
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization of para-aminobenzoic acid forms from specific solvents†
Mohammed Noorul
Hussain
 *a,
Luc
Van Meervelt
b and
Tom
Van Gerven
a
*a,
Luc
Van Meervelt
b and
Tom
Van Gerven
a
aProcESS division, Department of Chemical Engineering, KU Leuven, Leuven, Belgium. E-mail: mohammednoorul.hussain@uantwerpen.be
bDepartment of Chemistry, KU Leuven, Leuven, Belgium
First published on 13th February 2024
Abstract
para-Aminobenzoic acid (p-ABA) is a pharmaceutical compound with a challenging polymorph nucleation behavior. It has 4 polymorphs and multiple solvates. Among these forms, the α and the β form are subjects of interest for research in many studies. The α form nucleates predominantly in many solvents. The β form is known to nucleate in only 2 solvents. The reasoning for this was that the solvents, having a strong interaction with the COOH group, inhibit the dimerization of the COOH group leading to the nucleation of the β form. Following this reasoning, in this work, DMSO and DMF, which have strong interaction with the COOH group, were tested as solvents for anti-solvent crystallization experiments. Water was used as anti-solvent. The goal was to investigate the resultant polymorph nucleation under such specific solvent–solute interaction. Results showed that with DMSO as solvent at higher anti-solvent volume fraction the α form nucleated predominantly. At lower volume fractions there was no nucleation normally but addition of some seeds quickly led to β form nucleation. With DMF, a special result was seen where a new solvate between p-ABA and DMF was discovered. Single crystal XRD results confirmed that the formation of the solvate was a result of hydrogen bonding between the solute and the solvent. Overall, the results do confirm that solvents having strong interaction with the COOH group can lead to nucleation of forms other than the persistent form.
Introduction
Para-Aminobenzoic acid (p-ABA) is a pharmaceutical compound that exhibits a challenging polymorphic behavior. Its polymorphism has been a subject of investigation in numerous works in the literature, yet it is still seen as an under-studied and under-established substance.1 Until 2014, p-ABA was known to be having only two polymorphs, the α form (Fα) and β form (Fβ). However, in 2014 Benali-Cherif et al.2 reported the appearance of a new γ form (Fγ) and in 2019, Ward et al.3 discovered another form referred to as the δ form (Fδ). Further, a study published in 2020 by Cruz-Cabeza et al.4 revealed that p-ABA also forms solvates with acetone, dioxane and nitromethane solutions. Their study combined crystal structure prediction, crystallization experiments and a comprehensive survey of the Cambridge Structural Database (CSD – database of 1 million + crystal structures5) for investigating polymorphism in p-ABA. In this study, the solvates with acetone and dioxane were obtained through slow evaporation from acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water and dioxane:water solutions. The nitromethane solvate was obtained by Rosbottom et al.6 in cooling crystallization from ethanol and nitromethane solvent mixtures where the nitromethane presence was larger than 60 wt%. Fγ was crystallized from an aqueous solution that also contained a selenious acid additive.7 Fδ was obtained by transformation of Fα from water, ethanol:water and ethanol at high pressures of 0.3 GPa.3 While there are 4 polymorphs of p-ABA, the focus of research in the literature has been around Fα and Fβ, hence more information is available for these forms. The relationship between these two polymorphs is enantiotropic. The transition temperature between these two forms was reported to be 25 °C in works before 2011.8 In 2012, Hao et al.9 carried out a focused study on solvent-based transformation of these forms and determined the transition temperature to be 14 °C. Fα is the stable form above 14 °C and Fβ is the metastable form. The p-ABA molecule has an aromatic ring, an amine (NH2) functional group and a carboxylic acid (COOH) functional group. p-ABA is capable of both accepting and donating hydrogen bonds.1 In Fα, the carboxylic acid groups in p-ABA monomers form O–H⋯O–H hydrogen bonds to form carboxylic acid dimers. In Fβ, the N–H⋯O–H hydrogen bonds are formed between the carboxylic acid group of one monomer and the amino group of another monomer. Fα and Fβ both belong to the monoclinic crystal system. Fα has a morphology of fibrous needles whereas Fβ has a prismatic morphology. Fα vs. Fβ nucleation is an interesting case of kinetic vs. thermodynamic influence in polymorphic crystals. In various systems where two enantiotropically related polymorphs are involved, the meta-stable form is the kinetically favored form and the stable form is the thermodynamically favored form.10 In such compounds, for example, o-ABA, D-mannitol, eflucimibe, L-histidine, stavudine, BPT propyl ester, L-glutamic acid, famotidine etc., the kinetically favored meta-stable form nucleates at high supersaturation and the thermodynamically favored stable form nucleates at low supersaturation.11 A contrary behavior is observed in the case of p-ABA polymorphs.
:water and dioxane:water solutions. The nitromethane solvate was obtained by Rosbottom et al.6 in cooling crystallization from ethanol and nitromethane solvent mixtures where the nitromethane presence was larger than 60 wt%. Fγ was crystallized from an aqueous solution that also contained a selenious acid additive.7 Fδ was obtained by transformation of Fα from water, ethanol:water and ethanol at high pressures of 0.3 GPa.3 While there are 4 polymorphs of p-ABA, the focus of research in the literature has been around Fα and Fβ, hence more information is available for these forms. The relationship between these two polymorphs is enantiotropic. The transition temperature between these two forms was reported to be 25 °C in works before 2011.8 In 2012, Hao et al.9 carried out a focused study on solvent-based transformation of these forms and determined the transition temperature to be 14 °C. Fα is the stable form above 14 °C and Fβ is the metastable form. The p-ABA molecule has an aromatic ring, an amine (NH2) functional group and a carboxylic acid (COOH) functional group. p-ABA is capable of both accepting and donating hydrogen bonds.1 In Fα, the carboxylic acid groups in p-ABA monomers form O–H⋯O–H hydrogen bonds to form carboxylic acid dimers. In Fβ, the N–H⋯O–H hydrogen bonds are formed between the carboxylic acid group of one monomer and the amino group of another monomer. Fα and Fβ both belong to the monoclinic crystal system. Fα has a morphology of fibrous needles whereas Fβ has a prismatic morphology. Fα vs. Fβ nucleation is an interesting case of kinetic vs. thermodynamic influence in polymorphic crystals. In various systems where two enantiotropically related polymorphs are involved, the meta-stable form is the kinetically favored form and the stable form is the thermodynamically favored form.10 In such compounds, for example, o-ABA, D-mannitol, eflucimibe, L-histidine, stavudine, BPT propyl ester, L-glutamic acid, famotidine etc., the kinetically favored meta-stable form nucleates at high supersaturation and the thermodynamically favored stable form nucleates at low supersaturation.11 A contrary behavior is observed in the case of p-ABA polymorphs.
