
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developing a model-driven workflow for the digital design of small-scale batch cooling crystallisation with the antiviral lamivudine†
Thomas
Pickles
 a,
Chantal
Mustoe
a,
Christopher
Boyle
ab,
Javier
Cardona
abc,
Cameron J.
Brown
a and
Alastair J.
Florence
*a
a,
Chantal
Mustoe
a,
Christopher
Boyle
ab,
Javier
Cardona
abc,
Cameron J.
Brown
a and
Alastair J.
Florence
*a
aCMAC Future Manufacturing Research Hub, Technology and Innovation Centre, The University of Strathclyde, Glasgow, G1 1RD, UK. E-mail: alastair.florence@strath.ac.uk
bDepartment of Chemical and Process Engineering, University of Strathclyde, Glasgow, G1 1XJ, UK
cDepartment of Electronic and Electrical Engineering, University of Strathclyde, Glasgow, G1 1XW, UK
First published on 8th January 2024
Abstract
We present a workflow that uses digital tools to optimise the experimental approach and maximise the efficiency in achieving the required process parameters for a desired set of crystallisation responses, kinetics and objectives. Model-driven small-scale experiments can contribute to reducing time and material waste in the development of pharmaceutical crystallisation processes. The workflow presented here guides the development of a small-scale batch cooling crystallisation process via solubility measurements, particle shape and size determination, form identification and preliminary kinetic parameter estimation to make crystals that satisfy quality target parameters (for shape, size and solubility) for a given active pharmaceutical ingredient (API). The case study herein follows the development of a crystallisation process for lamivudine, an API used in the preventative treatment of human immunodeficiency virus (HIV). This work identifies ethanol as a suitable solvent, meeting the acceptable solubility parameters for industrially relevant processes and yielded the biorelevant form, form I. The target kinetic parameters that were measured included induction time, growth rate and nucleation rate for lamivudine in ethanol under a range of conditions as guided by experimental planning models. Data was collected as part of the development of a DataFactory platform in which experimental optimisation can be autonomously implemented and all measurements stored in a crystallisation parameter database. This database will have further value in informing model development and continuous crystallisation process design and optimisation. The model objective-driven development workflow identified the following conditions, 19.9 °C, 600 RPM and supersaturation of 1.70, as achieving the desired objective successfully in 80 polythermal and 28 isothermal experiments. Integration of the workflow alongside the optimisation algorithm within the automated DataFactory system will enable fully autonomous, rapid data collection for small-scale API crystallisation. Such autonomous systems could play vital roles in pharmaceutical development and manufacturing driving towards more efficient and sustainable practices via digital transformation.
1. Introduction
Crystallisation is widely used as a fundamental purification step in the primary processing of active pharmaceutical ingredients (API) and many other molecular materials that also imparts the physical and bulk properties to the material relevant to subsequent processing.1 The fundamental rate processes controlling crystallisation outcomes i.e. primary/secondary nucleation, crystal growth, agglomeration, phase transformations, and impurity rejection can all be measured using a range of established techniques2,3 but remain difficult to predict for any given combination of API solute, solvent system, equipment geometry and process parameters. Hence process development can involve extended experimental efforts to select the composition and process conditions under which desirable outcomes can be achieved and transfer this across scales. Thus, approaches to enable rapid, optimised selection of conditions that provide the thermodynamic and kinetic control to achieve desirable size, shape and form outcomes early in the development cycle are of considerable interest.Lamivudine, a nucleoside reverse transcriptase inhibitor used as an antiviral medication to treat and prevent human immunodeficiency virus (HIV) and hepatitis B virus (HBV),4,5 is also being investigated as a potential treatment for COVID-19.6 Lamivudine is currently produced using cooling crystallisation in methanol and (−) binol.7 There is limited prior literature describing the solubility of lamivudine and no kinetic parameters have been published. Past studies have shown that the dissolution rate of lamivudine is not substantially affected by changes in pH and that, subsequently, lamivudine exhibits good bioavailability in clinical usage.8 Good bioavailability is determined by the biopharmaceutical classification system (BCS) assignment of group III, noting it is on the border of group I for permeability.9 Lamivudine has been shown to produce three crystalline forms, form I, which is a 0.2 hydrate that exhibits needle geometry, and two anhydrous forms, form II, and III which typically adopt bipyramidal10 and needle morphologies, respectively.11 Existing patents state that form I and form II are preferred in solid oral dosing.12 Therefore, the objective of the crystallisation is the purification and management of physical attributes to ease downstream processing.
The DataFactory project at CMAC, The University of Strathclyde, involves the development of an autonomous data collection platform primarily focusing on API solubility and kinetic parameters.13 The work presented here lays the foundation for a robotic workflow that can be carried out without human supervision. The core experimental process will follow the steps discussed in this paper but be carried out robotically, i.e. by a Kuka KMR iiwa similar to the one used by Burger et al.14 When autonomous around-the-clock experiments are possible, the DataFactory will have the capacity to output data up to four times faster than human-led laboratory work due to operational hours increasing from 8–12 hours per weekday to 24 hours every day. A crystallisation parameter database is being developed to support the development of a suite of predictive tools in the form of a crystallisation classification system (CCS) that will accelerate the development of robust, sustainable crystallisation processes. This platform aligns with the wider need for more structured data management and curation adhering to findable, accessible, interoperable, and reusable (FAIR)15 data principles to support digital transformation in pharmaceutical and other process industries.
