated quantitatively. The coordination data for the atoms are based on the literature.13 | ||
The ion arrangements are very different in the lattice directions. The view from the [001] direction, Fig. 8(a), indicates the hexagonal arrangement of ions; the illustrated [Fe(CN)6]4− ions are located on the same lattice plane, whereas the La3+ ions are on two different La3+ planes, as indicated in Fig. 8(b). The views from the [010] and [1] directions (Fig. 8(b) and (c), respectively) correspond to the observations from the corner and the edge directions of the cross-sectional hexagon, respectively. In Fig. 8(b), [Fe(CN)6]4− ions are in a rectangular arrangement, but La3+ ions are in a parallelogram one; La3+ and [Fe(CN)6]4− ions are located on the same plane with a relatively narrow lattice spacing, as shown in Fig. 8(c). In Fig. 8(c), both La3+ and [Fe(CN)6]4− ions are in rectangular arrangements; La3+ ions and [Fe(CN)6]4− ions are on different planes, as shown in Fig. 8(b).
Moreover, the C–N–La bonds are bent in the KLa-HCF lattice, as illustrated in Fig. 8. According to the coordination data obtained by single crystal X-ray diffraction, the six CN− ions rectangularly coordinate to the center Fe2+ ion even in the hexagonal inverse NiAs structure of KLa[Fe(CN)6]·4H2O.13,20 As a result, the CN− ligands are not directed to the center of La3+ ions, different from the linear C–N–Fe configuration for Prussian blue, Fe4[Fe(CN)6]3·nH2O (cubic lattice, Fm3m or Pm3m).21 The structural features of KLa-HCF would cause irregular ion deposition and contribute to its dendric growth tendency and, thus, to the self-organized formation of its seven-rod bundle morphology.
KLa-HCF is a kind of PBA; PBAs are a group of substances in which other metal ions substitute either (or both) Fe ions for Prussian blue, K3xFeIII(4−x)[FeII(CN)6]3·nH2O (x = 0–1, typically x = 0). Though the present study has focused on the morphological interest of the lanthanum PBA, syntheses and applications of various PBAs have been widely studied owing to their functionality.22 The substituting metal ions studied are mainly d-block transition elements such as Co, Ni, Cr, or Mn; these PBAs have been of interest in magnetic, display, and battery materials, etc.
Lanthanoid substituting PBAs, Ln-HCF, including La-HCF, are expected as precursors of LnFeO3 perovskites, which can be converted from Ln-HCF by calcining at about 600 °C or higher depending on the kind of Ln.15,23 For example, perovskite-type LaFeO3 is expected to be applied to electrode materials for CO2 reduction in solid oxide electrolysis cells,24 catalysts for the oxygen evolution reaction,25etc. The SmFeO3 perovskite has been applied to gas sensors.26 On the other hand, on Ln-HCFs themselves, relatively small numbers of studies on microcrystal syntheses or applications seem to be reported despite the functionalities of lanthanoids.27–29 The present study expects to trigger studies on lanthanoid Prussian blue analogs.
Conclusions
Potassium lanthanum hexacyanidoferrate(II) microcrystals formed the seven-rod bundle morphology via the precipitation process under acidic conditions around pH 2 without a specific growth modifier. The constituent ion arrangement on the crystal lattice framework possibly originated from the self-organized formation of the morphology with the assistance of the growth kinetics.Author contributions
F. S. designed the study and prepared the manuscript. F. S. and R. N. synthesized and characterized the microcrystals. All the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Z. Li and Y. Nishinaka, Department of Materials Science, Chiba University, for the TG-DTA measurement. The XRD patterns were obtained at the Center for Analytical Instrumentation, Chiba University. This work was supported by JSPS KAKENHI Grant Number JP22K04946.Notes and references
- I. H. Leubner, J. Imaging Sci., 1985, 29, 219 Search PubMed; T. Sugimoto, Adv. Colloid Interface Sci., 1987, 28, 65 CrossRef CAS.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793 CrossRef CAS PubMed; J. Zhai, Y. Zhai, L. Wang and S. Dong, Inorg. Chem., 2008, 47, 7071 CrossRef PubMed; F. Shiba, R. Fujishiro, T. Kojima and Y. Okawa, J. Phys. Chem. C, 2012, 116, 3394 CrossRef; W. Zhu, K. Liu, X. Sun, X. Wang, Y. Li, L. Cheng and Z. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 11575 CrossRef PubMed.
- C. Li, Z. Hou, C. Zhang, P. Yang, G. Li, Z. Xu, Y. Fan and J. Lin, Chem. Mater., 2009, 21, 4598 CrossRef CAS; P. Li, Y. Liu, Y. Guo, X. Shi, G. Zhu and H. Zuo, Ceram. Int., 2015, 41, 6620 CrossRef; J. Zou, Q. Zhu, X. Li, X. Sun and J.-G. Li, J. Alloys Compd., 2021, 870, 159380 CrossRef.
- F. Shiba, T. Fujiwara, Y. Takeda and Y. Okawa, CrystEngComm, 2022, 24, 2958 RSC.
- I. Sunagawa, in Crystals: Growth, Morphology and Perfection, Cambridge University Press, Cambridge, 2005, ch. 4 Search PubMed.