Firstly, the stable form Fα nucleates dominantly and is even considered the kinetically favored form. Secondly, the meta-stable form Fβ nucleates at low supersaturations rather than at high supersaturations in only two solvents that it has been found to nucleate in so far.1,8
The key to understanding this behavior lies in understanding the dominant nucleation of Fα. The Fα structure is based on the dimers formed by the carboxylic acid groups. The carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is a much better hydrogen bond acceptor in comparison to the nitrogen in the amino (NH2) group. This is the reason why the dimerization is dominant leading to the formation of Fα. The solvent–solute interaction is one crucial aspect that can have an effect on this dimerization process and can assist in nucleating polymorphs selectively. Two studies specifically investigated the effect of solvent on the polymorphism of p-ABA. In the first work by Gracin and Rasmuson,1 an experimental investigation of cooling crystallization of p-ABA from 11 solvents was carried out. These solvents were ethanol, methanol, hexanol, acetone, acetic acid, acetonitrile, hexane, toluene, 2-propanol, water and ethyl acetate. It was possible to crystallize Fβ only from water and ethyl acetate. In water, Fβ crystallized when the supersaturation was less than 1.5 and only at temperatures below 20 °C. In ethyl acetate, Fβ crystallized in experiments below 15 °C. The supersaturation in this case was also low but was not exactly determined. The reason for this outcome was that the solvent (water or ethyl acetate) had strong interactions with the carboxylic acid group which inhibited the formation of the carboxylic acid dimers as in Fα. This inhibition was sufficient to allow the N–H⋯O–H bonding and nucleate Fβ. In the second work by Rosbottom et al., molecular modelling was carried out to screen the solute–solvent and solute–solute interactions in organic molecules.12 The modelling was carried out for p-ABA in acetonitrile, ethanol and water solutions. The modelling showed that water had the strongest interactions with the carboxylic acid group while acetonitrile had the weakest interactions with the carboxylic acid group which led to nucleation of Fα. Ethanol had strong interactions with all groups in the solute. This modelling confirmed that Fβ is formed in those solvents that have strong interaction behavior with, specifically, the carboxylic acid group of p-ABA. There are several articles in the literature that deal with crystallization of p-ABA.1,7,8,13 However, all these works are based on cooling crystallization with single solvents. There is only one example of anti-solvent crystallization of p-ABA, which is an experimental work by Garg and Sarkar.14 A crystallization system with ethanol as solvent and water as anti-solvent was investigated at isothermal temperature of 15 °C. Two aspects of this study are interesting. Firstly, the difficult-to-nucleate Fβ, was successfully nucleated. Secondly, Fβ nucleated again at low supersaturation (<1.06) and Fα nucleated at high supersaturations.
O) is a much better hydrogen bond acceptor in comparison to the nitrogen in the amino (NH2) group. This is the reason why the dimerization is dominant leading to the formation of Fα. The solvent–solute interaction is one crucial aspect that can have an effect on this dimerization process and can assist in nucleating polymorphs selectively. Two studies specifically investigated the effect of solvent on the polymorphism of p-ABA. In the first work by Gracin and Rasmuson,1 an experimental investigation of cooling crystallization of p-ABA from 11 solvents was carried out. These solvents were ethanol, methanol, hexanol, acetone, acetic acid, acetonitrile, hexane, toluene, 2-propanol, water and ethyl acetate. It was possible to crystallize Fβ only from water and ethyl acetate. In water, Fβ crystallized when the supersaturation was less than 1.5 and only at temperatures below 20 °C. In ethyl acetate, Fβ crystallized in experiments below 15 °C. The supersaturation in this case was also low but was not exactly determined. The reason for this outcome was that the solvent (water or ethyl acetate) had strong interactions with the carboxylic acid group which inhibited the formation of the carboxylic acid dimers as in Fα. This inhibition was sufficient to allow the N–H⋯O–H bonding and nucleate Fβ. In the second work by Rosbottom et al., molecular modelling was carried out to screen the solute–solvent and solute–solute interactions in organic molecules.12 The modelling was carried out for p-ABA in acetonitrile, ethanol and water solutions. The modelling showed that water had the strongest interactions with the carboxylic acid group while acetonitrile had the weakest interactions with the carboxylic acid group which led to nucleation of Fα. Ethanol had strong interactions with all groups in the solute. This modelling confirmed that Fβ is formed in those solvents that have strong interaction behavior with, specifically, the carboxylic acid group of p-ABA. There are several articles in the literature that deal with crystallization of p-ABA.1,7,8,13 However, all these works are based on cooling crystallization with single solvents. There is only one example of anti-solvent crystallization of p-ABA, which is an experimental work by Garg and Sarkar.14 A crystallization system with ethanol as solvent and water as anti-solvent was investigated at isothermal temperature of 15 °C. Two aspects of this study are interesting. Firstly, the difficult-to-nucleate Fβ, was successfully nucleated. Secondly, Fβ nucleated again at low supersaturation (<1.06) and Fα nucleated at high supersaturations.
The effect of solvent on selective polymorph crystallization has also been observed in other organic polymorphic systems. For example, Khoshkhoo and Anwar15 studied crystallization of sulphathiazole which has four different polymorphs (namely I–IV) with acetone, acetone/chloroform, n-propanol and water. Each solvent seemed to favor a different polymorphic outcome. Acetone was supporting the nucleation of form I and IV, while a mixture of acetone/chloroform supported the nucleation of form III as well. n-Propanol supported only form I while water was the only solvent to support form II. The reason for this behavior is attributed to the affinity that a particular solvent may have to certain faces of a polymorph. This would lead to adsorption of the solvent on these faces and thereby inhibit the deposition of solute molecules. In another case that of mefenamic acid as studied by Abdul Mudalip et al.16 different solvents led to nucleation of specific polymorphs. Solvents like ethyl acetate, ethanol, isopropyl alcohol, dimethylacetamide and acetone led to crystallization of form I in cooling crystallization experiments while using DMF produced pure form II crystals. No solvent entrapment was observed with any of the solvents chosen in this study.
Solvent selection in general is quite important for polymorphism as it can promote or inhibit specific polymorphs. It is a subject much worthy of shedding further light in to. Following reports in the literature, in the case of p-ABA, the solvent plays a very important role in nucleating its polymorphs. Thus further targeted studies with p-ABA (a compound of interest in the literature for such studies) as a model compound can help in understanding this relation more. Therefore, in this work, p-ABA was used as a model compound to investigate the effect of particular solvents on its polymorphism. Since the focus here is on breaking the carboxylic acid dimerization, the solvent systems of DMSO:water and DMF:water were selected as they were not tested before. DMSO and DMF are known to have strong interactions with the carboxylic acid group.13,17 Anti-solvent crystallization experiments were conducted in batch conditions. Experiments were conducted first without seeds and then some experiments were conducted with seeding. The goal was to investigate the polymorphic outcome under such specific solvent and solute interactions with the idea of unravelling more information about polymorph control during crystallization.