In this work, the target workflow builds on the stages and guidelines of previous process development workflows for pharmaceutical processes.2 This workflow integrates predictive models, automation and robotics to improve overall experimental efficiency and speed. The workflow also establishes and tests the logic necessary to enable an autonomous robotic data-collection platform to undertake the key tasks towards the selected experimental objective. Such autonomous platforms have the potential to accelerate development timescales and allow rapid process design.16 Autonomous development systems that can intelligently vary experimental conditions to achieve a target outcome based on real-time data also promote sustainability by reducing human error, minimizing waste and optimising resource utilisation, contributing towards a more efficient and sustainable pharmaceutical manufacturing sector.17
2. Experimental
In this study, images taken during the crystallisation experiments were collected using Technobis's Crystalline Reactor system at a rate of one image every five seconds. Each of the eight reactor vials heats and cools following its own temperature profile, and every reactor has an HD camera focused on the lower part of the vial. X-ray powder diffraction (XRPD) patterns were collected using a Bruker D8 Discover, and the data was visualized using DIFFRAC.EVA18 software from Bruker. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed simultaneously using a Netzsch STA 449 F1 Jupiter and data was analysed using Proteus Analysis.19 Solid and solvent dosing was carried out using a Zinsser Crissy GGXXL robotic platform, which recorded solid and liquid dosed masses with a precision of ±0.005 mg. The Zinsser platform was used to dose amounts in the range of 5 to 1000 mg of solid and 1 to 8 mL of solvent. Raman scans were collected using a Tornado Focused Non-Contact probe from a stirred suspension in 8 mL glass vials.The optimisation of crystallisation parameters to achieve the desired outcome was conducted in Modde 12.1 (Sartorius)20 following a full factorial design of experiment (DoE) plan with a multiple linear regression model (MLR). This model allows a 3-dimensional optimisation space i.e., 2 objectives and a combination of input parameters. Additional optimisation objectives can be added as constraints but will not explicitly feature as objectives of the optimisation algorithm. The experimental section and subsequent optimisation were performed in tandem as batch offline iterations. The initial DoE plan had high exploration across the whole design space and covered the corners, edges and centre points. Iteratively, the DoE experimental coverage got smaller to focus on exploiting the true optimum. Termination criteria for the optimisation loop was ±0.01 supersaturation and ±1 °C between MLR recommended experiments.
3. Workflow
In this section, each stage of the workflow, corresponding to the boxes in Fig. 1, is described in detail. The challenges associated with the application of this workflow are also discussed for each step.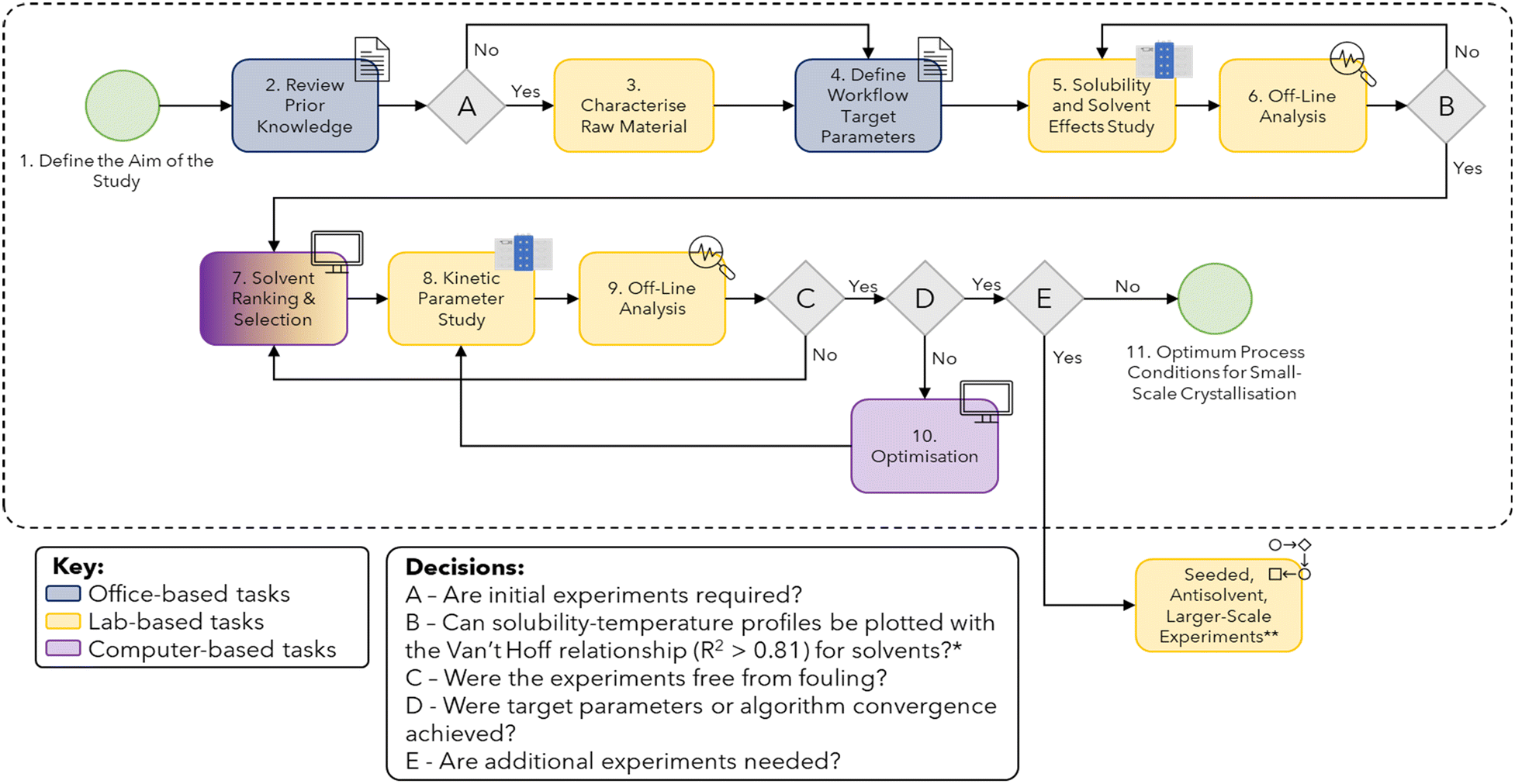 | ||
| Fig. 1 Workflow for small-scale batch cooling crystallisation data collection (for more details refer to Table S1 in the ESI†). *Minimum of 3–4 data points must be used for reliable estimates of R2 values. For some solvent systems, this may not be achievable as qualitative solubility can still be used to eliminate potential solvents. **Although not explicitly discussed in the case study of this paper, if the target parameters for the study are not met then further study can be done to explore seeded, antisolvent and larger-scale crystallisation. | ||
3.1. General considerations and challenges
The following section discusses the challenges associated with the generalisability of each step of the workflow. Each subsection corresponds to a step in the workflow.If prior solubility data is not available for a given system, initial experiments are conducted systematically or guided by predictive models.25 When conducting experiments systematically, exploring concentrations at the lower and upper bounds of the target range allows us to generate qualitative solubility classifications that guide subsequent experimental concentrations. For some APIs, the workflow-specific target parameters may not be met using a single solvent from the library chosen. In this scenario, a binary solvent screen is recommended as intermolecular interactions in binary solvent systems can alter molecular solubilities.26
The reliable measurement of the induction time of a solute in a given solution also requires multiple data points in order to properly sample the probability distribution.30 The number of experiments and/or cycles required to address this challenge can therefore be expensive in terms of time and data storage. In this study, five cycles were used to reduce measurement uncertainty. Fewer cycles would have resulted in a higher degree of uncertainty while more cycles would have risked exceeding the computational and temporary data storage limits available for a given experiment.