- T. Sugimoto, in Monodispersed Particles, Elsevier, Amsterdam, 2nd edn, 2019, ch. 3 Search PubMed.
- I. H. Leubner, R. Jagannathan and J. S. Berry, Photogr. Sci. Eng., 1980, 24, 268 Search PubMed; T. Sugimoto, J. Colloid Interface Sci., 1983, 91, 51 CrossRef CAS.
- S. Hamada, Y. Kudo and I. Ishiyama, Shikizai Kyokaishi, 1996, 69, 658 CAS.
- K. Chen, C. Sun, S. Song and D. Xue, CrystEngComm, 2014, 16, 5257 RSC.
- F. Shiba, M. Yuasa and Y. Okawa, CrystEngComm, 2018, 20, 4910 RSC.
- J. E. Maskasky, J. Imaging Sci., 1986, 30, 247 CAS.
- M. Otomo and Y. Wakamatsu, Bunseki Kagaku, 1968, 17, 764 CrossRef CAS.
- S. G. Duyker, G. J. Halder, P. D. Southon, D. J. Price, A. J. Edwards, V. K. Peterson and C. J. Kepert, Chem. Sci., 2014, 5, 3409 RSC.
- F. Shiba, U. Mameuda, S. Tatejima and Y. Okawa, RSC Adv., 2019, 9, 34589 RSC; F. Shiba, A. Yamamoto, Y. Shinta, U. Mameuda, Y. Tahara and Y. Okawa, RSC Adv., 2021, 11, 8767 RSC.
- F. Goubard and A. Tabuteau, J. Solid State Chem., 2002, 167, 34 CrossRef CAS; H. Aono, T. Nishida, M. Kurihara, M. Sakamoto and Y. Sadaoka, Ceram. Int., 2012, 38, 2333 CrossRef.
- W. A. Bentley and W. J. Humphreys, in Snow Crystals, Dover Publications, New York, 1962, pp. 50–73 Search PubMed.
- T. Kobayashi, Philos. Mag., 1961, 6, 1363 CrossRef CAS.
- W. F. Berg, Proc. R. Soc. London, Ser. A, 1938, 164, 79 CAS.
- E. Yokoyama and T. Kuroda, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 41, 2038 CrossRef CAS PubMed.
- G. W. Beall, D. F. Mullica and W. O. Milligan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1446 CrossRef.
- H. J. Buser, D. Schwarzenbach, W. Petter and A. Ludi, Inorg. Chem., 1977, 16, 2704 CrossRef CAS; F. Herren, P. Fischer, A. Ludi and W. Hälg, Inorg. Chem., 1980, 19, 956 CrossRef.
- A. Azhar, Y. Li, Z. Cai, M. B. Zakaria, M. K. Masud, Md. S. A. Hossain, J. Kim, W. Zhang, J. Na, Y. Yamauchi and M. Hu, Bull. Chem. Soc. Jpn., 2019, 92, 875 CrossRef CAS.
- S. Nakayama, M. Sakamoto, K. Matsuki, Y. Okimura, R. Ohsumi, Y. Nakayama and Y. Sadaoka, Chem. Lett., 1992, 2145 CrossRef CAS; E. Traversae, M. Sakamoto and Y. Sadaoka, J. Am. Ceram. Soc., 1996, 79, 1401 CrossRef.
- M. Pidburtnyi, B. Zanca, C. Coppex, S. Jimenez-Villegas and V. Thangadurai, Chem. Mater., 2021, 33, 4249 CrossRef CAS.
- H. J. Kim, S. H. Kim, S.-W. Kim, J.-K. Kim, C. Cao, Y. Kim, U. Kim, G. Lee, J.-Y. Choi, H.-S. Oh, H.-C. Song, W. J. Choi, H. Park and J. M. Baik, Nano Energy, 2023, 105, 108003 CrossRef CAS.
- H. Aono, M. Sato, E. Traversa, M. Sakamoto and Y. Sadaoka, J. Am. Ceram. Soc., 2001, 84, 341 CrossRef CAS.
- X. Liu, H. Xie and J. Mao, J. Electroanal. Chem., 2022, 911, 116228 CrossRef CAS.
- A. Zentko, M. Bokor, M. Lukáčová, M. Maryško, M. Mihalik, Z. Mitróová and M. Zentková, Phys. Status Solidi A, 2003, 196, 340 CrossRef CAS.
- V. S. Perera, L. D. Yang, J. Hao, G. Chen, B. O. Erokwu, C. A. Flask, P. Y. Zavalij, J. P. Basilion and S. D. Huang, Langmuir, 2014, 30, 11847 CrossRef CAS PubMed; M. Perrier, A. Gallud, A. Ayadi, S. Kennouche, C. Porredon, M. Gary-Bobo, J. Larionova, Ch. Goze-Bac, M. Zanca, M. Garcia, I. Basile, J. Long, J. de Lapuente, M. Borrasd and Y. Guari, Nanoscale, 2015, 7, 11899 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details for the composition evaluations and outer plane angle estimations. See DOI: https://doi.org/10.1039/d3ce01140b |
| This journal is © The Royal Society of Chemistry 2024 |