Materials and methods
Fα of para-aminobenzoic acid (p-ABA) from Alfa Aesar with 98+% purity was used as the starting material as it is the commercially available form. Anti-solvent crystallization experiments were carried out with two solvents – dimethyl sulfoxide (DMSO, 99% Fischer Scientific) and dimethylformamide (DMF, 99.8% Bio-Solve). Distilled water was used as anti-solvent. Batch experiments were conducted in all cases in 100 ml volume jacketed vessels. In the case of DMSO and DMF, solubilities had to be measured. This was done by adding Fα directly to the solvent in small amounts until no more compound would be dissolved. Since anti-solvent experiments had to be conducted below 20 °C, solubilities also had to be measured at these temperatures. The high viscosity of DMSO and the low temperature conditions made it very difficult to filter the slurries (crystals would dissolve during filtration) and obtain precise data. Crystal16 (Technobis, Benchtop Crystallization System) was tried for measuring solubility but that also presented problems to generate reliable data. Hence, solubility values were obtained through dissolution tests. These data helped in making an informed selection of the starting concentrations. They are only meant to be used as guides rather than for calculating supersaturations.Using DMSO and DMF as solvents, a solution was made by dissolving p-ABA in the solvent mixture of DMSO/DMF and water at much higher temperatures than the solubility temperature. This solution was cooled to the intended crystallization temperatures. The crystallization product was analyzed for its polymorphic form. Powder XRD (Bruker D2 Phaser Cu Kα – 1.54 Å), ATR-FTIR (Perkin-Elmer Spectrum 100 with UATR accessory), SEM (JEOL JSM 6010LV) and an endoscopic microscope (KKmoon 20× microscope) were used wherever possible to analyze the polymorphic form. The product from DMF experiments was additionally analyzed with single crystal XRD (Agilent SuperNova diffractometer with Eos CCD detector using Mo-Kα radiation).
Seed production
For seeded experiments, seeds of Fβ were added to the supersaturated solution to check whether seeding would produce Fβ or not. Fβ seeds were produced from batch cooling crystallization from an aqueous solution. Experiments were conducted first at 3 g kg−1, i.e., the solubility concentration at 15 °C in water. The temperature was brought down from 25 °C to 4 °C. There was nucleation at 4 °C which was clearly Fβ as confirmed by the microscope, ATR-FTIR, SEM and powder XRD. The collectable yield was very low at only 0.08 g (7%). To increase the yield, experiments were conducted at a higher starting concentration of 4.125 g kg−1. Crystals obtained from the prior experiment were added as seeds at a temperature of 18 °C. The seed quantity was around 0.08–0.12 g. Nucleation was clearly noticed at a temperature of 8 °C. The final temperature was dropped to 2.7 °C to extract maximum yield. The final mass of collected crystals from these experiments was 0.610 g (37%). Furthermore, a similar technique was applied in an experiment with a starting concentration of 4.9 g kg−1. The yield from this experiment was nearly 1 g (51.2%).For single crystal XRD analysis, large (mm size) single crystals were grown by slow anti-solvent crystallization experiments. A 90 g L−1 solution of p-ABA in DMF:water solution was prepared. The volume fraction (VF) of water was 0.5. This solution was placed in a refrigerator at 4.5 °C. Since there was no mixing, induction time was extremely high since there was no nucleation even after 24 h. Some seeds were added to this supersaturated solution which were allowed to grow for 72 h to produce sufficiently large single crystals (5–7 mm). The solvate nucleation was highly reproducible as confirmed by 4 duplicate experiments.
Results and discussion
Experiments with DMSO as solvent
Experiments were carried out with DMSO as solvent following the reasoning presented by Gracin and Rasmuson1 and Cruz-Cabeza et al.7 for nucleation of Fβ in water. The solvents that have strong interaction with the carboxylic acid group inhibit the formation of dimers as in Fα and promote nucleation of Fβ. Fig. 1 shows the bonding in Fα and Fβ. There is evidence from the literature that DMSO has strong interactions with the carboxylic acid group.13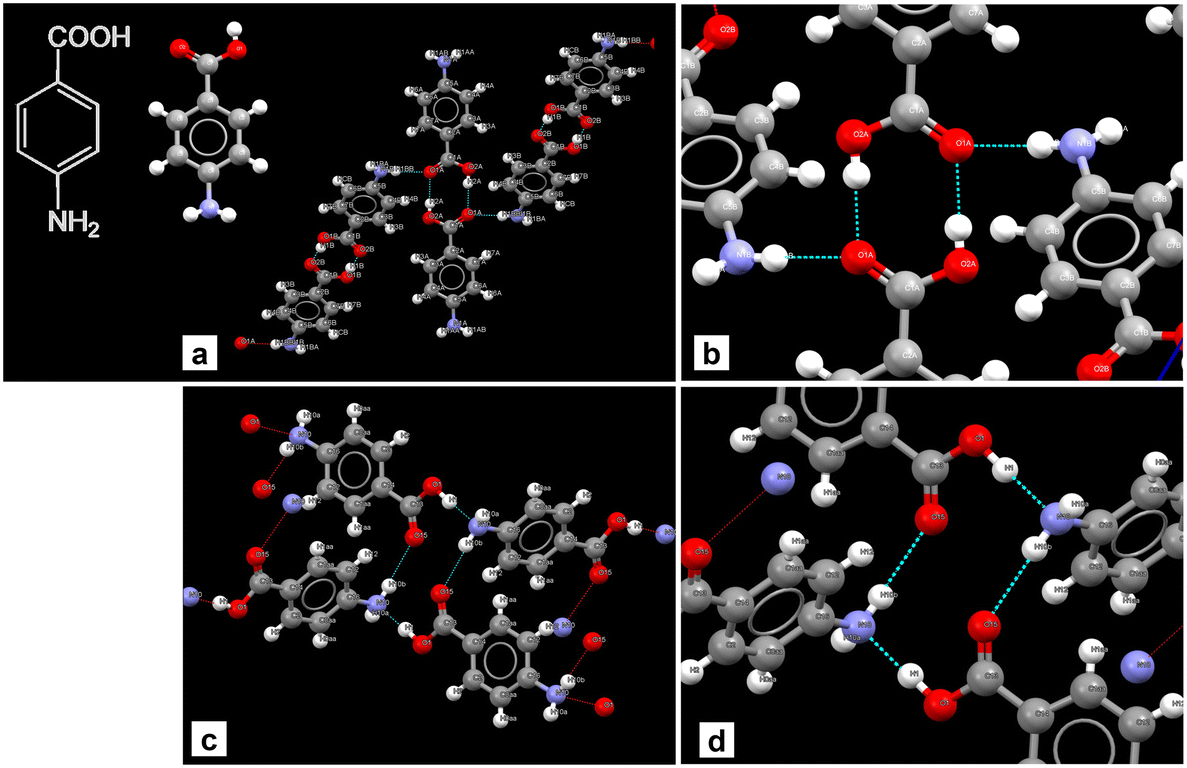 | ||
| Fig. 1 Crystal structures and bonding in Fα (CSD-AMBNAC19) and Fβ (CSD-AMBNAC08). a) Bonding in Fα showing centro-symmetric dimers of the COOH group in two p-ABA monomers, b) close up of the dimers, c) the alternative hydrogen bonding in Fβ between COOH group of one monomer and NH2 of another, and d) close up of the bonding. | ||
Approximate solubility data was measured for four anti-solvent volume fractions (VFs) at 15 °C. For an anti-solvent VF of 0.15, the approximate solubility measured was 944 g L−1. The freezing point of DMSO is above 15 °C (i.e. 18.5 °C), however, when mixed with water the freezing point decreases and crystallization experiments were possible at lower temperatures. Fig. 2 shows the crystallization experiments conducted as well as the solubilities. Beginning with a VF of 0.15, crystallization was carried out with a starting concentration of 944 g L−1, i.e., the solubility concentration. The temperature was lowered from 20 °C to 11 °C first and at a rate of 4 °C min−1. The supersaturated solution was stirred for 3 h. However, there was no crystallization. In the next experiment, the temperature was dropped from 20 °C to 8 °C, but still there was no crystallization after 3 h. The next experiment was conducted at a 0.19 VF. The solubility now decreases to 870 g L−1, and this was chosen as the starting concentration. In this crystallization experiment, a similar result was seen. There was no crystallization at temperatures of 11 °C and 8 °C. The next experiment was conducted at a 0.20 VF and three starting concentrations were tested which were 834 g L−1 (solubility concentration), 790 g L−1 and 750 g L−1. There was again no nucleation.