4. Results & discussion: workflow case study of lamivudine
In this section the individual steps of the workflow, as described in Table S1 of the ESI,† are presented using lamivudine as a test case. Note, the section headings below, are numbered 4.2.X, where X refers to the specific stages of the workflow illustrated in Fig. 1.4.1. Materials
Lamivudine (90 g, CAS ID: 134678-17-4) (Fig. 2) was purchased from Molekula Ltd as an off-white powder and form II. Lamivudine is a suspected teratogen. Therefore, dosing was performed in a fume hood and samples were transferred in capped vials. The solvents used for the cooling crystallisation were purchased from Sigma Aldrich, Fisher Scientific, VWR, and Alfa Aesar or prepared in-house (deionised water). | ||
| Fig. 2 Molecular structure of lamivudine. | ||
Lamivudine was chosen as the API of interest due to its useful pharmaceutical applications, and, in particular, its antiviral properties. There is also limited data in the literature for lamivudine solubility and crystallisation kinetics.34 Contextual data was collated for lamivudine from the British National Formulary (BNF) and National Health Service of dictionary of medicines and devices (NHS dm+d).35 The BNF recommends a dosage of 150 mg twice daily when used as a preventative treatment for HIV and HBV. The BNF and NHS dm+d shows current suppliers include Teva, Alliance and Milpharm. The developability classification23 of lamivudine was calculated using BASF Zoomlab36 and assigned a classification of group I, meaning that solubility and permeability are not expected to influence bioavailability.
The solvent library was chosen using the International Conference on Harmonization (ICH) residual solvent guidelines, safety, health and environment (SHE) ranking, cost, molecular descriptors, a principal component analysis (PCA) study in the literature37 and a clusterSim study in the literature.38 Relative locations of solvents on a PC1 vs. PC2 plot37 of 272 solvents were used to ensure the diversity of solvents. To do this, solvents were selected from all four quadrants of the plot published by Diorazio et al. An online tool, from the same study, allows users to visualise solvents in different ICH and SHE classifications in their relevant quadrant of the PCA. ClusterSim38 was then used to identify solvents with similar properties for our study. The solvents chosen from the PCA tool37 were selected using a multidimensional scaling (MDS) plot which also showed similar solvents that sit within the same cluster.38 While we ensured a diverse group of solvents was present in our selected solvents for this work, also choosing solvents from the same clusters allows us, and future users of the data provided, to compare experimental results within solvent families (i.e. alcohols) not just between solvent families to investigate any effects on crystallisation rate processes.
4.2. Key workflow tasks
Previous manufacturing routes consist of purification by crystallisation in methanol. This purification method allowed for the separation of the pharmaceutically active (−)-cis isomer.42
The optimisation objectives of the kinetic parameter estimation had target values of an induction time of 1 hour and a growth rate of 1 μm min−1. These target parameter values correspond to typical values that lead to operable conditions in larger-scale crystallisation processes.43 Additional parameter constraints were included in the kinetic parameter estimation including a D90 size distribution of 100 μm to 250 μm so that the resultant powder would be free-flowing.22
From a bioavailability and drug product perspective, there was no targeted preference between form I and form II as the both forms have similar oral bioavailability and are both used in commercial formulations.44 Form I and form III exhibited needle geometry, and thus would not satisfy the aspect ratio target parameter. Form III is also currently not used as drug product. Form II, therefore, is the most desirable crystal due to increased thermal stability and flow properties for downstream processing.
1. Heat to 10 °C below the solvent boiling point or 90 °C (whichever is lowest) at a rate of 0.5 °C min−1 and hold for 10 minutes.
2. Cool to 5 °C at a rate of 0.5 °C min−1 and hold for 10 minutes.
3. Repeat the cycle 2 more times.
The slow heating rate in the above temperature profile was chosen to reduce the error of the dissolution temperature reported (refer to Fig. S3 in the ESI†). The maximum temperature was set to ensure that boiling solvents were not handled, thus making the experiment safe. The slow cooling rate was also used to reduce the error in determining the cloud point, and each sample vial underwent this temperature cycle three times to check for anomalous results. The temperature profile was the same for all experiments and the stir rate was fixed at 600 RPM throughout.
The clear point, or the time at which full dissolution occurs, was defined as the temperature where no particles were observed in the collected images. In the literature, transmission data is used to identify the clear point.2 Transmissivity, however, often gives a clear point below the true value as transmission reached 100% even when a few particles are still present in the images.
Similarly, the cloud point, or the time at which primary nucleation first occurs, was defined as the temperature at which particles were first observed in the collected images rather than when transmissivity goes below 100% or a similar threshold. The meta-stable zone width (MSZW) is the difference between the clear and cloud points as defined by the image data.