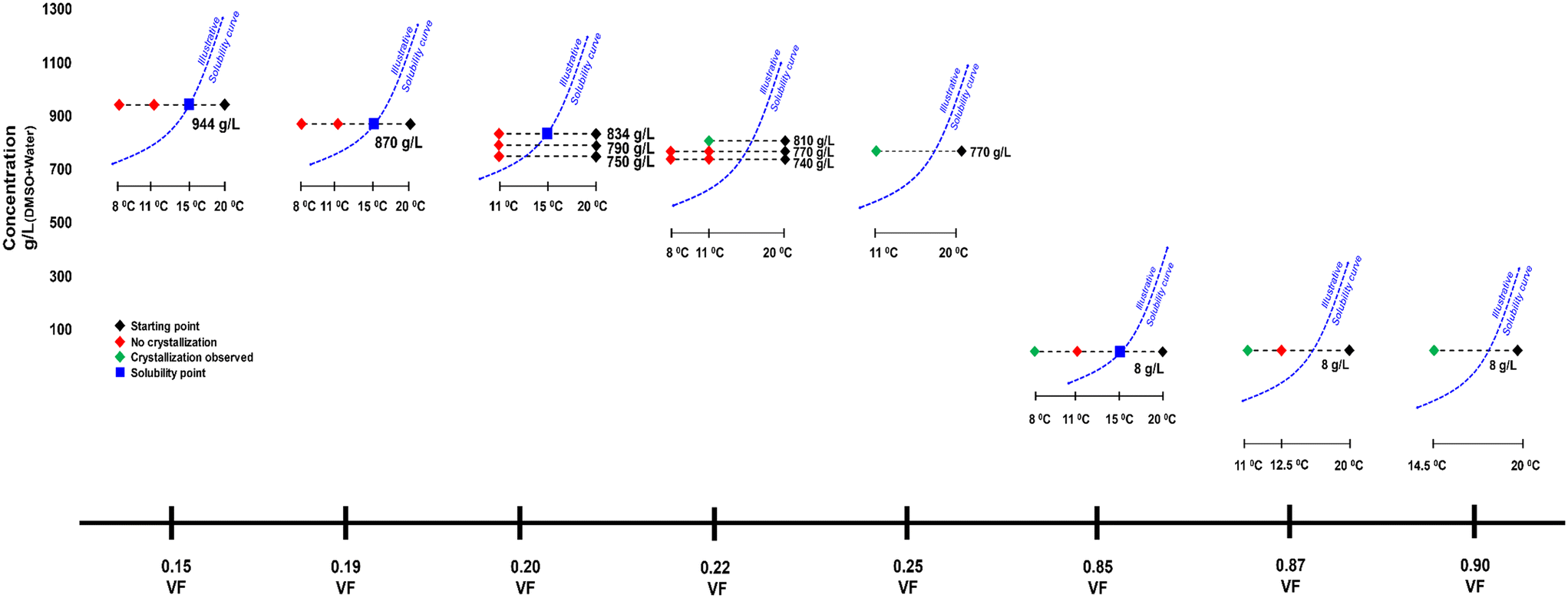 | ||
| Fig. 2 Description of crystallization experiments conducted with DMSO. VF refers to the volume fraction of anti-solvent water. The dotted blue curve is an illustrative solubility curve with respect to temperature. It is given as a guide to provide better visual context. | ||
In the next experiment where the VF was 0.22 and starting concentration was 810 g L−1 (solubility) there was nucleation of Fα within 100 min at 11 °C. At 770 g L−1 and 740 g L−1, there was still no crystallization even at the temperature of 8 °C. However, when the VF was increased to 0.25 at 770 g L−1 there was nucleation of Fα at 11 °C.
For a very high VF of 0.85 the solubility drops drastically to 8 g L−1. In an experiment carried out at a VF of 0.85 and a starting concentration of 8 g L−1 (i.e., solubility concentration), there was again no crystallization at 11 °C, but there was nucleation when the temperature was dropped to 8 °C. The crystallized form here was Fα. When the VF is increased to 0.87 at the same concentration, there was nucleation at 11 °C, but again the form was Fα. Further, at the same concentration when the VF is raised to 0.90, crystallization occurs during cooling at 14.5 °C.
It seemed that in the conditions at which the supersaturation was low (i.e., at low VFs) and Fβ could possibly nucleate, the induction time was extremely high leading to no nucleation. Hence, two experiments were conducted for 6 h to see whether there would be nucleation. At VFs of 0.15 and 0.85, their respective solubility concentrations experiments were conducted where the temperature was dropped to 11 °C with an expectation to nucleate Fβ. However, there was still no nucleation.
Seeded experiments (Fig. 3) were conducted next to initiate nucleation in these low supersaturation conditions that showed no nucleation. Experiments were conducted at four VFs of 0.15, 0.22, 0.85 and 0.87. At the 0.15 and 0.85 VFs, experiments were conducted at solubility concentrations of 944 g L−1 and 8 g L−1, respectively. At the 0.22 VF, experiments were conducted at starting concentrations of 740 g L−1 and 770 g L−1. As for the 0.87 VF, experiments were conducted at a starting concentration of 8 g L−1. In these experiments, 0.13 g of Fβ seeds were added, which is an extremely small amount compared to the dissolved mass. In almost all cases there was nucleation within few minutes of seed addition (Fig. 3). Instantaneous analysis of morphology under the endoscope indicated that the nucleated form was similar to the seeds, i.e., Fβ (Fig. 4). While, without seeding, there was no crystallization in these conditions, with seeding there was quick nucleation of the seeded form.
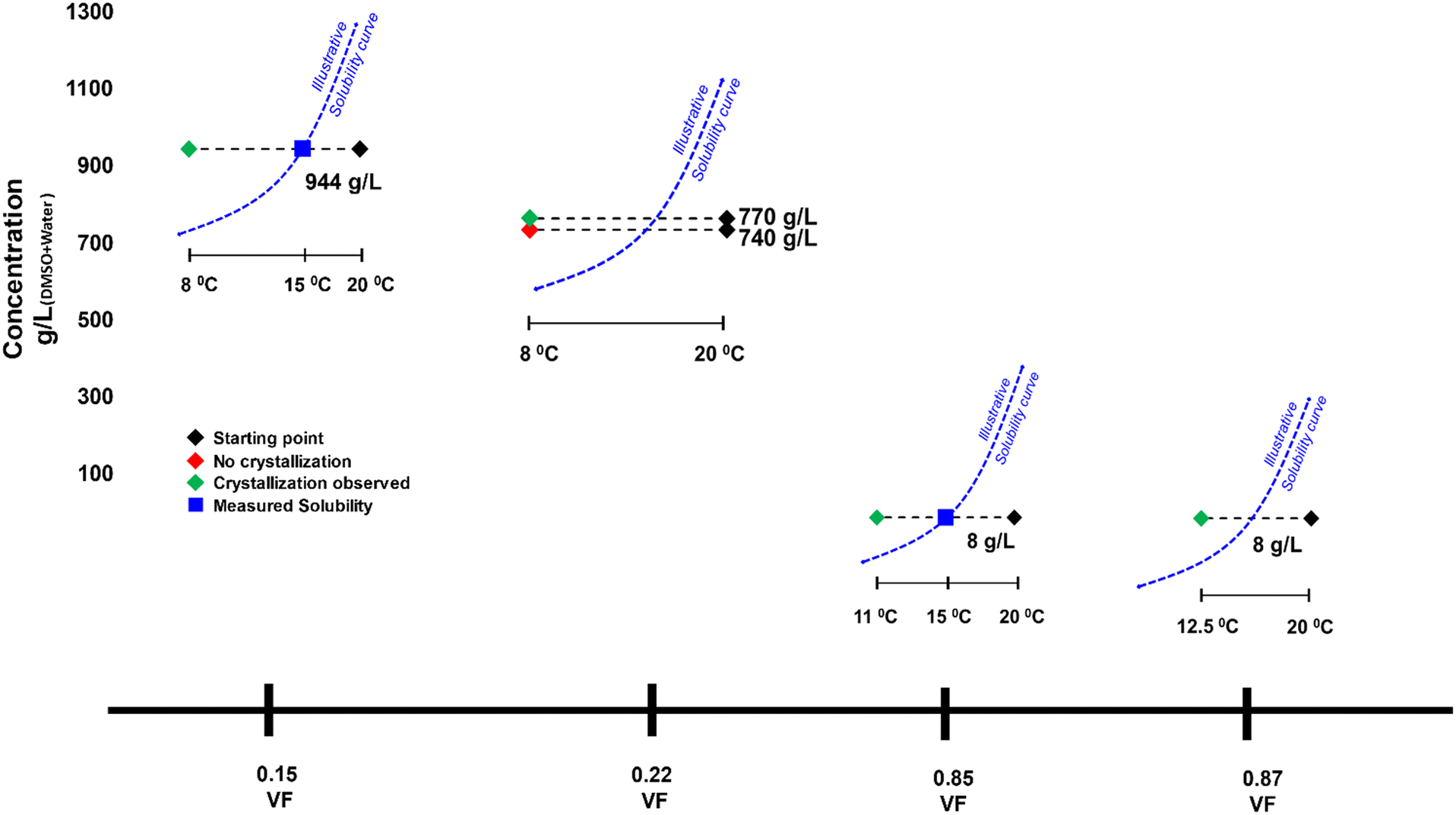 | ||
| Fig. 3 Description of seeded crystallization experiments conducted with DMSO. VF refers to the volume fraction of anti-solvent water. Dotted blue curve is an illustrative solubility curve with respect to temperature provided for better visual context. | ||
 | ||
| Fig. 4 Instantaneous endoscopic pictures from experiments with DMSO at different VFs to observe morphology. For non-seeded crystallization: a) 0.85 VF–8 g L−1, b) 0.22–810 g L−1, c) 0.25 VF–770 g L−1, d) 0.87 VF–8 g L−1, and e) 0.90 VF–8 g L−1, and for seeded experiments: f) 0.15 VF–944 g L−1, g) 0.85 VF–8 g L−1, h) 0.22 VF–770 g L−1, and i) 0.87 VF–8 g L−1. | ||
Experiments with DMF as solvent
DMF is another solvent that has strong interactions with the carboxylic acid group.17 With DMF as solvent, experiments were limited to only three volume fractions of anti-solvent: 0.85, 0.5 and 0.15. Beginning with the VF of 0.85, the solubility measured at this condition was 14.6 g L−1 at 15 °C. A crystallization experiment was conducted with a starting concentration of 14.6 g L−1. The temperature was lowered at 4 °C min−1 from 20 °C to 11 °C. Crystallization occurred within 2 h in this experiment. The form was analyzed using a microscope and ATR-FTIR and it was confirmed as Fα (ATR-FTIR results are shown in Fig. 5). In the next experiment a VF of 0.5 was used. The approximate solubility at this condition was measured as 90 g L−1 at 15 °C. The experiment was conducted again by cooling from 20 °C at the same concentration. There was crystallization in this case also and, interestingly, the microscopic analysis showed that the crystals to have a prismatic morphology. The morphology was completely different from the needle-like morphology of Fα and closer to that of Fβ. FTIR results (peaks at 3233, 3348 and 3449 cm−1) were even more surprising as the peaks obtained did not conform to either Fα (peaks at 3360 and 3460) or Fβ (peaks at 3275 and 3380 cm−1). This meant that a different form was being produced. Furthermore, a peculiar observation was made when an experiment was attempted at the anti-solvent VF of 0.15. In this experiment, it was even difficult to dissolve the compound as the added Fα was quickly transformed to this new prismatic form. It was clear that with an increase in the content of DMF the occurrence of this form and the transformation rate to this form increased. The transformation behavior was also seen with pure DMF as solvent. The crystals appeared to be prismatic under a microscope but to further observe the crystal morphology, SEM analysis was conducted.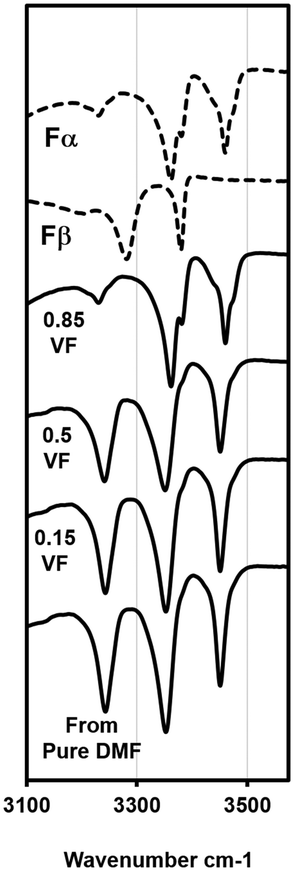 | ||