The solubility and solvent effects study determined that 11 of the 31 solvents were antisolvents (lamivudine solubility <0.005 g g−1 at elevated temperature) due to dissolution not being observed for vials with 0.005 g g−1 concentration of lamivudine. This concentration represents the lower limit of the Zinsser dosing platform's capabilities. The solubility profiles (Fig. 3) of lamivudine in the remaining 20 solvents showed that in 4 solvents, lamivudine was too soluble (lamivudine solubility >0.25 g g−1 at elevated temperature), in 13 solvents, lamivudine was not sufficiently soluble (lamivudine solubility <0.005 g g−1 at elevated temperature) and in the 3 remaining solvents, the solubility of lamivudine satisfied the solubility constraints. These 3 solvents were ethylene glycol, methanol and ethanol.
 | ||
| Fig. 3 The temperature-dependant solubility profile of lamivudine in 20 of the 31 solvents. The blue highlighted area shows solubility target parameters for low temperatures and the red highlighted area for high temperatures. | ||
Small differences were observed in the powder patterns for the crystals grown in methanol, water and 1-pentanol. TGA and Raman was used to confirm that form I was present.
No major fouling or agglomeration was observed under the crystallisation conditions tested in any of the solvents used with the exception of 4-methyl-2-pentanone in which aggregates of needle crystals formed.
Ultimately out of 31 solvents evaluated for crystallisation of lamivudine, ethanol was the only system to satisfy the target parameters for shape and solubility. Specifically, these experiments showed that the form was II, the crystal shape was bipyramidal, and the solubility exceeded 50 g L−1 at elevated temperature meeting all the objectives for this step.
1. Heat to 68 °C at a rate of 1 °C min−1 and hold for 10 minutes.
2. Cool to the experimental temperature at a rate of 10 °C min−1 with no stirring.
3. Hold at the isothermal point for 6 hours.
4. Repeat the cycle 2 more times.
The fast heating rate in the above temperature profile was chosen given that the dissolution point did not need to be measured; it was only necessary to ensure that dissolution was achieved. The maximum temperature was set at 68 °C to ensure that ethanol did not reach its boiling point and that solvent loss from evaporation was minimised. A fast crash cooling rate was used to achieve the isothermal temperature of interest as fast as possible to reduce the risk of nucleation occurring during the cooling ramp. The isothermal regime was held for 6 hours to capture crystallisations with longer induction times. Each sample vial underwent this temperature cycle three times to check for anomalous results and to get a median estimate for induction time. Induction times are reported as median values of these three measurements to reduce the impact of outliers. The temperature profile was the same for all experiments to minimise the variability across experiments. The stir rate was fixed at 600 RPM unless otherwise stated (refer to Fig. S5 in the ESI†).
Induction time, defined as the time between reaching isothermal conditions and the point at which crystals first start to nucleate, was determined using image data in this study, specifically using Helmli's mean. Helmli's mean45 is an image feature used in computer vision that has been found to be a good indicator of the presence of particles in in-line microscopy.46 Here, Helmli's mean is calculated with a window size of 5 pixels and thus termed HELM5. Induction time is estimated by measuring the time from temperature crash to where HELM5 rises above a threshold. Due to differences in particle habit, lighting, and solubilities, the threshold is not a fixed value but is chosen based on the range of HELM5 values obtained in each experiment. The threshold HELM5 value is taken as the 5th percentile of the range of values HELM5 obtained over the course of the experiment. Induction time estimation using HELM5 has been shown to have improved accuracies over other approaches.46,47
Finally, in this study image data was also used to determine the growth rate (the change in the particle size, over time). These parameters were determined using data spanning from slightly before the induction time (to account for potential errors in induction time estimate) until the time when the images became too crowded to identify single particles. Images were analysed to detect individual particles using the deep learning model “Mask R-CNN”.48 This model was adept at identifying individual objects in crowded situations and at multiple scales making it well-suited to the task of particle characterisation.49 Versions of the Mask R-CNN are available pre-trained on standard datasets which can be further trained (“fine-tuned”) to apply to new tasks. This transfer learning reduces the training time for new applications.
200 images from the Crystalline platform were chosen from various experiments to form a training set (which included both form I and form II of lamivudine amongst other APIs) based on their varied lighting conditions, particle habit, and particle solid loading. These images were manually annotated with annotations indicating the position and size of particles. These annotated images were supplied to a training algorithm to teach the Mask R-CNN model to detect particles on Crystalline images. The trained model was then applied to new images resulting in predicted particle locations, and a given confidence score. Predictions of confidence score less than 50% were discarded. The remaining particles were sized by fitting a rotated rectangle to the detected outline, minimising its area. The long side of this rectangle is referred to as particle length. Lengths were aggregated for an image and compiled in a histogram plot to determine the number-weighted-particle size distribution (PSD). Repeating across all images resulted in one PSD for every 5 seconds of the experiment during nucleation and growth. Quantiles and mean values of the PSDs were tracked over time. A linear fit was made to the mean size over time, and the gradient of the line gave an estimate of growth rate. The nucleation rate was estimated by tracking the number of detected particles for each image and again performing a linear fit of this number over time with the gradient of the line giving the number of new particles per unit time.