| Fig. 5 ATR-FTIR plots for experiments with the DMF–water system and standard curves for Fα and Fβ. Results at the 0.85 VF yields Fα, experiments with the VF of 0.5 and lower yield the new form (see Fig. S1 in the ESI† for full spectra). | ||
The SEM analysis (Fig. 6) showed that this new form was very different from both Fα and Fβ. However, there was no clear shape or morphology of the crystals. This led to a doubt whether this new form was even crystalline or not. To confirm this, an XRD analysis was conducted (Fig. 7). The first observation from this analysis was that there were sharp peaks, which confirm that the material is crystalline. The second observation was that the peaks of the diffraction pattern were different from those of Fα and Fβ. The diffraction patterns for the Fγ and Fδ were calculated from the Crystal Information Files (CIF) provided by Cruz-Cabeza et al.7 and Ward et al.3 using the Mercury 2020 software.18 These patterns were also different from that of the obtained crystals, confirming that it is not one of these polymorphs. A possibility now was that it is a solvatomorph (solvate) formed when the DMF molecules are incorporated in the p-ABA molecule leading to a new crystal structure. To completely confirm what the crystal is, and whether DMF is indeed being incorporated to form a solvate, further analysis with single crystal XRD was conducted. Hence, experiments were conducted to grow a large single crystal suitable for analysis.
 | ||
| Fig. 6 SEM pictures of different crystals. a) and c) show Fα and Fβ. b) and d) show the solvate. | ||
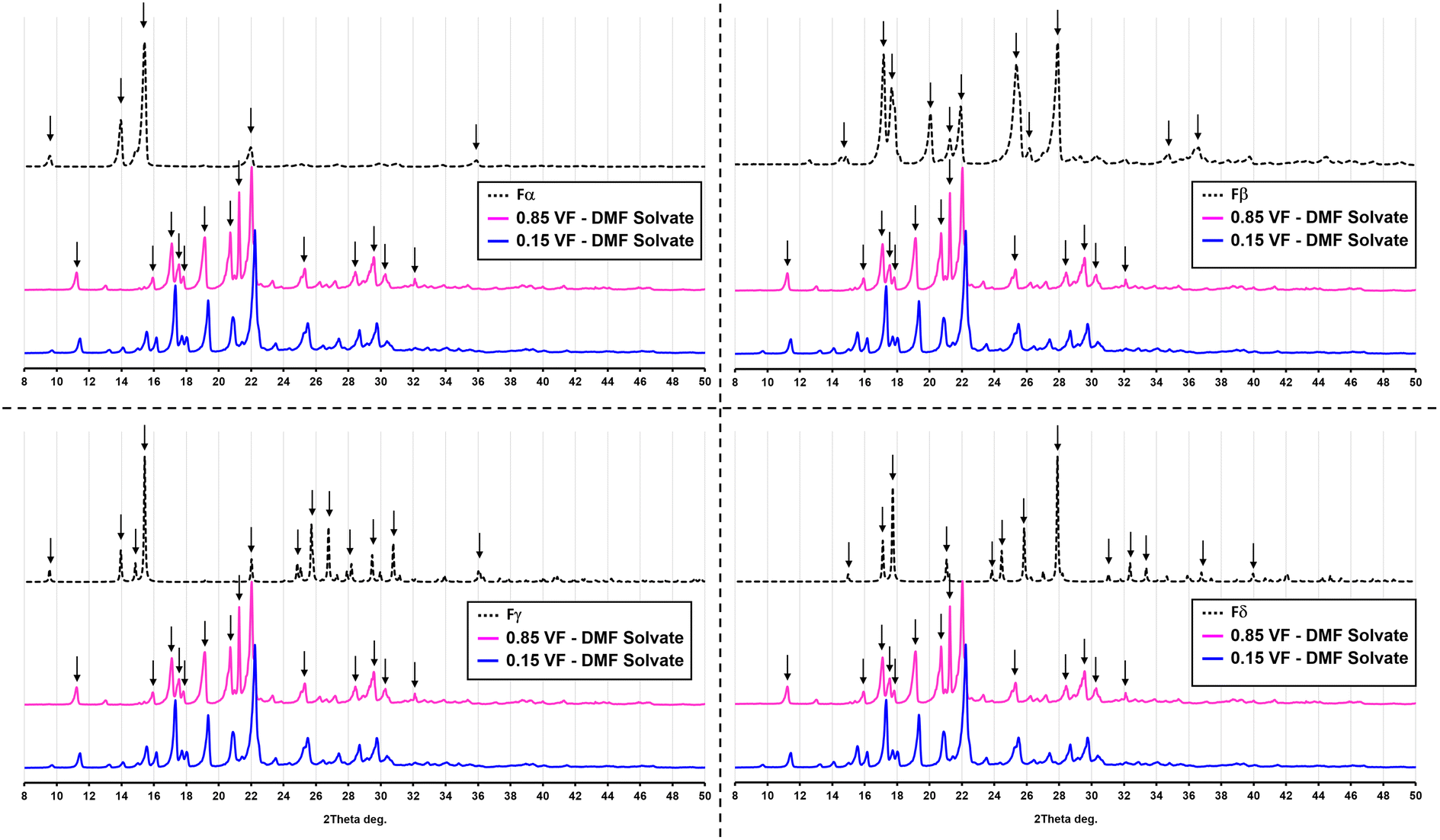 | ||
| Fig. 7 Powder XRD pattern of the solvate Sp-ABA/DMF in comparison with the four polymorphic forms of p-ABA. | ||
In initial trials to obtain single crystals, large single crystals of Fα were added to DMF and allowed to transform. The crystals obtained from this transformation were again prismatic but not of a particular shape. Also, these crystals were smaller than the required size for the analysis. Experiments were then conducted by placing a supersaturated solution in a refrigerator at 4.5 °C and adding seeds after the solution was allowed to cool down for at least an hour, so that the system is likely in a meta-stable zone. The crystals obtained from these experiments were larger and had a definitive ‘rhombus’-like shape. This shape was clearly different from Fα and Fβ. Eventually, crystals of 5–7 mm range were obtained, as shown in Fig. 8.
 | ||
| Fig. 8 Single crystal of the solvate Sp-ABA/DMF grown for single crystal XRD analysis. | ||
Single crystal XRD results confirmed that the crystal was a solvate of p-ABA and DMF, which will herein be referred to as Sp-ABA/DMF. Sp-ABA/DMF crystallizes in the monoclinic space group P21/n with a β angle of 110.772°, like the base crystal Fα (β angle is 93.670°). Sp-ABA/DMF resulted from the formation of a hydrogen bond between the hydrogen in the carboxylic acid group of p-ABA and the oxygen in DMF [graph set D1,1(2)]. In addition, the amino group forms two hydrogen bonds, one with a neighboring carboxylic acid group [graph set C1,1(8)] resulting in chain formation of p-ABA molecules, and one with a neighboring DMF molecule [graph set D1,1(2)]. Table 1 gives the details of the hydrogen-bond geometry. Fig. 9 shows the bonding structure from the crystal information file obtained from single crystal XRD analysis. From the figure it is clear that parallel chains of p-ABA molecules are linked by DMF molecules. The crystal packing shows no π⋯π or C–H⋯π interactions. It is noticed that this solvate does not stay stable when it is stored open to air at room temperature. The surface turns opaque white after one week in such conditions but stays as solvate in sealed containers or while dipped in solvent (Fig. 10). An ATR-FTIR analysis confirms that the solvate returns back to Fα in such de-solvation conditions (Fig. 11). SEM analysis shows that the opaque white color is a result of several pores formed on the surface of the crystal as the incorporated DMF evaporates (Fig. 11).
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| Symmetry codes: (i) ½ + x, ½ − y, −½ + z; (ii) ½ − x, −½ + y, ½ − z. | ||||
| N1–H1A⋯O1i | 0.81(3) | 2.16(3) | 2.958(3) | 174(2) |
| N1–H1B⋯O3ii | 0.87(2) | 2.34(2) | 3.193(3) | 169(2) |
| O2–H2⋯O3 | 0.87(3) | 1.76(3) | 2.608(2) | 165(3) |
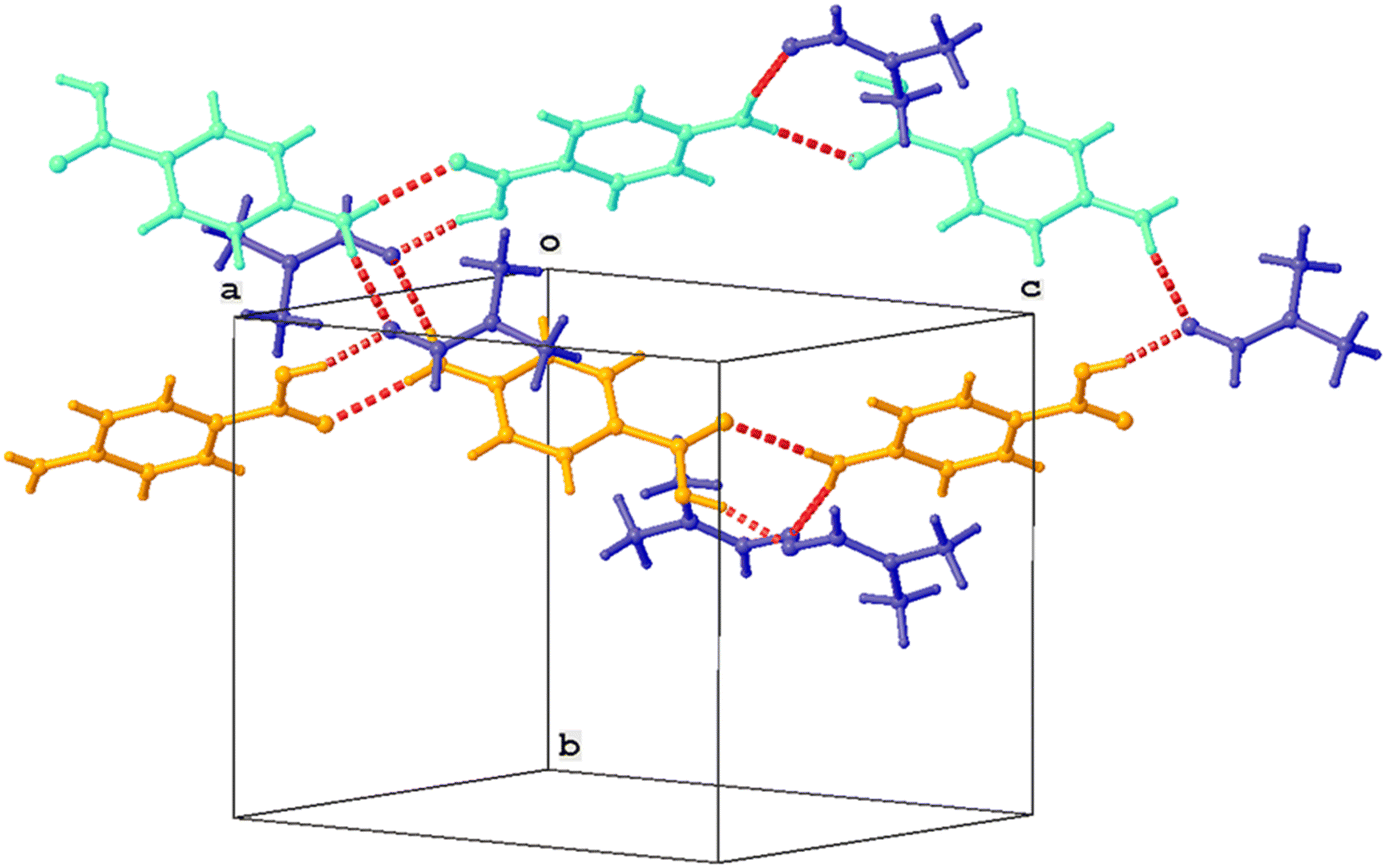 | ||
| Fig. 9 Partial crystal packing showing chain formation between p-ABA molecules (orange, cyan). Parallel chains are linked by DMF molecules (blue). Hydrogen bonds are shown as red dashed lines. | ||
 | ||
| Fig. 10 White, opaque single crystal (Sp-ABA/DMF) because of de-solvation in air. | ||
 | ||
| Fig. 11 Left) ATR-FTIR plots of Sp-ABA/DMF de-solvated in comparison with that of the transparent Sp-ABA/DMF crystal – it conforms to that of Fα indicating de-solvation. Right) SEM pictures of the surface of the a) transparent solvate crystal vs. b) and c) the opaque Sp-ABA/DMF crystal. | ||
The reason why DMF forms a solvate with p-ABA lies in the ability of the solvent molecule to form both weak and strong hydrogen bonds. During the nucleation stage, to form solute–solute aggregates, the solute–solvent interactions must be overcome. When solvents have a greater number of hydrogen bond donor and acceptor sites, it becomes difficult to overcome the solute–solvent interactions and the solvent is more likely to be incorporated in the crystal.19 DMF can form strong and weak hydrogen bonds with solute molecules and hence has a high ability to be incorporated and form solvates.17,19 DMF can form C–H, N–H and O–H hydrogen bonds with solute molecules. DMF has been shown to form such hydrogen bonds in compounds having the carboxylic acid group and result in the formation of the solvate. For instance, DMF forms solvates with terephthalic acid, pyromellitic acid and hemimellitic acid, each of which have multiple carboxylic acid groups.