An initial full factorial design screening experimental plan50 was created (Table 1, “initial screening”). The experimental bounds on isothermal temperature and supersaturation (SS = C/C*, where SS is the supersaturation, C is the solution concentration and C* is the equilibrium concentration at the given temperature) were dictated only by physical limitations of solvent operating temperatures and industrial process solubility requirements to explore as large a design space as possible.
| Run order | Isothermal temperature (°C) | Supersaturation | Median induction time (s) | The standard deviation of induction time (s) | Mean growth ratea (μm s−1) | The standard deviation of growth rate (μm s−1) |
|---|---|---|---|---|---|---|
| a Growth rate values are missing due to the image analysis being unable to detect particle growth likely due to the small sample size of images collected at the point of nucleation before the images became too convoluted. | ||||||
| Initial screening | ||||||
| 1 | 10 | 1.93 | 2778 | 374 | 0.0355 | 0.0052 |
| 2 | 25 | 1.67 | 3495 | 1073 | 0.0001 | 0.0213 |
| 3 | 25 | 1.56 | 3737 | 1496 | 0.0153 | 0.0200 |
| 4 | 40 | 1.84 | 1585 | 1555 | 0.0185 | 0.0173 |
| 5 | 10 | 1.42 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 380 380 |
2596 | 0.0263 | 0.0000 |
| 6 | 25 | 1.63 | 1591 | 1008 | 0.0287 | 0.0238 |
| 7 | 40 | 1.39 | 10017 |
8011 | ||
| MLR output: SS 1.70, temp 28.5 °C | ||||||
An MLR model was fitted to the initial results and the optimizer feature in Modde 12.1 was run with the stated target values for induction time (1 hour) and growth rate (1 μm min−1). The MLR model trained on the initial screening results returned process conditions of a supersaturation of 1.70 and an isothermal temperature of 28.5 °C. Similarly, a partial least squares (PLS) model was fitted to the data where an optimisation returned similar values of a supersaturation of 1.69 and an isothermal temperature of 29.5 °C.
All samples from each iteration of the optimisation loop were fingerprinted by XRPD (Fig. S6 in the ESI†) where results confirmed that all runs gave the desired form, form II. No fouling or aggregation was observed through image or observational analysis. The D90 of crystals detected by image analysis for the final run (run 7, optimisation iteration 3) was 105 μm. The aspect ratio was 0.62. Therefore, the kinetic estimation section of the workflow was completed as all target parameters achieved values within the designated range.
:1. The process conditions required for these crystal attributes were isothermal crystallisation from ethanol at 19.9 °C, 600 RPM and supersaturation of 1.70. The resulting crystals had a D90 of 105 μm and a bipyramidal crystal habit. The solubility of lamivudine in ethanol also satisfied all target constraints with a value of 10 g L−1 at low temperatures and a value greater than 50 g L−1 at elevated temperatures. This approach also enabled kinetic parameter estimations for lamivudine from ethanol under various process conditions (Tables 1 and 2). These optimum conditions can be visualised in the DataFactory Dashboad (Fig. 4).
| Run order | Isothermal temperature (°C) | Supersaturation | Median induction time (s) | The standard deviation of induction time (s) | Mean growth ratea (μm s−1) | The standard deviation of growth rate (μm s−1) |
|---|---|---|---|---|---|---|
| a Growth rate values are missing due to the image analysis being unable to detect particle growth likely due to the small sample size of images collected at the point of nucleation before the images became too convoluted. | ||||||
| Optimisation iteration 1 | ||||||
| 1 | 28.5 | 1.78 | 1524 | 81 | ||
| 2 | 30 | 1.77 | 1310 | 248 | ||
| 3 | 28.5 | 1.77 | 615 | 464 | 0.0186 | 0.0088 |
| 4 | 27 | 1.89 | 2000 | 374 | 0.0413 | 0.0309 |
| 5 | 28.5 | 1.8 | 2290 | 2023 | 0.0147 | 0.0000 |
| 6 | 30 | 1.68 | 4536 | 957 | 0.0740 | 0.0193 |
| 7 | 27 | 1.73 | 2460 | 895 | 0.0387 | 0.0000 |
| MLR output: SS 1.74, temp 19.6 °C | ||||||
| Optimisation iteration 2 | ||||||
| 1 | 19.5 | 1.75 | 3901 | 301 | 0.0875 | 0.0778 |