The occurrence of Sp-ABA/DMF was interesting not just for the fact that a new solvate was discovered but because it also provided additional information for controlling polymorphs in crystallization. The main inference that can be deduced is in the context of a solute–solvent relationship in p-ABA. Experimental studies that have obtained Fβ based their rationale on the interaction between the solvent and the carboxylic acid group of p-ABA. A large interaction inhibits the dimerization of the COOH group and enables the nucleation of Fβ. The results in this study do show that such solvents can inhibit the formation of Fα, which is quite difficult to avoid if not impossible in other solvents. However, the results do also show that such interactions might not always support the nucleation of Fβ but can also lead to formation of solvates. Additional details about the crystal and single crystal XRD analysis are provided in the ESI.†
For further studies, there is much scope in studying the de-solvation process of the crystal which can help in understanding the crystal. This can be undertaken via techniques like DSC/TGA studies or in situ XRD studies. These can unravel whether the de-solvation process is direct or involves intermediate polymorph changes where the polymorphs can be the known ones or new ones while also providing physicochemical properties of the phases. In addition to that, for further understanding of the solvate formation, computational methods such as the Hirschfield surface analysis can be employed which can provide more details about the intermolecular forces (hydrogen bonding, van der Waals forces etc.), thus a more fundamental insight in to the process.
Conclusions
In this work, batch, anti-solvent crystallization of p-ABA was conducted using 2 solvents – DMSO and DMF. The anti-solvent was water in all cases. The goal was to investigate p-ABA polymorph nucleation wherein the solvent has a specific interaction with the carboxylic acid group present in p-ABA. In p-ABA crystallization, the polymorph that nucleates dominantly is Fα. Therefore, the selection of DMSO and DMF was based on the rationale that solvents with strong interaction with the COOH group tend to support the nucleation of Fβ.In both DMSO and DMF cases, experiments were conducted at different anti-solvent volume fractions. With DMSO, it was noticed that experiments at low supersaturation had very high induction times and did not show any nucleation even in six hours. Experiments at higher supersaturation resulted in nucleation of Fα. However, when a small amount of Fβ seeds were added in low supersaturation experiments, there was nucleation of possibly Fβ in just a few minutes. In experiments conducted with DMF, a peculiar result was observed. When the content of DMF was low in anti-solvent experiments (anti-solvent volume fraction of 0.85) the nucleated form was Fα. But when DMF content was high (anti-solvent volume fraction of 0.5 and 0.15) there was nucleation of a totally new crystal. This crystal was determined as a solvate of p-ABA and DMF after SEM, ATR-FTIR, powder XRD and single crystal XRD analysis. Single crystal XRD analysis showed that this solvate (Sp-ABA/DMF) was formed as result of hydrogen bonding between the oxygen in DMF and the hydrogen in the COOH group of p-ABA. These results do confirm that solvents having strong interaction with the COOH group can lead to nucleation of forms other than the persistent form.
Notes
The ESI† provides further details of the crystal structure.Author contributions
Mohammed Noorul Hussain: conceptualization, methodology, investigation, data processing, and writing – original draft. Luc Van Meervelt: investigation, data processing, and writing – review & editing. Tom Van Gerven: conceptualization, methodology, resources, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from VLAIO (Catalisti) in the MMICAS project HBC.2020.2627. Prof. Luc Van Meervelt thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035.References
- S. Gracin and Å. C. Rasmuson, Cryst. Growth Des., 2004, 4, 1013–1023 CrossRef CAS.
- R. Benali-Cherif, R. Takouachet, E. Bendeif and N. Benali-Cherif, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 323–325 CrossRef CAS PubMed.
- M. R. Ward, S. Younis, A. J. Cruz-Cabeza, C. L. Bull, N. P. Funnell and I. D. H. Oswald, CrystEngComm, 2019, 21, 2058–2066 RSC.
- A. J. Cruz-Cabeza, E. Taylor, I. J. Sugden, D. H. Bowskill, S. E. Wright, H. Abdullahi, D. Tulegenov, G. Sadiq and R. J. Davey, CrystEngComm, 2020, 22, 7447–7459 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoota and S. C. Warda, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- I. Rosbottom, C. Y. Ma, T. D. Turner, R. A. O'Connell, J. Loughrey, G. Sadiq, R. J. Davey and K. J. Roberts, Cryst. Growth Des., 2017, 17, 4151–4161 CrossRef CAS.
- A. J. Cruz-Cabeza, R. J. Davey, I. D. H. Oswald, M. R. Ward and I. J. Sugden, CrystEngComm, 2019, 21, 2034–2042 RSC.
- S. Gracin, M. Uusi-Penttilä and Å. C. Rasmuson, Cryst. Growth Des., 2005, 5, 1787–1794 CrossRef CAS.
- H. Hao, M. Barrett, Y. Hu, W. Su, S. Ferguson, B. Wood and B. Glennon, Org. Process Res. Dev., 2012, 16, 34–41 CrossRef.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, Informa Healthcare USA, Inc., New York, 2009 Search PubMed.
- B. K. Hodnett and V. Verma, Processes, 2019, 7(5), 272 CrossRef CAS.
- I. Rosbottom, J. H. Pickering, R. B. Hammond and K. J. Roberts, Org. Process Res. Dev., 2020, 24, 500–507 CrossRef CAS.
- P. G. Takis, K. D. Papavasileiou, L. D. Peristeras, G. C. Boulougouris, V. S. Melissas and A. N. Troganis, Phys. Chem. Chem. Phys., 2017, 19, 13710–13722 RSC.
- R. K. Garg and D. Sarkar, J. Cryst. Growth, 2016, 454, 180–185 CrossRef CAS.
- S. Khoshkhoo and J. Anwar, J. Phys. D: Appl. Phys., 1993, 26, 90–93 CrossRef.
- S. K. A. Mudalip, M. R. A. Bakar, P. Jamal, F. Adam, R. C. Man, S. Z. Sulaiman, Z. I. M. Arshad and S. M. Shaarani, MATEC Web Conf., 2018, 150, 02004 CrossRef.
- S. H. Dale and M. R. J. Elsegood, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, 444–448 CrossRef PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. Nangia and G. R. Desiraju, Chem. Commun., 1999, 605–606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2279352. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01055d |
| This journal is © The Royal Society of Chemistry 2024 |