| 2 | 18 | 1.74 | 1050 | 199 | 0.0440 | 0.0000 |
| 3 | 18 | 1.64 | 2305 | 3314 | 0.0472 | 0.0414 |
| 4 | 19.5 | 1.7 | 1745 | 564 | 0.0136 | 0.0196 |
| 5 | 21 | 1.81 | 2715 | 1483 | 0.0497 | 0.0265 |
| 6 | 21 | 1.68 | 6080 | 7293 | 0.0287 | 0.0000 |
| 7 | 19.5 | 1.72 | 1070 | 458 | 0.0279 | 0.0030 |
| MLR output: SS 1.70, temp 19.5 °C | ||||||
| Optimisation iteration 3 | ||||||
| 1 | 21.5 | 1.68 | 2999 | 2132 | 0.0053 | 0.0014 |
| 2 | 21.5 | 1.62 | 2021 | 470 | 0.0015 | 0.0007 |
| 3 | 17.5 | 1.77 | 1986 | 803 | 0.0059 | 0.0005 |
| 4 | 19.5 | 1.72 | 1787 | 367 | 0.0028 | 0.0007 |
| 5 | 19.5 | 1.74 | 1506 | 986 | 0.0030 | 0.0018 |
| 6 | 17.5 | 1.6 | 11552 |
2063 | 0.0020 | 0.0017 |
| 7 | 19.5 | 1.71 | 1320 | 3079 | 0.0089 | 0.0077 |
| MLR output: SS 1.70, temp 19.9 °C | ||||||
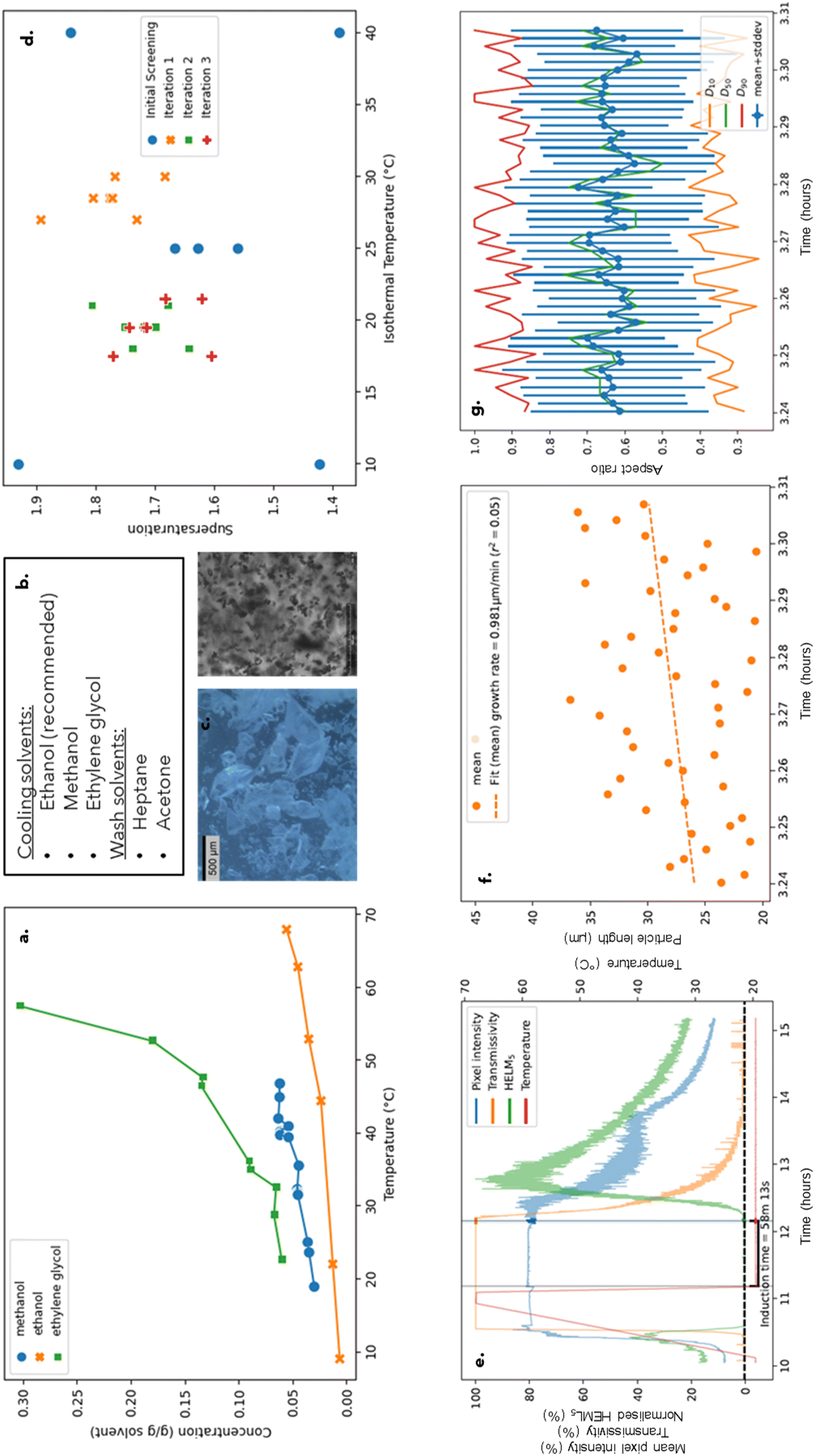 | ||
| Fig. 4 DataFactory dashboard showing thermodynamic and kinetic crystallisation parameters for the proposed optimum lamivudine recrystallisation – solubility profiles (a), solvent classification (b), microscopy and crystalline images (c), DoE design space (d) and induction time (e), growth rate (f) and aspect ratio over time (g) for the optimum run. | ||
5. Conclusions
Following workflows such as the one presented in this work can provide direction and reduce cross-researcher variability in experimental work, thus promoting reliable, reproducible and robust data generation. This work has demonstrated the usefulness of the proposed workflow to go from API selection to an optimised system for small-scale batch cooling crystallisation. By following the workflow, we have determined an alternative method to the current crystallisation process of lamivudine, specifically recrystallisation from methanol that results in the more desirable bipyramidal habit of form II. This case study yielded experimental parameters for lamivudine crystallisation that satisfied our target parameters for an operable isothermal crystallisation process from ethanol at 19.9 °C, 600 RPM and supersaturation of 1.70. Crystalline images and XRPD data confirmed that the resultant crystals were form II, the most stable form with desired downstream processing properties.This work also shows that a solubility and kinetic dataset could be generated using only 53 g of lamivudine and 1169 mL of solvents compared to previous methods that used 252 g of solute and 1448 mL of solvents.2 While less material was used in the work presented here, the reduction in material did not result in reduced information content as we collected approximately 7500 high-definition images with extracted thermodynamic and kinetic parameters and solution and solid-state data. Although droplet studies would result in significantly less material use, such thermodynamic and kinetic measurements would also not be possible. Furthermore, our contributions to the solubility and kinetic parameter literature data for lamivudine can also be used to inform future wet lab experiments and train and test improved solubility models.
Workflows such as this enable the future development of smarter experimental planning tools and predictive models, whereby model-driven experimental measurements can also be used to continuously improve models. Incorporating digital tools could help improve efficiency in crystallisation process development by reducing the amount of time and materials needed for the experimental sections of the workflow. This work has shown that the individual components, such as data analytics, optimisation and hardware, can be integrated and linked together in the logical, step-by-step manner necessary for achieving successful closed-loop automation.
This workflow can be applied to other APIs to identify industry-relevant, robust crystallisation conditions to inform the design of pharmaceutical manufacturing processes. Furthermore, applying this workflow to a wide range of APIs will enable the creation of a solubility and kinetics crystallisation parameter database that spans a varied chemical and crystallisation process space.
The availability of quantitative data on solubility and kinetic parameters in a wide range of APIs could aid laboratory-based researchers in optimising crystallisation processes to improve yield and crystal quality51via the application of ML models.22,25,52 Such FAIR data collection for solubility and kinetic parameters can lead to the development of a crystallisation parameter database in a structured format developing benefits analogous to the CSD or the Protein Data Bank (PDB). Workflows such as this will allow us to generate the standardised data that will be an invaluable resource for the predictive design of molecular crystallisation processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC Future Continuous Manufacturing and Advanced Crystallization Research Hub (Grant ref.: EP/P006965/1) for funding this work. The authors would like to acknowledge that this work was carried out in the CMAC National Facility support by UKRPIF (UK Research Partnership Fund) award from the Higher Education Funding Council for England (HEFCE) (Grant ref.: HH13054) We are grateful to the Core Project MDPC industrial steering team: Jorge Calderon de Anda (AstraZeneca) and Francesca Perciballi (AstraZeneca), Neda Nazemifard (Takeda), Vaclav Svoboda (Pfizer), Chris Burcham (Eli Lilly), and Jan-Sebastiaan Uyttersprot (UCB), as well as the observing members, especially Kevin Girard (Pfizer), for project guidance.References
- S. Vedantam and V. V. Ranade, Sadhana, 2013, 38, 1287–1337 CrossRef CAS.
- C. J. Brown, T. McGlone, S. Yerdelen, V. Srirambhatla, F. Mabbott, R. Gurung, M. L. Briuglia, B. Ahmed, H. Polyzois, J. McGinty, F. Perciballi, D. Fysikopoulos, P. MacFhionnghaile, H. Siddique, V. Raval, T. S. Harrington, A. D. Vassileiou, M. Robertson, E. Prasad, A. Johnston, B. Johnston, A. Nordon, J. S. Srai, G. Halbert, J. H. ter Horst, C. J. Price, C. D. Rielly, J. Sefcik and A. J. Florence, Mol. Syst. Des. Eng., 2018, 3, 518–549 RSC.
- S. G. Ring, P. Colonna, K. J. I'Anson, M. T. Kalichevsky, M. J. Miles, V. J. Morris and P. D. Orford, Carbohydr. Res., 1987, 162, 277–293 CrossRef CAS.
- R. K. Harris, R. R. Yeung, R. B. Lamont, R. W. Lancaster, S. M. Lynn and S. E. Staniforth, J. Chem. Soc., Perkin Trans. 2, 1997, 2653–2660 RSC.
- D. T. Y. Lau and D. Lau, Hepatology, 2000, 32, 828–834 CrossRef CAS PubMed.
- J. J. García-Trejo, R. Ortega and M. Zarco-Zavala, Front. Oncol., 2021, 11, 664794 CrossRef PubMed.
- B. N. Roy, G. P. Singh, D. Shrivastava, H. S. Jadhav, U. P. Aher and S. C. Deokar, An improved process for the manufacture of lamivudine, WO2011141805A2, 2011.
- K. Prakash, P. Narayana Raju, K. Shanta Kumari and M. Lakshmi Narasu, E-J. Chem., 2008, 5, 1159–1164 CrossRef CAS.
- S. Strauch, E. Jantratid, J. B. Dressman, H. E. Junginger, S. Kopp, K. K. Midha, V. P. Shah, S. Stavchansky and D. M. Barends, J. Pharm. Sci., 2011, 100, 2054–2063 CrossRef CAS PubMed.
- R. K. Harris, R. R. Yeung, R. B. Lamont, R. W. Lancaster, S. M. Lynn and S. E. Staniforth, Perkin Trans. 2, 1997, 2653–2660 RSC.
- R. Chadha, P. Arora and S. Bhandari, Int. Scholarly Res. Not., 2012, 2012, 671027 Search PubMed.
- B. P. Reddy, M. S. Reddy, P. V. Reddy and M. V. Kumari, Solid oral dosage forms of lamivudine, US8481554B2, 2009.
- T. Pickles, C. Mustoe, C. Brown and A. Florence, 2022 APS Special Issue, 2022, 7.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- M. D. Wilkinson, M. Dumontier, I. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. Da Silva Santos, P. E. Bourne, J. Bouwman, A. J. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. T. Evelo, R. Finkers, A. Gonzalez-Beltran, A. J. G. Gray, P. Groth, C. Goble, J. S. Grethe, J. Heringa, P. A. C. 'T Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. J. Lusher, M. E. Martone, A. Mons, A. L. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. Van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. A. Swertz, M. Thompson, J. Van Der Lei, E. Van Mulligen, J. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao and B. Mons, Sci. Data, 2016, 3, 160018 CrossRef PubMed.
- S. Knoll, C. E. Jusner, P. Sagmeister, J. D. Williams, C. A. Hone, M. Horn and C. O. Kappe, React. Chem. Eng., 2022, 7, 2375–2384 RSC.
- S. Guenat, P. Purnell, Z. G. Davies, M. Nawrath, L. C. Stringer, G. R. Babu, M. Balasubramanian, E. E. F. Ballantyne, B. K. Bylappa, B. Chen, P. De Jager, A. Del Prete, A. Di Nuovo, C. O. Ehi-Eromosele, M. Eskandari Torbaghan, K. L. Evans, M. Fraundorfer, W. Haouas, J. U. Izunobi, J. C. Jauregui-Correa, B. Y. Kaddouh, S. Lewycka, A. C. Macintosh, C. Mady, C. Maple, W. N. Mhiret, R. K. Mohammed-Amin, O. C. Olawole, T. Oluseyi, C. Orfila, A. Ossola, M. Pfeifer, T. Pridmore, M. L. Rijal, C. C. Rega-Brodsky, I. D. Robertson, C. D. F. Rogers, C. Rougé, M. B. Rumaney, M. K. Seeletso, M. Z. Shaqura, L. M. Suresh, M. N. Sweeting, N. Taylor Buck, M. U. Ukwuru, T. Verbeek, H. Voss, Z. Wadud, X. Wang, N. Winn and M. Dallimer, Nat. Commun., 2022, 13, 3559 CrossRef CAS PubMed.
- Bruker, DIFFRAC.EVA: software to evaluate X-ray diffraction data (Version 5.2), https://www.bruker.com/content/bruker/int/en/products-and-solutions/diffractometers-and-x-ray-microscopes/x-ray-diffractometers/diffrac-suite-software/diffrac-eva.html Search PubMed.
- Netzsch, Proteus, (https://analyzing-testing.netzsch.com/en/products/software/proteus) Search PubMed.
- Sartorius, Modde version 12.1, (https://www.sartorius.com/en/products/process-analytical-technology/data-analytics-software/support/knowledge-base/modde-121-550674) Search PubMed.
- F. L. Muller, M. Fielding and S. Black, Org. Process Res. Dev., 2009, 13, 1315–1321 CrossRef CAS.
- T. Deng, V. Garg, L. Pereira Diaz, D. Markl, C. Brown, A. Florence and M. S. A. Bradley, Int. J. Pharm., 2022, 628, 122309 CrossRef CAS PubMed.
- J. M. Butler and J. B. Dressman, J. Pharm. Sci., 2010, 99, 4940–4954 CrossRef CAS PubMed.
- M. Leane, K. Pitt and G. Reynolds, Pharm. Dev. Technol., 2015, 20, 12–21 CrossRef CAS PubMed.
- A. D. Vassileiou, M. Robertson, B. G. Wareham, M. Soundaranathan, S. Ottoboni, A. J. Florence, T. Hartwig and B. F. Johnston, Digital Discovery, 2023, 2, 356–367 RSC.
- J. Qiu, J. Albrecht and J. Janey, Org. Process Res. Dev., 2019, 23, 1343–1351 CrossRef CAS.
- J. Boetker, T. Rades, J. Rantanen, A. Hawley and B. J. Boyd, Mol. Pharmaceutics, 2012, 9, 2787–2791 CrossRef CAS PubMed.
- T. Vankeirsbilck, A. Vercauteren, W. Baeyens, G. Van der Weken, F. Verpoort, G. Vergote and J. P. Remon, TrAC, Trends Anal. Chem., 2002, 21, 869–877 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- C. Brandel and J. H. Ter Horst, Faraday Discuss., 2015, 179, 199–214 RSC.
- M. Schoenitz, L. Grundemann, W. Augustin and S. Scholl, Chem. Commun., 2015, 51, 8213–8228 RSC.
- A. D. Clayton, E. O. Pyzer-Knapp, M. Purdie, M. F. Jones, A. Barthelme, J. Pavey, N. Kapur, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, Angew. Chem., Int. Ed., 2023, 62(3), e202214511 CrossRef CAS PubMed.
- R. Moriconi, M. P. Deisenroth and K. S. Sesh Kumar, Mach. Learn., 2020, 109, 1925–1943 CrossRef.
- M. J. Jozwiakowski, N.-A. T. Nguyen, J. M. Sisco and C. W. Spancake, J. Pharm. Sci., 1996, 85, 193–199 CrossRef CAS PubMed.
- NHS, NHS (dm+d) (https://services.nhsbsa.nhs.uk/dmd-browser/).
- B. ZoomLab, https://www.basf.com/global/en/who-we-are/innovation/our-innovations/health_and_nutrition/zoomlab.html.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760–773 CrossRef CAS.
- A. Johnston, R. Bhardwaj-Miglani, R. Gurung, A. D. Vassileiou, A. J. Florence and B. F. Johnston, J. Chem. Inf. Model., 2017, 57, 1807–1815 CrossRef CAS PubMed.
- Y. Du, H. Zhang, J. Xue, W. Tang, H. Fang, Q. Zhang, Y. Li and Z. Hong, Spectrochim. Acta, Part A, 2015, 137, 1158–1163 CrossRef CAS PubMed.
- M. J. Jozwiakowski, N.-A. T. Nguyen, J. M. Sisco and C. W. Spancake, J. Pharm. Sci., 1996, 85, 193–199 CrossRef CAS PubMed.
- S. J. Mazivila, R. A. E. Castro, J. M. M. Leitão and J. C. G. Esteves da Silva, J. Pharm. Biomed. Anal., 2019, 169, 235–244 CrossRef CAS PubMed.
- B. N. Roy, G. P. Singh, D. Shrivastava, H. S. Jadhav, U. P. Aher and S. C. Deokar, An improved process for the manufacture of lamivudine, WO2011141805A2, 2011.
- D. A. Maldonado, A. Vassileiou, B. Johnston, A. J. Florence and C. J. Brown, Digital Discovery, 2022, 1, 621–635 RSC.
- B. Parthasaradhi Reddy, M. Srinivas Reddy, P. Venkateswar Reddy and M. Vanaja Kumari, Solid oral dosage forms of lamivudine, US8481554B2, 2009.
- F. S. Helmli and S. Scherer, Adaptive shape from focus with an error estimation in light microscopy, ISPA 2001 Proceedings of the 2nd International Symposium on Image and Signal Processing and Analysis, In conjunction with 23rd International Conference on Information Technology Interfaces IEEE Cat., Pula, Croatia, 2001, pp. 188–193, DOI:10.1109/ISPA.2001.938626.
- F. J. S. Doerr, C. J. Brown and A. J. Florence, Cryst. Growth Des., 2022, 22, 2105–2116 CrossRef CAS PubMed.
- T. Han, Y. Tang, X. Yang, Z. Lin, B. Zou and H. Feng, Remote Sensing, 2021, 13, 4918 CrossRef.
- K. He, G. Gkioxari, P. Dollar and R. Girshick, Mask R-CNN, arXiv, 2018, preprint, arXiv:1703.06870, DOI:10.48550/arXiv.1703.06870.
- C. Boyle, C. Brown, J. Sefcik and J. Cardona, Improved Particle Characterisation from in-Line PAT: Comparison of Deep Learning and White-Box Methods, Phoenix, Arizona, USA, 2022 Search PubMed.
- D. C. Montgomery, Design and analysis of experiments, John wiley & sons, 2017 Search PubMed.
- H. Chen, R. H. Chiang and V. C. Storey, Manag. Inf. Syst. Q., 2012, 1165–1188 Search PubMed.
- C. Xiouras, F. Cameli, G. L. Quilló, M. E. Kavousanakis, D. G. Vlachos and G. D. Stefanidis, Chem. Rev., 2022, 122, 13006–13042 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ce00897e |
| This journal is © The Royal Society of Chemistry 2024 |